
Coupling of an atmospheric pressure microplasma ionization source with an Orbitrap Fusion Lumos Tribrid 1M mass analyzer for ultra-high resolution isotopic analysis of uranium†
Edward D.
Hoegg
 a,
Simon
Godin
b,
Joanna
Szpunar
b,
Ryszard
Lobinski
b,
David W.
Koppenaal
c and
R. Kenneth
Marcus
*a
a,
Simon
Godin
b,
Joanna
Szpunar
b,
Ryszard
Lobinski
b,
David W.
Koppenaal
c and
R. Kenneth
Marcus
*a
aDepartment of Chemistry, Clemson University, Clemson, SC 29634, USA. E-mail: marcusr@clemson.edu
bCNRS, Institute for Analytical & Physical Chemistry of the Environment & Materials, UPPA, IPREM, UMR 5254, Helioparc 2, Av Pr Angot, F-64053 Pau, France
cPacific Northwest National Laboratory, EMSL, 902 Battelle Blvd, Richland, WA 99354, USA
First published on 28th May 2019
Abstract
The coupling of a combined atomic and molecular (CAM) ionization source, the liquid sampling-atmospheric pressure glow discharge (LS-APGD) microplasma to an Orbitrap Fusion Lumos Tribrid 1M mass spectrometer is described as a significant step towards the elimination of isobaric interferences in elemental/isotopic mass spectrometry. The developed setup permits broadband, elemental mass spectra to be easily acquired at a mass resolution (m/Δm) of 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. A new ion source housing and integrated control system provide enhanced stability and sensitivity of the microplasma ion source in comparison to the previous designs. Detailed evaluation of the ion sampling parameters provided insight into differences with other Orbitrap platforms leading to a 5-fold increase in resolution in elemental/isotopic analysis. The roles of data acquisition/ion processing parameters were evaluated with regards to the precision and accuracy of 235U/238U isotope ratios. Significantly, the precision of those measurements improves as a function of pre-set mass resolution up to a value of 1000000, where a precision of 0.086% RSD is obtained for uranium concentrations of 100 ng mL−1. Analytical response curves (log–log), acquired for the two uranium isotopes at a resolution of 120000 were linear (R2 = 0.9969) over more than 5 orders of magnitude, with limits of detection of 1 pg mL−1 for U. Based on 60 μL injection volumes, this represents a mass of 70 fg 235U. These figures of merit satisfy International Target Values (ITVs) for Measurement Uncertainties in Safeguarding Nuclear Materials set by the International Atomic Energy Agency (IAEA). Ultimately, and totally unique from beam-type instruments, the move to the higher-resolution Orbitrap platform provides all of the benefits of high mass resolution, with no penalties in sensitivity, precision, or dynamic range for uranium analysis.
000. A new ion source housing and integrated control system provide enhanced stability and sensitivity of the microplasma ion source in comparison to the previous designs. Detailed evaluation of the ion sampling parameters provided insight into differences with other Orbitrap platforms leading to a 5-fold increase in resolution in elemental/isotopic analysis. The roles of data acquisition/ion processing parameters were evaluated with regards to the precision and accuracy of 235U/238U isotope ratios. Significantly, the precision of those measurements improves as a function of pre-set mass resolution up to a value of 1000000, where a precision of 0.086% RSD is obtained for uranium concentrations of 100 ng mL−1. Analytical response curves (log–log), acquired for the two uranium isotopes at a resolution of 120000 were linear (R2 = 0.9969) over more than 5 orders of magnitude, with limits of detection of 1 pg mL−1 for U. Based on 60 μL injection volumes, this represents a mass of 70 fg 235U. These figures of merit satisfy International Target Values (ITVs) for Measurement Uncertainties in Safeguarding Nuclear Materials set by the International Atomic Energy Agency (IAEA). Ultimately, and totally unique from beam-type instruments, the move to the higher-resolution Orbitrap platform provides all of the benefits of high mass resolution, with no penalties in sensitivity, precision, or dynamic range for uranium analysis.
Introduction
The field of atomic mass spectrometry has been dominated for the last 3 decades by the inductively coupled plasma (ICP) ionization source,1,2 coupled to a diversity of mass analyzers, predominately quadrupole, but also sector field and recently time-of-flight. Quadrupoles provide low cost, robust platforms, that operate under conditions of unit mass resolution, though suffering from the impossibility of resolving potential isobaric (same nominal mass) interferences. In order to alleviate isobaric interferences, multi-quadrupole geometries have been implemented to impart chemical resolution prior to the mass analysis step.3,4 A number of successful applications have been developed using triple quadrupole systems, as a means of pre-sorting/selecting ions prior to identity altering chemical reactions within the second mass filter.5 In complex systems, quadrupole-based systems may not be sufficient to separate potential isobars based solely through chemical reactions, demanding the need for high resolution analysers. Likewise, quadrupole analyzers fall short in terms of the precision/accuracy requirements in many isotope ratio analyses.For those instances where greater mass resolving powers are required, or where isotope ratio measurements are to be improved, sector field (SF) geometries are the commercially available alternative platforms for ICP-MS.6–8 In this regard, mass-scanning and multi-collector arrangements can be employed to deliver mass resolving powers of m/Δm = 3000–10000. Scanning-mode instruments provide greater analytical flexibility in terms of multielement analysis, particularly where unknown species need to be identified. Multi-collector instruments are the instruments of choice for applications in isotope ratio mass spectrometry (IRMS).8 As a general rule, ICP-MS can very much be considered a mature technology, suitable for many applications, but not evolving to deliver measurements of greater performance with regards to directly alleviating isobaric interferences.
Over the last 25 years, there have been a couple of reports describing the interfacing of plasma sources to mass spectrometers of far greater mass resolution than SF platforms.9,10 Specifically, Eyler and co-workers9 interfaced an ICP source to a 7 T Fourier transform-ion cyclotron resonance (FT-ICR) instrument. To affect such a coupling, extensive increases in the vacuum pumping capacity and additions of ion transfer optics had to be implemented. In this sole publication, the authors demonstrated a mass resolving power of ∼10000, achieved for the separation of 41K and 40ArH. While the mass-resolving power demonstrated in both of these reports is far greater than achieved on SF instruments of that time (and indeed currently available), there were never commercial efforts to deliver such performance to the market. Considerable losses in sensitivity, the need for a large power ICP generator, large Ar consumption and Ar related interferences, have provided the impetus for the development of microplasma sources capable of analyte atomization and ionization to be coupled with ultrahigh resolution mass analysers such as FT-ICR or Orbitrap instruments.
