
Fluorescent aggregation-induced emission (AIE)-based thermosetting electrospun nanofibers: fabrication, properties and applications†
Weili
Li
 *a,
Yexin
Ding
a,
Mike
Tebyetekerwa
*b,
Yixiao
Xie
a,
Lin
Wang
a,
Hongkun
Li
c,
Rong
Hu
d,
Zhiming
Wang
d,
Anjun
Qin
d and
Ben Zhong
Tang
*de
*a,
Yexin
Ding
a,
Mike
Tebyetekerwa
*b,
Yixiao
Xie
a,
Lin
Wang
a,
Hongkun
Li
c,
Rong
Hu
d,
Zhiming
Wang
d,
Anjun
Qin
d and
Ben Zhong
Tang
*de
aSchool of Material Science and Engineering & National Demonstration Center for Experimental Materials Science and Engineering Education, Jiangsu University of Science and Technology, Zhenjiang 212003, P. R. China. E-mail: liweili@just.edu.cn
bResearch School of Electrical, Energy and Materials Engineering, College of Engineering and Computer Science, The Australian National University, Canberra, ACT 2601, Australia. E-mail: mike.tebyetekerwa@anu.edu.au
cState and Local Joint Engineering Laboratory for Novel Functional Polymeric Materials, Laboratory of Advanced Optoelectronic Materials, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, P. R. China
dGuangdong Innovative Research Team, State Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou 510640, P. R. China
eDepartment of Chemistry, Hong Kong Branch of Chinese National Engineering Research Center for Tissue Restoration and Reconstruction, The Hong Kong University of Science & Technology, Clear Water Bay, Kowloon, Hong Kong, P. R. China. E-mail: tangbenz@ust.hk
First published on 24th September 2019
Abstract
A curing process in nanofiber-based thermosetting polymers involves both rapid solvent evaporation and chemical cross-linking at the nanoscale, which altogether present a complicated scenario to study. In this paper, we employed aggregation-induced emission (AIE) phenomena with the help of fluorescence tests and scanning electron microscopy to study functional fluorescent AIE-based thermosetting nanofibers with reference to their fabrication, properties and possible applications. The properties that dictate the electrospinning of nanofibers were first studied together with their properties. Finally, we tested the obtained functional nanofibers as thermo-sensitive probes and chemosensors. These applications were possible courtesy of the restriction of intermolecular rotation (RIR) mechanism of the AIE luminogen (AIEgen) of TPE, which was successfully knitted onto the thermosetting polymer Epoxy. Its presence around the TPE-Epoxy structure dictates the fluorescence behaviors of the final composite depending on the material environment.
Introduction
Electrospinning is a widely known scalable technique used to prepare fibers with nano to micrometer sizes.1 These can be employed in a wide range of applications, including but not limited to; medicine, energy and environment, agriculture, electronics and many more. It is common and relatively easy to employ thermoplastic polymers such as polyacrylonitrile,2 polystyrene,3 polyvinyl alcohol,4 polycarbonate,5 polyethylene glycol,6 and polyvinylpyrrolidone7 to obtain and study electrospun nanofibers. This is due to their easy-to-obtain viscosity and ability to form reliable electrospinning solutions.8 Thermosetting polymers, due to their crosslinking, are also important. They have partly replaced structural components of steel on aircraft and satellites because of their excellent mechanical properties. Due to their insoluble nature, they are widely applied in adhesives, composite engineering plastics, anti-corrosive paints, and so on.9–12 However, their chemical reactivity and numerous crosslinks present a lot of uncontrollable factors during their preparation. Also, forming their spinnable solutions (if to made into fibers) is not a straightforward approach, and this partly explains why not so many researchers concentrate on their application in nano/microfibers via electrospinning.Beyond unmodified nanofibers, functional nanofibers with a wide range of unique applications can also be prepared by electrospinning. The spinning polymer can be doped with the necessary organic/inorganic nanoparticles,13 nanowires,14 proteins15 and many others to obtain new properties. In other cases, the polymer can be functional-polymerized by modifying its structural chemistry before spinning. Amongst the many multi-functional nanofibers, fluorescent nanofibers show speculative and promising potential in many areas due to their unique properties. A straightforward approach to prepare fluorescent nanofibers is by doping readily available organic or inorganic fluorescent powders into the spinning dope prior to electrospinning.16 However, these traditional fluorescent powders usually tend to present prominent fluorescence when in solution form, but not in the solid state because of the widely known aggregation-caused quenching (ACQ) effect.17–19 So, therefore, though the approach is straightforward, the resultant performance of the obtained fluorescent nanofibers is unreliable due to the ACQ effect. Hence, aggregation-induced emission (AIE), the opposite to ACQ fluorescence behavior, which was first demonstrated by Tang's group in 2001, could be utilized as a perfect solution to the problem in this scenario.20,21 As the fluorescent behavior of an AIE luminogen (AIEgen) is closely related to its intramolecular rotation, it has previously been proven that it could efficiently emit strong fluorescence, even when it is doped into other polymer materials in the solid state.22–24
In our previous work, we were able to knit fluorescent AIE TPE-2CH2Br onto thermosetting epoxy resin chains (Scheme 1).25 We were able to prove that the synthesized modified epoxy resin (having TPE-2CH2Br) could emit strong fluorescence according to the restriction of intermolecular rotation (RIR) mechanism. Indeed its fluorescence responsiveness to the external stimulus made it possible to be applied as a smart coating. In a similar direction, herein, fluorescent nanofibers (FNFs) based on the modified AIE-Epoxy polymer have been prepared via electrospinning. We systematically detail the preparation, properties and applications of the resultant modified fluorescent AIE-based thermosetting TPE-Epoxy nanofibers. The obtained results are provided in the following manner. (1) First, the electrospinning of the thermosetting fibers was studied to get good and reliable materials. (2) This was followed by examining the fluorescence properties of the resultant fibers, together with understanding the curing behavior and process of formation of thermosetting polymers at the nanoscale. (3) And finally, the functional fibers were applied and tested as thermo-sensitive probes and chemosensors.
