
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA miniaturized push–pull-perfusion probe for few-second sampling of neurotransmitters in the mouse brain†
Floris T. G.
van den Brink
a,
Thas
Phisonkunkasem
b,
Ashish
Asthana
a,
Johan G.
Bomer
a,
Arn M. J. M.
van den Maagdenberg
 bc,
Else A.
Tolner
bc and
Mathieu
Odijk
*a
bc,
Else A.
Tolner
bc and
Mathieu
Odijk
*a
aBIOS – Microdevices for Chemical Analysis group, MESA+ Institute for Nanotechnology, Techmed Centre, University of Twente, Hallenweg 15, 7522 NH Enschede, The Netherlands. E-mail: m.odijk@utwente.nl
bDepartment of Human Genetics, Leiden University Medical Center, Einthovenweg 20, 2333 ZC Leiden, The Netherlands
cDepartment of Neurology, Leiden University Medical Center, Einthovenweg 20, 2333 ZC Leiden, The Netherlands
First published on 5th March 2019
Abstract
Measuring biomolecule concentrations in the brain of living animals, in real time, is a challenging task, especially when detailed information at high temporal resolution is also required. Traditionally, microdialysis probes are used that generally have sampling areas in the order of about 1 mm2, and provide information on concentrations with a temporal resolution of at least several minutes. In this paper, we present a novel miniaturized push–pull perfusion sampling probe that uses an array of small 3 μm-wide sampling channels to sample neurotransmitters at a typical recovery rate of 61%, with a reduced risk of clogging. The added feature to segment the dialysate inside the probe into small water-in-decane droplets enables the detection of concentrations with a temporal resolution of a few seconds. Here we used the probe for in vivo recordings of neurotransmitter glutamate released upon electrical stimulation in the brain of a mouse to demonstrate the feasibility of the probe for real-time neurochemical brain analysis.
Introduction
One major challenge in neuroscience research is to obtain reliable information on the concentration of neurotransmitters and other signalling molecules, with a high temporal resolution, at specific locations in the brain.1 Such neurochemical knowledge can be used to unravel the complex dynamics of neuronal brain activity, not only in the healthy brain, but also in a disease context. Unfortunately, current neurochemical measurement approaches have technical limitations that hamper real-time, local assessment of these neurochemical processes.The analysis of neurochemical molecules, such as glutamate or acetylcholine, in brain extracellular fluid is traditionally done with microdialysis,2,3 push–pull perfusion sampling,4–6 or electrochemistry,7 for example with arrays of biosensor electrodes located on ceramic-based ‘Gerhardt’ probes8,9 or silicon-based micro-arrays.10 An overview of the benefits and disadvantages of the various methods is described in Table 1.

|
As discussed in a review by Kennedy,1 microdialysis techniques suffer from a low temporal (at least several minutes) and spatial (over ∼1 mm2) resolution. The smallest commercially available dialysis probes are ∼200 μm in diameter with a membrane tip length of 1 mm, resulting in a sampling area of 628![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 μm2 and therefore rather large compared to the size of the rodent brain.11 Alternatives for localized sampling are capillary push–pull perfusion sampling (PPPS) probes5,6 and direct sampling using a single capillary,12 typically with needle diameters between 90 and 360 μm and with sampling areas between 250 and 2000 μm2. The poor temporal resolution is caused by target molecules arriving at different points in time at the detector, depending of their position in the parabolic flow profile inside the transporting capillary, the so-called “Taylor dispersion effect”.13
000 μm2 and therefore rather large compared to the size of the rodent brain.11 Alternatives for localized sampling are capillary push–pull perfusion sampling (PPPS) probes5,6 and direct sampling using a single capillary,12 typically with needle diameters between 90 and 360 μm and with sampling areas between 250 and 2000 μm2. The poor temporal resolution is caused by target molecules arriving at different points in time at the detector, depending of their position in the parabolic flow profile inside the transporting capillary, the so-called “Taylor dispersion effect”.13
In an attempt to overcome some of these problems, capillary PPPS probes have been attempted that make use of microfluidic droplet generators.5,14,15 Combining microfluidic droplet generators – to compartmentalize the samples – with commercially available probes further improved the temporal resolution in microdialysis experiments.16,17 The major advantage of compartmentalization is that it limits Taylor dispersion, yet it comes at the cost of a more complex analysis afterwards. Segmented flow sampling has also been used in combination with electrospray ionization-mass spectrometry for the detection of acetylcholine in vivo.18 Furthermore, a microfabricated needle with integrated droplet generator was used for the collection and delivery of nanoliter volumes of liquid with a particularly high temporal resolution (averaged 3.4 seconds).19 To this end the fluorescent sample is pulled through 2.6 μm-wide ‘slits’ using negative outlet pressure, which produces a train of aqueous sample droplets that are captured in the oil phase that flows along the narrow channels.
Miniaturization is necessary to enable more localized measurements, which also should limit tissue damage inflicted during the measurement. As indicated in Table 1, push–pull types of probes can suffer from clogging,22 a problem that can be solved by sampling through a porous aluminium oxide membrane located on top of the microfabricated probe needle.25 Even more advanced, hybrid approaches that combine fluidic handling with electrical sensing for measurements of neuronal or ion activity, have been demonstrated in probes that combined EEG recordings for monitoring brain activity and drug dosing via microfluidic channels.26,27 A hybrid probe combining direct sampling and droplet compartmentalization, and EEG readout has been published recently by the group of Renaud,14 that allows tracking changes in local concentrations of trace metals, such as sodium, magnesium, potassium, calcium, copper and zinc, by laser-ablation – inductively coupled plasma – mass spectrometry.
In contrast to the sampling techniques described earlier, electrochemical probes have the advantage of high temporal and spatial resolution. However, the concept of electrochemical probes relies on selectivity towards one specific type of molecule that is typically provided by including enzymatic reactions, which prove difficult to keep stable, and impossible to periodically calibrate, once placed in the brain.28 Electrochemical probes have been used to measure a variety of ions and neurotransmitters such as glutamate,7,29 dopamine,30 and choline.10 Direct electrochemical detection can be used for e.g. dopamine. However, a more robust sensing mechanism is based on an enzymatic conversion of the respective neurotransmitter to obtain sufficient selectivity, followed by amperometric detection. Alternatively, silicon-based probes can be equipped with different electrochemical sensing areas that are functionalized using ionophores, achieving for example selectivity for K+ on one sensing area, while another sensing area can be made pH-sensitive, thus enabling simultaneous measurements of K+ and H+ concentrations.31,32 With such an ion-sensitive probe, in vivo K+ variations were recorded in a mouse brain during induction of cortical spreading depolarization (CSD), a strong wave of neuronal and glial cell depolarization.23,33
Here we present a microfabricated push–pull perfusion sampling (μPPPS) probe capable of sampling with a spatial resolution of ∼4000 μm2. In our probes, the probe tip (140 μm wide) is equipped with two rows of ten 3 μm-wide microfluidic sampling channels that act as a sort of ‘frit’ that allows molecules to be captured while larger extracellular matrix debris is kept outside. A novelty of our ‘frit’ design is that it allows for adjustment of the internal flow rate and pressure drop over these microfluidic sampling channels and tuning of mass transport between a more ‘diffusive’ versus a ‘convective’ mode. We designed two types of probes. In the first version (probe type 1), our microfabricated needles are equipped with a droplet generator inside the probe tip to capture dialysate in droplets immediately after it is sampled into the probe. This approach decouples sample collection from sample analysis, as the changes in concentration are stored in this ‘chemical memory’. In a second version of our probe (probe type 2) an external droplet generator is connected to a push–pull microprobe. Here, the dialysate and sample is combined with enzymes (glutamate oxidase and horseradish peroxidase) and a fluorophore precursor (Amplex Red) prior to droplet formation, in order to create a reaction mixture that produces a fluorescent signal (resorufin) that represents the amount of target neurotransmitters. A major benefit of our method compared to enzymatic electrochemical sensors, is that the enzymes and their reaction products are not in contact with brain tissue (thus preventing confounding effects of glutamate conversion at the site of measurement) while providing similar spatial and temporal resolution.
Sample analysis for both types of probes is done on-line, outside the sampling area, using a dedicated optical setup based on fluorescence detection. The optical filters and light source are tuned for the detection of resorufin, a fluorophore frequently used in commercially available assay kits for e.g. glutamate or acetylcholine. The outlet fused silica capillary of the μPPPS probe is mounted in front of a LED source and photo multiplier tube, enabling the on-line detection of resorufin at concentrations <1 nM.
