
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A novel coordination mode of κ1-N-Br-pyridylbenz-(imida, oxa or othia)-zole to Pt(II): synthesis, characterization, electrochemical and structural analysis†
Juan Nicasio-Collazo*ab,
Gonzalo Ramírez-Garcíaa,
Marcos Flores-Álamoc,
Silvia Gutiérrez-Granadosa,
Juan M. Peralta-Hernándeza,
José Luis Maldonadob,
J. Oscar C. Jimenez-Halla a and
Oracio Serrano*a
a and
Oracio Serrano*a
aDepartamento de Química, Universidad de Guanajuato, Cerro de La Venada S/N, CP 36040, Guanajuato, Gto, Mexico. E-mail: oraciosinh@ugto.mx
bResearch Group of Optical Properties of Materials (GPOM), Centro de Investigaciones en Óptica, AP 37000, León, Guanajuato, Mexico. E-mail: nicasio.collazo@cio.mx
cFacultad de Química, Universidad Nacional Autónoma de México (UNAM), Circuito Exterior S/N, Coyoacán, Cd. Universitaria, CP 04510, Ciudad de México, Mexico
First published on 7th May 2019
Abstract
Herein, three novel Pt(II) complexes with formula [trans-Pt(Br-PyBenz-X)(Cl)2(DMSO)] (1–3) having Br-pyridylbenz-(imida, oxa or othia)-zole (L1–3) derivatives as potential bidentate ligands, under an unusual κ1-N-coordination mode are reported. All compounds were obtained straightforwardly via reaction of corresponding LPB1–3 and [Pt(Cl)2(DMSO)2] (DMSO = dimethyl sulfoxide), at 100 °C in acetonitrile, respectively. 1–3 complexes were characterized by analytical and spectroscopic data: melting point, FT-IR, Raman, UV/Vis and NMR experiments. Cyclic voltammetry studies show an irreversible two-electron process at −0.50 and −0.51 V, which was ascribed to the Pt(II)/Pt(IV) couple, for complexes 2 and 3. The crystal structure of complex 2 was elucidated by single-crystal X-ray diffraction, where the platinum atom exhibits a square plane geometry, where LPB2 adopts an unusual mono-coordinated mode via an N-κ1-benzoxazole ring. According to DFT calculations the first N-coordination exchanging one DMSO molecule is favourable, while the second N-coordination is highly impeded.
Introduction
Benzimidazole and its derivatives are important heterocyclic compounds. These ligands have been found to exhibit biological activities and clinical applications.1 In the last decades, transition metal complexes with benzimidazole derivatives have shown attractive chemical properties2,3 and numerous applications in catalysis,4–6 biological activity,7 organic light-emitting diodes8,9 and photovoltaics.10 Furthermore, N,N′-donor chelating ligand pyridyl-benzimidazole has been used as a bipyridine analogue in a wide variety of complexes with different metal ions such as Ir(III),11,12 Ru(II),13 Pt(II),14 respectively. Barnham and Ruiz et al.11 reported that a series of Ir(III), Ru(II) and Pt(II) inhibited in vitro aggregation of the peptide fragment amyloid Aβ1–42, representing a potential use as therapeutic agents for Alzheimer's disease. Recently, Espino, Bolink and Orti et al.12 reported the synthesis of iridacycle complexes with pyridyl-imidazole ligands, which were used as electroluminescent materials in light-emitting electrochemical cells devices. Later, Wang et al.14 reported an elegant work on the synthesis of pyridyl-benzimidazole ligands, which display fluorescent emission in the purple/blue region at room temperature and phosphorescent emission in the blue/green region at 77 K. Fan et al.15 showed that W(0) species exhibit enhanced phosphorescence, at room temperature, from MLCT excited stated with quantum yields on the order of 10−3. Additionally, Housecroft and Bolink et al.16 reported an elegant synthesis of cyclometalated Ir(III) compounds having pyridyl-benzoxa and thiazole derivatives (LBP), giving an efficient and stable red-emission, with active layers lifetimes greater than 1000 h. Furthermore, Ward et al.13 described a series of Re-carbonyl, Pt-acetylide and Ru-bipyridine complexes with LBP ligands. Particularly, Pt species showed luminescence in the range 553–605 nm arising from the 3MLCT state, with emissive lifetime up to 500 ns and quantum yields up to 6% in CH2Cl2 at room temperature. Studies reported by DeStefano et al.17,18 on Pt- and Zn-LBP complexes showed that they exhibit a color blue luminescence in the solid state and in solution.In early reports, Westcott et al.7 showed that the reaction of [Pt(Cl2)(COE)]2 (COE = cis-cyclooctene) or K2PtCl4 with LPB ligands gives the expected [cis-Pt(Cl)2(LPB)] complexes (Chart 1a). Later, when benzimidazole fragment is in position three with respect to pyridine, the reaction lead to the monodentate [cis-Pt(Cl)2(LPB)2], where coordination to the metal occurs via the pyridine ring (Chart 1e). Part of our research has been focused on understanding the reactivity of 2-(6-bromopyridyl-2-yl)-1-phenylbenzimidazole, LPB1, ligand towards dialkyl–palladium complexes. A few years ago, we reported the synthesis of seven-membered palladacycles through selective C–Br bond activation of a Br-pyridine-benzimidazole derivative and concomitant C–C coupling bond, where no-innocent cyclooctadiene ligand (COD) plays an essential role (Chart 1b).19 Also, a five-membered palladacycles synthesis can be achieved when the COD is displaced by a phosphine ligand (Chart 1c). In 2018, we reported a direct synthesis on rollover palladacycles was achieved via C–H bond activation reaction in remote position on Br-pyridyl-benzothiazole (LPB2) by PdCl2, where the dimethylformamide plays an important role on the reaction pathway.20 However, to the best of our knowledge, well-characterized transition metal complexes containing LPB ligands showing a N-κ1-coordinated mode still relatively scarce in the literature.7 Here in, we report our DFT calculations and experimental results on the reactivity of Br-Pyridylbenz-(imida, oxa or othia)-zole ligands toward platinum complexes.