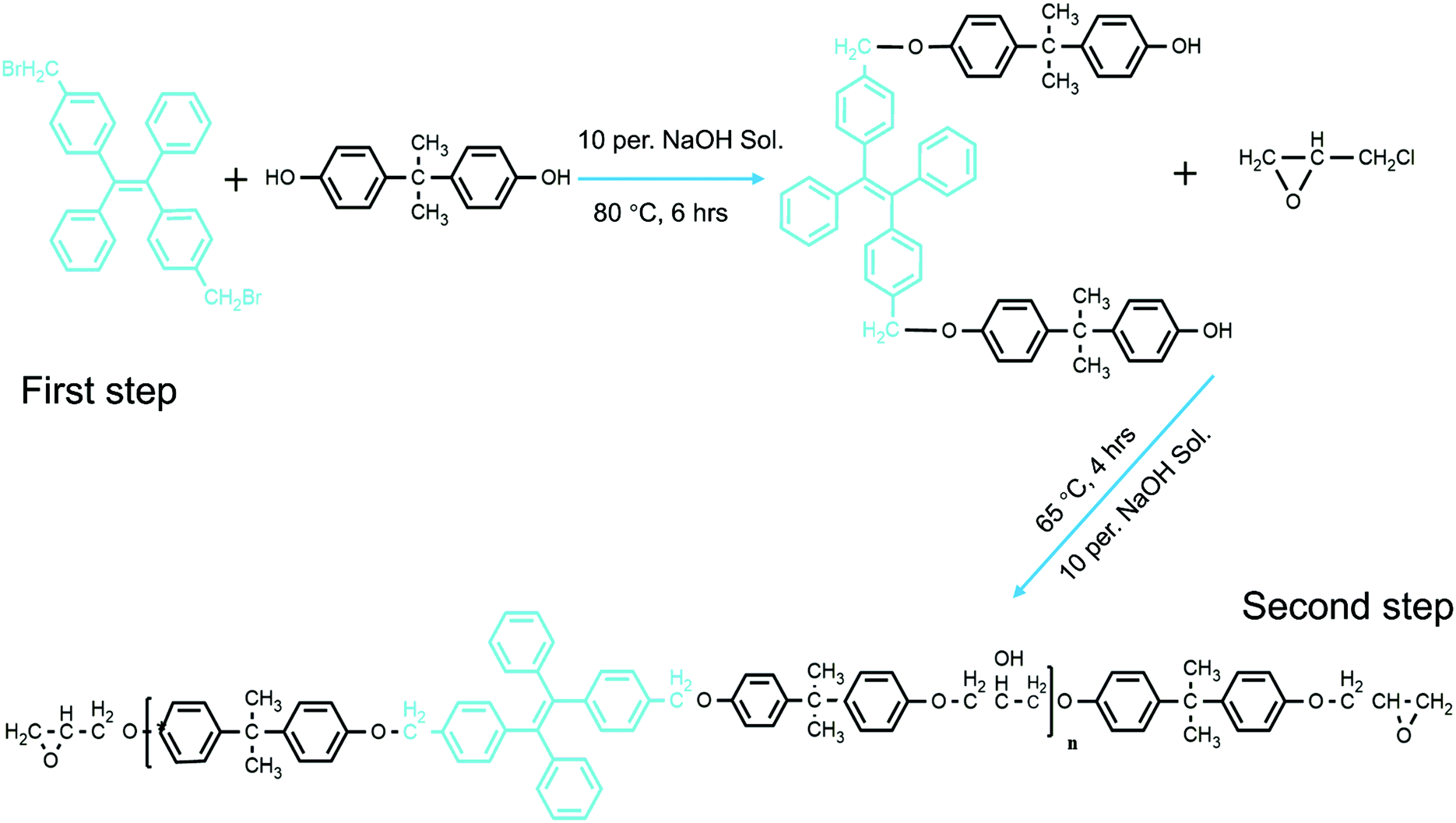 | ||
| Scheme 1 The synthetic route followed to obtain TPE-epoxy. | ||
Results and discussion
Effect of curing time and crosslinking on the morphology of electrospun FNFs
The preparation of thermosetting polymer-based nanofibers by electrospinning is a very complicated process. Therefore, different factors that control this process are better studied individually. First, crosslinking was thought of as a vital factor which would affect the preparation of the spinning solution. Thus, the curing process and time are significant towards obtaining suitable morphology of the nanofibers. The viscosity of the spinning solution was observed to vary with the pre-crosslinking time and the polymer concentration. The linear increase in viscosity of the spinning polymer with curing time (Fig. 1a) verified that TPE-Epoxy-20 could react with the curing agent at room temperature moderately. According to Fig. 1b, when the pre-crosslinking time was 1 h, the diameter of the nanofiber was very small with lots of polymer beads appearing due to the low molecular weight of the pre-crosslinked fluorescent resins. As the crosslinking reaction continued for 2 h and 3 h, the obtained nanofibers presented a smooth nanofiber shape with a uniform diameter. However, when the pre-crosslinking time reached 4 h, the high viscosity of the spinning solution proved to be detrimental to the electrospinning process, and the diameter of the resultant nanofibers became very thick with uneven morphology. See Fig. S1 (ESI†) for statistical diameter distribution of the nanofibers. | ||
| Fig. 1 Factors affecting the morphology and formation of TPE-Epoxy thermosetting electrospun nanofibers. (a) Pre-crosslinking time which tunes the viscosity of the spinning solution. According to figures in (b) from the SEM images it can be seen that the pre-crosslinking time plays a vital role in controlling the morphology of the electrospun nanofibers due to its ability to vary the viscosity of the spinning solution. In (c) the content of the polymer added to the spinning liquid is plotted against the viscosity of the resultant solution. It was found that this factor also had a direct effect on the nanofibers as shown in (d). Also the change in the type of TPE-Epoxy resin directly affected the viscosity (e) and hence the electrospinning process. In (f) it can be seen that with too much epoxy content nanofibers cannot be obtained. By changing the DMF:Acetone content as the spinning solvent, conductivity could be tuned as shown in (g), and this would give different nanofiber morphology as in (h). Too much acetone leads to formation of micro bubbles within the nanofiber membranes. For all the nanofibers, statistical diameter distributions have been analyzed and supplied in the ESI,† Fig. S1. (White scale bar is 10 μm and Black scale bar is 50 μm.) | ||