The introduction of Orbitrap technologies by Makarov and co-workers11 opened up the area of high resolution mass spectrometry to new adaptors by virtue of the relative compactness of the instruments and alleviating the need for a superconducting magnet. While there are a variety of Orbitrap designs and performance benchmarks, mass resolving powers of >100000 are fairly routine. Orbitraps are now a staple in the area of ultra-high resolution mass spectrometry, with the majority of applications lying in the vein of liquid chromatography (LC) detectors for biomacromolecule analysis.12,13 Based on previous efforts with ICP-FT-MS, Koppenaal and co-workers14 assembled an ICP-Orbitrap instrument looking to obtain ultrahigh mass resolution on a simpler platform, though substantial modification of the base spectrometer vacuum system were still required. From that instrument, the geochronologically-important 87Sr–87Rb isotope pair was near-baseline resolved, with an operating resolution of ∼370000 demonstrated.
In 2011, Marcus and Koppenaal15,16 proposed to couple the liquid sampling-atmospheric pressure glow discharge (LS-APGD) microplasma ionization source to an Orbitrap Exactive instrument. Unique to that coupling, no modifications to the commercial mass spectrometer vacuum system or ion optics were required. (A conceptually-similar, but practically different, approach has been described by Shelley and co-workers using a solution cathode glow discharge (SCGD).17,18) Since the earliest works, a number of studies have gone on to describe the operating characteristics of the coupling, with particular emphasis on its performance in applications to uranium isotope ratio measurements.19–23
The resolution achieved for the uranium isotopes on the previous Exactive and Q-Exactive instruments, including Q-Exactive Focus and Q-Exactive Plus instruments, has ranged between 70000 and 120000 depending on the instrument in question. The latter case was sufficient to isolate the nominal isobars of 236U16O2+ and 235U16O17O+.22 As more complex samples are analyzed, and lower concentrations are probed, that level of resolution may not be acceptable. Greater challenges may be particularly profound for f-elements, where diverse combinations of M+, MOx+, MOx(H2O)y+, etc. may be produced. The advent of the Orbitrap Fusion Lumos 1M Tribrid Mass Analyser with the ability to deliver mass resolving powers approaching m/Δm = 1000000 (specified at m/z = 200 Da)24 has a place to reveal isotopic fine structure in organic compounds but also opens new perspectives in inorganic analysis by atomic MS. As recently demonstrated with the LS-APGD:Tribrid coupling, the ability to achieve resolution of >1.7 M has allowed the separation of 87Sr and 87Rb.25 Particular examples of challenges in the area of nuclear forensics are the separation of 238U vs.238Pu, requiring a resolution of ≈195000 and 129I and 129Xe requiring a resolution of ≈635000. To be sure, routine resolution of >500000, as demonstrated here, can be usefully applied in a large number of instances of elemental/isotopic/molecular analyses.
The goals of this research were the operational improvement of the previously developed LS-APGD ion source, its coupling to the Orbitrap Fusion Lumos 1M Tribrid mass analyser, and a parametric optimization and the evaluation of analytical performance of the resulting system. We describe a new, integrated ion source assembly and control unit that has been developed to improve further both the analyte responsivities and measurement precision. Key ion sampling parameters were evaluated to understand their roles in ion throughput and spectral composition. Additionally, ion transient sampling parameters were evaluated to determine their effects on measurement precision. Ultimately, we address questions of how the quantitative aspects of elemental/isotopic analysis are influenced by the higher mass resolution measurement. Stated another way, do the obvious benefits of high mass resolution in terms of complex sample analysis come with sacrifices in terms of quantitative figures of merit as in beam-type instruments?
Experimental
In terms of the LS-APGD ion source, a major development was the design of an enclosed ion source housing and integrated control box. Fig. 1 is an expanded view of the source housing, with the essential operation components. The design and machining were performed by the Clemson University, Department of Physics machine shop. The assembly is designed to mount to any Orbitrap-based instrument, and mounts directly to the bulkhead of the ThermoScientific Orbitrap Fusion Lumos 1M in the place of the commercial electrospray ionization (ESI) source, including the necessary mounting mechanisms and interlocks. Specific details of the source housing design and components are included in the ESI.†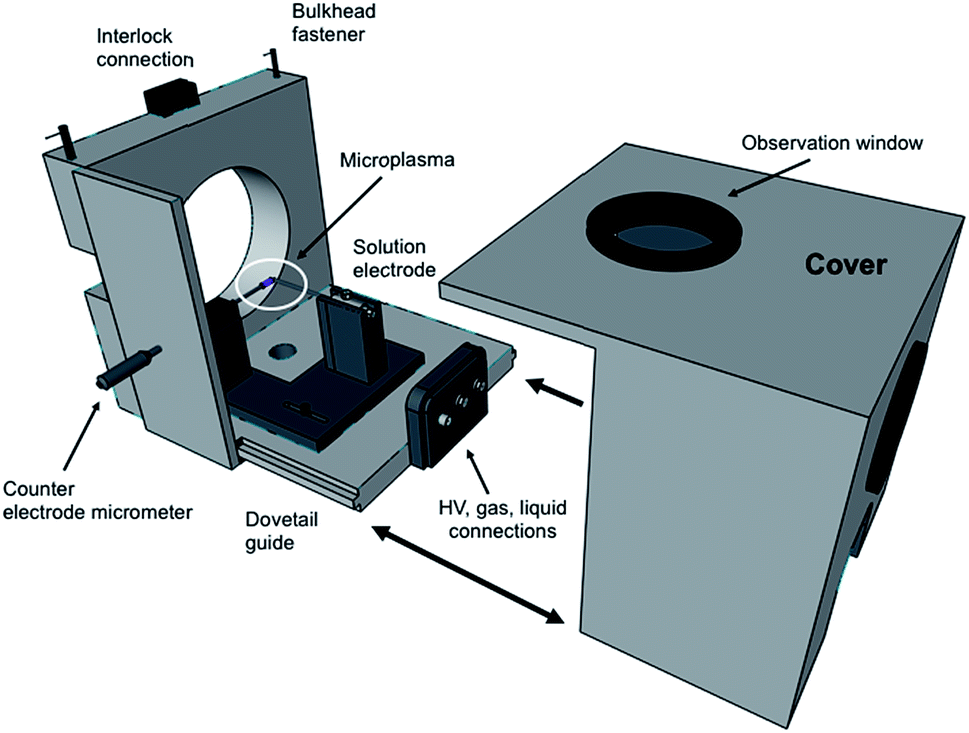 | ||
| Fig. 1 Expanded view of the new LS-APGD ion source housing and components. | ||
The control/utility box was designed based on input and experiences of the research team and assembled by GAA Custom Engineering, LLC (Benton City, WA). The self-contained unit is comprised of a 10 × 5 × 9′′ polycarbonate shell to which the syringe pump is mounted to the top, and the high voltage power supply and sheath gas mass flow controller are mounted within. A 7′′ diagonal display with resistive touchscreen is mounted on the front of the control box, providing input to vary each of the operational parameters. Specific details of the control/utility box design and components are included in the ESI.†
A ThermoScientific (San Jose, CA) Orbitrap Fusion Lumos 1M Tribrid instrument (located at CNRS in Pau, France) was operated without any modifications, other than the replacement of the as-delivered ESI source housing with that of the LS-APGD ion source housing. There are fundamental differences in the ion sampling and transport optics from the previous efforts on Exactive series Orbitrap instruments, which are described in detail in the ESI.† Each of these parameters was evaluated to optimize the performance of the present coupling. Other data acquisition parameters that were investigated included the number of microscans, number of scans, and the preset mass resolution. The overall Lumos operation was controlled under the TUNE software and data processing completed using the QualBrowser software.
