
Bistrifluoromethylated organocuprate [Ph4P]+[Cu(CF3)2]−: synthesis, characterization and its application for trifluoromethylation of activated heteroaryl bromides, chlorides and iodides†
He
Liu
and
Qilong
Shen
 *
*
Key Laboratory of Organofluorine Chemistry, Center for Excellence in Molecular Synthesis, Shanghai Institute of Organic Chemistry, University of Chinese Academy Sciences, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, P. R. China. E-mail: shenql@sioc.ac.cn
First published on 13th May 2019
Abstract
The synthesis and characterization of a bistrifluoromethylated organocuprate [Ph4P]+[Cu(CF3)2]− and its reactions with a variety of activated heteroaryl bromides, chlorides and iodides were described. These results showed that complex [Ph4P]+[Cu(CF3)2]− can serve as a trifluoromethylating reagent.
The trifluoromethyl group (–CF3) represents one of the privileged structural motifs in drugs as a result of its unique electronic and steric properties that can lead to higher metabolic stability and better pharmacokinetics.1 For this reason, an increasing number of trifluoromethylated new drug molecules have been on the market, including apalutamide (Erleada), elagolix (Orilissa), tecovirimat (TPOXX), and doravirine (Pifeltro) that were approved by the U.S. Food and Drug Administration (FDA) in 2018 (Fig. 1).2 Thus, it is generally accepted that the development of efficient methods for the preparation of the trifluoromethylated compounds is crucial for new drug discovery. Consequently, numerous elegant trifluoromethylative methods have been established in the past few decades.3
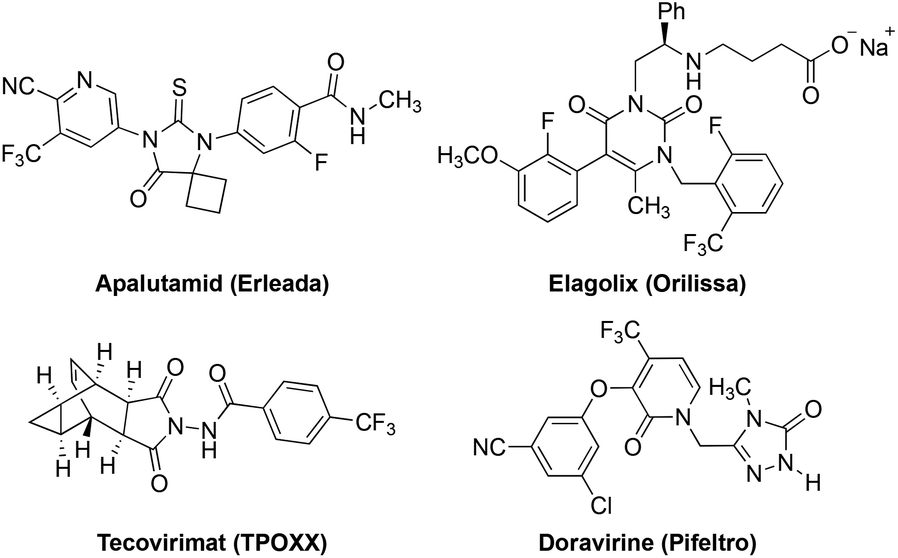 | ||
| Fig. 1 Drug molecules containing the CF3 group. | ||
Copper-mediated trifluoromethylation of aryl halides represents a classic method for the preparation of trifluoromethylarenes because of its high yields, low costs, and ease of operation.4 The first copper-mediated trifluoromethylation of aryl iodides using CF3I as the trifluoromethyl source was reported by McLoughlin and Thrower in 1965.5 It was proposed that reactive “CuCF3” was initially generated upon mixing copper with CF3I, which then reacted with aryl iodides to give trifluoromethylated arenes. Since then, a variety of methods for the generation of the active “CuCF3” from different trifluoromethyl sources such as CF3H,6 CF3I,7 Me3SiCF3,8 difluorocarbene precursors,9 electrophilic trifluoromethylating reagents10 and other trifluoromethyl metal reagents11 have been developed (Fig. 1, eqn (1)).