Next, the pre-crosslinking time was set to 2 h, and the concentration of the spinning polymer solution was varied between 50% and 65% to study its influence on the viscosity of the spinning solution, whose results are presented in Fig. 1c. The other different studied nanofibers prepared in this work are thoroughly described in Table S1 (ESI†). The viscosity of the spinning liquid is known to have a significant influence on the morphology of electrospun nanofibers.6,8 From Fig. 1d, when the polymer concentration was kept at 50 wt%, a spinning solution with low viscosity could be attained. This was unable to form a continuous jet in the high voltage electric field during electrospinning, and the obtained nanofibers had many polymer beads (see Fig. 1d (50 wt%)). With the viscosity of the spinning liquid increasing to 55 wt%, the bead formation on the nanofibers gradually dwindled. FNFs with perfect morphology were obtained by increasing the concentration to 60 wt%, whereas higher concentrations gave unordered and large diameter nanofibers. This is ascribed to the filaments’ ability to split after being spun from the spray nozzle declining dramatically, and the polymers thus could readily form thicker microfibers as seen in Fig. 1d (65 wt%).
The effect of epoxy value alone on the resultant nanofibers was also probed. The concentration of the spinning liquid was set to 60 wt%, with the pre-crosslinking time of 2 h. Different epoxy values of the fluorescent resin were applied to study their impact. According to Fig. 1e, the viscosity of the spinning liquid increased linearly with decreasing epoxy value of the fluorescent resin. For the TPE-Epoxy 51, the viscosity of the spinning solution was quite low, and the polymers jets would spray instead of forming fine filaments in the high-voltage electric field, which led to the formation of just a flat coating (Fig. 1f (TPE-Epoxy 51)). With epoxy value of the fluorescent resin decreasing, the viscosity of the spinning liquid increased. For the samples TPE-Epoxy 20 and TPE-Epoxy 12, nanofibers could be obtained which both showed good morphology with a uniform-size.
The solvent effect on the electrospinning process, of the polymer was also studied. The fluorescent resin (TPE-Epoxy-20) was dissolved into different solvents by changing the relative proportion of components. As is seen in Fig. 1g, the conductivity of the mixed solvents increased linearly with increasing content of DMF. A similar trend has been confirmed by another study.26 The resultant surface morphologies of FNF membranes are presented in Fig. 1h. Samples with DMF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acetone = 10:90 (by weight), presented uniform nanofiber morphology (1.94 ± 0.31 μm). When the content of DMF in DMF:acetone was set to 30:70, the obtained nanofiber got thinner (1.41 ± 0.41 μm). Generally, with the content of DMF increasing and acetone decreasing, the obtained diameter of the nanofiber lowered along with more polymer gels appearing on the surface of the membrane. This is due to the relatively high conductivity of DMF solvent. However, though the solution conductivity becomes better, the viscosity is being raised by the presence of more DMF (whose η is 0.92 mPa s). This ultimately affects a natural electrostatic drawing process, and hence the formation of beads and bubbles in the fiber membranes. In addition, DMF has a high boiling point (154 °C), and it is reckoned that its extra residues can sit on the membranes which can lead to the dissolving of the nanofibers and emergence of a large gel.
