
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The pivotal role of MBD4–ATP7B in the human Cu(I) excretion path as revealed by EPR experiments and all-atom simulations†
Zena
Qasem‡
a,
Matic
Pavlin‡
b,
Ida
Ritacco
b,
Lada
Gevorkyan-Airapetov
a,
Alessandra
Magistrato
 *b and
Sharon
Ruthstein
*a
*b and
Sharon
Ruthstein
*a
aChemistry Department, Faculty of Exact Sciences, Bar-Ilan University, 529002, Israel. E-mail: Sharon.ruthstein@biu.ac.il
bCNR-IOM at SISSA, via Bonomea 265, 34135, Trieste, Italy. E-mail: alessandra.magistrato@sissa.it
First published on 12th June 2019
Abstract
Copper's essentiality and toxicity require a meticulous mechanism for its acquisition, cellular distribution and excretion, which remains hitherto elusive. Herein, we jointly employed electron paramagnetic resonance spectroscopy and all-atom simulations to resolve the copper trafficking mechanism in humans considering the route travelled by Cu(I) from the metallochaperone Atox1 to the metal binding domains 3 and 4 of ATP7B. Our study shows that Cu(I) in the final part of its extraction pathway is most likely mediated by binding of Atox1 monomer to MBD4 of ATP7B. This interaction takes place through weak metal-stabilized protein–protein interactions.
Significance to metallomicsCombined use of EPR measurements and MD simulations succeeded in showing that the Cu(I) extrusion path is most likely mediated by Atox1 binding to MBD4 of ATP7B. Efficient Cu(I) trafficking must rely on a subtle balance of transient interactions and appropriate conformational selection of the metallochaperone and its partner ATPase, which only MDB4 satisfies. In addition, this research stresses the significance of monitoring the structural flexibility of a biological system in solution, and of integrating these data with atomic-level information, in order to disclose the exact role of a protein in biological pathways. |
Introduction
Copper is an essential, yet toxic, metal ion for cells. Hence, its free amount must be strictly limited. In human cells, one of the pathways tightly controlling the in-cell copper (Cu(I)) concentration involves three proteins: (i) the Ctr1 Cu(I) transporter, which reduces dietary Cu(II) (accumulated from the blood-stream) to Cu(I) and transfers it to its cytoplasmic domain,1–3 (ii) the metallochaperone Atox1, which delivers Cu(I) ion from Ctr1 to ATP7A/B in the Golgi,4,5 and (iii) ATP7A/B, which either transports Cu(I) to the secretory pathway, where these ions are incorporated into copper-dependent enzymes, or exports it out of the cell.6,7 Hence, these transporters, by establishing transient protein–protein interactions, evolved to tightly regulate Cu(I)'s cellular needs while minimizing its toxic effects. Pathological mutations in ATP7A and ATP7B proteins alter normal Cu(I) distribution, leading to severe human pathologies (Menkes and Wilson's disease, neurodegeneration, and cancer).8 At the end of the transport route, Atox1 interacts with the N-terminal domain of ATP7A/B, which contains six metal-binding domains (MBDs) connected by linkers. Each MBD (∼7 kDa) has a ferredoxin-like fold with a compact βαββαβ structure and a conserved metal-binding motif MXCXXC (X stands for any amino acid), located in the solvent-exposed β1–α1 loop (also called a Cu loop), which binds Cu(I) ions. Atox1 is a small soluble cytosolic Cu(I) receptor, structured in the same βαββαβ motif of MBDs from ATP7A/B.9,10 The structural similarity between the chaperone and the target proteins is a hallmark of Cu(I) transporters.In the last decade, distinct biophysical tools have been used to characterize the interaction between Atox1 and the six MBDs of ATP7A/B, sometimes leading to contradictory conclusions.11–13 Nuclear magnetic resonance (NMR) studies disclosed that the six MBDs can be differentiated into two units, comprising MBD1–3 and MBD5–6, whereas MBD4 serves as a linker between them.11,14,15 Cu(I) binding to MBD1–4 stimulates its transport by ATP7A and presumably facilitates Cu(I) trafficking.16 The structures of MBD3 and MBD4, and their relative spatial arrangement were also solved by NMR.17,18 NMR studies and classical molecular dynamics (MD) simulations indicated that Atox1 can bind to MBD4, but not to MBD3.4,19 Single-molecule FRET (smFRET) experiments succeeded in reporting a dynamic situation, where Atox1 can coordinate both MBD3, MBD4, and the two domains simultaneously, MBD3–4.20 In these experiments Atox1 interacted with MBDs as a monomer.5,16 Notably, different structural models of the Atox1–MBD heterodimer were suggested (Fig. 1): (i) Atox1 interacts “face-to-face” with MBD with the two α helices of each protein pointing to each other. Here, one Atox1 monomer interacts with one MBD at a time. (ii) A “back-to-face” model, where Atox1 is sandwiched between two MBDs. Namely, Atox1 points to one MBD with its α1 helix, while interacting with the other via its β2 and β3 sheets.17,20 In this scenario, the identity of the MBD and its interaction mode with Atox1 remain elusive.
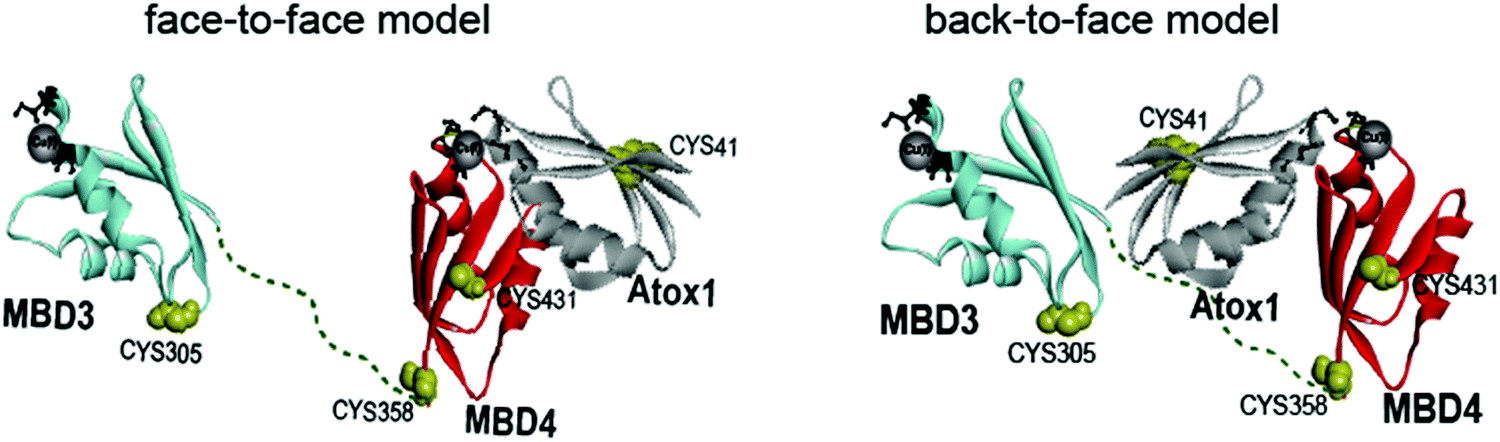 | ||
| Fig. 1 Schematic representation of the face-to-face and back-to-face models between Atox1 and MBD3–4.20 | ||
In the last few years we have investigated the Cu(I) trafficking mechanism by electron paramagnetic resonance (EPR) spectroscopy. This technique complements NMR and smFRET studies, owing to its sensitivity to the proteins’ conformational flexibility in solution. For example, EPR experiments recently showed that Atox1 can accommodate distinct conformations, depending on the interacting partner protein,21–23 and that it interacts as a homodimer with the Ctr1 intracellular domains. Conversely, all-atom simulations have the potential to rationalize at the atomic-level resolution the spectroscopic findings. Aiming at clarifying the way and the identity of the MBD of ATP7B involved in the Cu(I) excretion path, we concomitantly employed EPR spectroscopy and all-atom MD simulations.