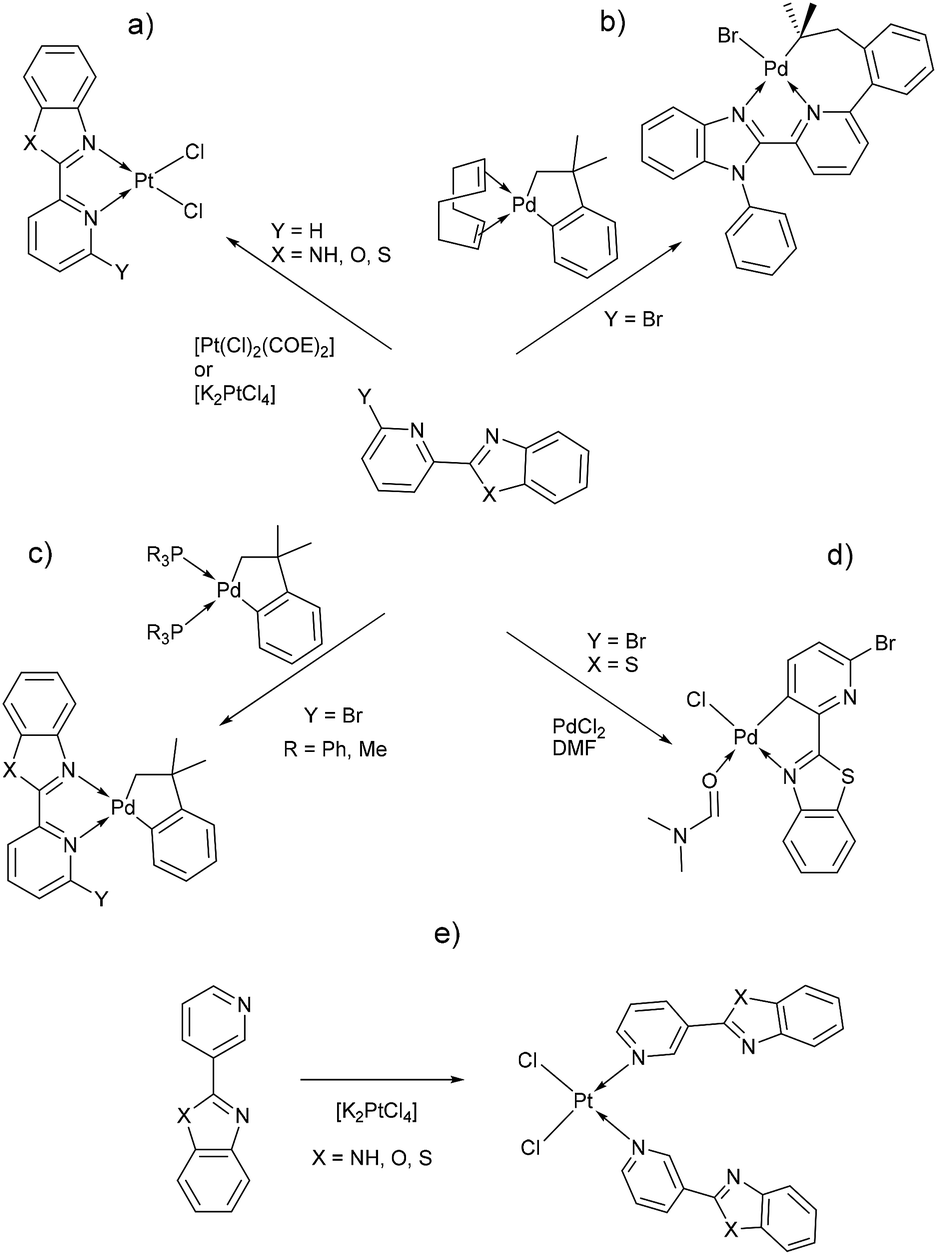 | ||
| Chart 1 | ||
Results and discussion
Our first attempts were addressed to obtain a dialkyl–platinum complexes, through a stochiometric reaction of [Pt(COD)(Me)2] with LPB1–3, using CH2Cl2 as solvent at room temperature. However, under these conditions the reaction do not give the desired compound, instead, the starting materials are recovered. The replaced of dimethyl for an chloride–platinum complexes with general formula [PtCl2(COE)2] or [PtCl2(COD)] does not show a positive effect on the reactivity towards LPB1–3, according to the NMR analysis of the proton and platinum spectra. This suggest, that bromine atom into the LPB1–3 ligands plays an interesting role on the complex formation according to Westcott report's (Charts 1a and e). Later, when PPh3 was added to the reaction mixture, an spontaneous formation of complex [PtCl2(PPh3)2] takes place in quantitative yield, according to the 31P and 195Pt NMR experiments.21 Given the low reactivity of organoplatinum species, we carried out the reaction with Pt coordination species like [PtCl2(DMSO)2] or [PtCl2(THT)2] (DMSO = dimethyl sulfoxide; THT = tetrahydro-thiophene) with LPB1–3, retrieving the starting materials even under drastic reaction conditions (150 °C). Similar results have been described by Zucca et al.22On the other hand, the use of K2PtCl4 with one equivalent of LPB2 in DMSO, did not show any reactivity under temperatures below 100 °C. When the temperature was increased to 150 °C, the 1H NMR spectrum showed only signals assigned to the free ligand. Nevertheless, the 195Pt NMR spectrum showed a new signal at −3526 ppm, differing from K[PtCl3(DMSO)] or [PtCl2(DMSO)2] signals which appeared at −2959 and −3450 ppm, respectively.23
Later, a stochiometric reaction of [PtCl2(COD)] with LPB2, in THT solution at 150 °C during a week, gave a yellow solid in a yellow solution. 195Pt NMR spectrum showed a signal at −3360 ppm, suggesting the formation of an analogous specie. Similar results were obtained when [PtCl2(COD)] complex was used as platinum source in solution of THT at 150 °C of temperature. Given the mentioned scarce reactivity, we decided to study the solvent effect of CH2Cl2, CHCl3, isopropyl alcohol or 2-ethoxyethanol at temperatures below to 100 °C, using [PtCl2(DMSO)2] as platinum source and one equivalent of LPB2; however, under these conditions the starting materials were recovered in quantitative amounts.
To our delight, when the reaction was carried out, in acetonitrile at 100 °C, it leads to a pale yellow solid (Scheme 1). Temperature plays an important role, since a complex was not produced when the mixture was at temperatures below to 60 °C. Also, the solvent effect is not trivial since formation of metallic platinum was observed when isopropyl alcohol or 2-ethoxyethanol were used instead of acetonitrile.