:acetone = 10:90 (by weight), presented uniform nanofiber morphology (1.94 ± 0.31 μm). When the content of DMF in DMF:acetone was set to 30:70, the obtained nanofiber got thinner (1.41 ± 0.41 μm). Generally, with the content of DMF increasing and acetone decreasing, the obtained diameter of the nanofiber lowered along with more polymer gels appearing on the surface of the membrane. This is due to the relatively high conductivity of DMF solvent. However, though the solution conductivity becomes better, the viscosity is being raised by the presence of more DMF (whose η is 0.92 mPa s). This ultimately affects a natural electrostatic drawing process, and hence the formation of beads and bubbles in the fiber membranes. In addition, DMF has a high boiling point (154 °C), and it is reckoned that its extra residues can sit on the membranes which can lead to the dissolving of the nanofibers and emergence of a large gel.
Properties and formation behavior of electrospun thermosetting FNFs
For the thermosetting-based polymer nanofibers, we found out that the obtained cross-section morphology was different if compared to thermoplastic polymers previously obtained in similar circumstances. Here, an FNF-2 (i.e. TPE-Epoxy-20, DMF:acetone is 10:90, 60 wt% concentration, 2 h crosslinking time, see Table S1, ESI†) sample was taken as the example, and studied with high resolution FESEM. Its surface showed flat and smooth morphology, and it was rather peculiar to observe a core/sheath structure (Fig. 2a), which can only be obtained by core–shell electrospinning with thermoplastic-based polymer nanofibers. We have proposed a schematic to explain the observation (Fig. 2b). It might be caused by the complicated curing process involved with thermosets. There are two proposed steps herein which the thermoset nanofibers are predicted to undergo during the curing process: (1) solvent evaporation and (2) thermal crosslinking. During the first process, the skin of the nanofiber gradually forms on solution evaporation and the outer layer tends to dry more rapidly than the inner core of the nanofibers, which could generate an internal stress for the uncured TPE-Epoxy resin in the inner core of the nanofiber. As time passes, the inner TPE-Epoxy resin is squeezed out to relieve the internal stress during the thermal crosslinking process, and thus the core/sheath morphology of the nanofiber is formed.
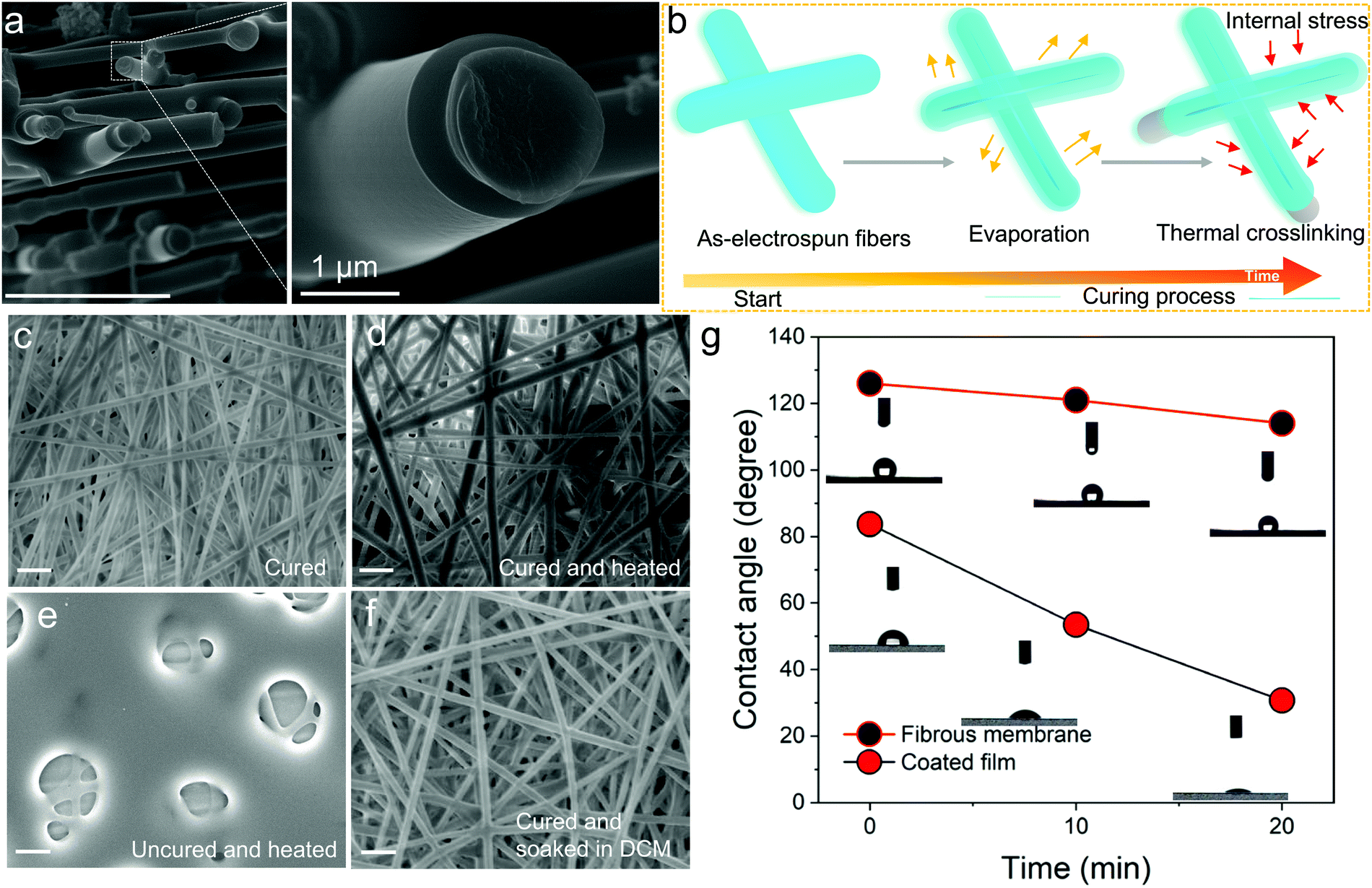 | ||