Here we first describe the development and characterization of the two types of newly developed μPPPS probes34 using fluorescein and glutamate, and then show the capability of our approach to measure – in an in vivo experiment in a mouse – glutamate levels in the brain in relation to electrically evoked neuronal network activity.
Design
Probe design
The operating principle of the μPPPS probe is illustrated in Fig. 1. The probe is operated using a dialysate inlet, a continuous phase inlet (in this case decane) and a droplet outlet. Dialysate flow is infused in the probe tip at a constant flow rate. A concentration gradient develops across the microfluidic sampling channels and molecules will diffuse inwards to be collected by the dialysate flow. This series of 10 sampling channels (3 μm wide, 20 μm height), spanning a length of 100 μm on both sides of the probe tip, reduces the risk of probe clogging and provides a reasonable spatial resolution along the length of ∼100 μm. Note that the sampling channels are small open microfluidic channels providing transport of ions into the probe, which is different from porous materials such as membranes or hydrogels. The resulting diffusion coefficients of transported ions are therefore equally high as in bulk media. The cross-sectional area through which molecules enter the probe is 1200 μm2. Note however, that there are two rows of 10 sampling channels on each side of the tip, spatially separated by 140 μm. Two types of probes were fabricated. Probe type 1 has a droplet generator in the needle of the probe (see Fig. 1, and 5A). Probe type 2 uses an external glass chip droplet generator (Fig. 5D–F) that was used to enable addition of an enzymatic assay kit to detect neurotransmitter (glutamate). A large hydraulic resistance in the form of a narrow channel (probe type 1: channel width 5 μm, channel height 20 μm, and 1 mm channel length; probe type 2: channel width 20 μm, channel height 20 μm, and 1 cm channel length) is present between the tip and the droplet generator to avoid the continuous phase (decane) reaching the tip in case of small imbalances in flow in practical applications. For probe type 1, following sample collection (from tissue) and the hydraulic resistor, the organic continuous phase comes in contact with the dialysate sample inside a curved T-junction to create a train of water droplets that preserves concentration fluctuations in time, providing a temporal resolution of just a few seconds, while enabling both on-line and off-line analysis without losing temporal information.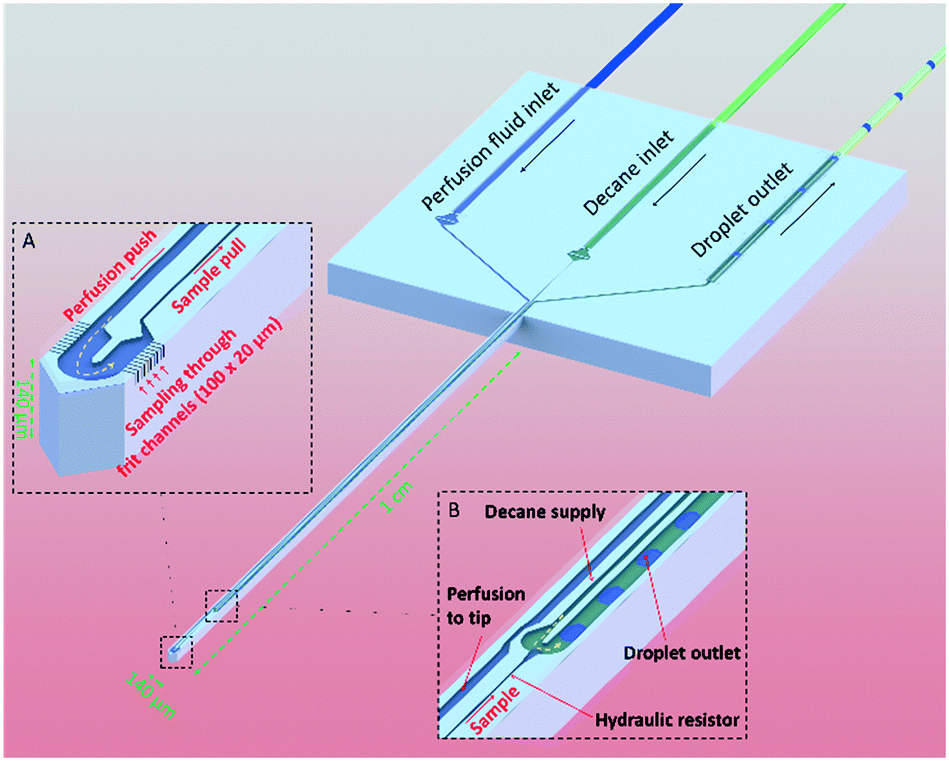 | ||
| Fig. 1 Schematic visualization of the μPPPS principle. Sample from tissue is collected through small sampling channels (see sampling tip inset, A). A push–pull configuration inside the needle ensures rapid transport of the sample to the external measurement equipment. A droplet generator is either integrated in the shaft of the probe (see droplet generator inset, B; probe type 1) or located externally (probe type 2), to prevent dispersion and to maintain a good temporal resolution. | ||
To promote efficient sample collection, a low negative pressure is applied to the droplet outlet. Ideally, the push–pull probe is operated in such a way that the push and pull flows are equal, which prevents withdrawing of liquid from or introducing liquid to the measurement volume (i.e. a sample cup, or mouse brain tissue). In practice, this is difficult to realize and the operating conditions are determined in such a way that a small inward flow is established in order to establish a stable sampling process.
An electrical equivalent circuit is used to determine the appropriate flow and pressure conditions to operate the probe and to predict the inward flow rate. Hydraulic resistances of the microfluidic channels and connected tubing, together with the flow and pressure sources are modelled in a circuit as shown in S1.† From this schematic, it can be calculated that an outlet pressure of −80 kPa, a continuous phase flow of 200 nL min−1 and perfusion flow of 50 nL min−1 give rise to an inwards flow rate of 126 nL min−1.
Numerical simulations
To predict the recovery rate of both types of our μPPPS probes, simulations were run on a 3D-model (see S10†) of the probe using COMSOL Multiphysics (version 5.3). Flow velocity (u) and pressure (p) variables were calculated using the Stokes flow approximation of the Navier–Stokes equation (creeping flow physics model) (1) and the continuity eqn (2), after which concentration (c) distributions were calculated using the steady-state convection–diffusion equation (transport of diluted species physics model, with convection and without migration) (3):| μ∇2u − ∇p = 0 | (1) |
| ∇·u = 0 | (2) |
| u·∇c − D∇2c = 0 | (3) |
The viscosity is used from water at 293 K and the mass transport is studied of a substance with D = 0.9 × 10−9 m2 s−1, equal to the value for monosodium glutamate.35 Boundary conditions are a perfusion inflow of 25 to 100 nL min−1, and an outlet pressure between −10 kPa and −80 kPa vs. atmospheric pressure (1 bar or 101.32 kPa). Note that this outlet pressure is set right after the tip of the needle using a laminar outflow boundary condition, including the small hydraulic resistance, but not including the droplet generator, or connecting tubing towards the detection area. Pressure around the probe tip was set to 1 bar (atmospheric pressure). All other boundaries carry a no-slip condition (u = 0). An arbitrary concentration (c0) of 1 mM was defined at the boundaries around the probe tip and of 0 mM at the perfusion inlet. We have validated that any arbitrary concentration will lead to identical results for the recovery rate, as we do not take any adsorption effects into account. The concentration gradient normal to the outlet is set to zero and the flux normal to all other boundaries is zero.
The concentration distributions at the tip are shown in Fig. 2, for a perfusion flow rate (QH2O) of 50 nL min−1 and an outlet pressure of either (C) −10 kPa or (D) −80 kPa. In the plot it can be seen that the sampling channels promote a significant uptake of compounds around the probe tip. To quantify this uptake, the recovery rate (the ratio between sampled concentration, and external concentration) is calculated at the channel outlet. The calculated values are plotted in Fig. 2A for varying perfusion flow rates and outlet pressures. As can be expected, the recovery rate increases if the negative outlet pressure is increased or the perfusion flow rate is decreased. To measure concentration changes at a reasonable temporal resolution, a certain inward flow is needed to provide a sufficient recovery rate. For example, the recovery rate under conditions similar to those discussed above (i.e. with a perfusion flow rate of 50 nL min−1 and an outlet pressure of −80 kPa) is 88%.