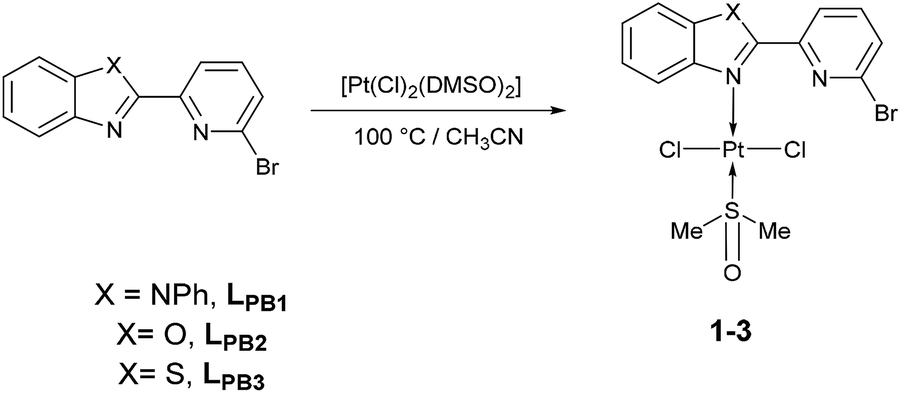 | ||
| Scheme 1 Synthetic route of [trans-PtCl2(DMSO)(LPB1–3)], 1–3, species. | ||
Unfortunately, all compounds show very low solubility in common organic solvents, the NMR experiments were carried out in DMSO-d6. All experimental measurements were recorded immediately, after DMSO-d6 addition, in order to avoid ligand exchange between Pt(II) complexes and DMSO-d6.24,25 Thus, under our conditions, only complexes 2–3 were soluble enough. The 1H NMR spectrum of 2 revealed a signal at 2.54 ppm with integrates to 6 protons, the signal was assigned to methyl protons, giving a singlet corresponding to DMSO fragment. In the region of 7.47 to 8.36 ppm seven signals appeared, assigned to aromatic protons corresponding to the LPB2 free fragment. 13C{1H} NMR spectrum showed a characteristic signal for the methyl group in 40.4 ppm assigned to the coordinate DMSO. In the aromatic region, four signals appeared in 120.5, 122.9, 130.6 and 140.9 ppm assigned to the carbons of phenyl fragment, and three signals in 111.5, 125.3 and 126.6 ppm assigned to the pyridyl fragment, according to 2D NMR experiments. Finally, the 195Pt NMR gave an intense signal at −3455 ppm, without any other significative signals, assuming the formation of only one platinum complex. The 1H NMR spectrum of complex 3 shows a singlet at 2.54 ppm, which is assigned to the methyl groups in the DMSO fragment. In the aromatic region between 7.53 to 8.35 ppm, seven well defined signals are observed and allocated into two sets: (i) three signals corresponding to the pyridine fragment (7.85, 7.99 and 8.35 ppm) and four signals for benzothiazole fragment (7.53, 7.59, 8.12 and 8.19 ppm). 13C{1H} NMR spectrum shows a signal at δ 40.4 ppm for methyl group of DMSO. Additionally, the signals at 119.8, 130.3 and 141.1 ppm are assigned to C–H for pyridine fragment and the signals at 122.7, 123.5, 126.3 and 126.9 are assigned to C–H in benzothiazole fragment, according to the 2D NMR 1H–13C. In good agreement with the latter, the 195Pt NMR spectrum shows one signal at −3455 ppm.
UV-Vis spectra of the LPB1–3 ligands showed a maximum absorption peak between 313–321 nm (Fig. S1†), which could be assigned to π → π* absorption (spin allowed S0 → 1IL transition).26 A hypochromic effect was observed in all UV-Vis spectra of 1–3, attributed to the Pt coordination towards the LPB1–3 ligands, respectively. Additional experimental evidence onto Pt-complexes synthesis was confirmed via melting point (decomposition to black solid), IR and Raman spectroscopies. Thus, the ligand bands of the complexes are slightly shifted with respect to free ligands. The main IR and Raman bands of complexes 1–3 are listed in ESI (see Table S1†). Identification of the ν(Pt–N), ν(Pt–Cl) and ν(Pt–S) at ca. 319, 330 and 283 cm−1 bands were compared among them and in relation to the [PtCl2(DMSO)2] complex.27–29
Additionally, suitable crystals of complex 2 were grown from slow evaporation of acetonitrile solution. Fig. 1 shows the molecular structure of compound 2. The Pt(II) ion in [trans-PtCl2(L2)(DMSO)] exhibited distorted square plane geometry around Pt center where the ligand LPB2 was coordinated to the metal through a N atom in benzoxazole unit and the N atom of Br-pyridine remained free. The chlorine atoms adopted trans configuration, and the remaining position was occupied by the sulfur atom of the DMSO molecule coordinated to the metal center. Westcott et al.7 have reported the synthesis and characterization of cis-dichloroplatinum complexes containing mono coordinated piridynbenzimidazole, oxazole and thiazole derivatives were the coordination to metal center occurs via pyridine ring. To the best of our knowledge, our results demonstrated the influence of Br atom avoiding pyridin's nitrogen coordination to Pt metal center.