| Fig. 2 Properties and formation behavior of the TPE-Epoxy thermosetting electrospun nanofibers. (a) Magnified FESEM image and the cross section of the fibers. (b) Schematic illustration of the phenomena responsible for the core shell-type of morphology observed in (a). Solvent evaporation and thermal crosslinking during curing are highly responsible. (c) SEM images of the surface morphology of fibers on curing, (d) curing after being heated for 30 min at 100 °C, (e) uncured fiber membranes after being heated and (f) cured fibers after being dipped into DCM solution. (g) Comparison between contact angle measurement of electrospun fibers and flat coating based on a TPE-Epoxy polymer. (Scale bar 10 μm.) | ||
Besides the non-conventional morphology, the prepared thermoplastic nanofiber membrane showed some other unique features. For example, when the cured FNF-2 samples (Fig. 2c) were heated for 30 min at 100 °C (Fig. 2d). The nanofiber is seen to keep its original appearance, morphology and shape. While, for the uncured nanofiber membranes the mesh was disorganized and melted (Fig. 2e). This is attributed to the absence of three-dimensional networked structures in the later samples which would otherwise give nanofibers with better resistance to heat. In addition, the cured FNF-2 samples could still retain their morphology after being soaked into DCM (Fig. 2f), while the uncured FNF-2 dissolved into this solution. In a nutshell, the prepared TPE-epoxy nanofibers presented excellent thermo-tolerance and solvent resistance properties, and these properties are important for any material for a wide range of applications.
Water contact angle measurements were carried out to determine if the obtained nanofibers were hydrophobic or hydrophilic. This would help us to apply them in suitable environments later. From the results, the nanofiber membranes showed excellent hydrophobicity due to their high porosity and rough overall surface morphology which created a lotus effect. As is summarized in Fig. 2g, the contact angle reached 126° for FNF-2, which is much higher than that of the flat coating based on TPE-Epoxy-20 obtained by spin-coating (83.7°). Moreover, the FNF-2 demonstrated persistent hydrophobic property. The contact angle remained slightly stable at 114° after 20 min, while, the contact angle of the thin-film coating rapidly fell to 30.7°.
Fluorescence properties of FNFs
FNF-2 samples which showed good morphology were employed to study the modified nanofibers’ fluorescent properties. At first, the membrane was placed under UV light (365 nm), and could emit strong blue fluorescence (Fig. 3a) which can be easily explained according to the RIR effect brought about by the presence of the AIEgen in epoxy. This observation was also confirmed by confocal fluorescence microscopy (Fig. 3b). The uniform luminescence behavior verified