Materials and methods
Cloning, expression and purification of ATP7B3–4 metal binding domains
The ATP7B3–4 metal binding domains’ (MBD3–4) gene was first amplified by PCR using primers containing specific ATP7B MBD3–4 sequences and flanking regions corresponding to the expression vector sequences pTYB12.The forward primer of ATP7B MBD3–4:
5′-GTTGTACAGAATGCTGGTCATATGAGACCTTTATCTTCTGCTAAC-3′
Reverse primer of ATP7B MBD3–4:
5′-GTCACCCGGGCTCGAGGAATTTCAGTGGTTTCCAAGAGGGTTAGT-3′
This amplicon was cloned into the pTYB12 vector by restriction-free cloning.24
pTYB12 is a cloning and expression vector that allows the overexpression of the ATP7B MBD3–4 as a fusion to a self-cleavable intein tag. The self-cleavage activity of the intein allows the release of ATP7B MBD3–4 from the chitin-bound intein tag. The clone was expressed in E. coli strain Origami 2. The starter from stock glycerol was grown at 37 °C to an optical density of 0.5–0.6 (OD600) using terrific broth (TB) medium supplemented with ampicillin and tetracycline as selection factors, then induced with 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) at 18 °C overnight. In the next step, bacteria were harvested by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 min. Then, the pellet was resuspended in lysis buffer (25 mM Na2HPO4, 150 mM NaCl, 20 mM PMSF, 1% Triton, and pH 8.8) and sonicated (10 min of pulse 30 s of 40% amplitude). Finally, the lysate was centrifuged at 14500 rpm for 30 min, and the supernatant was kept.
000 rpm for 30 min. Then, the pellet was resuspended in lysis buffer (25 mM Na2HPO4, 150 mM NaCl, 20 mM PMSF, 1% Triton, and pH 8.8) and sonicated (10 min of pulse 30 s of 40% amplitude). Finally, the lysate was centrifuged at 14500 rpm for 30 min, and the supernatant was kept.
Following the expression of the fusion protein ATP7B MBD3–4-intein, there is a crucial need to purify the native ATP7B MBD3–4. Therefore, the lysate was loaded on the chitin bead column allowing the ATP7B MBD3–4-intein to bind to the resin via its chitin-binding tag. Then, the resin was washed with 50-column volumes of lysis buffer. Next, 5 ml dithiothreitol (50 mM DTT) was added and incubated for 48 h at 4 °C to perform a self-cleavage of the intein. As the final step, elution fractions were collected from the column using the chitin column buffer (pH = 8.8) and checked by 14% tricine SDS-PAGE.
Spin labeling
The spin-labeling process (Scheme S1 of the ESI†) was performed in the presence of Cu(I) ions in order to prevent spin-labeling of cysteine residues that are involved in Cu(I) binding. Before labeling, 10 mM DTT was added to the protein solution and mixed overnight at 4 °C. DTT was dialyzed out using 1 kDa dialysis cassettes (Pierce). Next, 0.25 mg of S-(2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methanesulfonothioate (MTSSL, TRC) dissolved in 15 μl dimethyl sulfoxide (DMSO, Bio lab) was added to 0.75 ml of 0.01 mM protein solution (20-fold molar excess of MTSSL). The protein solution was then vortexed overnight at 4 °C. The free spin label and Cu(I) ions were removed by several dialysis cycles over 4 days. The mass of the spin-labeled protein was confirmed by a mass spectrometer, and the concentration was determined by a BCA assay. The final spin-labeled protein concentration was between 0.01 and 0.03 mM.Atox1 expression, purification and spin labeling is similar to the expression of ATP7B and was described in a series of publications.21–23
Cu(I) addition
After spin-labeling and all the dialysis steps were carried out for removal of free spin labels from the solution, no Cu(I) ions were found in the apo-protein solution. This was verified by adding 0.1 mM KCN to the solution and acquiring the CW-EPR spectra. When no Cu(I) ions are in the solution, the spectra of the apo-protein in the presence and absence of KCN is similar and different from those of the holo-protein.For EPR measurements: Cu(I) (tetrakis(acetonitrile) copper(I) hexafluorophosphate) was added to the protein solution under nitrogen gas to preserve anaerobic conditions. No Cu(II) EPR signal was observed at any time.
Q-band DEER measurements
The DEER experiment π/2(νobs) − τ1 − π(νobs) − t′ − π(νpump) − (τ1 + τ2 − t′) − π(νobs) − τ2 − echo was carried out at 50 ± 0.2 K on a Q-band Elexsys E580 spectrometer (equipped with a 2 mm probe head). A two-step phase cycle was employed on the first pulse. The echo was measured as a function of t′, whereas τ2 was kept constant to eliminate relaxation effects. The observer pulse was set at 60 MHz higher than the pump pulse. The durations of the observer π/2 and π pulses were 40 ns each. The duration of the π pump pulse was 40 ns as well, and the dwell time was 20 ns. The power of the 40 ns π pulse was 20.0 mW. The value τ1 was set to 200 ns and τ2 to 1200 ns. Observer frequency: 33.83 GHz; pump frequency: 33.89 GHz; magnetic field: 12030 G. The samples were measured in 1.6 mm capillary quartz tubes (Wilmad-LabGlass). The data were analysed using the DeerAnalysis 2016 program, with Tikhonov regularization.25 We optimized the regularization parameter in the L curve by examining the fit of the time domain signal. The modulation depth for the two spin systems was about 6%.
Model building and molecular dynamics simulations
First, we prepared MBD3 and MBD4, both in the apo and holo states. The models were built based on their NMR structure in the apo form (PDB ID 2ROP).18 To create the Cu(I)-bound systems, we superimposed both MBDs to one monomer of the Atox1 homodimer X-ray structure, which contains a Cu(I) bound between the two monomers (PDB ID 1FEE).15 Since MBD3–4 are structurally very similar to Atox1 and share the same MXCXXC motif, we also adopted the same position of Cu(I), observed in Atox1, for MBD3/4.Next, we built 6 additional systems, in which Cu(I)-bound Atox1 formed dimers with apo MBD3 and 4, considering their Cys268/370 and Cys271/373 residues either in the protonated or deprotonated form. Namely, the cysteine (Cys) residues binding Cu(I) in Atox1 were considered in a deprotonated form, whereas the ones on MBD3/4 were considered either in protonated or deprotonated forms. Conversely, in the simulations of the apo systems, all Cys residues were protonated. Protonation states of other ionizable residues were controlled with propKa.26
All models were relaxed by performing classical MD (cMD) simulations using an Amber parm14SB-ILDN force field for treatment of the protein.27 For the Cu(I) and Cys residues coordinating it, we used parameters from Op’t Holt and Merz.28 Monomeric systems were neutralized by addition of 8, 7, 1 and 0 Na+ ions for holo MBD4, apo MBD4, holo MBD3 and apo MBD3, respectively. The systems containing Atox1–MBD3/4 complexes were neutralized by the addition of 8, 1, 10, 3, 7 and 0 Na+ ions for holo Atox1–MBD4 with protonated C370 and C373, holo Atox1–MBD3 with protonated Cys268 and C271, apo Atox1–MBD4, apo Atox1–MBD3, holo Atox1–MBD4 with deprotonated C370 and C373 and holo Atox1–MBD3 with deprotonated C268 and C271. All systems were solvated in explicit water using the TIP3P model.29 The resulting models counted ∼25000 and ∼50000 to 58000 atoms for the two monomers and the two heterodimers, respectively.
Berendsen barostat and Langevin thermostats were used for controlling pressure and temperature.30,31 Particle mesh Ewald has been used to treat long-range electrostatics, and the time step in simulations was 2 fs. We used the Amber18 code32 cuda program. After a careful equilibration, based on the geometry optimization and gentle heating in the NVT ensemble, 200 ns of cMD simulations were performed.