The respective discharge parameters and electrolytic solution makeup were held constant throughout the studies, with specific values derived from previous works19 and confirmed during a cursory evaluation performed at the start of this study (liquid flow = 30 μL min−1, sheath gas flow = 0.5 L min−1, and discharge current = 30 mA). Additional specific details are provided ESI.† Research grade helium (Linde, St. Priest, France) was employed as the sheath gas in the LS-APGD source operation. The electrolytic solution for the basic characterization in the elemental analysis mode was 2% HNO3 (Fisher Optima, Pittsburgh, PA). The aqueous, primary test mixture was composed of Rb, Ag, Ba, Pb, and U, all at a concentration of 0.5 μg mL−1, prepared from primary solutions from High Purity Standards (Charleston, SC). The U test solution can be traced to a U isotopic reference material (CRM 129a, New Brunswick Laboratory, Argonne, IL), provided by High Purity Standards and employed in all studies directed towards characterizing the isotope ratio measurement performance. The certified 235U/238U value for the CRM is 0.0072614.
Results and discussion
The much higher mass resolution of the Orbitrap Fusion Lumos 1M Tribrid results from both changes in the Orbitrap cell geometry as well as improved electronics and longer transient sampling times (effecting ion sampling rates and potential susceptibility to drift). Critical ion sampling (optics) and data acquisition parameters (microscans per scan) are interrogated as to their effects on analyte responsivity, isotope ratio values, and their precision.Optimization of ion sampling conditions
In comparison with the previously described coupling with the Exactive version of the Orbitrap, there are fundamental differences in how ions are sampled in the Lumos implementation. Most importantly, the parameters effecting the initial de-clustering of the desired metal ions, in particular removing solvent-related ligands, and eventual transport of ions to the C-trap assembly are quite different as an ion funnel assembly is applied here versus an S-lens optical element. Therefore, a directed optimization exercise was undertaken to understand the roles of those in-source dissociation and ion funnel rf focusing voltages. Following is a brief discussion of these results while the full results can be found in the ESI.†In-source collisional dissociation, affected by the potential applied to the ion transfer capillary (ITC), has long been proven to be an essential tool for removal of loosely-bound solvent species in ESI- and APCI-MS.26,27 The same has been found in the sampling of the LS-APGD microplasma. Using the multielement test solution at a concentration of 500 ng mL−1, the role of the ITC potential on the response of the test elements was evaluated. The general effect was an increase in analyte ion responses up to 80 volts, reflecting the removal of solvent molecules to yield bare metal ions (ESI Fig. S2†). Beyond the 80 V level, it is believed that a general loss in throughput occurs. Notably different from the other test elements, the response for UO2 shows a continuous increase. In this case, the degree of solvent molecule attachment to the UO2+ ion is far more extensive than the other metal ions, and the level of de-clustering continues at higher voltages. Finally, the response of the “spectral background”, which takes the general form of (H2O)nH+, shows a significant decrease with an increase in dissociation energy. In this case, it is clear that these cluster species are indeed effectively reduced using in-source CID.
In addition to optimizing the CID energy, the ion funnel was optimized to effect transport of analyte ions to the mass analyzer. Originally described by Smith et al.,28 the role of the funnel is to focus a spatially dispersed ion cloud (i.e., exiting the ITC) into the primary entrance aperture of the mass analyzer. Each of the ion species increases in response to rf potential to a very broad maxima occurring between ∼100–125 V (ESI Fig. S2†). The parallel responses, and broad range of tunability ensure a level of experimental robustness to minor variations in operation conditions between determinations.
Basic spectral characteristics
Having determined the most beneficial ion source operation and MS sampling conditions, the mass spectrometric aspects of the Lumos 1M were evaluated. The operational mass resolution was set at 500000, as the use of the ultimate 1000000 value would limit the mass range to exclude the lower-mass Rb isotope as the higher-mass elements were desired. Simply, high resolution operation is limited to specific frequency widths at a given time. By operating at a lower resolving power, the operating pressure of the HCD cell was able to be increased without appreciable degradation in the achieved resolution. As such for these experiments the HCD pressure was optimized at 0.012 Torr N2 and the operating potential of the HCD was optimized to 100 eV, similar to those values used in previous work.19Fig. 2 is a broadband mass spectrum of a multielement test mixture composed of Rb, Ag, Ba, Pb, and U, all at concentrations of 500 ng mL−1 in 2% HNO3. Each isotopic packet is labeled with the system-calculated mass resolution for those species. Consistent with previous studies, uranium was observed most prominently as a molecular species, UO2+. Operation at the highest resolution values, by definition, occurs within smaller ion frequency windows than displayed here. As depicted, the mass resolution specification of 500000 at m/z = 200 Da is realized for the Pb species, with higher values attained for the lower-mass analytes. The spectrum here is clearly dominated by the signals of the analyte species, with relatively minute amounts of background species seen. Based on past experience, and confirmed here, these are predominately solvent cluster ions composed of water molecules and nitric acid. Seen as well is the signature of BaOH+, which is 16% of the atomic Ba ion signal. The level of resolution attained here for the BaOH ions would allow resolution of those hydroxides from potential isobars of all of the lanthanide elements as well as lanthanide-oxide related species in that spectral region.