further that TPE molecules were well knitted and dispersed in the polymer matrix homogeneously instead of being self-aggregated in one area. When the PL of FNF-2 was compared to the thin-film coating of TPE-Epoxy-20, it was found that FNF-2 had a higher PL intensity and quantum yield (Fig. S2, ESI†). FNF-2 and the thin-film coating had a quantum yield of 17.96% and 5.51%, respectively. To explain this observation, we predict that the stretched state of the polymer nanofiber induced during electrospinning results in the orientation of the polymer chains. This consequently constrains the intramolecular rotation of TPEs and thus the high observed PL and quantum yield in the nanofibers. In addition, since the nanofibers were stacked layer by layer during electrospinning, this facilitates the nano and microporous morphology within the membranes capable of absorbing more UV light, and hence enhanced fluorescence intensity.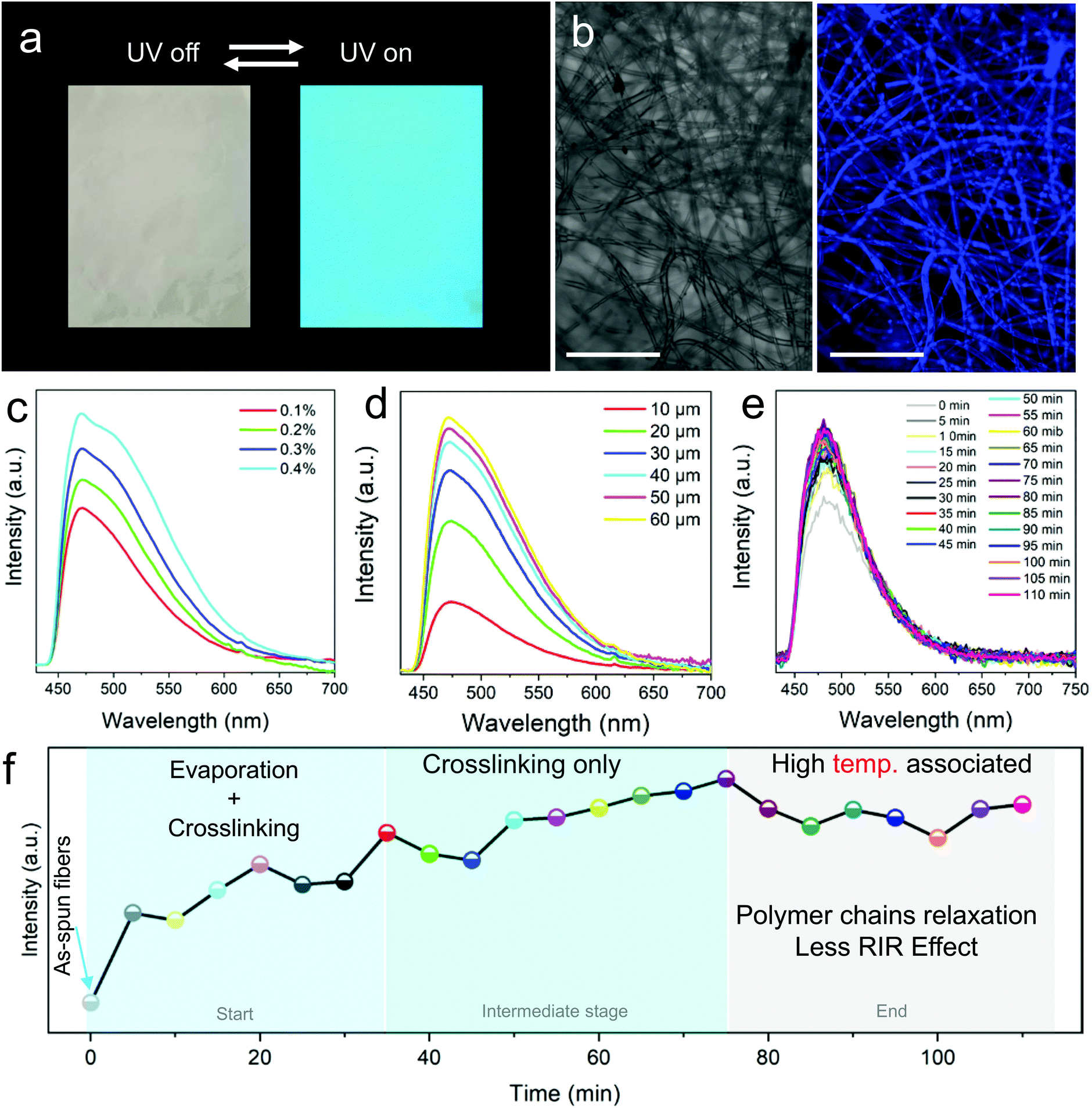 | ||
| Fig. 3 Fluorescence property and curing theory of thermosetting Epoxy-TPE nanofibers. (a) Naked eye fluorescence of nanofiber membranes under UV light (365 nm). The arrows show switch on and off. (b) Relatively high spatial resolution images of nanofibers viewed under a confocal fluorescence microscope. (Exact image before and after UV excitation (405 nm).) PL intensity of the nanofibers with respect to (c) changing TPE AIEgen doping percentages, (d) thickness of the membranes and (e) curing time. The observed PL intensity changes in (e) are further used to explain the complicated curing process of TPE-Epoxy thermosetting nanofibers in (f). (Scale bar 100 μm.) | ||
Furthermore, different FNFs were obtained by varying the content of TPE-2CH2Br to find out the optimum doping value required for AIE to obtain the fluorescent nanofibers. In Fig. 3c, the peak position of the PL spectra of different obtained FNF samples were located at around 475 nm, while their intensities increased linearly with the content of TPE-2CH2Br, which is a typical AIE behavior (Fig. S3, ESI†).27,28 On the other hand, the effect of membrane thickness on the fluorescent property of FNFs was studied. Interestingly, they didn’t present a linear trend (Fig. 3d). For the FNF with the thickness lower than 30 μm, the fluorescence intensity increased linearly with the thickness rapidly. However, with the membrane thickness gradually increasing, the enhancement in the fluorescence intensity slowed gradually. This can be ascribed to the opaque characteristics of the FNF, as the membrane thickness increased to a very high value such that it could not permit enough UV light to reach its bottom.