A cluster analysis was performed with the cpptraj tool of Ambertools18 on the cMD trajectory. The most representative frames extracted from the MD trajectory were used as starting structures for QM/MM (i.e., quantum mechanics (Born Oppenheimer)/molecular mechanics) MD simulations, using the CP2K code.33,34 This method treats part of the system at the QM level, usually the metal binding portion of the system, whereas the remaining part of the protein, the solvent and the counter ions are treated at the MM level. In these simulations we considered the QM region Cu(I) and side chains of residues coordinating it (C268 and C271 in MBD3, C370 and C373 in MBD4, and C12 and C15 and K60 in Atox1). The QM region here is treated at the density functional theory (DFT) level with the BLYP exchange–correlation functional35,36 by employing a dual Gaussian-type/plane waves basis set (GPW).37 In particular, we used a double-ζ (MOLOPT) basis set38 along with an auxiliary PW basis set with a density cutoff of 400 Ry and Goedecker–Teter–Hutter (GTH) pseudopotentials.39,40 This level of theory has often been used in successful QM/MM MD simulations of biomolecules.33,41–45 The dangling bonds between the QM and MM regions were saturated by using capping hydrogen atoms. All QM/MM MD simulations were performed by using an integration time step of 0.5 fs in the NVT ensemble. All systems were initially optimized, heated to 300 K in 2 ps, and equilibrated at 300 K without constraints for 5 ps by using a Nosé–Hoover thermostat.
During the QM/MM MD simulations, we observed that the Cu(I) coordination changed from tetrahedral (as in the crystal structure) to linear bi-coordination. Therefore, we changed the force field parameters for classical MD (by changing the reference bond lengths and angle values of the Cu(I) coordinating residues, whereas the spring constants were kept at the same values as in ref. 28) to match the QM/MM MD geometry and we performed an additional 200 ns-long cMD simulation starting from a representative snapshot taken from the QM/MM MD simulations for all systems.
Cluster analysis using a hierarchical agglomerate approach was performed on all systems as described before (on frames from 20 to 200 ns from simulations for all systems) and additional RMSD and RMSF analyses (again on frames ranging from 20 to 200 ns for all systems) were performed using Ambertools18. The cutoff for clustering was 2.5 Å for all Atox1–MBD3/4 complexes, whereas for monomeric MBD3 and MBD4 it was 2.0 Å and 1.5 Å, respectively.32 The electrostatic potential surface of each system was calculated based on the corresponding structure of the highest populated cluster using the PDB2PQR webserver.46 Figures were done with the Chimera1.12 software.47
Results
Pulsed EPR experiments (such as double electron–electron resonance (DEER)) can measure distances between paramagnetic probes in the nm range.48,49 The combination of pulsed EPR with site-directed spin labelling (SDSL), where an electron spin is introduced into diamagnetic proteins, is widely used in biophysical research.50 The spin label, commonly attached to cysteine (Cys) residues, is nitroxide, and the nitroxide-labelled Cys residue is hereafter termed R1 (Scheme S1, ESI†). Among the seven Cys residues present in MBDs3–4, four of them form the Cu(I) binding sites, and therefore could not be spin labelled in the presence of Cu(I) ions. Hence, spin labelling was limited to C305 (located in the loop between β3 and α2 of MBD3), and C358 and C431 (located in β1 and β4 of MBD4, respectively). MBD3–4 and Atox1 proteins were expressed, purified and spin-labeled. Fig. 2 presents the DEER distance distribution functions for the WT–MBD3–4, MBD3–4_C431A mutant, and MBD3–4_C305A mutant (see Fig. S1 for DEER raw data, ESI†). The latter mutations were introduced to monitor the distance distributions relative only to the two remaining cysteine residues, which are spin-labelled. Hence, C305A and C431A are employed to detect the C431R1–C358R1 and C305R1–C358R1 distance distributions in MBD4 and MBD3–4, respectively. Owing to the low concentration of the protein, the DEER time scale was acquired up to 1.2 ms; thus we could not detect the distance between C431R1 and C305R1, which is around 5.0 nm. Superposition of the distance distribution of C431A with that of C305A agrees well with that of the WT protein (Fig. S1, ESI†), confirming that the structural and dynamical features of MBD3–4 are insensitive to the C431R1–C305R1 distance contribution. The DEER data indicate that both the C305R1–C358R1 and C431R1–C358R1 mean distances slightly increase upon Cu(I) binding. Additionally, metal binding widens the distribution function of the C305A mutant (i.e., the C431R1–C358R1 distance distribution) with respect to the C431A mutant (i.e., the C305R1–C358R1 distance distribution). Thus, the spin labels at C431R1 are more dynamic, underlying an enhanced motion when Cu(I) binds to MBD4. Consistently, the flexibility of β4 also increases upon Cu(I) binding to Atox1.21 Continuous-wave (CW) EPR experiments support pulsed EPR data, and detect large amplitude motions for the MBD3–4_C305A mutant upon Cu(I) binding (Table S1, Fig. S2 and S3, ESI†). Hence, metal binding induces internal protein rearrangements that also affect protein flexibility.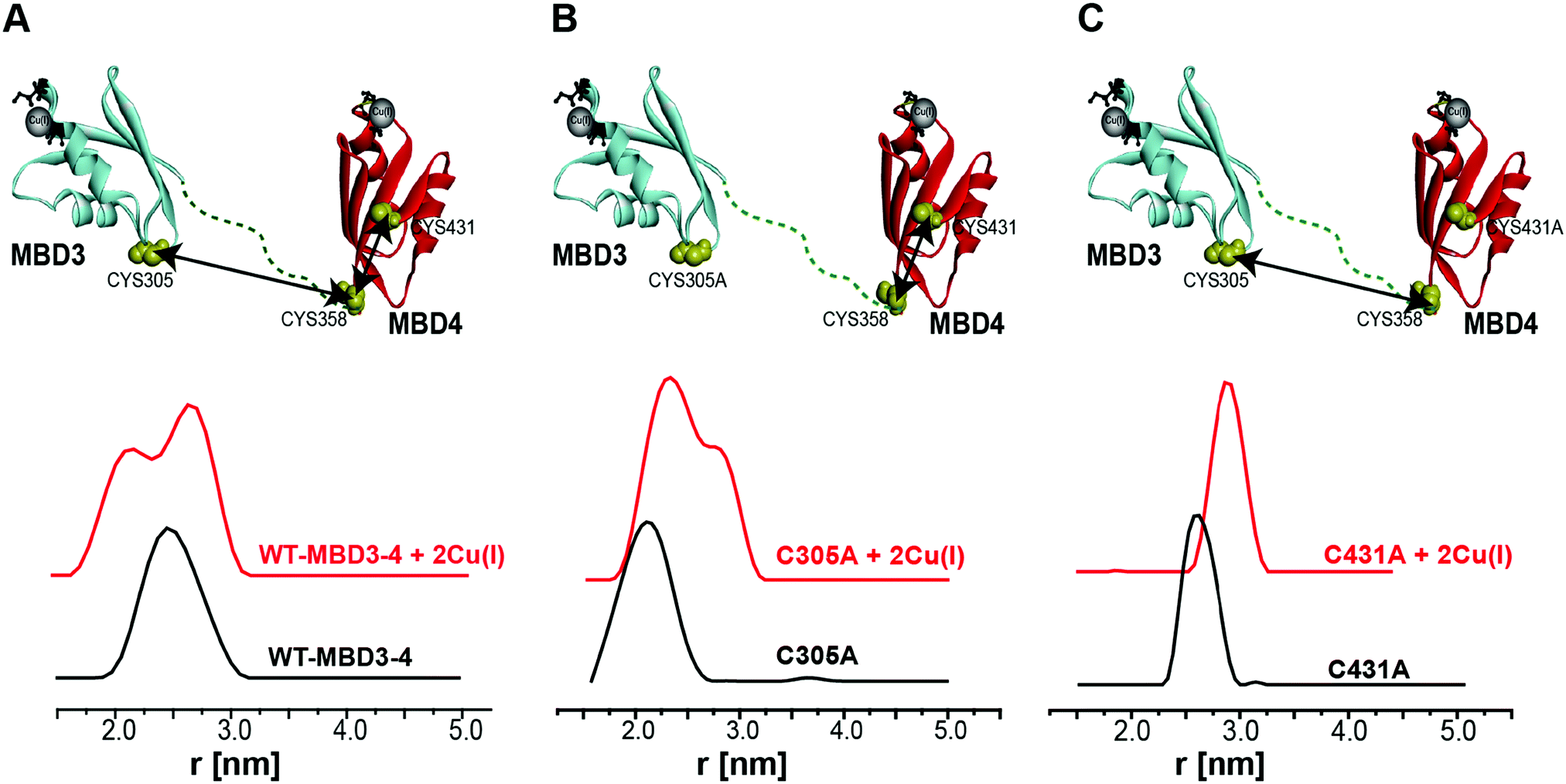 | ||