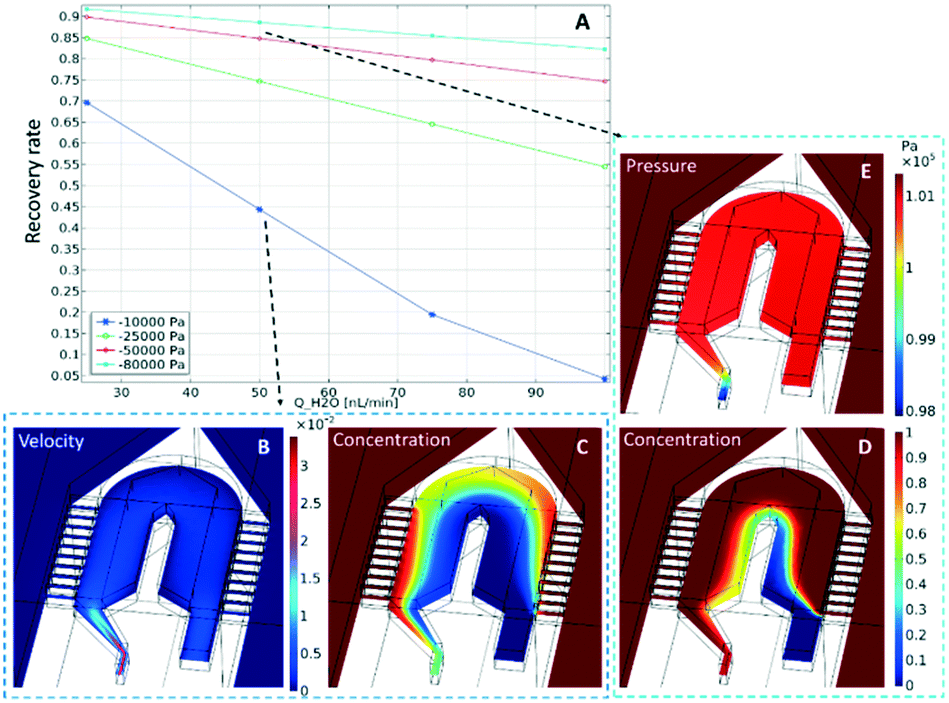 | ||
| Fig. 2 COMSOL simulations to determine (A) the recovery rate of the μPPPS probe tip as a function of flow rate (x-axis) and outlet pressure (legend). Velocity profile (m s−1) (B) and concentration profile (C) using 50 nL min−1 and −10 kPa outlet pressure. Pressure distribution (E) and concentration profile (D) for a flow rate of 50 nL min−1 and −80 kPa outlet pressure. Note that the pressure outside the probe is set to 1 bar (101.32 kPa). | ||
Methods
Probe fabrication
The μPPPS probes were fabricated in the Nanolab cleanroom of the MESA+ Institute of Nanotechnology, following the steps as illustrated in Fig. 3.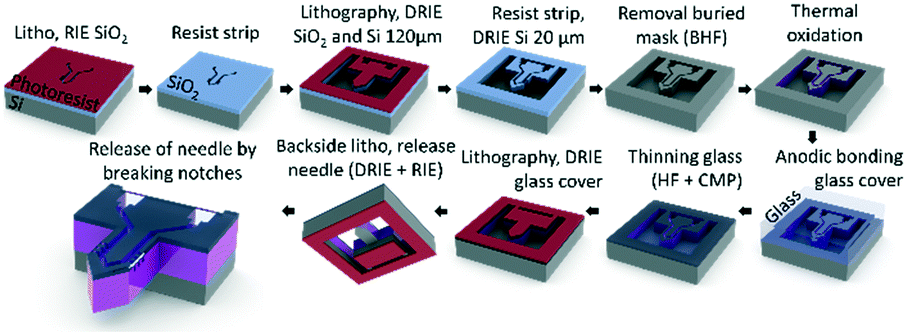 | ||
| Fig. 3 Schematic overview of the fabrication process of the μPPPS probe. Note that the probe and layer thicknesses are not drawn to scale. See text for a full description, and Table S6† for a more detailed process flow. | ||
Silicon wafers are provided with a thermal SiO2 layer, which was used as a buried mask that is patterned with the fluidic channel structures. Following a second lithography step, fluidic inlets and outlets (140 μm) and the channel structures (20 μm) were etched in two consecutive deep reactive ion etching (DRIE) steps. The delicate sampling channel structures are protected by a thin thermally grown oxide layer to prevent damage during the last DRIE step to release the probe. The channels were covered with a 500 μm thick borosilicate glass wafer by anodic bonding, etched with 25% hydrogen fluoride (HF) and subsequently polished back to ∼20 μm thickness using chemical mechanical polishing. In the last two steps, the probes were released by etching slits through the glass and the silicon layers surrounding the structures. The probes were kept in place within the wafer with two small silicon notches that allow one to carefully detach a probe from the substrate using tweezers. A more detailed process flow is included in Table S6.†
Prior to their characterization, the probes were mounted on a micro-milled polystyrene substrate featuring recessed placeholders for the probe and capillaries to simplify alignment using cyanoacrylate glue (Pattex – super gel, Henkel, Nieuwegein, Netherlands). Some 2.5 cm of 30 μm inner diameter (ID)/130 μm outer diameter (OD) fused silica capillary tubing was glued (type NOA83H, Norland Optical Adhesives, Cranbury (NJ), USA) in the probe inlets and outlet. The NOA83H glue is UV-curable, which was used to cure the glue before it enters the microfluidic channels by capillary forces. Subsequently, the cyanoacrylate was added on this joint to add additional bonding strength. Curing of the cyanoacrylate was sped up using a spray can of CA accelerator (Zip Kicker, ZAP, Ontario (CA), USA). The other end of the 130 μm OD tubing was subsequently glued by repeating the glue step with NOA83H and cyanoacrylate to 30 cm of 150 μm ID/360 μm OD-fused silica capillary tubing. Finally, the 360 μm OD-fused silica capillary was fixed to the polystyrene substrate using epoxy (Loctite EA M-31CL, Henkel, Nieuwegein, Netherlands). See Fig. 5A for a photo of the assembled device.
A different probe type 2 was constructed for the glutamate level measurements, because an additional inlet was needed to supply the chemicals for an enzyme-based assay. This device is shown in Fig. 5D. In this case, a push–pull microprobe without internal droplet generator is connected to a dialysate inlet. The outlet of this probe is connected with a 2.5 cm long glass capillary of 30 μm ID/130 μm OD to an external borosilicate glass droplet generator. This droplet generator contains a Y-junction where the dialysate + sample flow is combined with the enzymes and fluorophore precursor. Droplets were generated from this mixture in decane at a T-junction, and the droplet contents were subsequently mixed in a meandering channel. In the first step, 100 μm channels in this droplet generator were wet-etched (25% HF, etch rate ∼0.9 μm min−1). In the second step, 380 μm inlets were wet-etched for the glass capillaries to be glued in place.
Chemicals
Sampling with the μPPPS probes was done by generating aqueous droplets in decane as the continuous phase. Decane has relatively low values for gas solubility compared to e.g. fluorinated alkanes, which prevents gas bubbles being formed inside the probe due to the negative outlet pressures.36,37In vitro characterization was done using fluorescein that was prepared in a 10 mM stock solution and titrated in de-ionized water to sample from varying concentrations. Water was purified using an Elga PureLab flex purification system. Water and decane were filtered using a 0.22 μm pore size filter prior to use. Decane, resorufin sodium salt and fluorescein sodium salt were obtained from Sigma-Aldrich GmbH (Steinheim, Germany).For in vivo recordings a sterile minimal perfusion fluid (MPF) solution, consisting of 140.3 mM NaCl, 3.0 mM KCl, 1.0 mM MgCl2·6H2O2 and 1.2 mM CaCl2·2H2O2 in MilliQ and filtered with a 0.2 μM sterilizing filter, was used for the ‘push’ flow. Glutamate measurements were performed using an Amplex Red glutamic acid/glutamate oxidase assay kit (Thermo Fisher Scientific, Breda, The Netherlands). With this method, L-glutamic acid is oxidized by glutamate oxidase to produce α-ketoglutarate, H2O2 and NH3. Subsequently, H2O2 reacts with Amplex Red in a 1:1 ratio catalysed by horseradish peroxidase to generate resorufin, a highly fluorescent product.