Several groups have tried to elucidate the structure of the reactive “CuCF3” species. For example, in 1986, Burton and coworkers observed a new peak with a chemical shift at −28.8 ppm in 19F NMR spectra when CuBr was treated with CF3CdX in DMF at −50 °C. Upon warming to room temperature, two new peaks at −32.3 and −35.5 ppm showed up. Then, these 19F NMR signals were assigned to [(CF3)Cu]·L (L = metal halide), and the cuprate CdI+[(CF3)2Cu]− and [Cu(CF3)4]− (Fig. 1, eqn (2)).12 In 2008, Kolomeitsev observed similar phenomena when CuBr was treated with CF3SiMe3 and KF in DMF–DMI at 0 °C. Accordingly, these 19F NMR signals were assigned to [CF3Cu]·KBr (−28.8 ppm), [(CF3)2Cu]−K+ (−32.4 ppm) and [(CF3)4Cu]−K+ (−35.7 ppm) (Fig. 1, eqn (3)).13 Likewise, Yagupolskii also observed similar 19F NMR signals when 2.0 equivalents of CuBr were treated with Zn(CF3)Br·DMF at room temperature.14 These studies also showed that [(CF3)4Cu]− was generated from oxidation of two other species and is not reactive for trifluoromethylation. Yet, the role of the proposed [Cu(CF3)]·KBr or [Cu(CF3)2]− in trifluoromethylation remains elusive. In 2008, Vicic isolated an ate-type organocopper(I) complex [(SIMes)2Cu]+[Cu(CF3)2]− and found that it was not reactive for trifluoromethylation of aryl iodides (Fig. 1, eqn (4)). However, the cuprate could be converted to neutral species [(SIMes)CuCF3] that was able to react with aryl iodides to give the trifluoromethylated arenes in good yields.15
Interestingly, previous studies showed that the peak in 19F NMR spectra corresponding to bistrifluoromethylated cuprate [Cu(CF3)2]− disappeared when the in situ generated trifluoromethylated copper mixtures were allowed to react with aryl iodides to afford the trifluoromethylated arenes in high yields. It was proposed that [Cu(CF3)2]− might serve as the reservoir for the formation of active “CuCF3”. We were, thus, curious to know whether and how the active “CuCF3” was generated from bistrifluoromethylated cuprate [Cu(CF3)2]−. We envisaged that if a stable bistrifluoromethylated cuprate [Cu(CF3)2]− with an ammonium or phosphonium counter cation could be prepared, these questions might be addressed. Herein, we reported the preparation and characterization of stable bistrifluoromethylated organocuprate [Ph4P]+[Cu(CF3)2]−1. Further studies showed that low conversion for the formation of desired trifluoromethylated arene was observed for the reaction of [Ph4P]+[Cu(CF3)2]−1 with phenyl iodide at 80–100 °C, as expected. Interestingly, upon addition of 1.0 equivalent of CuI, [Ph4P]+[Cu(CF3)2]−1 was able to react with aryl iodides in almost quantitative yield.16 In addition, the active “CuCF3” generated from [Ph4P]+[Cu(CF3)2]−1 and CuI was highly reactive to allow the trifluoromethylation of activated heteroaryl bromides and chlorides.
We initially attempted to prepare the bistrifluoromethylated cuprate by the reaction of CuCl with excess Me3SiCF3 (3.5 equiv.) in the presence of excess KF (6.5 equiv.) at room temperature. The reaction was closely monitored by 19F NMR spectroscopy. It was found that a peak at −27.8 ppm showed up after 15 min. Efforts to isolate this species failed. Interestingly, this peak disappeared after 30 min and a new peak at −31.4 ppm, along with two small peaks at −84.3, −117.3 ppm that corresponded to a [CuC2F5] species, showed up. Addition of tetraphenylphosphonium chloride Ph4PCl to the mixture and further stirring for 30 min led to the isolation of a stable bistrifluoromethylated cuprate [Ph4P]+[Cu(CF3)2]−1 as a white solid in 74% yield (Fig. 2, eqn (5)).