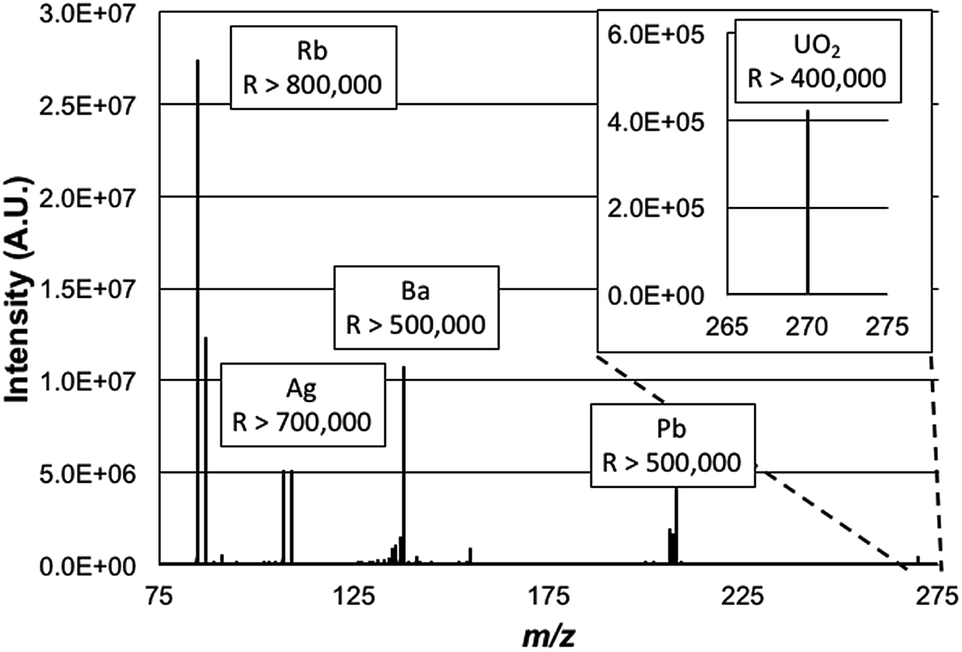 | ||
| Fig. 2 Representative mass spectrum of a multi-element solution containing 500 ng mL−1 each of Rb, Ag, Ba, Pb and U. The spectrum was taken in ‘full scan’ mode using a resolution setting of 500000, specified for m/z = 200 Da. Conditions: discharge current = 30 mA, solution flow rate = 30 μL min−1, and sheath gas flow rate = 0.5 L min−1. | ||
Evaluation of new ion source housing and integrated control system
Most atmospheric pressure ionization sources are actually operated in contained, controlled environments, that operate at ambient pressure. This is true for ESI, APCI, most ADI, and even ICP sources. The reasons for enclosing sources are straight forward; including safety, environmental control, and stability. To this end, the new source assembly was implemented in this study along with the use of the integrated controller box. The control box is designed explicitly to provide safety and stability in terms of the chosen hardware components and software control.To assess the efficacy of the cube enclosure, a 500 ng mL−1 multielement test mixture was continuously fed to the plasma and the variability of the summed total ion chromatogram (TIC) for the signals of the elements determined over a 15 min time frame determined. The mass analyzer was operated at a resolution set at 120000 at m/z = 200 Da. Data sets were taken for the case of the source cover removed and in place, across 3 ion sampling distances. The sampling distances were varied as a means to assess potential influences of ambient air disturbances in the net signals and stability. As seen in Fig. 3, the use of the closed cube apparatus has a pronounced impact on both the net ion signals and the temporal stability of those signals. As might be expected, there is a marked improvement in stability, with the variability decreasing by ∼43% at the normal, closest sampling distance. More surprisingly, the net signal intensity is seen to increase by ∼2× upon enclosing the plasma. The reason for this increased analytical response likely lies in the very different environment surrounding the plasma upon enclosure. In the case of the closed (though not hermetically-sealed) system, there exist a flow of the He sheath gas surrounding liquid electrode (0.5 L min−1) and a steady stream of water vapor through the evolution of the liquid electrolyte (30 μL min−1). Thus, there is surely a net flow of these gases out of the cell, to the exclusion of ambient nitrogen egressing into the plasma region. As such, the potential deleterious effects of N2 on the energetics of the plasma may be negated, thus the higher analytical responsivity. Likewise, any local pressure variations and their perturbation of local aerodynamics are minimized. In addition to more robust operation, there may be some component of enhanced ion transport to, and through, the vacuum/ion sampling orifice as the pressure within the ion source housing is slightly above ambient.
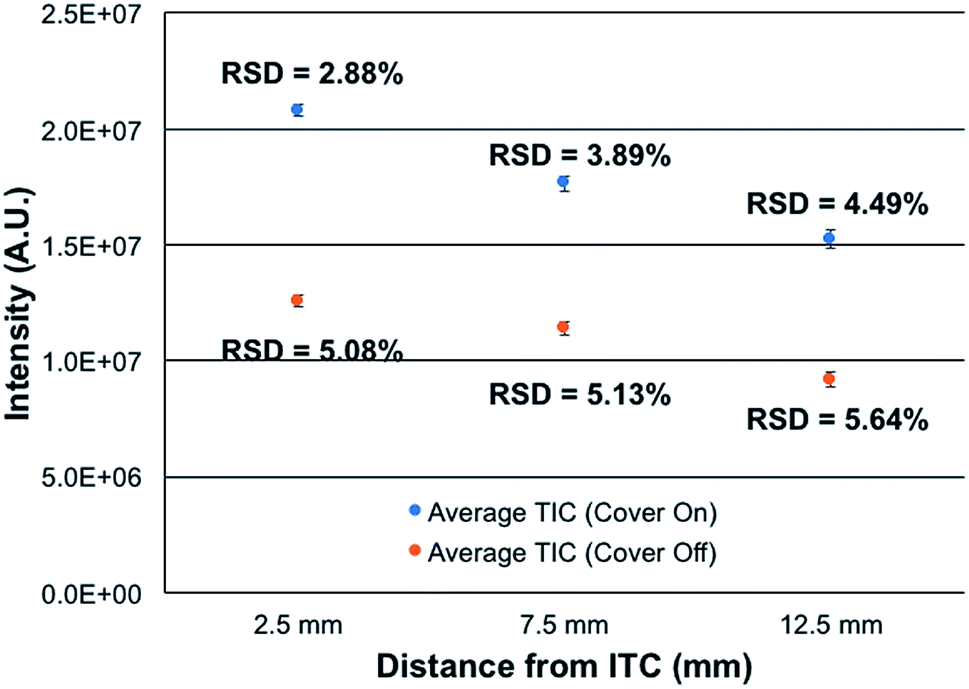 | ||
| Fig. 3 Effect of enclosing LS-APGD plasma source on signal intensity and stability as a function of LS-APGD sampling distance. The intensity was calculated from the total ion chromatogram collected over 15 minutes while directly infusing a multi-element solution containing 500 ng mL−1 each of Rb, Ag, Ba, Pb and U. The data was taken using a resolution of 120000, specified for m/z = 200 Da. Conditions: discharge current = 30 mA, solution flow rate = 30 μL min−1, and sheath gas flow rate = 0.5 L min−1. | ||
The spatial differences seen in both the signal intensity and the plasma stability are not surprising. Previous efforts on Exactive Orbitrap platforms and ion trap instruments reflect similar spatial variations in analyte intensity. In the case of the Orbitrap,16 steady increases in analyte signal-to-background (S/B) response (∼10× overall) were seen across sampling distances from ∼5–12.5 mm, followed by a dramatic drop of >80% at 15 mm. The passage through a sharp spatial maximum was attributed to the competing processes affecting desolvation, ionization, and recombination. The same phenomenon was seen when sampling the microplasma with a quadrupole ion trap.29 The monotonic decreases in intensity as a function of distance depicted in Fig. 4 are very minor, less than 25% across the 2.5–12.5 mm sampling range. There is far less sensitivity to positioning, which bodes well in general operation robustness. While not documented, there may be different gas dynamics/electrostatics involved on the ion sampling on the Lumos 1M than the Exactives, which provide better ion transfer to/through the entrance aperture. Ultimately, the new closed ion source housing and integrated controller system deliver better quantitative performance than the previous, open sources. Additionally, the lack of a critical dependence on ion sampling position makes the physical operation far more forgiving from the point of view of the operator.