| Fig. 2 (A) MBD3–4 NMR structure (PDB 2ROP) showing the three cysteine (Cys) residues accessible for spin labelling (represented in yellow), and the Cu(I) sites in grey. C431 is missing from the PDB structure and was modelled for representation purposes. The black arrows mark the measured distances by DEER. The DEER distance distribution functions for WT–MBD3–4 (spin-labelled at three different positions) (C431R1, C358R1, and C305R1) in the absence (black line) and presence of Cu(I) (red line). (B) DEER distance distribution functions for MBD3–4_C305A mutant (corresponding to the C358R1–C431R1 distance), and (C) MBD3–4_C431A mutants (corresponding to the C305R1–C358R1 distance) in the absence (black line) and presence of Cu(I) (red line). | ||
By using DEER measurements, we also explored the transient interactions occurring between Atox1 and MBD3–4. Atox1 is a dimer in solution, and when spin-labelled at C41R1, a bimodal distance distribution function23 is observed (Fig. 3). Previously, by using various distance distribution constraints, we computed two distinct conformational states of the Atox1 homodimer, which we called closed and open conformations.21 The distribution around 4.2 nm corresponds to a closed conformation, which agrees with that of PDB ID 3IWX,51 while the distribution around 2.3 nm corresponds to an open conformation not yet trapped by either NMR or X-ray crystallography.21 Interestingly, when adding spin-labelled Atox1 to MBD3–4 solution, the distribution around 4.2 nm disappears, suggesting that in the presence of MBD3–4, Atox1 no longer exists as a homodimer. Cross-linking experiments also support this conclusion (Fig. S4, ESI†). Furthermore, the additional peak appearing at 3.4 ± 0.3 nm (a grey area in Fig. 3) can be assigned to the formation of the MBD3–4–Atox1 complex (corresponding to the distance between C41R1 of Atox1 and C431R1/C358R1/C305R1 of MBD3–4), irrespective of Cu(I) addition. Moreover, in the presence of Cu(I), the distance distributions are narrower, implying that the MBD3–4–Cu(I)–Atox1 complex is less dynamic, and, possibly, more tightly bound.17 Hence, in contrast to other experimental findings, our results indicate that a complex between MBD3–4 and Atox1 can also form in the absence of Cu(I), even though the metal contributes to rigidification and, possibly, stabilization of the adduct. Cross-linking experiments (Fig. S4, ESI†) confirmed that Atox1 monomer binds to MBD3–4 by interacting with only one of the two domains. To identify the MBD implicated in Atox1 binding, we performed DEER experiments on MBD3–4_C305A in which only MBD4 is spin-labelled. As a result, if Atox1 interacts exclusively with MBD4, no change in the distance distribution function for the spectra of WT and C305A–MBD3–4 should occur. Indeed, similar broad distance distribution function (between 1.5 and 3.5 nm) detected in the two cases confirms that Atox1 probably interacts only with MBD4 (Fig. 3). In the holo state, the distributions are narrower, and therefore it is more accurate to compare between the two states. There, the width of the bimodal distribution of the holo C305A in the presence of Atox1 is similar to the holo MBD3–4 in the presence of Atox1 (Fig. 3 and Fig. S1, ESI†), confirming that no interaction between C305R1 and C41R1 exists. It is important to note, that if Atox1 was sensitive to MBD3, a distance between C41R1 of Atox1 and C305R1 should be between 2.5 nm to 3.5 nm, and therefore should be detectable. Moreover, for holo C305A, the population of the distribution around 3.4 nm (corresponds to MBD4–Atox1 interaction) is almost equal to the distribution around 2.3 nm that corresponds to the distance measured in MBD4 (C358R1–C431R1) (Fig. 3 and Fig. S1, ESI†). For WT–MBD3–4, two distances are being measured (C305R1–C358R1, C358R1–C431R1) while for MBD34–C305A, only one distance is being measured between C431R1–C358R1. The fact that in WT–MBD3–4 the ratio between 2.3 nm to 3.4 nm distributions is about two, and for MBD34–C305A is equal, suggests that Atox1 interacts with MBD4 in a way that only one distance can be detected between C41R1 of Atox1 to MBD4 (C358R1/C431R1). Additionally, the lack of MBD3 sensitivity to the interaction with Atox1 may indicate that the latter interacts with MBD4 in a face-to-face manner rather than in the face-to-back mode (the sandwich model) evoked by smFRET, since only in this scenario no interaction between Atox1 and MBD3 occur.20
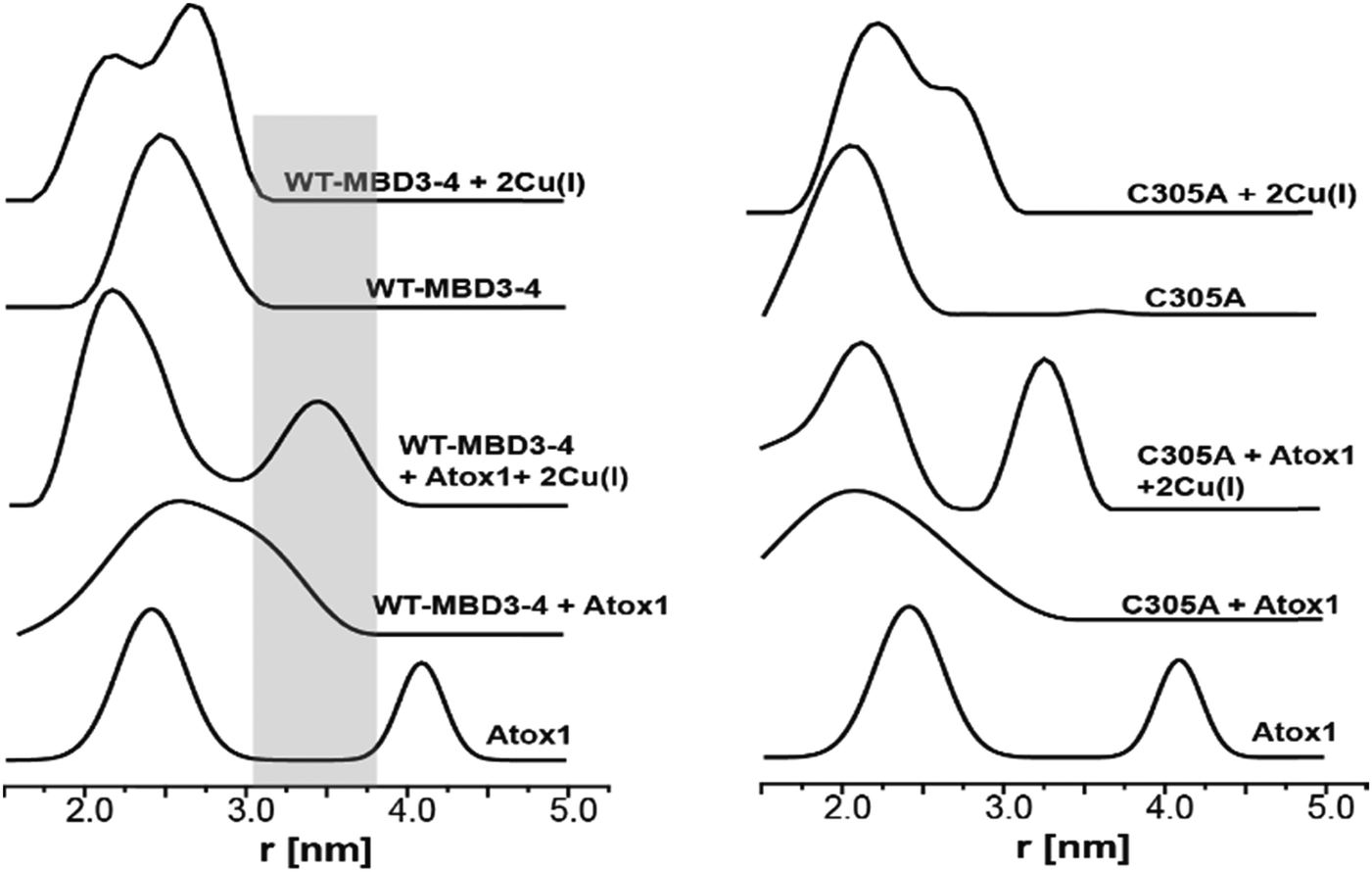 | ||
| Fig. 3 DEER distance distribution functions for WT–MBD3–4 and MBD3–4_C305A as a function of spin-labelled Atox1 and Cu(I). The grey area marks the distance distribution raised owing to the interaction between Atox1 and MBD4. | ||
Building on these experimental findings, we carried out all-atom simulations to assess how Cu(I) binding affects the structural and dynamical properties of MBD3/4 monomers and those of the Atox1–MBD3/4 heterodimers. To this end, we performed 200 ns-long cMD simulations in explicit solvent.