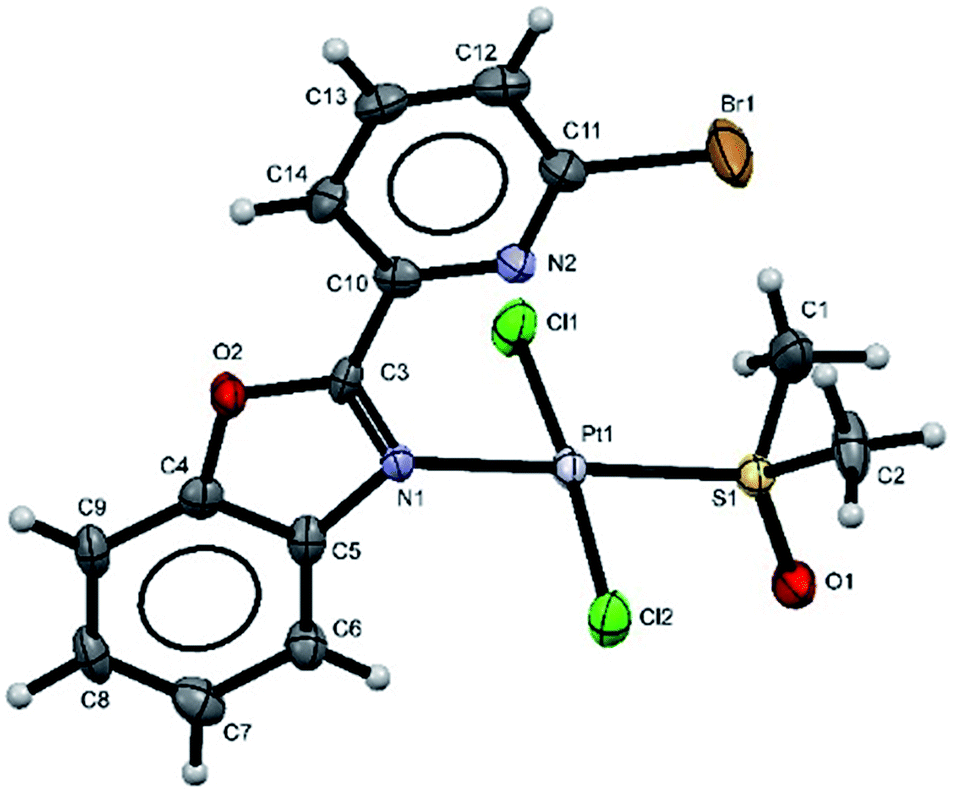 | ||
| Fig. 1 The molecular structure of complex 2 (the thermal ellipsoids are drawn at 60% probability). Selected bond lengths [Å] and angles (°); Cl(1)–Pt(1) 2.2990(12); Cl(2)–Pt(1) 2.3022(12); N(1)–Pt(1) 2.051(3); Pt(1)–S(1) 2.2108(11); N(1)–Pt(1)–S(1) 179.52(10); N(1)–Pt(1)–Cl(1) 87.00(10); S(1)–Pt(1)–Cl(1) 93.47(4); N(1)–Pt(1)–Cl(2) 89.68(10); S(1)–Pt(1)–Cl(2) 89.84(4); Cl(1)–Pt(1)–Cl(2) 174.85(5). | ||
The redox properties of all compounds were studied by cyclic voltammetry with a golden electrode in anhydrous DMF solution of 0.1 mol L−1 NBu4BF4 as the supporting electrolyte (Fig. 2).
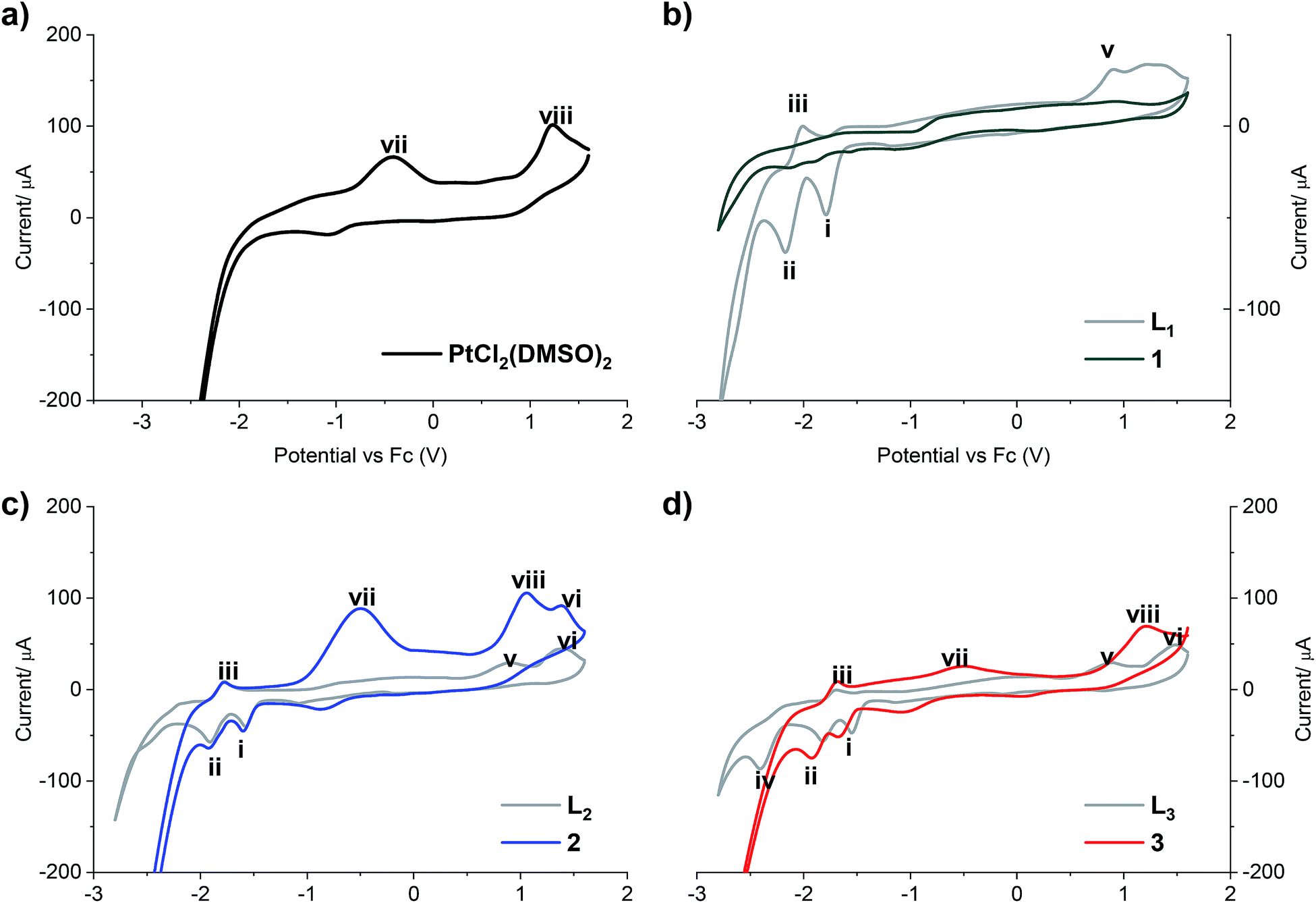 | ||
| Fig. 2 Cyclic voltammograms of (a) PtCl2(DMSO)2; (b) LPB1 free ligand and complex 1; (c) LPB2 free ligand and complex 2; (d) LPB3 free ligand and complex 3. Scan rate 300 mV s−1 cathodic direction. | ||
Two reduction steps occur in all the analyzed free ligands and for complexes 2 and 3 (Fig. 2, peaks i and ii). First, the wave (i) is assigned to the irreversible reductive cleavage of the C–Br bond in the halopyridine moiety (see Table S2† for potential values).30 The wave (ii) is associated to wave (iii), and this couple is assigned to one-electron reversible redox transition response. This reduction/oxidation process is attributed to the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of the benz(imida, oxa or othia)zole rings, giving rise to stable radical anions in which nitrogen has the negative charge and the resulting carbon radical is stabilized by the adjacent heteroatom and the pyridine ring.31
C bond of the benz(imida, oxa or othia)zole rings, giving rise to stable radical anions in which nitrogen has the negative charge and the resulting carbon radical is stabilized by the adjacent heteroatom and the pyridine ring.31
An additional cathodic peak was observed at −2.4 V only when the free LPB3 ligand was analyzed (Fig. 2d). This irreversible reduction is expected to occur on the pyridine ring.32 However, we were unable to observe this wave for other ligands at the encompassed potentials in the electroactive window. It could be attributed to the strongest donating character of the benzothiazole, favoring the pyridine reduction with respect to the analogous benzoxazole or benzimidazole compounds, which include most electronegative heteroatoms. This shift towards more negative potentials was also observed by Kapturkiewicz et al.26 for unsubstituted 2-(2-pyridyl)benzazole ligands. In the case of complex 3, the mentioned peak at −2.4 V disappears due to the coordination of the N at the pyridine with the metal center of Pt(II) (Fig. 2d). The peak (v), present in the cyclic voltammograms of all the free ligands at potentials between 887 and 916 mV is attributed to the irreversible oxidation of the nitrogen atom at the azole ring, which also coordinates with the platinum center, and in consequence it was not noticed when the complexes were