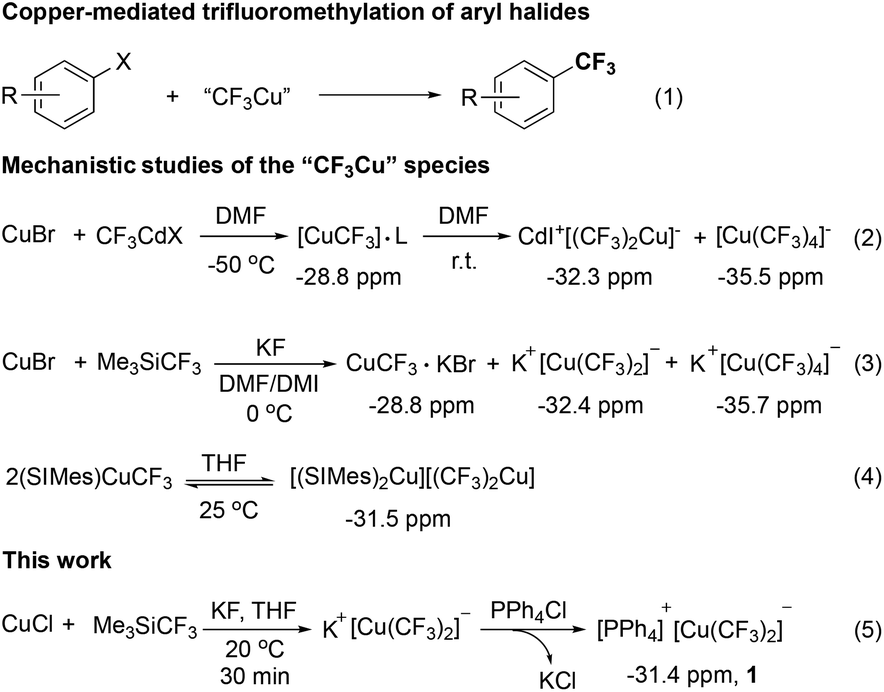 | ||
| Fig. 2 Trifluoromethylation of aryl halides with trifluoromethylated copper and the pursuit of the active “CF3Cu” species. | ||
Complex [Ph4P]+[Cu(CF3)2]−1 was fully characterized by 1H, 19F and 31P NMR spectroscopy, as well as elemental analyses. The 19F NMR spectrum for the CDCl3 solution of 1 showed a chemical shift of δ −31.4 ppm, which is almost the same as that for [(SIMes)2Cu]+[Cu(CF3)2]−. The structure was unambiguously confirmed by X-ray diffraction of its single crystal (Fig. 3). The X-ray structure revealed that the bond length of two identical Cu–CF3 bonds is 1.930(3) Å, which is shorter than that from [(SIMes)2Cu]+[Cu(CF3)2]− (1.970(6) Å).15
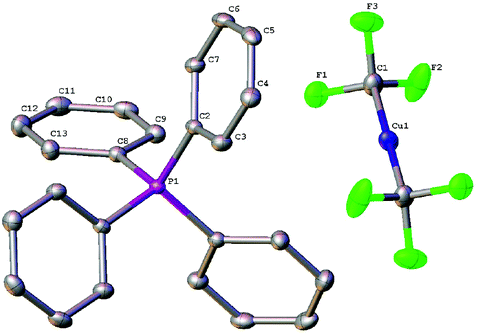 | ||
| Fig. 3 ORTEP diagram of 1. Hydrogens are omitted for clarity. | ||
Complex 1 is soluble in several common solvents such as CH2Cl2, CDCl3, THF, DMF and DMSO, but has poor solubility in solvents such as toluene, dioxane and diethyl ether. In the absence of air, a solution of complex 1 in DMF or THF was stable for at least 24 h without decomposition. In the presence of air, however, the solution turned blue within 5 min and no fluorine signal was observed by 19F NMR. Presumptively, a trifluoromethylated copper(II) species was generated.
Having developed a general protocol for the preparation of bistrifluoromethylated cuprate, we next explored the reactivity of [Ph4P]+[Cu(CF3)2]−1 with aryl iodides. The reaction of 4-methyl-iodobenzene with 1.2 equivalents of [Ph4P]+[Cu(CF3)2]−1 in DMF was chosen as a benchmark to test its reactivity. Notably, the formation of trifluoromethylarene was low (18% yield) when the reaction was conducted at 80 °C for 3.0 h. On increasing the reaction temperature to 100 °C, the formation of the desired product was observed in 62% yield after 3 h. These results confirmed Vicic's observation that the reaction of cuprate [Cu(CF3)2]−Q+ with aryl iodides was not effective.15 These results also suggest that, to generate the active “CuCF3”, an additive that is able to convert the cuprate to the active species is necessary. To our delight, when 1.0 equiv. of CuI was added, the trifluoromethylated product 4-methyl-trifluoromethylbenzene was obtained in 81% yield after 3 h at 80 °C and in 93% yield after 3 h at 100 °C. Using other copper salts such as CuBr, CuCl or CuBr2, decreasing the amount of CuI to 0.8 equiv. or conducting the reaction at lower temperature (80–90 °C) led to slightly lower 77–88% yields (see Table S1 in the ESI† for details).