All MBD monomers reached structural stability within a few tens of ns, with MBD4 deviating the least from the initial structure and displaying the lowest flexibility in both the holo and the apo forms, as compared with MBD3 (Fig. S5 and Table S2, ESI†). In contrast, Cu(I) binding partially rigidifies the structure of MBD3 (Fig. S5, ESI†), while destabilizing important structural motifs: (i) the Cu(I) loop, containing the C268 (Cu(I)-binding residue), frequently loses its initial conformation (Fig. 4, Fig. S6 and Table S2, ESI†); (ii) helix α1, containing the second Cu(I)-binding residue, C271, starts unfolding (Fig. 4 and Fig. S6, ESI†); (iii) M266, a conserved residue of Cu(I) loop, frequently becomes solvent exposed in MBD3. In contrast, M368 remains packed inside the hydrophobic pocket in MBD4, contributing to the protein's structural stability (Fig. 4, Fig. S6A, B and Table S2, ESI†).
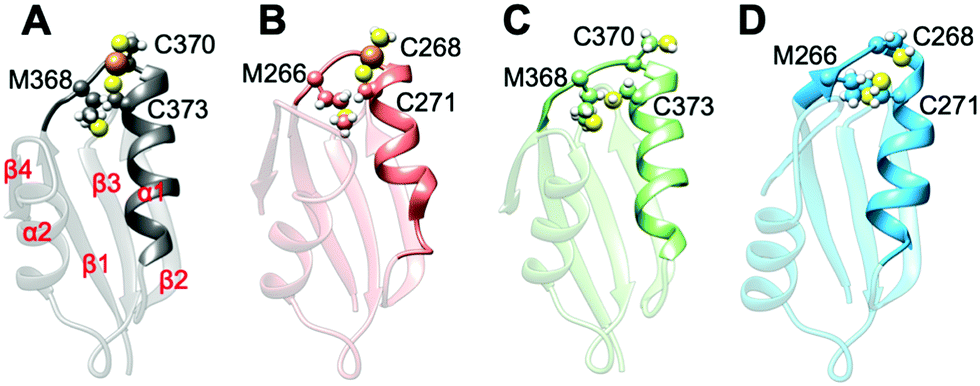 | ||
| Fig. 4 Structures of the most populated clusters of MBD3/4. (A) Holo MBD4. Secondary structure motifs α1, α2 and β1–4 are labelled. (B) Holo MBD3. (C) Apo MBD4. (D) Apo MBD3. Cu(I) is shown as an orange sphere; sulphur and hydrogen atoms are in yellow and white, respectively. Carbon atoms are shown in the colour scheme of the protein. The Cu loop and α1 are shown in opaque, whereas the rest of the protein is shown as transparent new cartoon representation. | ||
In both MBD3 and 4, β4 changes its conformation upon Cu(I) binding, consistent with the DEER data presented in Fig. 2. The DEER data showed a broadening in the distance distribution function, which was detected for C305A, but not for C431A, suggesting that β4 becomes more flexible upon Cu(I) coordination. Thus, MBD4 appears to retain a pre-organized tertiary structure suitable for receiving Cu(I) along its excretion path, whereas Cu(I) binding to MBD3 alters important structural motifs (Fig. 4B, D, and Fig. S6A, B, ESI†), possibly impacting on an efficient Cu(I) transport.
Next, classical MD simulations were carried out on the apo and holo Atox1 bound to apo MBD3 or 4 to inspect the structural stability and dynamical properties of the resulting complexes.
In the apo Atox1–MBD3/4 adducts, C12/15@Atox1, C268/271@MBD3, and C370/373@MBD4, which form the Cu(I) binding sites, respectively, were protonated. The apo complex involving MBD4 was stable during our simulations thanks to hydrogen (H)-bond engaging K60@Atox1 to C373 (Fig. 5E, Fig. S7, and Table S3, ESI†). Surprisingly, also the apo Atox1–MBD3 complex was stable during the simulation, even if its structure significantly differs from that of the apo Atox1–MBD4. Here, K60@Atox1 establishes only hydrophobic interactions with C268@MBD3 (Fig. 5F, Fig. S7 and Table S3, ESI†). Thus, in contrast to experiments, cMD simulations predict that even the apo adduct involving MBD3 is stable.
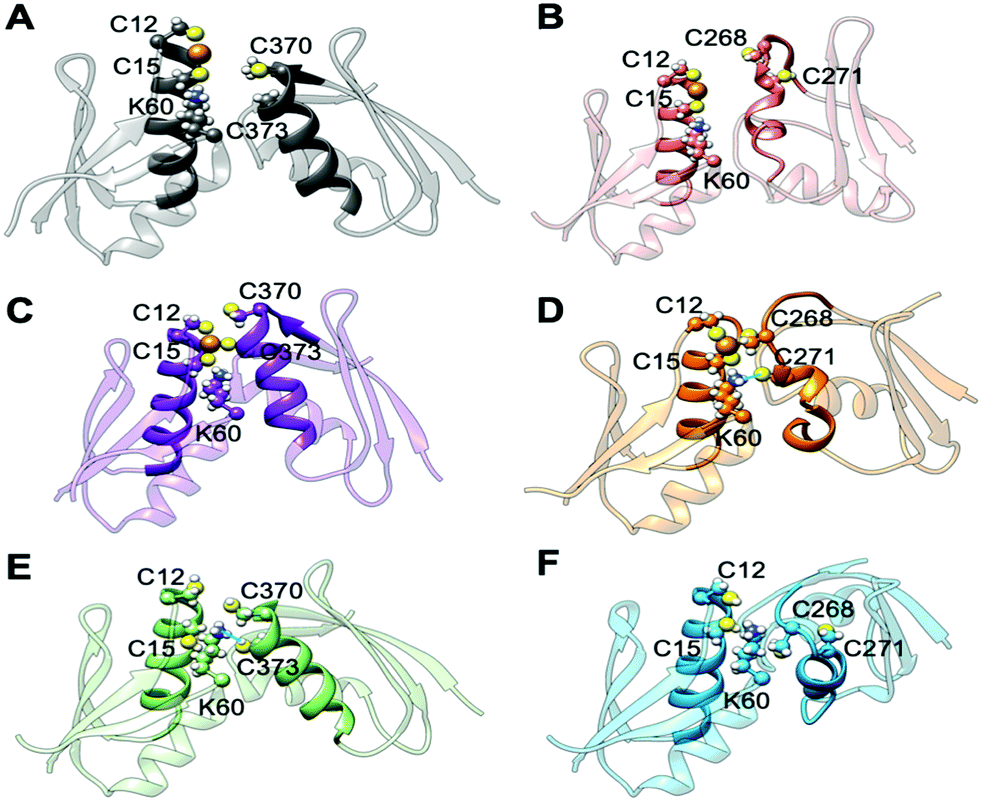 | ||
| Fig. 5 Representative structures of holo and apo Atox1 in complex with MBD3/4. (A) Holo Atox1–MBD4. (B) Holo Atox1–MBD3. (C) Holo Atox1–MBD4 with deprotonated C370 and C373. (D) Holo Atox1–MBD3 with deprotonated C268 and C271. (E) Apo Atox1–MBD4. (F) Apo Atox1–MBD3. Cu(I) is shown as an orange sphere; sulphur, nitrogen, and hydrogen atoms are in yellow, blue and white, respectively. Carbon atoms are shown in the colour scheme of the protein. The Cu loop and α1 in Atox1 and MBD3/4 are shown in opaque, whereas the rest of the protein is shown as transparent new cartoon. The H-bond in D and E is denoted as a thick blue line. | ||
When simulating the holo adducts, force field-based MD, which relies on predefined empirical parameters, does not allow one to simulate directly bond breaking and formation, and hence the rearrangements of the metal's coordination sphere, which may occur upon Cu(I) binding and during its delivery from Atox1 to MBD3/4.52 Therefore, in the simulations of the holo adducts, we needed to assess a priori the Cu(I) coordination bonds, keeping these unvaried during the MD simulations. In the face-to-face model of adducts that we considered here, Cu(I) was bound to Atox1, mimicking, in this manner, the short-lived encounter complex between the two proteins.17 In the first simulations, C268/271 and C370/C373 of MBD3 and 4, respectively, were kept protonated. Surprisingly, Cu(I) binding to Atox1 affected the structural flexibility of the heterodimer involving MBD4, resulting in the two most populated cluster structures as extracted from the MD trajectory (Table S3, ESI†). Among these, the structure of the most representative cluster appears suitable for Cu(I) trafficking, even though the distance between Cu(I), coordinated to Atox1, and C370/C373 of the MBD4 metal binding site, which should receive Cu(I) along the trafficking route, is too large to allow Cu(I) binding (Fig. 5A, 6C and Table S3, ESI†). This is most likely due to the neutral state of C370/C373, which does not allow these residues to establish strong electrostatic interactions with Cu(I). The H-bond between K60@Atox1 and C373@MBD4, stabilizing the apo heterodimer, vanishes here (Fig. 5A). In the corresponding complex with MBD3 α1, an essential structural element starts unfolding (Fig. 5B), and the distance between Cu(I) and C268/C271 is even larger than in MBD4 (Fig. 6D), making Cu(I) transfer to MBD3 even more unlikely than to MBD4. Moreover, the binding of Cu(I) to Atox1 significantly increased per-residue flexibility across the whole heterodimer (Fig. 6B).