 | ||
| Fig. 4 Effect of increasing the number of microscans making up each scan function on the accuracy and precision of the 235U/238U isotope ratio measurement. Each data point represents 3 acquisitions, each of which consisted of 10 scans. CRM 129a test solution = 100 ng mL−1. The data was taken using a resolution of 120000, specified for m/z = 200 Da. Conditions: discharge current = 30 mA, solution flow rate = 30 μL min−1, and sheath gas flow rate = 0.5 L min−1. | ||
Relationship between number of microscans/scans and 235U/238U precision and accuracy
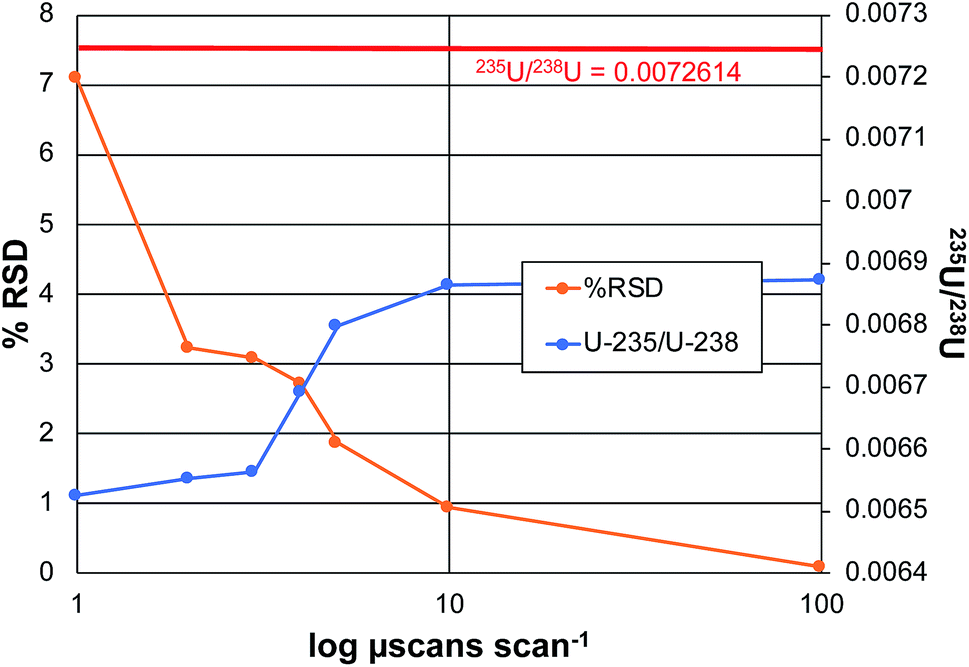
In the realm of quantitative elemental analysis, and furthermore in the field of isotope ratio mass spectrometry, the ability to make determinations with a high level of certainty is a fundamental goal. The practical question is how many times (or how long) must measurements be made to achieve optimum precision and accuracy. In the context of Orbitrap mass analyzers, the number of measurements is determined by the number of samplings of the ion beam, in the form of individual ion packets injected from the C-trap to the analyzer. Each injection produces a detected ion transient (termed a microscan), which can be Fourier-transformed to yield a mass spectrum. In practice, the Excalibur data system processes some number of microscans, which are then averaged prior to performing the Fourier transform to create a scan. The product mass spectrum from each scan can then be processed with other scans to form acquisition sets. In this way, one can take sufficient numbers (n) of microscans/scans/acquisitions to reach the targeted analytical metrics. Based on normal (e.g. poisson) statistics, the signal-to-noise (S/N) ratios should improve in proportion to the number measurements taken, n1/2. Of course, there are practical limitations relative to the cost/time-benefits with increasing the number of measurements.To this point in the method development, the coupling of the LS-APGD with Exactive Orbitrap analyzers, the critical data acquisition parameters have been investigated and optimized as they pertain most specifically to uranium determinations,20–23 but other elements have been characterized as well.19 As a general rule, precision and accuracy improve with number of microscans making up each scan, and the number of scans employed per acquisition. While the n-dependence on precision is expected, accuracy in the case of the disparate 235U/238U ratio (∼0.007), benefit is gained by virtue of gaining greater signal levels for the lower-abundance isotope. Fig. 4 depicts the attained precision and determined isotope ratios for the 235U/238U pair as a function of the number of microscans making up each scan for the case of analyzing CRM 129a, at a concentration of 100 ng mL−1. The precision reflects the variability of the isotope ratio for n = 3 acquisitions, each composed of 10 scans. The left-hand portion of the plot reflects scans composed of 1, 3, 5, 7, 8, and 10 microscans.
As seen, the precision improves dramatically with the number of microscans, almost in direct inverse proportion to a level of ∼1% RSD. Increasing the number to 100 microscans lowers the variability to 0.086%. This protocol requires 15 min for n = 3 measurement cycles; comparing well with those of TIMS and ICP-MS on sector-field instruments as described previously.22 In terms of a possible n1/2 dependence, an increase in total number of microscans from 10 to 1000 should improve the precision by a factor of 10; in fact the improvement is closer to 100×. As a point of comparison, earlier efforts on the Exactive Orbitrap platform yielded a precision of 3.9% RSD for 10 scans of 10 microscans each, for a 5 μg mL−1 CRM 129a sample. To be clear, given the length of transients required to attain the working resolution of 120000 on the Lumos 1M instrument, the present experiment took ∼5 min vs. 3 min for the 25000 resolution Exactive for 10 microscans per scan. A more practical comparison in terms of total analysis time, involved an Exactive with acquisitions composed of 100 scans, of 10 microscans each.23 For that 1 μg mL−1 sample, a value of 0.115% RSD was attained. Other studies on Q-Exactive Plus and Q-Exactive Focus instruments, using a resolution of 120000 and 70000 respectively, yielded virtually the same level of precision seen here, at 0.082% RSD for a 0.5 μg mL−1 concentration. Thus, the Lumos platform yields precision on par with other Orbitraps, but with the potential to likely do far better as each scan on this instrument can include up to 5000 microscans versus 10, albeit at a cost of time. As the S/N values are quite high (above the shot-noise limit), increasing the number of microscans/scans should provide a n1/2 improvement (at least).
The second metric of importance in an isotope ratio determination is accuracy. In all forms of IR-MS, there is some form of mass-related bias in the determinations, and so the use of reference materials as a means of making bias corrections is standard practice. That said, the more accurate the initial raw measurement, the less the dependence on ex post facto corrections. As seen in Fig. 4, there is a definitive increase in the computed 235U/238U value as the number of microscans per scan increases, approaching the certified value. The reason for this improvement is straight forward. As the number of microscans that are co-added increases, the level of the low abundance 235U signal increases at a greater rate than the background that is automatically subtracted across the spectrum based on the software threshold levels. This relationship between the 235U signal and the background has been well documented across all of the works using the Orbitrap analyzer.19,20,23 In all, the range in the amount of bias of the error (∼5.5% relative), and the error in the absolute value at the maximum (∼5% relative), are relatively small versus other mass analyzers. Greater accuracy would of course be realized through the use of an external standard.