Measurement setup
The microprobe was mounted in a custom-made holder and connected to a syringe pump (Nemesys, Cetoni GmbH, Korbussen, Germany) to infuse dialysate and decane. A pressure pump (MFCS-EZ, Fluigent, Paris, France) was used to apply negative pressure for sampling. For the electrical merging of droplets, a waveform generator (Agilent 33250A, Santa Clara, USA) was connected to an amplifier with an amplification of 100 V/V (Trek model 2210, Trek Inc., Lockport (NY), USA). To monitor the applied signal, an Agilent DSO3062A oscilloscope (Santa Clara, USA) was installed. For droplet merging pictures, a stereomicroscope with a PTGREY grasshopper 3 camera (FLIR Integrated Imaging Solutions GmbH, Ludwigsburg, Germany) was used. The droplets recorded inside the probe needle were recorded with an inverted Olympus microscope (Hamburg, Germany) with the same grasshopper 3 camera.The optical setup consists of a high-power LED (M530L3, Thorlabs, Munich, Germany) controlled by a LED driver (LEDD1B, Thorlabs). LED power was measured using a silicon photodiode (S121C, Thorlabs) connected to a power meter (PM100USB, Thorlabs). The LED was focused at a measurement tube, which is a 50 μm ID/360 μm OD glass capillary tube from which the polyimide coating is locally removed. Alignment and focus of the LED beam was verified using a CMOS camera (DCC1545M, Thorlabs). For fluorescence measurements, a right-angle mirror (DFM/M-P01, Thorlabs) was used to reflect the beam towards a photomultiplier tube (PMT) (H7422, Hamamatsu, Herrsching am Ammersee, Germany). The PMT signal was amplified with 1 V μA−1 using an amplifier unit (C7319, Hamamatsu) before being collected by a National Instruments myDAQ board (Woerden, Netherlands). See Fig. S2A† for more information on the measurement setup and a schematic overview of the optics.
In vivo recordings
For the in vivo experiments in a mouse brain, first, the fluorescent signal intensity was calibrated in vitro using a 0 μM, 5 μM and 10 μM glutamate solution, since the expected glutamate levels in the mouse cortex are between approximately 2 and 5 μM.24 Since auto-oxidation of Amplex Red and deterioration of glutamate oxidase are known problems with this assay kit, this calibration procedure was performed every day. Moreover, with these concentration steps the time delay between sampling and detection is measured as well, allowing one to link the timing of measured fluorescence signal to the cortical activity changes occurring in the brain.Subsequently, the mouse was prepared for in vivo surgery for measuring cortical glutamate levels (Fig. S5B†). Animals used for the experiments were male C57BL/6J mice (Harlan, Venray, The Netherlands), kept under standard housing conditions with a 12 hour light/dark cycle with free access to water and food. All animal experiments were approved by local (LUMC Authority of Animal Welfare) and national (Central Authority for Scientific Procedures on Animals) ethical committees in accordance with recommendations of the European Communities Council Directive (2010/63/EU). All efforts were made to minimize animal suffering. Mice were anesthetized with isoflurane (induction 4%; maintenance 1.5 to 2%; in pressurized air) and placed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA, USA) on a homoeothermic heating pad (Stoelting, Wood Dale, IL, USA) with continuous monitoring and maintenance of the core temperature (37.0 ± 0 .5°C). A blood pressure transducer (AD Instruments, Inc., Colorado Springs, CO, USA) was connected to PE-10 tubing for a catheter (BD Intramedic, VWR, Radnor, PA, USA) placed in the right femoral artery for continuous blood pressure monitoring, and for regular blood sample collection for assessing systemic physiology based on plasma pH, pCO2 and pO2 values as assessed by blood-gas analysis, using a Rapidlab blood gas analyzer 248 (Siemens Healthcare Diagnostics, the Hague, Netherlands). After exposing the skull bone, cortical windows were made over the right cortex using a dental drill (Fine Science Tools Inc., Foster City, CA, USA), in the presence of cooled saline to prevent overheating. Three windows were drilled for placement of i) a concentric Pt/Ir bipolar stimulation electrode with 125 μm tip diameter (FHC Inc., Bowdoin, ME, USA) for transcallosal electrical stimulation in the left somatosensory cortex (location in mm: 1.5 mm posterior/1.5 mm lateral from bregma and 0.3 mm ventral from dura), ii) a glass capillary electrode (FHC Inc., Bowdoin, ME, USA) filled with 150 mM NaCl for AC-potential recordings in the right somatosensory cortex (0.5 mm anterior/1.5 mm lateral from bregma and 0.3 mm ventral from dura), iii) the μPPPS probe in the right somatosensory cortex (0.5 mm anterior/2.5 mm lateral from bregma and 0.4 mm ventral from dura. See Fig. 4 for a schematic representation. AC-potential signals were used to record local field potentials from the cortex (“electrocorticogram”, ECoG), that were measured with respect to an Ag/AgCl reference electrode placed subcutaneously at the neck and amplified 10× (Molecular Devices, Sunnyvale, CA, USA). Data were recorded and analysed off-line using LabChart Pro (AD Instruments). An external output of the ECoG signal was also connected to the second channel of the National Instruments myDAQ board and recorded simultaneously with output of the PMT (glutamate-related fluorescence signal). The trigger output used for electrical stimulation was recorded simultaneously with signals from the glass recording electrode and the PMT (fluorescence signal), enabling synchronization of the electrical stimulation, evoked cortical activity changes and glutamate measurements. After filling the system with a fresh enzyme mixture and prior to probe insertion, the MPF flow was established at 100 nL min−1 to form a droplet around the tip and avoid immediate clogging of the probe by the extracellular matrix. After probe insertion, to a depth of 0.4 mm, the decane flow (500 nL min−1), enzyme mixture flow (200 nL min−1) and outlet pressure (−50 kPa) were applied.
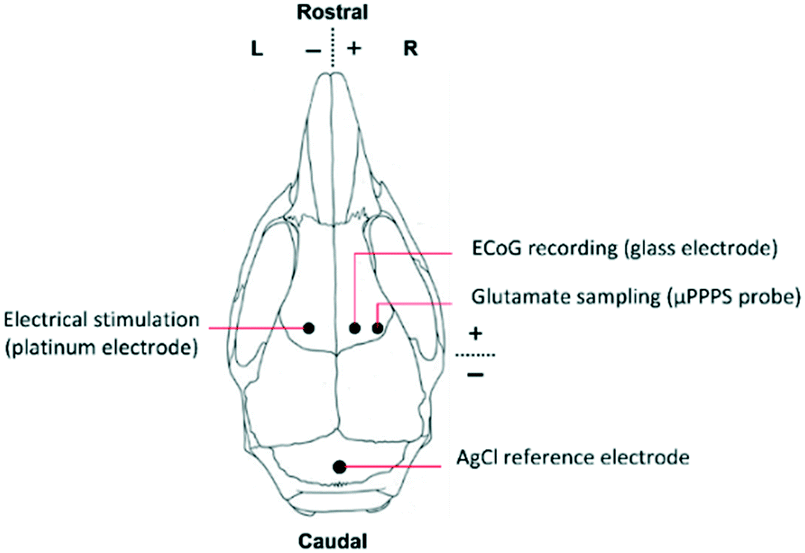 | ||
| Fig. 4 Schematic representation of a mouse skull indicating locations of the μPPPS probe in relation to the stimulation site, ECoG and reference electrode during in vivo experiments in an anesthetized mouse. | ||
After setting up this experiment, a recovery and baseline recording period of 15 minutes was observed, or longer in case signals had not stabilized. Next, transcallosal electrical stimulation was performed using the bipolar microelectrode on the left hemisphere (contralateral to the EcoG recording side) and a battery-driven isolated WPI stimulator (A395 isolated WPI stimulator) while measuring EcoG and glutamate signals in the right hemisphere as indicated above. Single-pulse stimulation (biphasic 1 ms pulses, 500 μs negative – 500 μs positive, i.e. ‘cathodic first’) was performed at 70% of the intensity yielding a maximum cortical response (which was 3.15 mA for the two mice) at 0.1 Hz, 6 stimuli. The block of 6 stimuli was repeated 3 times, at 10 minute interval. Current output intensity was monitored separately by a custom-made isolated current meter. The 3.15 mA stimulation intensity used for assessing the transcallosal cortical responses corresponds to a current density of ∼0.26 μA μm−2, given an exposed stimulation electrode surface area of approximately 12200 μm2.