analyzed. Furthermore, the wave (vi), observed when LPB2 and LPB3 ligands were analyzed, can be attributed to the irreversible oxidation of O and S, respectively (Fig. 2c and d). Such wave was not detected for LPB1 due to the presence of the phenyl substituents attached to the N. It should be noted that 1 shows very limited solubility in DMF. The absence of any peaks in voltammogram for complex 1 (Fig. 2b) is due to the immediate settling to the bottom of the cell when dispersed in solvent. Peak (vii) is only detected in the cyclic voltammograms of [PtCl2(DMSO)2], due an irreversible Pt(II)/Pt(IV) oxidation. These experiments show that the coordination between the 2 or 3 and the platinum center does not have any effects on the oxidation state of the metal, remaining as Pt(II). Finally, the peak (viii) is also observed only when the [PtCl2(DMSO)2] and its complexes where analyzed and it is assigned to the irreversible two-electron oxidation of the chloride anions.
Finally, we have performed theoretical calculations using the M06-L density-functional in combination with triple-ζ quality basis sets for carrying out geometry optimizations (see ESI† for further details). We have expressed energy values at the (SMD:DMSO)M06-L/def2-tzvpp level for discussing our results herein. Initially, we investigated the coordination mode for complexes [trans-PtCl2(DMSO)(LBP1–3)] (1–3). Since penta-coordinate moieties were reduced to well-known tetracoordinate complexes (Fig. 3a and b), we obtained that those of mode A were more stable than those of mode B for about 4 to 9 kcal mol−1 (see ESI†).
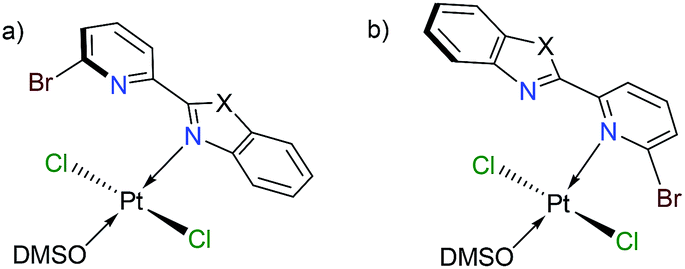 | ||
| Fig. 3 Platinum complexes having ligand LBP1–3 in a mono-coordination mode κ1-N. | ||
We conducted NBO calculations at the same level of theory, for complexes containing LPB2 ligand, aiming to understand this preference for coordination. We found that the N → Pt donation and back-donation are stronger for mode A stabilizing by 9.63 and 6.21 kcal mol−1, respectively, than for mode B according to our analysis (See ESI†). Although there is an increase in the Br → Pt donation in mode B stabilizing by 10.54 kcal mol−1 than for mode A, there is an increasing steric hindrance between Br and Cl vicinal atoms which deforms the square planar geometry of the metal centre. Furthermore, we calculated the reaction energies for the exchange of the N,N-donor ligand and solvent molecules in [PtCl2(DMSO)2] as shown in Table 1. The first N-coordination exchanging one DMSO molecule is favourable because of the negative values indicating exothermicity (and exergonicity) where the second N-coordination releasing the second DMSO is highly impeded. This is in line with our experimental evidence. The effect of substituents X = NPh, O or S; is also significant: heteroatoms with a more activating effect on the benz(imida, oxa or othia)zole ring make more available the lone-pair of the nitrogen that donates to the platinum.