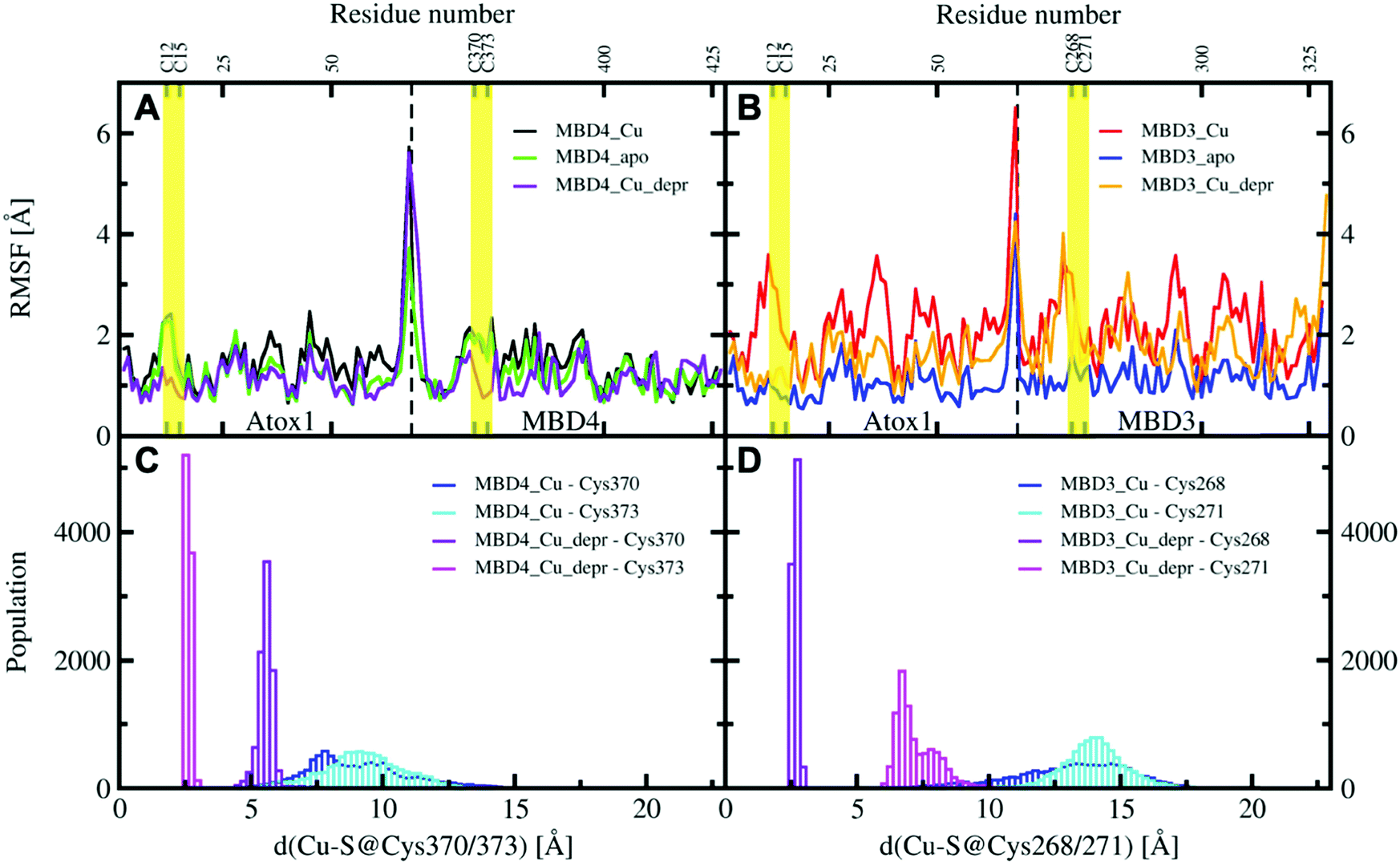 | ||
| Fig. 6 Per-residue RMSF plots of MBD4, (A) during the 200 ns of MD simulations (black – holo Atox1–MBD4, protonated Cys370 and 373; green – apo Atox1–MBD4; violet – holo Atox1–MBD4, deprotonated Cys370 and 373) and MBD3 (B) during the 200 ns of cMD simulations (red – holo Atox1–MBD3, protonated Cys268 and 271; blue – apo Atox1–MBD3; orange – holo Atox1–MBD3, deprotonated Cys268 and 271). Regions flanking the Cu(I)-binding residues in Atox1 and MBD3/4 are marked in yellow. Distribution of distances between Cu(I) cysteine residues of MBD4 (C) and MBD3 (D) in the complexes of holo Atox1 with MBD3/4 considering for the latter the protonated and deprotonated cysteine residues (blue – distance between Cu(I) and protonated S@Cys370/268; cyan – distance between Cu(I) and protonated S@Cys373/271; violet – distance between Cu(I) and deprotonated S@Cys370/268; magenta – distance between Cu(I) and deprotonated S@Cys373/271). | ||
Cu(I) delivery from Atox1 to MBD4/MBD3 would require the formation of direct interactions between Cu(I) and the receiving cysteine residues of the metal binding site. Since these distances are larger in the complex involving MBD3, a possible Cu(I) delivery from Atox1 to protonated MBD3 may be more unlikely than to MBD4.
However, during Cu(I) transport it is likely that C370/C373 will spontaneously deprotonate or their deprotonation will be co-adjusted by Cu(I) binding. In order to assess how the protonation of Cys residues of the MBD3/4's Cu(I) binding motif could impact on the stability of the adducts, we finally considered the holo Atox1–MBD3/4 complexes with deprotonated (depr) C268/271@MBD3 and C370/373@MBD4 residues. This allows mimicking a more advanced step in the Cu(I) delivery path at which the formation of a tri-coordinated state may take place. We remark, however, that an explicit coordination bond between the Cu(I) and Cys residues of the metal binding site is absent in cMD runs. Thus, the interactions between Cu(I) and C268/271 and C370/373 of MBD3/4, respectively, are purely based on electrostatics.53
In these simulations, the flexibility of the Atox1–Cu(I)–MBD4depr complex lowered, especially in the Cu(I)-binding region of both proteins, resulting in a predominant structure in the MD trajectory with Cu(I) bound by two Cys residues of Atox1 and strongly interacting with C373 of MBD4 (Fig. 5C, 6A and Table S3, ESI†). Conversely, deprotonation of C268 and C271 of MBD3 increases the flexibility of the Atox1–Cu(I)–MBD3depr complex, determining a split of the trajectory into three major clusters, bearing Cu(I) bound by two Cys residues from Atox1 and strongly interacting with C268 of MBD3. Per-residue flexibility of all regions of this heterodimer was lower compared to the neutral Atox1–Cu(I)–MBD3 complex, while being still higher than in the Atox1–Cu(I)–MBD4depr complex (Fig. 6A and B). As explained above, in order to have Cu(I) transfer the cysteine residues of the metal binding site should also lie at a favorable distance from Cu(I) in order to coordinate Cu(I). While C268@MBD3 and C373@MBD4, both lie at comparable distances from Cu(I), the C370@MBD4 is remarkably closer than C271@MBD3. Hence, Cu(I) delivery may be more likely towards MBD4 (Fig. 6C and D).