The raw glutamate signal obtained from the PMT was processed using Matlab (version R2017a, Mathworks Inc., Natick, MA, USA), first to remove all data points below the baseline background fluorescence, second, to apply a moving averaging filter of 500 points (equal to 0.5 seconds). The AC ECoG signal recorded on the second channel of the MyDAQ was obtained from the glass electrode using a 3rd order Butterworth band-pass filter with cut-offs at 0.5 and 200 Hz.
Results and discussion
Probe fabrication
Photos of mounted probes are shown in Fig. 5A and D, while SEM pictures of the probe without its glass cover are shown in Fig. 5K–M.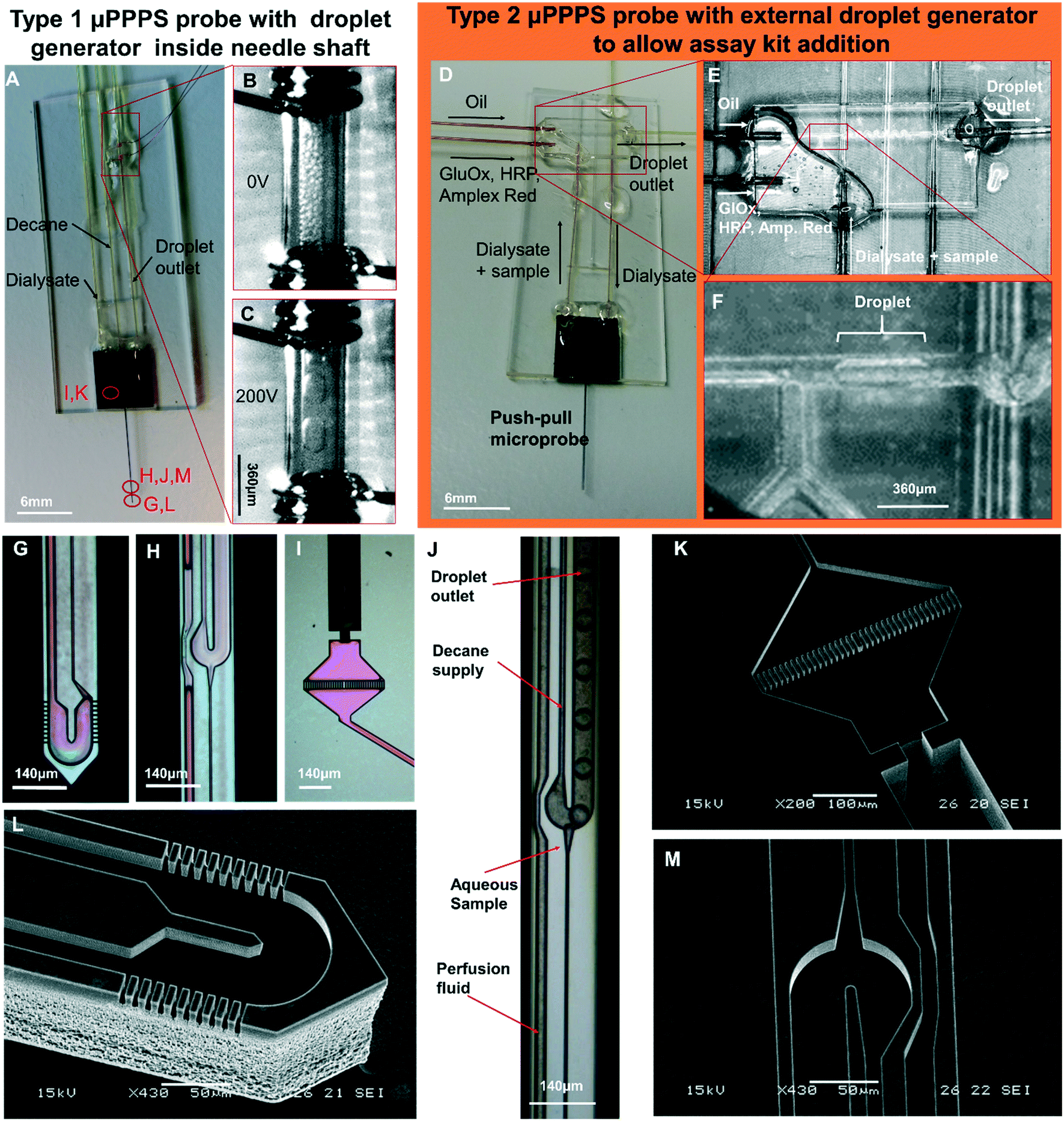 | ||
| Fig. 5 Push–pull microprobes. A: μPPPS probe type 1 with internal droplet generator located inside the shaft of the needle. B and C: Enlarged view of the electrode pair (spaced 1 mm) used for merging microdroplets (see Movies S9A and B†). Merging occurs as soon as the microdroplets are transferred from the 30 μm inner diameter outlet capillary to the 150 μm I.D. transport capillary. Droplets are visible inside the transport capillary before (B) and after (C) high-voltage is switched on (200 Vpp, 10 Hz square wave). D: μPPPS probe type 2 with external glass chip droplet generator to allow the addition of an assay kit consisting of enzymes and a fluorescent indicator to detect neurotransmitter (glutamate). E: Photo of the external droplet generator used for probe type 2 (see Movie S8†). F: Close-up of the plug-shaped droplets forming inside the external droplet generator. G: μPPPS probe type 1 tip (sampling channels are identical for probe types 1 and 2). H: Droplet generator inside needle (probe type 1). I: Inlet of the probe containing a pillar array filter. A 130 μm O.D. fused silica capillary is glued into the inlet channel. J: Droplet formation for probe type 1 (see Movie S7†). K: SEM photo of the inlet filter and capillary connecting channel. L: SEM photo of the tip of the μPPPS probe type 2, with the dialysate entry, return channel, and sampling channels visible. M: SEM photo of the droplet generator inside the needle (probe type 1). Microfluidic channel structures are all 20 μm deep, the capillary connecting channel is 140 μm deep. | ||
Electrodes connected to the transport capillary to fuse droplets to prevent dispersion of smaller droplets can be seen in Fig. 5A–C. Microscopy images of key features of the probe are shown in Fig. 5G–I. The purple coloring in the microscopy images arises from the silicon oxide passivation layer inside the channels. In Fig. 5G and L, the tip of the needle with its sampling channels is shown. Fig. 5H and J shows an inset of the middle part of the needle.
The delivery channel of perfusion fluid to the tip can be seen at the left, with the T-shaped droplet generator on the right. The narrow channel leading towards the droplet generator from the bottom side of the image is the hydraulic resistor that prevents the continuous phase (decane) from entering the tip if small imbalances in flow rate occur. At the dialysate and continuous phase inlet, small filters made of rectangular pillars with 5 μm spacing are included to prevent clogging of the probe as shown in Fig. 5I and K.
Droplet formation
First the possibility to generate droplets was tested for the two probe types (see Fig. 5J for type 1, and 5E and F for type 2). In Fig. 5J, droplet formation is visualized for a probe with the droplet generator embedded in the needle tip. In this experiment, the flow rate of oil was set to 200 nL min−1, perfusion with demineralized water was set to 50 nL min−1, and the outlet pressure was set to −50 kPa. From the high-speed video (400 frames per second (fps)), it was estimated that droplets were generated at a rate of 80 fps (see Movie S7†).For the second probe type, a push–pull probe is connected to an external droplet generator that enables the mixing of a sample containing the neurotransmitter glutamate with glutamate oxidase (GlOx), horseradish peroxidase (HRP) and a fluorophore precursor (Amplex Red) to obtain a glutamate concentration-dependent fluorescent signal. This arrangement is shown in Fig. 5D–F. Here, the flow rate of decane was set to 500 nL min−1, the flow rate of the GlOx/HRP/Amp. Red mixture was set to 200 nL min−1, perfusion of DI water was set to 100 nL min−1 and the outlet pressure was set to −50 kPa. Droplets are generated on average at a rate of 1 drop per second (see Movie S8†).
As soon as the microdroplets generated in probe type 1 are transferred from the outlet capillary to the much wider transport capillary, they form a dense packing at first (see Fig. 5B), but due to the parabolic flow velocity profile, droplets moving along the centerline will be faster than those closer to the wall again causing dispersion. Therefore, measurements will get distorted because the order of the droplets moving through the optical detector will be different from the order in which they were generated. As a result, the temporal resolution goes down to ∼1 minute, as shown in Fig. S3A.† To solve this, a pair of electrodes was installed right after the outlet capillary in order to electrically merge microdroplets into larger ones that fit the dimensions of the transfer capillary, thereby forming a stable segmented flow and preserving the temporal resolution of the concentration variations in the sample (see Fig. 5C).