|
||||
|---|---|---|---|---|
| X= | NC6H5 | O | S | NH |
| a ΔH and ΔG are in kcal mol−1. | ||||
| ΔHR1 | −6.2 | −2.0 | −1.9 | −5.7 |
| ΔGR1 | −4.0 | −0.4 | 0.4 | −3.5 |
| ΔHR2 | 17.6 | 18.4 | 16.6 | 16.1 |
| ΔGR2 | 5.6 | 6.7 | 4.9 | 4.2 |
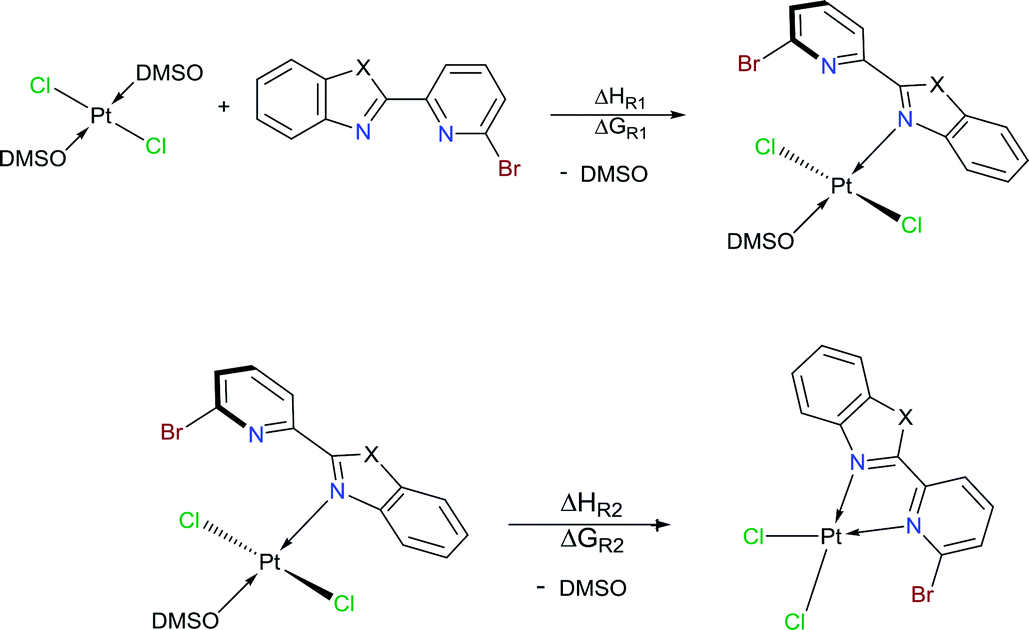
Moreover, according to our calculations, the use of a more labile ligand like cyclooctene in order to promote its exchange with the second N-coordination (see eqn (S1)†) will not favour the reaction (ΔHR = 18.0 kcal mol−1; ΔGR = 6.0 kcal mol−1). We also corroborate that reported reactions using [PtCl4]2− for obtaining the desired bidentate complexes (see eqn (S2)†) avoiding DMSO as solvent give exothermic (exergonic) reactions (ΔHR = −14.8 kcal mol−1; ΔGR = −17.4 kcal mol−1).
Conclusions
In summary, we have demonstrated that reactivity of potential bidentate 2-(6-Br-2-pyridyl)-benz-(imida, oxa or othia)-zole ligands (LPB1–3) with an equivalent of [PtCl2(DMSO)2] complex, in acetonitrile at 100 °C, yields the complexes with general formula [trans-PtCl2(LPB1–3)(DMSO)], 1–3, respectively. LPB1–3 fragment ligands are bonded to the platinum center under an unusual N-κ1-coordination mode. Furthermore, cyclic voltammetry studies show an irreversible two-electron process at −0.50 and −0.51 V, which was ascribed to the Pt(II)/Pt(IV) couple, for complexes 2 and 3. According to performance theoretical calculation the first N-coordination exchanging one DMSO molecule is favorable, under our experimental conditions, and the second N-coordination releasing the second DMSO is highly impeded. Finally, we demonstrated that effect of solvent, temperature, the bromine atom at the auxiliary ligands and platinum complexes source kind, plays an important role in the formation on this unusual mononuclear species.Experimental
General considerations
Manipulations of air and moisture compounds were performed under nitrogen atmosphere using standard Schlenk techniques. Acetonitrile was dried over 3 Å molecular sieves before use. K2PtCl4 was purchased from Pressure Chemical Co (USA). All materials were used as received from commercial suppliers without further purification. Compounds LPB1,19 LPB2,26 LPB3,33–35 and [Pt(Cl)2(DMSO)2]36 were prepared according to the literature. NMR spectra were recorded on a Bruker Ultra Shield 500 and Bruker AV-400 MHz spectrometer at room temperature. 1H and 13C{1H} chemical shift (δ) are given in part per million (ppm) relative to tetramethylsilane (TMS) and the solvent resonance were used as internal standards and 195Pt relative to K2PtCl4 (Solution in D2O). Elemental analysis experiments were carried out on 2400 CHNS/O Series II System PerkinElmer. IR spectra were recorded on a PerkinElmer Spectrum 100 FT-IR infrared spectrometer using KBr pellets. Raman spectra were recorded on a Renishaw InVia microscope Raman spectrometer. Raman measurements were acquired with an excitation line provided by a diode laser at 457 nm, by means of a 20× microscope's objective with a 0.4 numerical aperture; 2 s of integration time, and 20 accumulations were selected. UV-Vis absorption spectrum was collected using a spectrophotometer Perking Elmer, lambda 900, Waltham, MA, USA. Cyclic voltammetry studies were conducted at room temperature in a BASi Epsilon-EC potentiostat/galvanostat with included software system. A gold working electrode (2 mm), a platinum wire auxiliary electrode, and Ag/AgCl reference electrode were used in a standard three-electrode configuration. The measurements were carried out in 5 mL of DMF solutions of the compound (3.0 × 10−3 mol L−1) containing 0.05 mol L−1 tetrabuthylammonium tetrafluoroborate (NBu4BF4) as the supporting electrolyte. X-ray crystallographic studies were collected at Oxford Diffraction Gemini “A” diffractometer.Synthesis of ligands
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) eluent, yielding a brown powder (753 mg, 2.15 mmol). Yield 80%. Mp = 112–114 °C. 1H NMR (CDCl3, 500 MHz): δ = 8.16 (d, 1H, J = 7.7 Hz), 7.90 (d, 1H, J = 8.0 Hz), 7.58 (t, 1H, J = 7.8 Hz), 7.55–7.46 (m, 4H), 7.41–7.26 (m, 7H), 7.21 (d, 1H, J = 8.0 Hz) ppm. 13C{1H} NMR (CDCl3, 126 MHz): δ = 149.96, 148.77, 142.69, 140.76, 138.76, 138.06, 137.58, 129.31, 128.50, 128.18, 127.71, 124.42, 123.45, 123.02, 120.38, 111.21 ppm. IR (KBr)
:1) eluent, yielding a brown powder (753 mg, 2.15 mmol). Yield 80%. Mp = 112–114 °C. 1H NMR (CDCl3, 500 MHz): δ = 8.16 (d, 1H, J = 7.7 Hz), 7.90 (d, 1H, J = 8.0 Hz), 7.58 (t, 1H, J = 7.8 Hz), 7.55–7.46 (m, 4H), 7.41–7.26 (m, 7H), 7.21 (d, 1H, J = 8.0 Hz) ppm. 13C{1H} NMR (CDCl3, 126 MHz): δ = 149.96, 148.77, 142.69, 140.76, 138.76, 138.06, 137.58, 129.31, 128.50, 128.18, 127.71, 124.42, 123.45, 123.02, 120.38, 111.21 ppm. IR (KBr) ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 3042, 1595, 1577, 1557, 1501, 1435, 1389, 1328, 1267, 1200, 1151, 1117, 1072, 988, 846, 797, 760, 738, 699, 656, 624, 573 cm−1. = 3049, 1576, 1549, 1509, 1421, 1315, 1249, 1154, 1120, 996, 988, 864, 794, 757, 643, 531 cm−1. Mp = 182–184 °C. = 3092, 3062, 3045, 1578, 1541, 1475, 1452, 1427, 1408, 1349, 1281, 1247, 1238, 1159, 1131, 1111, 1085, 1075, 1005, 983, 929, 891, 818, 759, 738, 649, 623 cm−1.