The crosslinking that occurs during curing of the thermoset-nanofibers cannot easily be explained or tracked. However, we can employ the RIR effect in the fluorescent nanofibers to study this complicated phenomenon. After being fabricated by an electrospinning process, the FNF-2 samples were heated to 60 °C and the PL spectra were obtained every 5 min (Fig. 3e). During the curing process, a 3D cross-linked network structure could be constructed as the crosslinking reaction between the fluorescent resin and curing agent occurs. This would restrict the intramolecular rotation of TPE molecules and hence enhance the sample's PL intensity. As seen in Fig. 3e, the peak position of the PL spectra was located at around 480 nm. Whereas the PL intensity varied with the curing time. The curing process could be divided into three stages as described in Fig. 3f as constructed from Fig. 3e. In the first stage (0 min to 35 min), PL intensity grew rapidly due to the process that combines the evaporation of residual solvents and the crosslinking reaction. In the second stage, which is the intermediate stage (35 min to 75 min), PL intensity grew slowly as there was only crosslinking process taking place. As time passed, after crosslinking process completion, the relaxation of the polymer chain at a relatively high temperature might weaken the intramolecular rotation of TPE and thus cause a decrease in the PL intensity of nanofibers. A similar observation was reported previously in thin-films.29
Fluorescence responsiveness of the prepared FNFs
For reliability of the fabricated nanofibers, stability is paramount. So, first the fluorescence stability of the FNFs was studied prior to understanding their responsiveness to the environment. From the results in Fig. 4a, we confirmed that FNFs were stable even after 5 weeks of room condition storage. It is believed that, since the TPE was well knitted onto the cross-linked polymer chains via chemical bonds, the surrounding cross-linked polymer chains might have played a protective role. The intramolecular motion of a TPE molecule could be restricted for a long time and the nanofibers could keep good fluorescence stability. | ||
| Fig. 4 Fluorescence responsiveness and stability of the prepared TPE-Epoxy fibers. PL intensity of the nanofibers with respect to (a) storage time and (b) testing temperatures. (c) PL recycling behaviors of nanofibers when the membranes are exposed to high and low temperatures in the thermal cycling test measurement. (d) PL behaviors of nanofibers and thin film coating (Fig. S5, ESI†) in saturated DCM steam during different soaking times. (e) PL behaviors of nanofibers when exposed to different concentrations of nitrobenzene and the immersion time (f). | ||
As the movement of polymer chain segments is closely related to the external environment, the intramolecular motions of embedded TPE may be affected by it. FNF-2 samples were placed in the environment with temperatures ranging from 20 °C to 100 °C and PL obtained at each temperature (Fig. 4b). Changes in fluorescence were observed, which can be explained as follows. At room temperature, stiff polymer chain segments limited the intramolecular rotation of a TPE molecule, which resulted in the absorbed energy being annihilated through radiation decay mainly, and then the nanofibers emitted efficiently under UV light. However, with the testing temperature increasing, this gradually loosened the polymer chains hence weakening the constraint of TPE and thus expanded the intramolecular rotation space. The progressive intramolecular motions could consume the absorbed energy, which led to weak fluorescence emission of the AIEgen in the rubbery state of the polymers at high temperature. The observed fluorescence-temperature dependence was reversible and could be recycled and repeated many times according to Fig. 4c. Thus, these nanofibers can be used as thermos-sensitive probes. Similar responses to temperature were demonstrated with thin-films as shown in Fig. S4 (ESI†).
AIEgens are promising materials for fluorescent chemosensors due to their unique fluorescence mechanism; when they are knitted onto the polymer chains via chemical bonds, their response and sensitivity to the analytes might be amplified with the surrounding polymer chains. With this virtue, the prepared FNFs were probed for possible use as fluorescent chemosensors. At first, FNF-2 was immersed into saturated DCM vapor during the fluorescence testing. Its PL intensity decreased quickly, and it fell by nearly half in the first 5 min (Fig. 4d). After 1 h, the fluorescence of the FNFs was very faint. Although TPE AIEgens are knitted onto the cross-linked polymer chains and their intramolecular motions are suppressed to some degree, DCM is a good solvent for both the polymer chains and knitted AIEgens. The continuous penetration VOC facilitated the swelling of cross-linked nanofibers, which gradually weakened its bonding of TPE to Epoxy, thus the observed quenching. The recovered intramolecular motions of TPE therefore massively consume its energy of the excited state. Compared with similar polymers, but when applied as a thin coating, the sensitivity of the nanofiber membrane to VOC was better due to their morphology presenting a relatively high surface area (see Fig. S5 for comparison, ESI†).
The FNFs were also employed to detect the effect of nitrobenzene towards them. Nitrobenzene is an extremely toxic chemical (Threshold Limit value 5 mg m−3) which can even be easily absorbed through human skin. To avoid the influence of solvent, nitrobenzene was first dissolved into petroleum ether (PE) as the fluorescence property of the nanofiber membrane remained stable in the solution (Fig. S6, ESI†). At first, nanofibers were placed in PE with different concentration of nitrobenzene for 20 min. From Fig. 4e, significant decline in PL intensity was observed at 0.1 wt%. Then, the concentration of nitrobenzene was set at 0.1 wt%, and the PL spectrum varied with the immersion time. According to Fig. 4f, the fluorescence of nanofibers significantly weakened at 30 s. As time went by, the PL intensity declined further. Microporous morphology of the nanofibers might help trap the analyte molecules and thus facilitate the PL quenching process. In addition, the fluorescence annihilation resulted in a static quenching mechanism, in which the electron-rich polymer strands bind to the electron-deficient analytes and hence tend to be in a non-emissive or dark state, hence the observed changes in PL. These response results altogether signify that the fluorescent nanofibers can be employed as optical and real-time sensors in different environments.
Conclusions
In this paper, a series of thermosetting polymer based nanofibers with fluorescence properties were obtained by electrospinning. As it has been demonstrated that when the inter/intra molecular motion (i.e., rotation, vibration, bending, etc.) of the phenyl ring of the TPE is restricted, strong fluorescence emission occurs. Based on this feature, we systematically studied the electrospinning process, and curing behavior of thermosetting polymer-based electrospun nanofibers. According to RIR, the curing process in thermoset nanofibers occurs distinctively in 3 major stages. And in the three stages, first the evaporation and crosslinking occurs between 0 min and 35 min, this is followed by an intermediate crosslinking stage, occurring between 35 min and 75 min, and finally the relaxation of polymer chains for the last 35 minutes of the overall curing process. In addition, according to the variation tendency of PL spectra, the response of the FNFs to ambient temperature, and chemical vapours was studied. This study provides new ideas for studying the electrospinning process and opens up a new research avenue in the development of smart nanofibers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the National Natural Science Foundation of China (Grant No. 51673088).References
- J. Xue, T. Wu, Y. Dai and Y. Xia, Chem. Rev., 2019, 119(8), 5298–5415 CrossRef CAS PubMed.
- H. Hou, J. J. Ge, J. Zeng, Q. Li, D. H. Reneker, A. Greiner and S. Z. D. Cheng, Chem. Mater., 2005, 17, 967–973 CrossRef CAS.
- K. H. Lee, H. Y. Kim, H. J. Bang, Y. H. Jung and S. G. Lee, Polymer, 2003, 44, 4029–4034 CrossRef CAS.
- M. Tebyetekerwa, Z. Xu, W. Li, X. Wang, I. Marriam, S. Peng, S. Ramkrishna, S. Yang and M. Zhu, ACS Appl. Energy Mater., 2017, 1, 377–386 CrossRef.
- A. Welle, M. Kröger, M. Döring, K. Niederer, E. Pindel and I. S. Chronakis, Biomaterials, 2007, 28, 2211–2219 CrossRef CAS PubMed.
- M. Tebyetekerwa, X. Wang, I. Marriam, P. Dan, S. Yang and M. Zhu, Mater. Lett., 2017, 209, 400–403 CrossRef CAS.
- Q. Yang, Z. Li, Y. Hong, Y. Zhao, S. Qiu, C. Wang and Y. Wei, J. Polym. Sci., Part B: Polym. Phys., 2010, 42, 3721–3726 CrossRef.
- W. E. Teo and S. Ramakrishna, Nanotechnology, 2006, 17, R89 CrossRef CAS PubMed.
- L. S. Penn and T. T. Chiao, in Handbook of Composites, ed. G. Lubin, Springer, US, Boston, MA, 1982, pp. 57–88, DOI:10.1007/978-1-4615-7139-1_5.
- C. Ma, G. Xie, X. Zhang, L. Yang, L. Yang, H. Liu, W. Ke and Y. Wei, J. Mater. Chem. B, 2016, 4, 8009–8015 RSC.
- Z. Hu, D. Zhang, F. Lu, W. Yuan and Y. Huang, Macromolecules, 2018, 51(14), 5294–5303 CrossRef CAS.
- I. Marriam, F. Xu, M. Tebyetekerwa, G. Yang, L. Wei, X. Liu and Y. Qiu, Composites, Part A, 2018, 110, 1–10 CrossRef CAS.
- K. Friedemann, T. Corrales, M. Kappl, K. Landfester and D. Crespy, Small, 2012, 8, 144–153 CrossRef CAS PubMed.
- C. L. Zhang, K. P. Lv, N. Y. Hu, L. Yu, X. F. Ren, S. L. Liu and S. H. Yu, Small, 2012, 8, 2935 CrossRef CAS.
- S. Wang, Y. Zhang, H. Wang, G. Yin and Z. Dong, Biomacromolecules, 2009, 10, 2240–2244 CrossRef CAS PubMed.
- X. He, L. Tan, X. Wu, C. Yan, C. Dong, X. Meng and F. Tang, J. Mater. Chem., 2012, 22, 18471 RSC.
- B. Liu, H. Nie, X. Zhou, S. Hu, D. Luo, D. Gao, J. Zou, M. Xu, L. Wang and Z. Zhao, Adv. Funct. Mater., 2016, 26, 776–783 CrossRef CAS.
- R. Hu, N. L. C. Leung and B. Z. Tang, Chem. Soc. Rev., 2014, 43, 4494–4562 RSC.
- W. Li, J. Wang, Y. Xie, M. Tebyetekerwa, Z. Qiu, J. Tang, S. Yang, M. Zhu and Z. Xu, Prog. Org. Coat., 2018, 120, 1–9 CrossRef CAS.
- J. Luo, Z. L. Xie, J. Lam, L. Cheng, H. Chen, C. Qiu, H. Kwok, X. Zhan, Y. Q. Liu, D. B. Zhu and B. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- A. Qin, J. W. Y. Lam and B. Z. Tang, Prog. Polym. Sci., 2012, 37, 182–209 CrossRef CAS.
- Y. Yuan, J. Li, L. He, Y. Liu and H. Zhang, J. Mater. Chem. C, 2018, 6, 7119–7127 RSC.
- W. Li, Z. Qiu, M. Tebyetekerwa, J. Zhang, Y. Wang, T. Gao, J. Wang, Y. Ding and Y. Xie, Prog. Org. Coat., 2019, 127, 8–15 CrossRef CAS.
- W. Yao, M. Tebyetekerwa, X. Bian, W. Li, S. Yang, M. Zhu, R. Hu, Z. Wang, A. Qin and B. Z. Tang, J. Mater. Chem. C, 2018, 6, 12849–12857 RSC.
- D. S. Gill, A. N. Sharma and H. Schneider, J. Chem. Soc., Faraday Trans. 1, 1982, 78, 465–474 RSC.
- Y. Hong, J. W. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- W. Yao, M. Tebyetekerwa, X. Bian, W. Li, S. Yang, M. Zhu, R. Hu, Z. Wang, A. Qin and B. Z. Tang, J. Mater. Chem. C, 2018, 6, 7003–7011 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qm00342h |
| This journal is © the Partner Organisations 2019 |