Based on separate measurements of the generation rate and volume of the small and merged droplets, it is estimated that about 50 droplets are merged into one bigger droplet (see Movies S9A and B†). Although the segmented flow was restored by this merging step, theoretically this modification will reduce the temporal resolution only by the ratio of the original and merged droplet generation rates, which is estimated to be a factor of 50.
Optical detection
The uPPPS probes are designed to sample neurotransmitter from the brain, which will be detected using fluorescence spectroscopy. Our optical setup is designed for resorufin detection, which is a fluorescent dye used in a variety of commercially available assay kits (see S2† for more details).To demonstrate the possibility to detect resorufin at a sufficiently low concentration, the optical setup is first characterized with respect to the limit of detection for resorufin sodium salt. Fig. 6 shows a linear response in the lower concentration range of resorufin from 2 to 20 nM. Analysis of the recorded signal from the photomultiplier tube (PMT) reveals that the limit of detection (LOD) of this system can be estimated to be about 2 nM, which corresponds to 3 times the standard deviation of the blank value (see Fig. S2B†). Note that this LOD is reported for resorufin to determine the lower limitations of the optical setup. Basal glutamate levels in the brain are typically 3 orders of magnitude larger fluctuating around a few μM.38
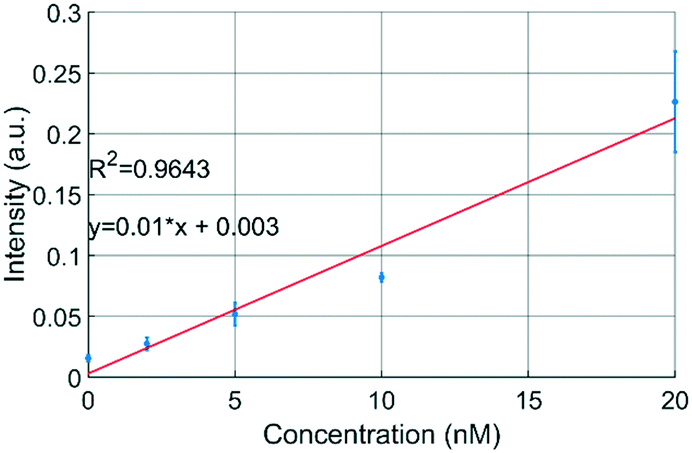 | ||
| Fig. 6 Fluorescence measurements with resorufin sodium salt demonstrate that concentrations <2 nM can be measured using the developed optical detection setup (see text for details). Resorufin was injected directly into the optical detection capillary (n = 3). | ||
In vitro sampling
Next, the sampling capability of probe type 1 was verified using fluorescein sodium salt. An aqueous stock solution of 10 mM fluorescein was prepared and a titration experiment was done in a 10 mL sample cup with a magnetic stirrer (500 rpm). Flow rates of 200 nL min−1 decane, 50 nL min−1 perfusion with demineralized water were used, and the outlet pressure was set to −80 kPa. Stepwise addition of 200 μL of stock solution with intervals of 100 seconds resulted in the fluorescence intensity plot of Fig. 7A. LED power was set to 300 μW. | ||
| Fig. 7 In vitro sampling from a 10 mL sample cup. A: Stepwise addition of fluorescein sampled with a probe type 1 having a droplet generator embedded in the needle. B: Relation between fluorescent readout and fluorescein concentration (red fitting curve) (n = 3), extracted from the green plateaus from the titration curves of Fig. 7A and S3B.† The blue line indicates a calibration curve with fluorescein directly infused into the detection capillary. C: Stepwise addition of glutamate sampled with a probe type 2 having an external droplet generator in which glutamate oxidase, horseradish peroxidase and Amplex red are added. In the presence of glutamate, Amplex red is converted to highly fluorescent resorufin. The peaks at 2.5 min (Fig. 7A) and 7.5 min (Fig. 7B) are residual fluorophore resulting from earlier conducted staircase experiments. D: Relation between fluorescent readout and glutamate concentration, extracted from the green plateaus indicated in Fig. 7B. | ||
For glutamate measurements using the type 2 probe, a commercial assay kit based on glutamate oxidase and the fluorescent marker resorufin was used. The design of the μPPPS probes does not allow for the addition of the enzymes and amplex red to the droplets, and adding these to the dialysate flow before the sampling area is undesirable for obvious reasons. Therefore, the push–pull probe with external droplet generator was fabricated, which allows one to add the components of the assay kit to the sample flow just before the droplet-generating T-junction. Analogous to the fluorescein titration experiment, a 10 mL sample cup was used with 1× PBS and a magnetic stirrer (500 rpm). A stepwise addition of 20 μL of 1 mM L-glutamate with intervals of 100 seconds resulted in the fluorescence intensity plot of Fig. 7C. Flow rates were set to 500 nL min−1 decane, 200 nL min−1 GlOx/HRP/Amp. Red mixture, 100 nL min−1 perfusion with sterile perfusion fluid and an outlet pressure of −50 kPa was used. A typical titration curve for fluorescein is indicated in Fig. 7A, and is representative of comparable curves obtained from a total of n = 3 experiments (see S3B†). From this curve it can be seen that a sudden increase of approximately 180 μM in concentration can be measured within 3 seconds. This delay of 3 seconds can be explained in part by the time needed for the added fluorescein to mix in the sample cup, and in part by the Taylor dispersion occurring in the needle between sampling channels and droplet generator. The relation between fluorescein concentration and fluorescent readout is calculated by an algorithm that cuts off any data-point below a minimum threshold that is 80% of the maximum plateau value. Comparing the intensity values measured for the droplets (n = 3) to a calibration curve recorded at the same LED power (see Fig. 7B), the recovery rate is estimated at 61%. However, as can be seen from the insets in Fig. 7, the fluorescent signal from the droplets never reach a plateau value. It is therefore not unlikely to assume that the real recovery rate might be even higher.
Glutamate titration using the probe type 2 (Fig. 7C) showed a slower response, roughly double the rise time observed with fluorescein. In addition to the time required for the glutamate to mix in the sample cup, the longer transfer distance between the tip of the probe and the external droplet generator causes some extra Taylor dispersion that adds to the total response time. Nonetheless, our results indicate that the compartmentalization in droplets are a significant improvement compared to the several minute time scale results shown using traditional dialysis probes combined with either flow injection analysis,20 or fluorescent readout in continuous flow.21
The relation between glutamate concentration and fluorescent readout is calculated in an identical manner as for the fluorescein experiments. The algorithm cuts off any data-point below a minimum threshold that is 80% of the maximum plateau value. Next, the mean of each plateau (green lines in Fig. 7C) is calculated, as shown in Fig. 7D. The sensitivity is 0.38 a.u. μM−1. Additional experiments (n = 2 in S4†) showed similar results, albeit with slightly different sensitivities of 0.45 and 0.30 a.u. μM−1. Linearity was good with R2 values between 0.9918 and 0.9969. A small offset is present at zero concentration, this could be due to known issues of self-formation of resorufin in the absence of glutamate with the Amplex Red assay kit.39 Based on the calibration curves shown in Fig. 7D and S4† it is expected that our setup should be sensitive enough to detect both basal levels (few μM) and bursts of glutamate (∼40 μM) in vivo.24,38 Compared to the fluorescein titrations, reproducibility was lower for experiments conducted with the glutamate assay kit, which was largely due to variable enzyme activities across (purchased) batches. The glutamate assay kit is sensitive to small changes in temperature during the droplet transfer, and to a lesser degree small variations in timing between the start of the reaction and the optical readout. Moreover, if resorufin is in direct contact with the walls of the channel adsorption can occur, leading to drift in the fluorescence intensity measurements.