= 3042, 1595, 1577, 1557, 1501, 1435, 1389, 1328, 1267, 1200, 1151, 1117, 1072, 988, 846, 797, 760, 738, 699, 656, 624, 573 cm−1. = 3049, 1576, 1549, 1509, 1421, 1315, 1249, 1154, 1120, 996, 988, 864, 794, 757, 643, 531 cm−1. Mp = 182–184 °C. = 3092, 3062, 3045, 1578, 1541, 1475, 1452, 1427, 1408, 1349, 1281, 1247, 1238, 1159, 1131, 1111, 1085, 1075, 1005, 983, 929, 891, 818, 759, 738, 649, 623 cm−1.General procedure for the synthesis of [trans-Pt(Cl)2(Ln)(DMSO)]
In a sealed tube was charged with LPBn (0.120 mmol), [Pt(Cl)2(DMSO)2] (0.120 mmol) and acetonitrile (15 mL). The reaction mixture was heated at 100 °C for 24 h and cooled down slowly to room temperature. Thus, a solid precipitate was observed. The solid was filtered and washed with Et2O giving the platinum complexes in good yield. All complexes were obtained in gram scale with good yields (up to 70%) and were characterized by FTIR, Raman, NMR, UV-Vis spectroscopies and melting point.) = (3091, 3064, C–H) 1595, 1582, 1557, 1500, 1456, 1445, 1407, 1389, 1333 (C–N), 1265, 1208, 1167, 1152, 1122 (SO) cm−1.) = (3037, 3009, C–H) 2991, 2926, 2917, 1534, 1401, 1354, 1305 (S–C), 1299 (C–N), 1156, 1133, 1020 (SO) cm−1.) = (3037, 3009, C–H), 2992, 2926, 2917, 1401, 1384, 1305 (S–C), 1299 (C–N), 1156, 1133, 1020 (SO) cm−1.X-ray crystallography
X-ray powder diffraction experiments were acquired in Diffractometer Model D2 PHASER-Bruker. Single crystal of complex was mounted on a glass fiber and analyzed with an Oxford Diffraction Gemini “A” diffractometer with a CCD area detector, a sealed tube X-ray source (λMoKα = 0.71073 Å), and a graphite monochromator. The double pass method of scanning was used to exclude any noise. The collected frames were integrated with the CrysAlis Pro and CryAlis RED software package (Agilent Technologies); analysis of the integrated data did not reveal any decay. Final cell parameters were determined by a global refinement of reflections; data were corrected for absorbance by analytical numeric absorption correction,37 employing a multifaceted crystal model based on expression upon the Laue symmetry using equivalent reflection. Structure solution and refinement were carried out with SHELXS-2014 and SHELXL-2014;38,39 Mercury CSD 39 and the WinGX suite were used for molecular graphics and presentation of single-crystal diffraction data.40,41 Full-matrix least-squares refinement was carried out by minimizing (Fo2 = Fc2)2. All non-hydrogen atoms were refined anisotropically. Crystallographic data have been deposited at the Cambridge Crystallographic Data Center as ESI† number CCDC 1849220.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors thank to the Laboratorio Nacional UG-CONACYT, Martín Olmos and Christian Albor for their technical assistance. J. N.-C. thanks the Postdoctoral fellowship from Universidad de Guanajuato (UG-DAIP-Excelencia Académica 2015). O. S. is gratefully to UG for financial support this research (DAIP-Excelencia Académica 2015, grant 002/2015, and DAIP-2018). J. L. M. thanks Ce-MIE-Sol 207450/27 (México) and CONACyT-SENER B-S-69369 (Bilateral grant Mexico-England), Fondo Sectorial CONACYT-SENER-Sustentabilidad energética.Notes and references
- W. Akhtar, M. F. Khan, G. Verma, M. Shaquiquzzaman, M. A. Rizvi, S. H. Mehdi, M. Akhter and M. M. Alam, Eur. J. Med. Chem., 2017, 126, 705–753 CrossRef CAS PubMed.
- B. Pashaei, H. Shahroosvand, M. Graetzel and M. K. Nazeeruddin, Chem. Rev., 2016, 116, 9485–9564 CrossRef CAS PubMed.
- J. S. Pap, A. Draksharapu, M. Giorgi, W. R. Browne, J. Kaizer and G. Speier, Chem. Commun., 2014, 50, 1326–1329 RSC.
- A. O. Ogweno, S. O. Ojwach and M. P. Akerman, Appl. Catal., A, 2014, 486, 250–258 CrossRef CAS.
- M. Zhang, R. Gao, X. Hao and W.-H. Sun, J. Organomet. Chem., 2008, 693, 3867–3877 CrossRef CAS.