The discovery that the addition of CuI could activate the bistrifluoromethylated cuprate [Ph4P]+[Cu(CF3)2]−1 prompted us to study whether complex 1 could serve as a general trifluoromethylating reagent. Most of the previously reported trifluoromethylation methods with in situ generated [CuCF3] focused mainly on reactions with aryl iodides; we, instead, would like to explore the reaction of [Ph4P]+[Cu(CF3)2]−1 with more challenging, less reactive substrates such as heteroaryl bromides and chlorides6,10 considering the importance of the trifluoromethylated heteroarene in the pharmaceutical and agrochemical industries.17 In addition, heteroaryl bromides and chlorides are much cheaper and commercially available than heteroaryl iodides.
A quick survey of the reaction conditions for the reaction of [Ph4P]+[Cu(CF3)2]−1 with 5-phenyl-2-bromopyridine in the presence of 1.0 equiv. of CuI disclosed that the reaction in DMF occurred smoothly after 3.0 h at 100 °C to give the corresponding 5-phenyl-2-trifluoromethylpyridine in 96% yield, as determined by 19F NMR spectroscopy with less than 5 mol% of the pentafluoroethylated side product (see Table S2 in the ESI† for details). The reaction conditions were general for a variety of activated heteroaryl bromides. As summarized in Scheme 1, activated ortho-bromo-substituted heteroarenes pyridine (2a–d), pyridazine (2h), pyrimidine (2i), quinoline (2k,m), quinoxaline (2n), and triazine (2p) underwent trifluoromethylation smoothly to generate the corresponding trifluoromethylated heteroarenes in high yields. In particular, substrates with bromo-substitution at the meta position of the nitrogen of the heteroarenes such as 3-bromopyridine derivatives (2f–g), 5-bromopyrimidine (2j) and 7-bromopyrido[2,3-b]pyrazine (2o) typically require 2.0 equiv. of [Ph4P]+[Cu(CF3)2]−1 and 2.0 equiv. of CuI to ensure full conversion and moderate yields. Likewise, reactions with a few less electron-poor bromo-substituted heteroarenes such as pyrazole (2q–r), oxadiazole (2u) and imidazo[1,2-a]pyridine (2s) also required the use of excess reagents and CuI for full conversion, while the reaction of several other types of bromo-substituted heteroarenes such as benzothiazole (2v), and thiazole (2w–x) could also achieve high yields with one equivalent of complex 1 and CuI.
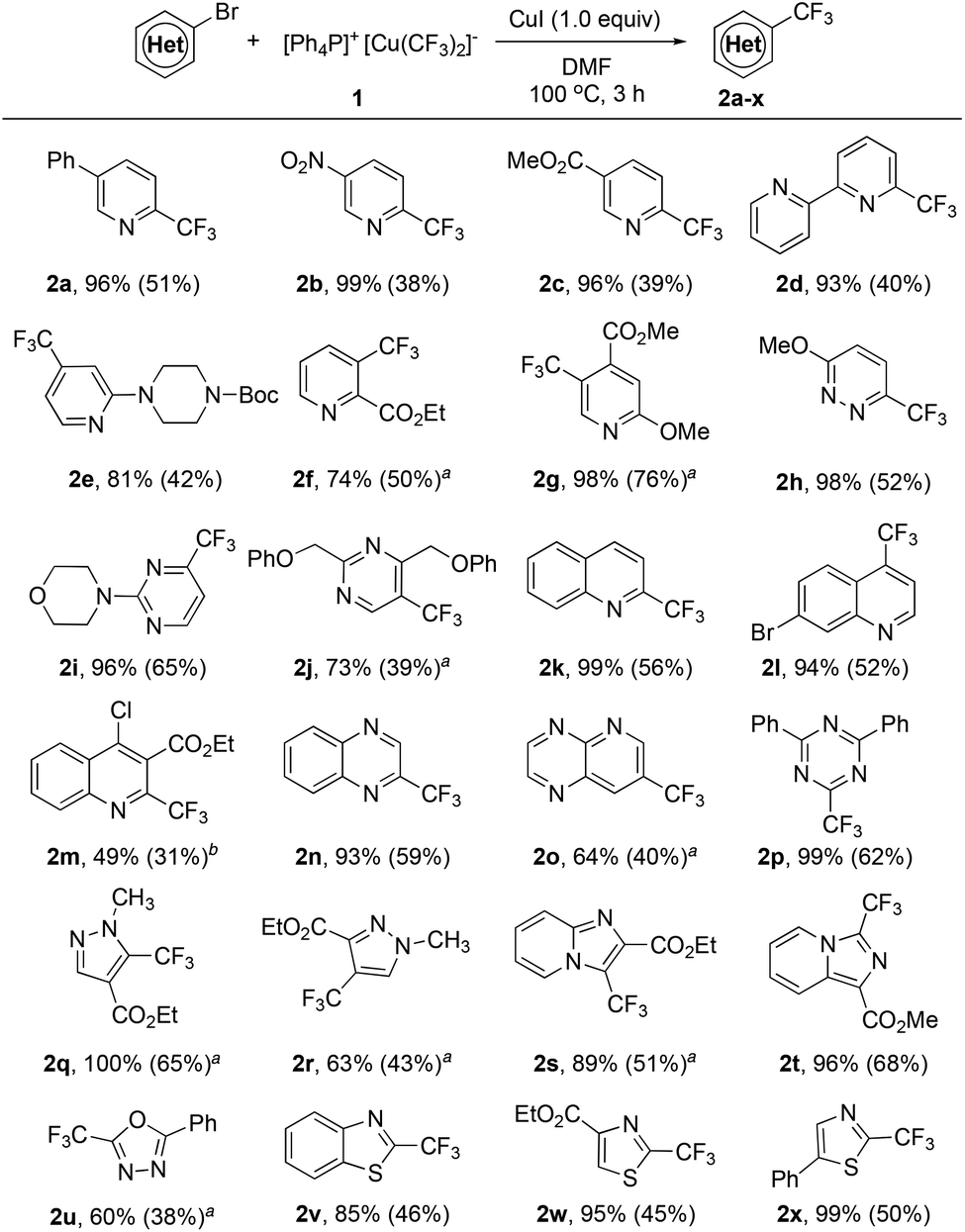 | ||