In vivo recordings
To demonstrate that the μPPPS probes are not only working in an in vitro environment, but are also functional in in vivo experiments, we used probe type 2 with external droplet generator to record changes in glutamate levels in the brain of an anesthetized mouse. As a proof of principle, such changes were recorded in the right sensorimotor cortex while cortical activity was evoked in the right sensorimotor cortex using transcallosal electrical stimulation in the left hemisphere40,41 (see Fig. 8). It has been demonstrated by others that increased cortical network activity is paralleled by increased extracellular levels of glutamate in vivo.42,43 Experiments were successfully performed in two mice, in which an electrical stimulation paradigm was repeated three times. Each stimulation paradigm consisted of a set of 6 biphasic 1 ms pulses at 70% of the maximum cortical response, delivered at 0.1 Hz. The resulting glutamate responses, as well as the recordings from a standard glass electrode are shown in Fig. 8. A simple staircase recording, similar to what is shown in Fig. 7 was conducted prior to insertion of the probe in the brain to check for proper functioning of the setup. From the delay observed between in vitro addition of glutamate and time of detection, the delay time was estimated to be approximately 4.25 minutes. This approximate delay time was used in post hoc analysis for shifting the droplet trace to the left (in case of the example in Fig. 8 by a shift of 4.25 minutes). Fig. 8 shows representative traces from one of the mouse experiments, with subsequent electrical stimulation of brain activity and related changes in fluorescent signal, as measured by the μPPPS probe, indicative of altered cortical glutamate levels. In the shown example, fluorescent signals from the sampled droplets increased following cortical electrical stimulation, either directly (at 10 and 17 min), or with a delay (25 min), in line with pronounced cortical burst activity (see inset). Observations are in line with expected rises in cortical glutamate levels during the evoked cortical activity.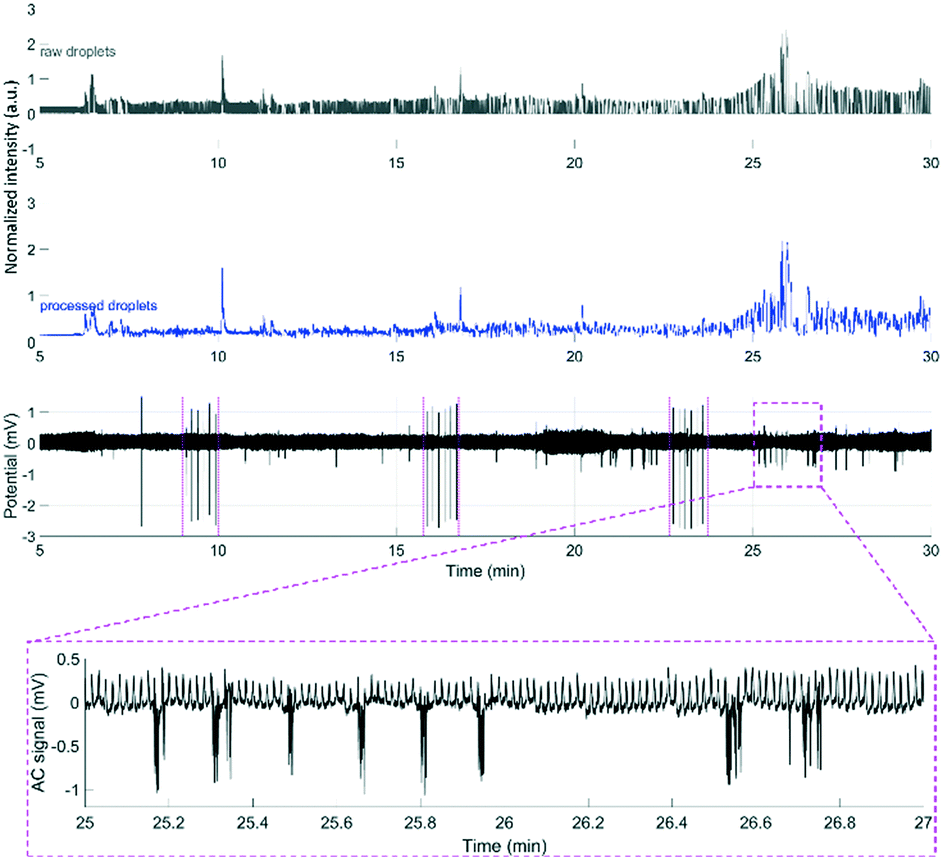 | ||
| Fig. 8 Example recordings of glutamate during an in vivo experiment using the μPPPS probe with external droplet generator for measuring neurotransmitter glutamate changes during evoked cortical activity by electrical stimulation on the left sensorimotor cortex (periods including the stimulus pulses, visible on the ECoG trace, are indicated by magenta-colored dashed lines around 9, 17 and 23 minutes) in an anesthetized mouse. See Fig. 6 for a schematic of the experimental approach. Shown are the normalized raw optical droplet read-out (grey) from the right sensorimotor cortex, the processed droplet signal (blue), and alternating current (AC) (black) local field potential “electrocorticogram” (ECoG) recordings from a glass electrode inserted in superficial layers of the right sensorimotor cortex. For the first two stimulation sessions, the glutamate signal shows a transient increase directly following cortical stimulation, whereas in the third session the glutamate signal increased with a delay, in line with a pronounced burst of cortical activity that is evident on the AC ECoG recording (see additional inset showing details of the AC response at higher time resolution). | ||
Conclusions and outlook
Here we have demonstrated the design and fabrication of a miniaturized push–pull perfusion sampling probe with a width of 140 μm and a height of 140 μm. Molecules (including neurotransmitters) enter the probe through 20 small sampling channels (20 × 3 μm2) located at the end of the probe, effectively providing a sampling area of approximately 1200 μm2. A recirculating, U-shaped channel in the tip of the needle, operated in a push–pull-like fashion ensures fast transport of sample to the in-house developed external measurement setup. The detection method used for the in vitro and in vivo glutamate experiments in this study is based on fluorescence spectroscopy using a sensitive photomultiplier tube, and (in combination with μPPPS probes of type 2) the mixing of externally generated droplet samples with glutamate oxidase and fluorophore precursor Amplex Red. The detection limit for the fluorescent reaction product of the Amplex Red assay kit, resorufin, in our experiments was ∼2 nM. Immediately after sampling, the dialysate is encapsulated into water-in-decane droplets to prevent dispersion, and to enhance the temporal resolution. In vitro experiments using a solution containing fluorescein demonstrated a recovery rate of 61%, while step-wise changes in concentration could be measured within 3 seconds. As proof of principle, transient changes in brain glutamate levels were recorded in vivo in the cortex of anesthetized mice that were electrically stimulated on the contralateral cortical hemisphere. These experiments demonstrated a correlation between evoked cortical network activity and enhanced glutamate release, as indicated by rises in fluorescent signal.Whereas our in vivo pilot data are encouraging, there is still a challenge with the μPPPS probe and fluorescent measurement approach. Small variations in droplet generation rate (in the order of a couple of seconds at most), which could occur due to technical changes in the fluidic system, will cause small fluctuations in the timing between brain activity changes and our detection of droplet signals, which can result in a larger cumulative variation due to the relatively long time between sampling and detection (4–5 minutes). This issue can be solved by detecting neurotransmitters immediately inside the body of the probe. This could be done optically (as we used in our study for detection outside of the probe), but, based on unpublished results from our team, it should also be possible to measure glutamate by electrochemical means inside droplets. In that scenario, inclusion of a separate channel for calibration fluids would allow periodic recalibration of such electrochemical measurements. An additional benefit offered by switching to a different detection method is to prevent the issues observed with the glutamate assay kit obtaining reproducible activity, which make the glutamate assay kit not suitable for quantitative measurements.
In our current design we did not include additional functionality (e.g. measuring electrical activity) to the glass cover of the probe during fabrication. As we have shown in previous work,23 it would be relatively straightforward to also include AC- and/or DC-EEG electrodes to create a hybrid probe that combines microfluidic sampling with brain activity recordings. Moreover, it would be feasible in the future to also include waveguides to enable optogenetic stimulation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
STW is gratefully acknowledged for funding this VENI project (grant no. 13607). The Netherlands Organ-on-Chip Initiative (NOCI), a Gravitation project (024.003.001) is acknowledged, funded by the Netherlands Wetenschaps Organisatie (NWO). We thank Jan van Nieuwkasteele for help with optics and camera recordings, Hans de Boer for assistance with the optical suitcase and probe positioner, Vasilis Papadimitriou for help with capturing the droplet formation on video, Karin Groothuis-Oudshoorn (University of Twente) and Jelle Goeman (Leiden University Medical Center) for their assistance with data statistics. The authors would like to thank Dr. Pieter Roelfsema (Dutch Brain Institute) and Gunnar Flik (Charles River Laboratories, Groningen) for fruitful discussions.References
- R. T. Kennedy, Curr. Opin. Chem. Biol., 2013, 17, 860–867 CrossRef CAS.