Relationship between mass resolution and 235U/238U precision and accuracy
In all forms of dispersive spectrometry (e.g. optical and mass), increases in spectral resolution (obtained by narrowing slits, etc.) are typically accompanied by coincident decreases in analyte signal levels and signal/background. In the case of Fourier transform spectrometers, though, enhanced resolution is attained by increased (longer) sampling of the relevant signals. Indeed, there was no observed change in the product ion signal intensities as resolution was increased over the course of these experiments. Likewise, it is generally true that greater numbers of complete spectra can be acquired per unit time. Thus, improvements in precision are usually realized in Fourier transform spectrometry; this practical consequence is referred to as Fellgett's advantage.30 Thus, while counterintuitive on some level, greater precision is expected as resolution is increased using the same spectrometer, in this case by increases in the ion transient observation time.
Fig. 5 presents the role of the designated spectral resolution (defined at m/z = 200 Da) on the obtained precision of the 235U/238U measurement as well as their absolute values. A 100 ng mL−1 concentration of CRM 129a was provided to the plasma on a continuous basis, with the values presented computed for triplicate (n = 3) acquisitions composed on 10 scans of 10 microscans, each. (This set of acquisitions corresponds to the vertical line in Fig. 4.) As can be seen, the % RSD for the measurements decreases almost monotonically from a value of ∼6.7 to 0.16% across the data set. Of course, the relevant control parameter here is the relative difference in measurement times across this range of resolution values; i.e., reflective of n. In this case, the transient/acquisition times required for increasing the resolution from 30000 to 1000000 is 5×. This translates to a realized improvement in precision by a factor of ∼40×, far more than anticipated. It is not immediately clear as to why the improvement is so large, but it may be the result of an added multiplicative advantage as the two isotopes are sampled at the same time (simultaneously). Thus, there is no sacrifice to precision performance for the sake of improved qualitative performance (i.e., resolution); only a sacrifice in throughput/time.
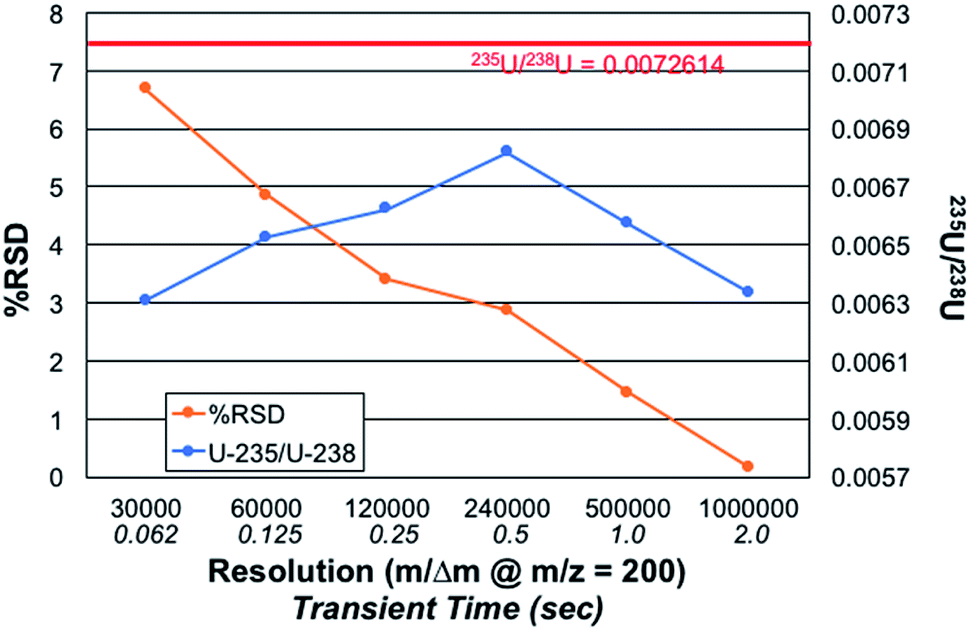 | ||
| Fig. 5 Effect of increasing mass resolution setting (specified at m/z = 200 Da) on the accuracy and precision of the 235U/238U isotope ratio measurement. Each data point represents 3 acquisitions, each of which consisted of 10 scans of 10 microscans. CRM 129a test solution = 100 ng mL−1. Conditions: discharge current = 30 mA, solution flow rate = 30 μL min−1, and sheath gas flow rate = 0.5 L min−1. | ||
The role of mass resolution on the measurement accuracy is complex. As described relative to the role of the number of microscans on accuracy, where greater number of measurements (n) improve S/B, there is a bias against small signals in the case of processing longer transients. The 235U/238U value is initially seen to increase with resolution, until such a point (m/Δm > 240000) whereas diminishing returns are realized. A fundamental bias is imparted in the processing of longer transients as the lower abundance (very much lower in the case of 235U) fall into the noise faster than intense signals. Considered another way, intense signals continue to contribute to the signal transients, while lesser ones cease to contribute. Thus, it is easy to see why the 235U/238U turns to lower values. Herein lies a different level of experimental control that is necessary when performing quantitative analyses via FT-MS methods. Such issues, so long as acknowledged and accounted for, should still allow for high quality quantitative analysis.
Assessment of calibration quality for uranium determinations
One of the earliest observations in the use of three-dimensional trapping mass analyzers was the limited total number of ions (generally stated as 106) that could be accumulated prior to degradation of mass spectrometric quality.31,32 This point was intended to infer that the dynamic range across a large number of species in a given spectrum was limited. Orbitrap control parameters limit the number of injection ions to ∼5 M ions, but a value of 500000 ions has been implemented here to minimize any space charge effects. With the use of various means of limiting the mass range of ions which are injected into an Orbitrap analyzer and the digitization range as well, high sensitivity and dynamic range were realized in the coupling of the LS-APGD and a Q-Exactive Plus Orbitrap.21
The quantification of uranium presents a unique opportunity as the analysis involves isotopes which differ in abundance by more than 2 orders of magnitude. Plotted in Fig. 6 are the overlaid log–log response curves of the 235U and 238U isotopes for total elemental concentrations ranging from 1–1000 ng mL−1 of CRM 129a. The data are the result of n = 3, 60 μL injections of each solution, with the spectra recorded at a mass resolution setting of 120k. As can be seen in the regression data, the linear fit is very well correlated, with the error bars of each concentration lying within the symbols. The linearity observed here covers over 5 orders of magnitude. While there are always questions of the excess weighting of high concentrations in log–log functions in particular, fitting of each of the isotopic responses yields R2-values of >0.99. Limits of detection can be computed based on the response curve characteristics as LOD = 3 (σx)/m, where σx represents the standard deviation of responses of either the blank or the deviation of the signal for the lowest point on the calibration curve and m is the slope of the response function. In the case of the blank-injection variability, the concentration LOD is 1 pg mL−1, with the mass-based value being 70 fg of U. Given the peculiarities of the background correction methods employed in the Excalibur data system, is it advisable to also consider the LODs based on the deviation of the signal at the lowest point on the response curve as another point of reference. In this case, the concentration detection limit is 6 pg mL−1, with the mass-based value being 380 fg of U. These two values are appreciably lower than found on the Q-Exactive Plus platform, which provide a lowest-point LOD of 30 pg mL−1.20 In that case, mass filtering before C-trap injection and extensive use of the HCD dissociation greatly aided in reducing background contributions. The latter was not applied on this Lumos 1M instrument. Thus, here again is evidence of the more efficient ion sampling efficiency (and perhaps throughput) of the Fusion Lumos 1M Tribrid instrument in comparison to the other instruments.
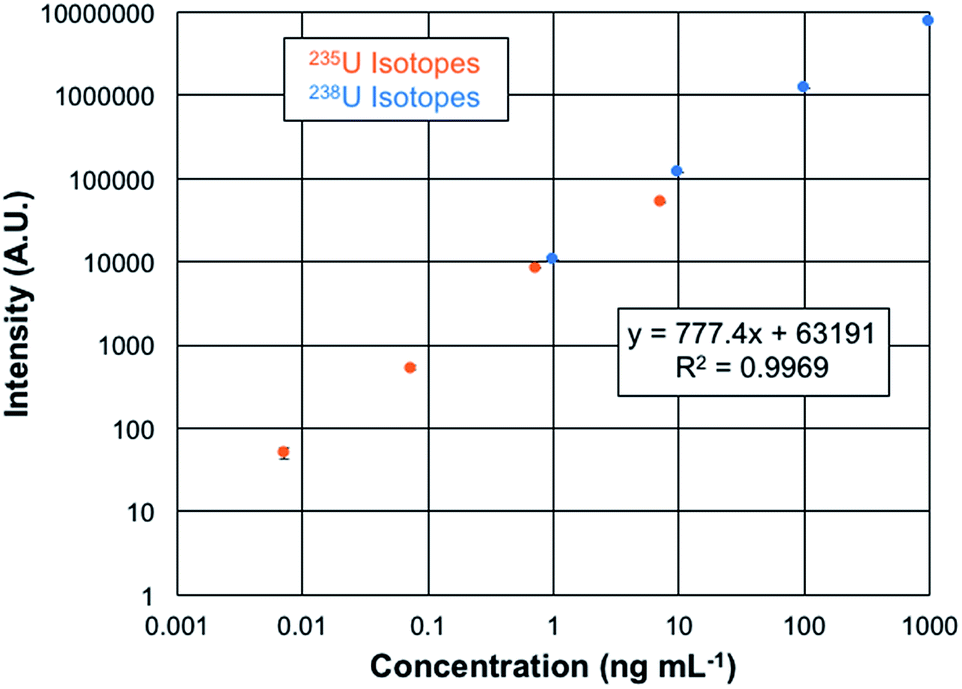 | ||
| Fig. 6 Uranium isotopic response curve for 235U and 238U obtained for serial dilutions of CRM 129a. Each data point consists of three 60 μL injections with responses collected using 10 microscans. A resolution of 120000 was used for these measurements. Conditions: discharge current = 30 mA, solution flow rate = 30 μL min−1, and sheath gas flow rate = 0.5 L min−1. Sodium citrate (100 μg mL−1) was used as the electrolytic solution to minimize sample carryover. | ||
Conclusions
The coupling of the LS-APGD microplasma to an Orbitrap Fusion Lumos 1M Tribrid mass spectrometer provides a unique combination of mass resolution, precision, and sensitivity, that should be of value across many areas of elemental/isotopic analysis. Implementation of a new ion source housing and integrated control system provides transitioning from the normal ESI source to the microplasma in a matter of minutes. Values of m/Δm > 400000 are readily obtained across the entirety of the relevant mass range (broad-band) and are unprecedented for elemental/isotopic analysis. As such, the extensive chemical methods often required to alleviate isobaric interferrents when using quadrupole- or SF-based platforms are not required. The level of precision obtained for these uranium isotope determinations (0.086% RSD) meets the random uncertainty component of 0.20% for natural uranium in the International Target Value (ITVs) for Measurement Uncertainties in Safeguarding Nuclear Materials set by the International Atomic Energy Agency (IAEA).33 While not employed here, use of external standards to bracket isotopic ratios and correct for any mass biases would further improve the isotope ratio performance. The elemental/isotopic detection limits (1 pg mL−1) are suitable for many projected applications. The ability to achieve quantitative responses of >5 orders of magnitude is demonstrated. Different from other high-resolution analyzer approaches, the quantitative aspects of elemental/isotopic analysis are not sacrificed for the sake of mass resolution.
Future efforts will seek to expand on the elemental/isotopic suite of analytes that can benefit from enhanced mass resolution and improvements in precision that bring the capabilities more in line with applications such as geochronology. Among those cases are the recently demonstrated separation of the 87Sr:87Rb pair, with a mass resolution of 1.7 M.25 In addition to increasing the number of measurements (n) and resolution as demonstrated here, it is believed that substantial improvements in precision may be found through optimization of the automatic gain control (AGC) and ion injection times. Much work remains in terms of developing quantitation methodologies, as quantitative methods have not been a prime area of interest the instrument's normal fields of application. Not to be forgotten as a unique aspect of this coupling is the ability of the LS-APGD to operating in modes that allow either atomic or molecular mass spectrometry using the same ionization source.34–36 We refer to this concept as a combined atomic and molecular (CAM) ionization source. The application of the LS-APGD as a CAM ionization source holds great promise in the areas of elemental speciation and metallobiochemistry, where neither the standard ICP nor ESI sources have been used in such a manner.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding for the collaborative project taking place at the IPREM facility in Pau, France was provided by the Environment and Energy Initiative (E2S) and the French National Research Agency (MARSS ANR 11-EQPX-0027 project). Support for the development of the LS-APGD ionization source at Clemson University was provided by the Defense Threat Reduction Agency, Basic Research Award #HDTRA1-14-1-0010; support for development of the LS-APGD ionization source at PNNL was provided by PNNL Laboratory Directed Research & Development (LDRD) funds.References
- Inductively Coupled Plasma Mass Spectrometry, ed. A. Montaser, Wiley-VCH, Weinheim, 1998 Search PubMed.
- D. Beauchemin, Inductively Coupled Plasma Mass Spectrometry Methods, Academic Press Ltd-Elsevier Science Ltd, London, 2017 Search PubMed.