| Scheme 1 Scope of the trifluoromethylation of activated heteroaryl bromides with reagent 1. Reaction conditions: Heteroaryl bromide (0.50 mmol), [Ph4P]+[Cu(CF3)2]−1 (0.50 mmol), CuI (0.50 mmol) in DMF (5.0 mL) at 100 °C for 3 h; yields were determined by 19F NMR with fluorobenzene as an internal standard; isolated yields shown in the parenthesis; a1.0 mmol 1 and 1.0 mmol CuI were used; bDi-trifluoromethylated product 2y was also isolated (see the ESI† for details). | ||
Encouraged by the excellent reactivity and broad substrate scope for the reaction of heteroaryl bromides with complex [Ph4P]+[Cu(CF3)2]−1 in the presence of 1.0 equiv. of CuI, we next attempted to extend the substrate scope to heteroaryl chlorides. Initially, the reaction of 2-chloropyridine with complex 1 in the presence of 1.0 equiv. of CuI was studied. However, the formation of the desired 2-trifluoromethylpyridine was not observed under various conditions. Notably, during the study of the substrate scope for heteroaryl bromides, we observed a ditrifluoromethylated side product 2y for the reaction of ethyl 2-bromo-4-chloroquinoline-3-carboxylate (Scheme 1, 2m). Presumptively, the presence of an ortho-ester group significantly enhances the reactivity of the C–Cl bond in heteroaryl chloride. The same accelerating effect of an ortho-ester group of chloropyridine was previously observed by Grushin and coworkers.6 Nevertheless, reported yields for the reaction of ethyl 2-chloronicotinate or other derivatives were moderate (20–60% yields). We thus studied the reaction of ethyl 2-chloronicotinate with complex 1 in the presence of CuI. To our delight, the desired trifluoromethylated product was formed in 80% after 3.0 h at 100 °C (see Table S3 in the ESI† for details). Furthermore, this ortho-ester directed trifluoromethylation of heteroarene is general. As summarized in Scheme 2, heteroaryl chlorides with an ortho-ester group (3b, 3d, 3f–h, 3m–n) reacted with reagent 1 in high yields. In addition, it was found that other electron-withdrawing groups such as nitro (–NO2) (3a, 3e, 3i, 3j, 3l) and acetyl (–COCH3) groups (3c) can also act as the directing group to facilitate the trifluoromethylation process. Interestingly, a few strong electron-poor heteroaryl chlorides could be directly trifluoromethylated even without the assistance of these directing groups (2n, 2p, 3k), which suggests that the electron properties of the heteroarenes would greatly affect the consequence of this reaction. To the best of our knowledge, the current protocol represents the first general method for copper-mediated trifluoromethylation of activated heteroaryl chlorides.