- T. I. F. H. Cremers, M. G. de Vries, K. D. Huinink, J. P. van Loon, M. v d Hart, B. Ebert, B. H. C. Westerink and E. C. M. De Lange, J. Neurosci. Methods, 2009, 178, 249–254 CrossRef CAS.
- C. J. Watson, B. J. Venton and R. T. Kennedy, Anal. Chem., 2006, 78, 1391–1399 CrossRef.
- N. A. Cellar, S. T. Burns, J. C. Meiners, H. Chen and R. T. Kennedy, Anal. Chem., 2005, 77, 7067–7073 CrossRef CAS.
- T. R. Slaney, J. Nie, N. D. Hershey, P. K. Thwar, J. Linderman, M. A. Burns and R. T. Kennedy, Anal. Chem., 2011, 83, 5207–5213 CrossRef CAS PubMed.
- S. Kottegoda, I. Shaik and S. A. Shippy, J. Neurosci. Methods, 2002, 121, 93–101 CrossRef.
- S. Qin, M. van der Zeyden, W. H. Oldenziel, T. I. F. H. Cremers and B. H. C. Westerink, Sensors, 2008, 8, 6860–6884 CrossRef CAS.
- B. K. Day, F. Pomerleau, J. J. Burmeister, P. Huettl and G. A. Gerhardt, J. Neurochem., 2006, 96, 1626–1635 CrossRef CAS PubMed.
- J. J. Burmeister, K. Moxon and G. A. Gerhardt, Anal. Chem., 2000, 72, 187–192 CrossRef CAS.
- O. Frey, T. Holtzman, R. M. Mcnamara, D. E. H. Theobald, P. D. Van Der Wal, N. F. De Rooij, J. W. Dalley and M. Koudelka-hep, Biosens. Bioelectron., 2010, 26, 477–484 CrossRef CAS PubMed.
- Brainlink, https://www.brainlink.nl/our-products/p/normal-cns-probes/1, (accessed 23 November 2018).
- R. T. Kennedy, J. E. Thompson and T. W. Vickroy, J. Neurosci. Methods, 2002, 114, 39–49 CrossRef CAS.
- G. Taylor, Proc. R. Soc. A, 1953, 219, 186–203 CAS.
- G. Petit-Pierre, P. Colin, E. Laurer, J. Déglon, A. Bertsch, A. Thomas, B. L. Schneider and P. Renaud, Nat. Commun., 2017, 8, 1239 CrossRef PubMed.
- T. Ngernsutivorakul, D. J. Steyer, A. C. Valenta and R. T. Kennedy, Anal. Chem., 2018, 90, 10943–10950 CrossRef CAS.
- M. Wang, T. Slaney, O. Mabrouk and R. T. Kennedy, J. Neurosci. Methods, 2010, 190, 39–48 CrossRef CAS PubMed.
- M. Wang, N. D. Hershey, O. S. Mabrouk and R. T. Kennedy, Anal. Bioanal. Chem., 2011, 400, 2013–2023 CrossRef CAS PubMed.
- P. Song, N. D. Hershey, O. S. Mabrouk, T. R. Slaney and R. T. Kennedy, Anal. Chem., 2012, 84, 4659–4664 CrossRef CAS PubMed.
- S. Feng, G. Liu, L. Jiang, Y. Zhu, E. M. Goldys and D. W. Inglis, Appl. Phys. Lett., 2017, 111, 183702 CrossRef.
- J. B. Gramsbergen, J. Skjøth-Rasmussen, C. Rasmussen and K. L. Lambertsen, J. Neurosci. Methods, 2004, 140, 93–101 CrossRef CAS PubMed.
- A. Morales-Villagrán, K. Pardo-Peña, L. Medina-Ceja and S. López-Pérez, J. Neurochem., 2016, 139, 886–896 CrossRef PubMed.
- W. H. Lee, T. R. Slaney, R. W. Hower and R. T. Kennedy, Anal. Chem., 2013, 85, 3828–3831 CrossRef CAS.
- M. Odijk, E. J. J. van der Wouden, W. Olthuis, M. D. D. Ferrari, E. A. A. Tolner, A. M. J. M. M. J. M. van den Maagdenberg and A. Van Den Berg, Sens. Actuators, B, 2015, 207, 945–953 CrossRef CAS.
- K. N. Hascup, E. R. Hascup, F. Pomerleau, P. Huettl and G. A. Gerhardt, J. Pharmacol. Exp. Ther., 2008, 324, 725LP–731 CrossRef.
- W. H. Lee, T. Ngernsutivorakul, O. S. Mabrouk, J. T. Wong, C. E. Dugan, S. S. Pappas, H. J. Yoon and R. T. Kennedy, Anal. Chem., 2016, 88, 1230–1237 CrossRef CAS PubMed.
- Y. Kim, H. J. Lee, D. Kim, Y. K. Kim, S. H. Lee and E. Yoon, in Transducers conference, 2013, pp. 876–879 Search PubMed.
- R. Rathnasingham, D. R. Kipke, S. C. Bledsoe and J. D. McLaren, IEEE Trans. Biomed. Eng., 2004, 51, 138–145 CrossRef PubMed.
- M. Perry, Q. Li and R. T. Kennedy, Anal. Chim. Acta, 2009, 653, 1–22 CrossRef CAS.
- E. R. Hascup, K. N. Hascup, M. Stephens, F. Pomerleau, P. Huettl, A. Gratton and G. A. Gerhardt, J. Neurochem., 2010, 115, 1608–1620 CrossRef CAS.
- M. D. Johnson, R. K. Franklin, M. D. Gibson, R. B. Brown and D. R. Kipke, J. Neurosci. Methods, 2008, 174, 62–70 CrossRef CAS.
- J. Voipio, M. Pasternack and K. Macleod, in Microelectrode techniques, The Plymouth workshop handbook, ed. D. Ogden, The company of Biologists, Cambridge UK, 2nd edn, 1994, pp. 275–316 Search PubMed.
- E. Bakker, P. Bühlmann and E. Pretsch, Chem. Rev., 1997, 97, 3083–3132 CrossRef CAS.
- J. P. Seymour, F. Wu, K. D. Wise and E. Yoon, Microsyst. Nanoeng., 2017, 3, 16066 CrossRef.
- F. van den Brink, A. Asthana, J. Bomer, E. Tolner, A. van den Maagdenberg and M. Odijk, 21st Int. Conf. Miniaturized Syst. Chem. Life Sci., 2017, pp. 215–216 Search PubMed.
- A. C. F. Ribeiro, M. M. Rodrigo, M. C. F. Barros, L. M. P. Verissimo, C. Romero, A. J. M. Valente and M. A. Esteso, J. Chem. Thermodyn., 2014, 74, 133–137 CrossRef CAS.
- L. N. Mizerovsky and K. P. Smirnova, Russ. Chem. Bull., 2010, 59, 673–676 CrossRef CAS.
- A. M. A. Dias, R. P. Bonifácio, I. M. Marrucho, A. A. H. Pádua and M. F. Costa Gomes, Phys. Chem. Chem. Phys., 2003, 5, 543–549 RSC.
- J. J. Burmeister and G. A. Gerhardt, Anal. Chem., 2001, 73, 1037–1042 CrossRef CAS PubMed.
- B. Zhao, F. A. Summers and R. P. Mason, Free Radical Biol. Med., 2012, 53, 1080–1087 CrossRef CAS PubMed.
- G. Tsenov and P. Mareš, Physiol. Res., 2007, 56, 485–491 CAS.
- E. A. Tolner, D. W. Hochman, P. Hassinen, J. Otáhal, E. Gaily, M. M. Haglund, H. Kubová, S. Schuchmann, S. Vanhatalo and K. Kaila, Epilepsia, 2011, 52, 104–114 CrossRef PubMed.
- M. H. Millan, T. P. Obrenovitch, G. S. Sarna, S. Y. Lok, L. Symon and B. S. Meldrum, Epilepsy Res., 1991, 9, 86–91 CrossRef CAS PubMed.
- L. Medina-Ceja, K. Pardo-Peña, A. Morales-Villagrán, J. Ortega-Ibarra and S. López-Pérez, BMC Neurosci., 2015, 16(1), 11 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8lc01137k |
| This journal is © The Royal Society of Chemistry 2019 |