- S. D. Tanner, V. I. Baranov and D. R. Bandura, Spectrochim. Acta, Part B, 2002, 57, 1361–1452 CrossRef.
- E. Bolea-Fernandez, L. Balcaen, M. Resano and F. Vanhaecke, J. Anal. At. Spectrom., 2017, 32, 1660–1679 RSC.
- N. Yamada and J. Takahashi, Bunseki Kagaku, 2018, 67, 249–279 CrossRef CAS.
- N. Bradshaw, E. F. H. Hall and N. E. Sanderson, J. Anal. At. Spectrom., 1989, 4, 801–803 RSC.
- N. Jakubowski, L. Moens and F. Vanhaecke, Spectrochim. Acta, Part B, 1998, 53, 1739–1763 CrossRef.
- D. C. Baxter, I. Rodushkin and E. Engstrom, J. Anal. At. Spectrom., 2012, 27, 1355–1381 RSC.
- K. E. Milgram, F. M. White, K. L. Goodner, C. H. Watson, D. W. Koppenaal, C. J. Barinaga, B. H. Smith, J. D. Winefordner, A. G. Marshall, R. S. Houk and J. R. Eyler, Anal. Chem., 1997, 69, 3714–3721 CrossRef CAS.
- R. K. Marcus, P. R. Cable, D. C. Duckworth, M. V. Buchanan, J. M. Pochkowski and R. R. Weller, Appl. Spectrosc., 1992, 46, 1327–1330 CrossRef CAS.
- Q. Z. Hu, R. J. Noll, H. Y. Li, A. Makarov, M. Hardman and R. G. Cooks, J. Mass Spectrom., 2005, 40, 430–443 CrossRef CAS.
- A. Michalski, E. Damoc, J. P. Hauschild, O. Lange, A. Wieghaus, A. Makarov, N. Nagaraj, J. Cox, M. Mann and S. Horning, Mol. Cell. Proteomics, 2011, 10, 11 CrossRef.
- M. Bantscheff, S. Lemeer, M. M. Savitski and B. Kuster, Anal. Bioanal. Chem., 2012, 404, 939–965 CrossRef CAS PubMed.
- D. W. Koppenaal, A. Carado, C. J. Barinaga, C. D. Quarles, R. K. Marcus, A. Graham, S. J. Ray and G. M. Hieftje, Presented in part at the 2012 Winter Conference on Plasma Spectrochemistry, Tucson, AZ, 2012 Search PubMed.
- R. K. Marcus, C. D. Quarles Jr, C. J. Barinaga, A. J. Carado and D. W. Koppenaal, Anal. Chem., 2011, 83, 2425–2429 CrossRef CAS PubMed.
- C. D. Quarles Jr., A. J. Carado, C. J. Barinaga, D. W. Koppenaal and R. K. Marcus, Anal. Bioanal. Chem., 2012, 402, 261–268 CrossRef.
- A. J. Schwartz, J. T. Shelley, C. L. Walton, K. L. Williams and G. M. Hieftje, Chem. Sci., 2016, 7, 6440–6449 RSC.
- A. J. Schwartz, K. L. Williams, G. M. Hieftje and J. T. Shelley, Anal. Chim. Acta, 2017, 950, 119–128 CrossRef CAS PubMed.
- E. D. Hoegg, C. J. Barinaga, G. J. Hager, G. L. Hart, D. W. Koppenaal and R. K. Marcus, J. Am. Soc. Mass Spectrom., 2016, 27, 1393–1403 CrossRef CAS.
- E. D. Hoegg, C. J. Barinaga, G. J. Hager, G. L. Hart, D. W. Koppenaal and R. K. Marcus, J. Anal. At. Spectrom., 2016, 31, 2355–2362 RSC.
- E. D. Hoegg, R. K. Marcus, G. J. Hager, G. L. Hart and D. W. Koppenaal, J. Anal. At. Spectrom., 2018, 33, 251–259 RSC.
- E. D. Hoegg, B. T. Manard, E. M. Wylie, K. J. Mathew, C. F. Ottenfeld and R. K. Marcus, J. Am. Soc. Mass Spectrom., 2019, 30, 278–288 CrossRef CAS PubMed.
- E. D. Hoegg, R. K. Marcus, D. W. Koppenaal, J. Irvahn, G. J. Hager and G. L. Hart, Rapid Commun. Mass Spectrom., 2017, 31, 1534–1540 CrossRef CAS PubMed.
- Orbitrap Fusion Lumos Tribrid Mass Spectrometer, ThermoScientific, http://planetorbitrap.com/orbitrap-fusion-lumos, 2018.
- E. D. Hoegg, S. Godin, J. Szpunar, R. Lobinski, D. W. Koppenaal and R. K. Marcus, J. Am. Soc. Mass Spectrom., 2019 DOI:10.1007/s13361-019-02183-w.
- R. D. Smith, J. A. Loo, R. R. O. Loo, M. Busman and H. R. Udseth, Mass Spectrom. Rev., 1991, 10, 359–451 CrossRef CAS.
- E. Rosenberg, J. Chromatogr. A, 2003, 1000, 841–889 CrossRef CAS.
- S. A. Shaffer, K. Q. Tang, G. A. Anderson, D. C. Prior, H. R. Udseth and R. D. Smith, Rapid Commun. Mass Spectrom., 1997, 11, 1813–1817 CrossRef CAS.
- L. X. Zhang, B. T. Manard, S. Konegger-Kappel and R. K. Marcus, Anal. Bioanal. Chem., 2014, 406, 7497–7509 CrossRef CAS PubMed.
- A. G. Marshall and F. R. Verdun, Fourier Transforms in NMR, Optical, and Mass Spectrometry: A User's Handbook, Elsevier, Amsterdam, 1990 Search PubMed.
- Practical Aspects of Ion Trap Mass Spectrometry, ed. R. E. March and J. F. J. Todd, CRC Press, Boca Raton, FL, 1995 Search PubMed.
- FT-ICR/MS: Analytical Applications of Fourier Transform Ion Cyclotron Resonance Mass Spectrometry, ed. B. Asamato, VCH Publishers, NY, NY, 1991 Search PubMed.
- K. Zhao, M. Penkin, C. Norman, S. Balsley, K. Maryer, P. Peerani, C. Pietri, S. Tapodi, Y. Tsutaki, M. Boella, G. Renha and E. Huhn, International Target Values 2010 for Measurement Uncertainties in Safeguarding Nuclear Materials, 2010 Search PubMed.
- L. X. Zhang and R. K. Marcus, J. Anal. At. Spectrom., 2016, 31, 145–151 RSC.
- R. K. Marcus, B. T. Manard and C. D. Quarles, J. Anal. At. Spectrom., 2017, 32, 704–716 RSC.
- L. X. Zhang, B. T. Manard, B. A. Powell and R. K. Marcus, Anal. Chem., 2015, 87, 7218–7225 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ja00154a |
| This journal is © The Royal Society of Chemistry 2019 |