Indeed, C271@MBD3 frequently H-bonded to K60@Atox1 and Atox1–Cu(I)–MBD3depr also manifested a loss of α1's secondary structure content (Fig. 5D, Fig. S8C, D and Table S3, ESI†), possibly as a result of this interaction. Therefore, and based on the experimental results, it is tempting to suggest that the loss of this structural motif may adversely affect the formation of the Atox1–Cu(I)–MBD3depr complex.
Nevertheless, cMD simulations show that, although the orientation of MBD3 is possibly disfavored to accept Cu(I) delivery from Atox1, the Atox1–MBD3 adducts are all stable, in contrast to experimental results.
Conclusions
In summary, the combined use of EPR measurements and MD simulations shows that Atox1 can interact with MBD3–4 in the apo form, even if the Atox1–MBD4 heterodimer appears to be more consistent with a faster metal transport. The EPR data suggest that Atox1 prefers to bind to MBD4 in a monomeric state, and not to MBD3. MD simulations reveal that in both holo encounter complexes only MBD4 retains complete structural integrity and the distances between Atox1–Cu(I) and the cysteine residues of the metal binding site of MBD4 are at shorter distances than those of MBD3, considering them either in the protonated or deprotonated form, suggesting that MBD4 may be more likely involved in the transport route. Moreover, Cu(I) binding also has an impact on the fold and on the flexibility of MBD3, which makes its attendance in the metal extrusion path more unlikely. However, in contrast to experiments, which did not probe isolated Atox1–MBD3 and Atox1–MBD4 systems, cMD simulations suggest that all heterodimers involving MBD3 and MBD4 are stable.EPR evidence also indicates that the Atox1 monomer binds to MBD4 in a face-to-face manner and that it is not sandwiched between MBD3 and MBD4. This mechanism is strikingly different from the acquisition path where Ctr1 delivers Cu(I) to the Atox1 homodimer,20 suggesting that the Atox1 homodimer breaks only upon interaction with ATP7B. Our outcomes propose that the Cu(I) extrusion path is most likely mediated by Atox1 binding to MBD4 of ATP7B. This is most probably due to (i) reduced flexibility of the Atox1–Cu(I)–MBD4depr heterodimer, and (ii) partial unfolding of the metal binding loop and its flanking residues, upon metal binding to MBD3 and upon interaction with Atox1.
Since Cu(I) transport is believed to occur via the formation of a metastable transient intermediate, maintaining and selecting a conformation which allows an optimal interaction between the metallochaperone and its partner ATPase is required for efficient Cu(I) extrusion.
Despite their structural similarity, our outcomes reveal that separate MBDs in ATP7B play distinct roles and emphasise that Atox1 carefully regulates the in-cell Cu(I) concentration by adopting a conformation and a monomeric/dimeric structure specific to its interacting protein.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Israel and Italian Ministry of Science grant financed to SR and AM and by the ERC-STG grant no. 754365 given to SR. AM thanks the Italian Association for Cancer Research (MFAG grant no. 17134). MP thanks CINECA for computational resources (ISCRA no. HP10BXE6H9). IR thanks AIRC for financial support through a ‘Gianni Bonadonna’ fellowship. AM thanks S. Fabris, S. Piccinin and A. Vanossi for useful discussions.References
- S. Schwab, J. Shearer, S. E. Conklin, B. Alies and K. L. Haas, Sequence proximity between Cu(II) and Cu(I) binding sites of human copper transporter 1 model peptides defines reactivity with ascorbate and O2, J. Inorg. Biochem., 2016, 158, 70–76 CrossRef CAS.
- Y. Shenberger, O. Marciano, H. Gottlieb and S. Ruthstein, Insights into the N-terminal Cu(II) and Cu(I) binding sites of the human copper transporter CTR1, J. Coord. Chem., 2018, 71, 1985–2002 CrossRef CAS.
- Y. Shenberger, A. Shimshi and S. Ruthstein, EPR spectroscopy shows that the blood carrier protein, human serum albumin, closely interacts with the N-terminal domain of the copper transporter, Ctr1, J. Phys. Chem. B, 2015, 119, 4824–4830 CrossRef CAS PubMed.
- A. Rodriguez-Granillo, A. Crespo, D. A. Estrin and P. Wittung-Stafshede, Copper-Transfer Mechanism from the Human Chaperone Atox1 to a Metal-Binding Domain of Wilson Disease Protein, J. Phys. Chem. B, 2010, 114, 3698–3706 CrossRef CAS PubMed.
- D. Strausak, M. K. Howie, S. D. Firth, A. Schlicksupp, R. Pipkorn, G. Multhaup and J. F. B. Mercer, Kinetic analysis of the interaction of the copper chaperone Atox1 with the metal binding sites of the Menkes protein, J. Biol. Chem., 2003, 278, 20821–20827 CrossRef CAS PubMed.
- S. Lutsenko, Human copper homeostasis: a network of interconnected pathways, Curr. Opin. Chem. Biol., 2010, 14, 211–217 CrossRef CAS PubMed.
- A. C. Rosenzweig, Copper Delivery by Metallochaperone Proteins, Acc. Chem. Res., 2001, 34, 119–128 CrossRef CAS.
- P. S. Donnelly, Z. Xiao and A. G. Wedd, Copper and Alzheimer's disease, Curr. Opin. Chem. Biol., 2007, 11, 128–133 CrossRef CAS PubMed.
- M. E. Portnoy, A. C. Rosenzweig, T. Rae, D. L. Huffman, T. V. O'Halloran and V. C. Culotta, Strcuture-Function Analyses of the ATX1 Metallochaperone, J. Biol. Chem., 1999, 274, 15041–15045 CrossRef CAS PubMed.
- A. C. Rosenzweig, D. L. Huffman, M. Y. Hou, A. K. Wernimont, R. A. Pufahl and T. V. O'Halloran, Crystal structure of the Atx1 metallochaperone protein at 1.02 A resolution, Structure, 1999, 7, 605–617 CrossRef CAS.
- C. H. Yu, N. Yang, J. Bothe, M. Tonelli, S. Nokherin, N. V. Dolgova, L. Braiterman, S. Lutsenko and O. Y. Dmitriev, The metal chaperone Atox1 regulates the activity of the human copper transporter ATP7B by modulating domain dynamics, J. Biol. Chem., 2017, 292, 18169–18177 CrossRef CAS PubMed.
- A. Badarau, S. J. Firbank, A. A. McCarthy, M. J. Banfield and C. Dennison, Visualizing the metal-binding versatility of copper trafficking sites, Biochemistry, 2010, 49, 7798–7810 CrossRef CAS PubMed.
- D. L. Huffman and T. V. O'Halloran, Function, structure, and mechanism of intracellular copper trafficking proteins, Annu. Rev. Biochem., 2001, 70, 677–701 CrossRef CAS PubMed.
- D. Achila, L. Banci, I. Bertini, J. Bunce, S. Ciofi-Baffoni and D. L. Huffman, Structue of human Wilson protein domains 5 and 6 and their interplay with domain 4 and the copper chaperone HAH1 in copper uptake, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5729–5734 CrossRef CAS PubMed.
- A. K. Wernimont, D. L. Huffman, A. L. Lamb, T. V. O'Halloran and A. C. Rosenzweig, Structural basis for copper transfer by the metallochaperone for the Menkes/Wilson disease proteins, Nat. Struct. Biol., 2000, 7, 766–771 CrossRef CAS PubMed.