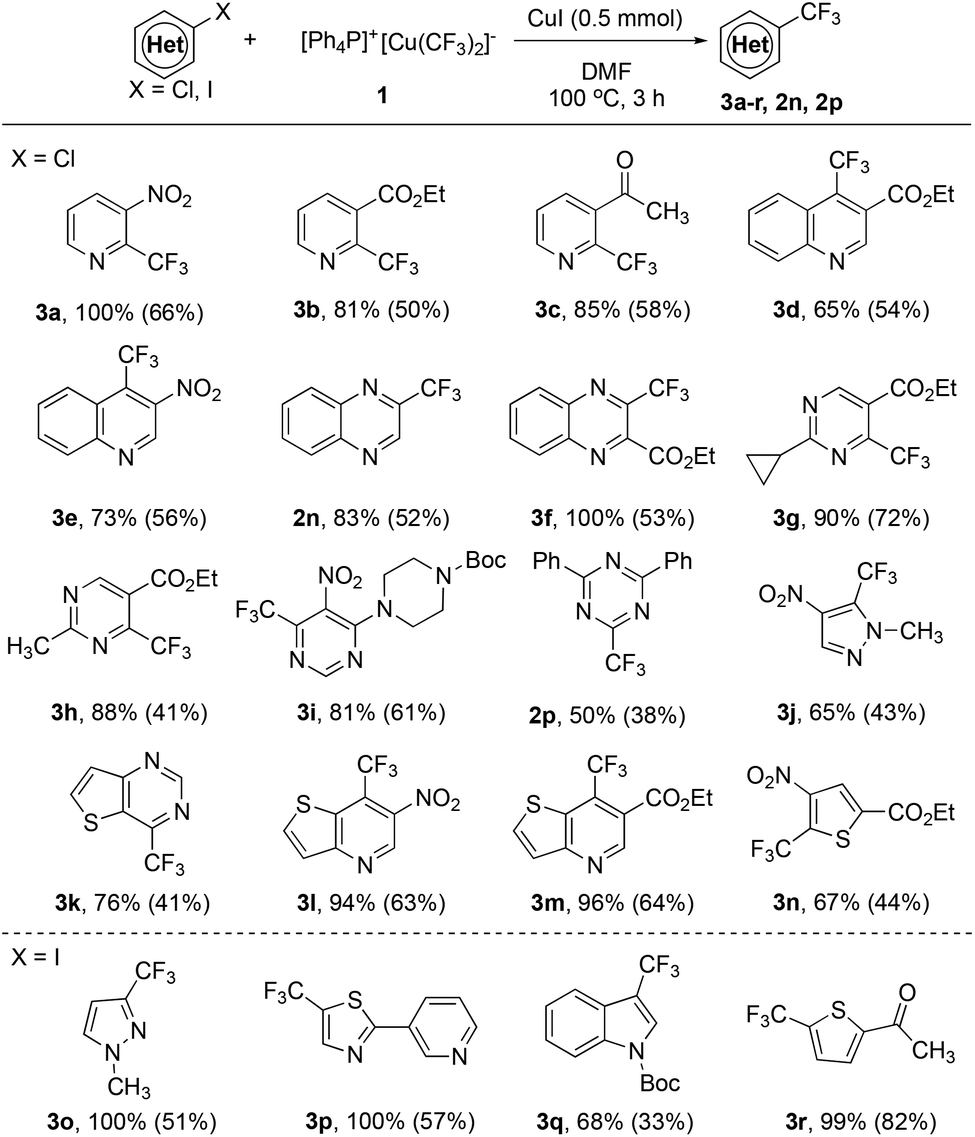 | ||
| Scheme 2 Scope of the trifluoromethylation of activated heteroaryl chorides and heteroaryl iodides with reagent 1. Reaction conditions: Heteroaryl chloride or iodide (0.50 mmol), [Ph4P]+[Cu(CF3)2]−1 (0.60 mmol), CuI (0.50 mmol) in DMF (5.0 mL) at 100 °C for 3 h; yields were determined by 19F NMR with fluorobenzene as an internal standard; isolated yields shown in the parenthesis. | ||
Finally, we also tried the reaction of complex 1 with electron-rich heteroaryl iodides. It was found that reactions of Boc-protected 3-iodoindole or 2-iodo-5-acetylthiophene and challenging substrates such as 3-iodo-1-methyl-1H-pyrazole or 5-iodo-2-(pyridin-3-yl)thiazole were all performed under the optimized conditions to give the corresponding trifluoromethylheteroarenes in moderate-excellent yields (Scheme 2, 3o–r).
To demonstrate the applicability of the current trifluoromethylating protocol, we tried to synthesize a few trifluoromethylated pharmaceutical intermediates and medicinally important compounds. As shown in Fig. 4, compounds 4 and 5, core structures of Retagliptin and Sitagliptin (Januvie), drugs for the treatment of diabetes mellitus type 2, were generated in 94% and 52% yields, respectively. Likewise, the trifluoromethylated derivative of Linagliptin, an antidiabetic agent, was synthesized in 91% yield. Finally, the trifluoromethylated imiquimod, a medication that acts as an immune response modifier to treat genital warts, was obtained in 71% yield.
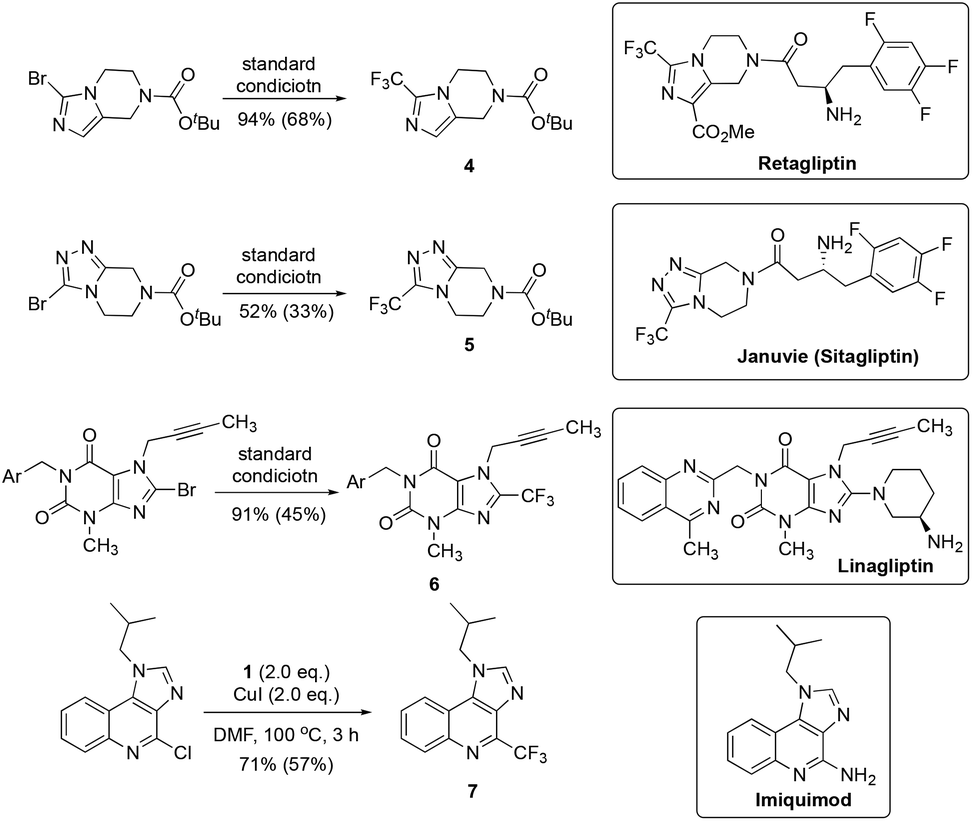 | ||
| Fig. 4 Application of the trifluoromethylation protocol for the preparation of trifluoromethylated pharmaceutical intermediates and medicinally important compounds. | ||
Conclusions
In summary, our curiosity toward the role of the trifluoromethylated cuprate in copper-mediated trifluoromethylation led to the discovery of a new trifluoromethylating reagent bistrifluoromethylated cuprate [Ph4P]+[Cu(CF3)2]−1. In the presence of 1.0 equivalent of CuI, this ate-type complex reacted with a variety of activated heteroaryl bromides and chlorides that are less expensive and readily commercially available in high yields. Further expanding the scope of the reaction and mechanistic studies of the copper-mediated trifluoromethylation are currently underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support from the National Natural Science Foundation of China (21625206, 21632009, 21572258, 21572259, and 21421002) and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB20000000) is greatly appreciated.Notes and references
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC.
- (a) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed; (b) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529 CrossRef CAS PubMed; (c) J. Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Acena, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422 CrossRef PubMed; (d) https://www.fda.gov/drugs/developmentapprovalprocess/druginnovation/ucm592464.htm .
- For selected recent reviews on trifluoromethylation methods, see: (a) J.-A. Ma and D. Cahard, Chem. Rev., 2008, 108, PR1 CrossRef CAS PubMed; (b) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS PubMed; (c) C. Alonso, E. M. Marigorta, G. Rubiales and F. Palacios, Chem. Rev., 2015, 115, 1847 CrossRef CAS PubMed.
- For selected reviews on copper-mediated trifluoromethylation: (a) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475 CrossRef CAS PubMed; (b) T.-F. Liu and Q. Shen, Eur. J. Org. Chem., 2012, 6679 CrossRef CAS.
- (a) V. C. R. McLoughlin and J. Thrower, U.S. Patent3408411, 1968 Search PubMed; (b) V. C. R. McLoughlin and J. Thrower, Tetrahedron, 1969, 25, 5921 CrossRef CAS.
- (a) A. Lishchynskyi, M. A. Novikov, E. Martin, E. C. Escudero-Adan, P. Novak and V. V. Grushin, J. Org. Chem., 2013, 78, 11126 CrossRef CAS PubMed; (b) K. P. S. Cheung and G. C. Tsui, Org. Lett., 2017, 19, 2881 CrossRef CAS PubMed.
- Y. Kobayashi and I. Kumadaki, Tetrahedron Lett., 1969, 47, 4095 CrossRef.
- (a) H. Urata and T. Fuchikami, Tetrahedron Lett., 1991, 32, 91 CrossRef CAS; (b) M. Oishi, H. Kondo and H. Amii, Chem. Commun., 2009, 1909 RSC.
- Selected examples for trifluoromethylation using difluorocarbene precursors: (a) Q.-Y. Chen and S.-W. Wu, J. Chem. Soc., Chem. Commun., 1989, 705 RSC; (b) J. G. MacNeil Jr. and D. J. Burton, J. Fluorine Chem., 1991, 55, 225 CrossRef; (c) M. M. Kremlev, A. I. Mushta, W. Tyrra, Y. L. Yagupolskii, D. Naumann and A. Möller, J. Fluorine Chem., 2012, 133, 67 CrossRef CAS.
- (a) C.-P. Zhang, Z.-L. Wang, Q.-Y. Chen, C.-T. Zhang, Y.-C. Gu and J.-C. Xiao, Angew. Chem., Int. Ed., 2011, 50, 1896 CrossRef CAS; (b) Y.-F. Liu, X.-X. Shao, L. Lu and Q. Shen, Org. Lett., 2015, 17, 2752 CrossRef CAS PubMed.
- (a) N. V. Kondratenko, E. P. Vechirko and L. M. Yagupolskii, Synthesis, 1980, 932 CrossRef CAS; (b) D. J. Burton and D. M. Wiemers, J. Am. Chem. Soc., 1985, 107, 5014 CrossRef CAS.
- (a) D. M. Wiemers and D. J. Burton, J. Am. Chem. Soc., 1986, 108, 832 CrossRef CAS; (b) M. A. Willert-Porada, D. Burton and N. C. Baenziger, J. Chem. Soc., Chem. Commun., 1989, 1633 RSC.
- A. Kuett, V. Movchun, T. Rodima, T. Dansauer, E. B. Rusanov, I. Leito, I. Kaljurand, J. Koppel, V. Pihl, I. Koppel, G. Ovsjannikov, L. Toom, M. Mishima, M. Medebielle, E. Lork, G.-V. Roeschenthaler, I. A. Koppel and A. A. Kolomeitsev, J. Org. Chem., 2008, 73, 2607 CrossRef CAS PubMed.
- M. M. Kremlev, W. Tyrra, A. I. Mushta, D. Naumann and Y. L. Yagupolskii, J. Fluorine Chem., 2010, 131, 212 CrossRef CAS.
- G. G. Dubinina, J. Ogikubo and D. A. Vicic, Organometallics, 2008, 27, 6233 CrossRef CAS.
- During the preparation of the manuscript, Maseras, Perez-Temprano and their coworkers described that the addition of excess CuI significantly accelerates the reaction of in situ generated (Cs)[Cu(CF3)2] from reaction of (Cs)[Ag(CF3)2] with CuI with aryl iodide, see: S. M. de Salinas, A. L. Mudarra, C. Odena, M. M. Belmonte, J. Benet-Buchholz, F. Maseras and M. H. Perez-Temprano, Chem. – Eur. J., 2019 DOI:10.1002/chem.201900496.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 19F and 13C NMR spectra of compounds 1, 2a–x, 3a–r, 4–7 as well as X-ray structure of complex 1. CCDC 1911058. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qo00527g |
| This journal is © the Partner Organisations 2019 |