- S. O. Ojwach, A. O. Ogweno and M. P. Akerman, Catal. Sci. Technol., 2016, 6, 5069–5078 RSC.
- X.-F. He, C. M. Vogels, A. Decken and S. A. Westcott, Polyhedron, 2004, 23, 155–160 CrossRef CAS.
- J.-H. Zhao, Y.-X. Hu, H.-Y. Lu, Y.-L. Lü and X. Li, Org. Electron., 2017, 41, 56–72 CrossRef CAS.
- J. Hu, C. Zhao, T. Zhang, X. Zhang, X. Cao, Q. Wu, Y. Chen, D. Zhang, Y. Tao and W. Huang, Adv. Opt. Mater., 2017, 5, 1700036 CrossRef.
- W.-K. Huang, C.-M. Lan, Y.-S. Liu, P.-H. Lee, S.-M. Chang and E. W.-G. Diau, J. Chin. Chem. Soc., 2010, 57, 1151–1156 CrossRef CAS.
- G. S. Yellol, J. G. Yellol, V. B. Kenche, X. M. Liu, K. J. Barnham, A. Donaire, C. Janiak and J. Ruiz, Inorg. Chem., 2015, 54, 470–475 CrossRef CAS PubMed.
- M. Martinez-Alonso, J. Cerda, C. Momblona, A. Pertegas, J. M. Junquera-Hernandez, A. Heras, A. M. Rodriguez, G. Espino, H. Bolink and E. Orti, Inorg. Chem., 2017, 56, 10298–10310 CrossRef CAS PubMed.
- N. M. Shavaleev, Z. R. Bell, T. L. Easun, R. Rutkaite, L. Swanson and M. D. Ward, Dalton Trans., 2004, 0, 3678–3688 RSC.
- Q. D. Liu, W. L. Jia and S. Wang, Inorg. Chem., 2005, 44, 1332–1343 CrossRef CAS PubMed.
- J. J. Lee, C. P. Yap, T. S. Chwee and W. Y. Fan, Dalton Trans., 2017, 46, 11008–11012 RSC.
- C. D. Ertl, C. Momblona, A. Pertegas, J. M. Junquera-Hernandez, M. G. La-Placa, A. Prescimone, E. Orti, C. E. Housecroft, E. C. Constable and H. J. Bolink, J. Am. Chem. Soc., 2017, 139, 3237–3248 CrossRef CAS PubMed.
- M. R. DeStefano and D. K. Geiger, Acta Crystallogr., Sect. C: Struct. Chem., 2016, 72, 491–497 CrossRef CAS PubMed.
- M. R. DeStefano and D. K. Geiger, Acta Crystallogr., Sect. C: Struct. Chem., 2017, 73, 697–702 CrossRef CAS PubMed.
- J. Nicasio-Collazo, E. Alvarez, J. C. Alvarado-Monzon, G. Andreu-de-Riquer, J. O. Jimenez-Halla, L. M. De Leon-Rodriguez, G. Merino, U. Morales, O. Serrano and J. A. Lopez, Dalton Trans., 2011, 40, 12450–12453 RSC.
- M. E. Tovar-Ramírez, R. Ramírez-Zúńiga, J. Nicasio-Collazo, K. Wrobel, J. G. Alvarado-Rodríguez, K. Wrobel and O. Serrano, ChemistrySelect, 2018, 3, 4133–4139 CrossRef.
- L. Rigamonti, C. Manassero, M. Rusconi, M. Manassero and A. Pasini, Dalton Trans., 2009, 0, 1206–1213 RSC.
- L. Maidich, G. Dettori, S. Stoccoro, M. A. Cinellu, J. P. Rourke and A. Zucca, Organometallics, 2015, 34, 817–828 CrossRef CAS.
- L. G. Marzilli, Y. Hayden and M. D. Reily, Inorg. Chem., 1986, 25, 974–978 CrossRef CAS.
- D. S. Martin, Inorg. Chim. Acta, Rev., 1967, 1, 87–97 CrossRef CAS.
- R. J. Mureinik, Coord. Chem. Rev., 1978, 25, 1–30 CrossRef CAS.
- R. Czerwieniec, A. Kapturkiewicz, J. Lipkowski and J. Nowacki, Inorg. Chim. Acta, 2005, 358, 2701–2710 CrossRef CAS.
- E. D. Risberg, J. Mink, A. Abbasi, M. Y. Skripkin, L. Hajba, P. Lindqvist-Reis, É. Bencze and M. Sandström, Dalton Trans., 2009, 0, 1328–1338 RSC.
- I. A. Efimenko, A. P. Kurbakova, Z. D. Matovic and G. Ponticelli, Transition Met. Chem., 1994, 19, 640–642 CrossRef CAS.
- B. Morzyk-Ociepa, K. Szmigiel, I. Turowska-Tyrk, M. Malik-Gajewska, J. Banach and J. Wietrzyk, Polyhedron, 2018, 153, 88–98 CrossRef CAS.
- M. S. Mubarak and D. G. Peters, J. Electroanal. Chem., 1997, 425, 13–17 CrossRef CAS.
- E. K. Beloglazkina, I. V. Yudin, A. G. Majouga, A. A. Moiseeva, A. I. Tursina and N. V. Zyk, Russ. Chem. Bull., 2006, 55, 1803–1809 CrossRef CAS.
- A. A. Isse, G. Berzi, L. Falciola, M. Rossi, P. R. Mussini and A. Gennaro, J. Appl. Electrochem., 2009, 39, 2217–2225 CrossRef CAS.
- Y.-W. Wu, J.-T. Wang, C. Jiang, W.-X. Chai and L. Song, Asian J. Chem., 2015, 27, 1529–1531 CrossRef CAS.
- H.-S. Jung and S.-H. Kim, Bull. Korean Chem. Soc., 2014, 35, 280–282 CrossRef CAS.
- S. Kottukulam Subran, S. Banerjee, A. Mondal and P. Paira, New J. Chem., 2016, 40, 10333–10343 RSC.
- R. Romeo, L. M. Scolaro, V. Catalano and S. Achar, Inorg. Synth., 2007, 32, 153–158 CrossRef.
- R. C. Clark and J. S. Reid, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 887–897 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1849220. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra01856e |
| This journal is © The Royal Society of Chemistry 2019 |