- L. Banci, I. Bertini, F. Cantini, N. Della-Malva, M. Migliardi and A. Rosato, The Different Intermolecular Interactions of the Soluble Copper-binding Domains of the Menkes Protein, ATP7A, J. Biol. Chem., 2007, 282, 23140–23146 CrossRef CAS PubMed.
- L. Banci, I. Bertini, V. Calderone, N. Della-Malva, I. C. Felli, A. Pavelkova and A. Rosato, Copper(I)-mediated protein-protein interactions result from suboptimal interaction surfaces, Biochem. J., 2009, 422, 37–42 CrossRef CAS PubMed.
- L. Banci, I. Bertini, F. Cantini, A. C. Rosenzweig and L. A. Yatsunyk, Metal Binding Domains 3 and 4 of the Wilson Disease Protein: Solution Structure and Interaction with Copper(I) Chaperone HAH1, Biochemistry, 2008, 47, 7423–7429 CrossRef CAS PubMed.
- K. Arumugam and S. Crouzy, Dynamics and Stability of the Metal Binding domains of the Menkes ATPase and their interaction with metallochaperone HAH1, Biochemistry, 2012, 51, 8885–8906 CrossRef CAS PubMed.
- A. M. Keller, J. J. Benitez, D. Klarin, L. Zhong, M. Goldfogel, F. Yang, T.-Y. Chen and P. Chen, Dynamic Multi-Body Protein Interactions Suggest Versatile Pathways for Copper Trafficking, J. Am. Chem. Soc., 2012, 134, 8934–8943 CrossRef CAS PubMed.
- A. R. Levy, M. Turgeman, L. Gevorkyan-Aiapetov and S. Ruthstein, The structural flexibility of the human copper chaperone Atox1: insights from combined pulsed EPR studies and computations, Protein Sci., 2017, 26, 1609–1618 CrossRef CAS PubMed.
- A. R. Levy, M. Nissim, N. Mendelman, J. Chill and S. Ruthstein, Ctr1 Intracellular Loop Is Involved in the Copper Transfer Mechanism to the Atox1 Metallochaperone, J. Phys. Chem. B, 2016, 120, 12334–12345 CrossRef CAS PubMed.
- A. R. Levy, V. Yarmiayev, Y. Moskovitz and S. Ruthstein, Probing the Structural Flexibility of the Human Copper Metallochaperone Atox1 Dimer and Its Interaction with the CTR1 C-Terminal Domain, J. Phys. Chem. B, 2014, 118, 5832–5842 CrossRef CAS.
- A. Erijman, A. Dantes, R. Bernheim, J. M. Shifman and Y. Peleg, Transfer-PCR (TPCR): a highway for DNA cloning and protein engineering, J. Struct. Biol., 2011, 175, 171–177 CrossRef CAS PubMed.
- G. Jeschke, Distance measurements in the nanometer range by pulse EPR, ChemPhysChem, 2002, 3, 927–932 CrossRef CAS.
- M. H. Olsson, C. R. Sondergaard, M. Rostkowski and J. H. Jensen, PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions, J. Chem. Theory Comput., 2011, 7, 525–537 CrossRef CAS.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- B. T. Op't Holt and K. M. Merz Jr., Insights into Cu(I) Exchange in HAH1 using Quantum Mechanical and Molecular Simulations, Biochemistry, 2007, 46, 8816–8826 CrossRef PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of simple potential functions for simulating liquid water, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, Molecular dynamics with coupling to an external bath, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- G. S. Grest and K. Kremer, Molecular dynamics simulation for polymers in the presence of a heat bath, Phys. Rev. A, 1986, 33, 3628–3631 CrossRef CAS.
- D. A. Case, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham, T. A. Darden, R. E. Duke, T. J. Giese, H. Gohlke, A. W. Goetz, N. Homeyer, S. Izadi, P. Janowski, J. Kaus, A. Kovalenko, T. S. Lee, S. LeGrand, P. Li, C. Lin, T. Luchko, R. Luo, B. Madej, D. Mermelstein, K. M. Merz, G. Monard, H. Nguyen, H. T. Nguyen, I. Omelyan, A. Onufriev, D. R. Roe, A. Roitberg, C. Sagui, C. L. Simmerling, W. M. Botello-Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu, L. Xiao and P. A. Kollman, AMBER 2016, University of California, San Francisco, 2016 Search PubMed.
- P. Vidossich and A. Magistrato, QM/MM Molecular Dynamics Studies of Metal Binding Proteins, Biomolecules, 2014, 4, 616–645 CrossRef PubMed.
- J. Hutter, M. Iannuzzi, F. Schiffmann and J. VandeVondele, cp2k: atomistic simulations of condensed matter systems, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 15–25 CAS.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, QUICKSTEP: fast and accurate density functional calculations using a mixed Gaussian and plane waves approach, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS.
- J. VandeVondele and J. Hutter, Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases, J. Chem. Phys., 2007, 127, 114105 CrossRef PubMed.
- S. Goedecker, M. Teter and J. Hutter, Separable dual-space Gaussian pseudopotentials, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 1703–1710 CrossRef CAS.
- C. Hartwigsen, S. Goedecker and J. Hutter, Relativistic separable dual-space Gaussian pseudopotentials from H to Rn, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 3641–3662 CrossRef CAS.
- E. Brunk and U. Rothlisberger, Mixed Quantum Mechanical/Molecular Mechanical Molecular Dynamics Simulations of Biological Systems in Ground and Electronically Excited States, Chem. Rev., 2015, 115, 6217–6263 CrossRef CAS.
- P. Campomanes, U. Rothlisberger, M. Alfonso-Prieto and C. Rovira, The Molecular Mechanism of the Catalase-like Activity in Horseradish Peroxidase, J. Am. Chem. Soc., 2015, 137, 11170–11178 CrossRef CAS.
- L. Casalino, G. Palermo, U. Rothlisberger and A. Magistrato, Who Activates the Nucleophile in Ribozyme Catalysis? An Answer from the Splicing Mechanism of Group II Introns, J. Am. Chem. Soc., 2016, 138, 10374–10377 CrossRef CAS.
- M. G. Quesne, T. Borowski and S. P. de Visser, Quantum Mechanics/Molecular Mechanics Modeling of Enzymatic Processes: Caveats and Breakthroughs, Chemistry, 2016, 22, 2562–2581 CrossRef CAS.
- J. Sgrignani, M. Iannuzzi and A. Magistrato, Role of water in the puzzling mechanism of the final aromatization step promoted by the human aromatase enzyme. Insights from QM/MM MD simulations, J. Chem. Inf. Model., 2015, 55(10), 2218–2226 CrossRef CAS PubMed.
- J. Sgrignani and A. Magistrato, The Structural Role of Mg2+ Ions in a Class I RNA Polymerase Ribozyme: A Molecular Simulation Study, J. Phys. Chem. B, 2012, 116(7), 2259–2268 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, UCSF Chimera–a visualization system for exploratory research and analysis, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- M. Pannier, S. Veit, A. Godt, G. Jeschke and H. W. Spiess, Dead-time free measurement of dipole-dipole interactions between electron spins, J. Magn. Reson., 2000, 142, 331–340 CrossRef CAS PubMed.
- A. D. Milov, A. G. Maryasov and Y. D. Tsvetkov, Pulsed Electron Double Resonance (PELDOR) and its Applications in Free-Radicals Research, Appl. Magn. Reson., 1998, 15, 107–143 CrossRef CAS.
- L. Columbus and W. L. Hubbell, A new spin on protein dynamics, Trends Biochem. Sci., 2002, 27, 288–295 CrossRef CAS PubMed.
- A. K. Boal and A. C. Rosenzweig, Crystal structures of cisplatin bound to human copper chaperone, J. Am. Chem. Soc., 2009, 131, 14196–14197 CrossRef CAS PubMed.
- P. Vidossich and A. Magistrato, QM/MM Molecular Dynamics Studies of Metal Binding Proteins, Biomolecules, 2014, 4, 616–645 CrossRef PubMed.
- J. Sgrignani, L. Casalino, F. Doro, A. Spinello and A. Magistrato, Can multiscale simulations unravel the function of metallo-enzymes to improve knowledge-based drug discovery?, Future Med. Chem., 2019, 11, 771–791 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9mt00067d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |