
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRole of location, season, occupant activity, and chemistry in indoor ozone and nitrogen oxide mixing ratios†
Shan
Zhou
 a,
Cora J.
Young
b,
Trevor C.
VandenBoer
b and
Tara F.
Kahan
*ac
a,
Cora J.
Young
b,
Trevor C.
VandenBoer
b and
Tara F.
Kahan
*ac
aDepartment of Chemistry, Syracuse University, Syracuse, New York 13244, USA
bDepartment of Chemistry, York University, Toronto, Ontario M3J 1P3, Canada
cDepartment of Chemistry, University of Saskatchewan, Saskatoon, Saskatchewan S7N 5C9, Canada. E-mail: tara.kahan@usask.ca
First published on 21st June 2019
Abstract
Understanding the oxidizing environment indoors is important for predicting indoor air quality and its impact on human health. We made continuous time-resolved measurements (30 s) of several oxidants and oxidant precursors (collectively referred to as oxidant*): ozone (O3), nitric oxide (NO), and NO2* – the sum of nitrogen dioxide (NO2) and nitrous acid (HONO). These species were measured in three indoor environments – an occupied residence, a chemistry laboratory, and an academic office – in Syracuse, New York, during two seasons in 2017 and 2018. Oxidant* levels differed greatly between the residence, the lab and the office. Indoor-to-outdoor ratios (I/O) of O3 were 0.03 and 0.67 in the residence and office; I/ONO (I/ONO2*) were 11.70 (1.26) in the residence and 0.13 (1.70) in the office. Little seasonal variability was observed in the lab and office, but O3 and NO2* levels in the residence were greater in spring than in winter, while NO levels were lower. Human activities such as cooking and opening patio doors resulted in large changes in oxidant* mixing ratios in the residence. In situ chamber experiments demonstrated that the increase in O3 and NO2* levels during door-open periods was due to a combination of physical mixing between indoor and outdoor air, gas-phase production of NO2 from O3–NO chemistry, and heterogeneous formation of HONO on indoor surfaces. Our results also highlight the importance of chemistry (with NO, alkenes, and surfaces) in O3 mixing ratios in the residence, especially during door-open periods.
Environmental significanceOzone is often considered to be the most important oxidant indoors, but few recent measurements have been made in residences. We made time-resolved, continuous measurements of ozone in a residence and a university over two seasons (spring and fall/winter). Ozone levels were much lower in the residence (<1 ppbv) than in the university lab or office (∼22 ppbv). Three implications of this work are: (1) ozone measurements made in non-residential buildings may not be generalizable to residences; (2) differences in oxidant levels in residential and non-residential buildings are amplified by common human activities in residences (especially cooking and opening windows); and (3) oxidants cannot be treated in isolation, as their levels are often interdependent. |
1 Introduction
Ozone (O3) exposure is a major public health concern. Many studies have reported associations between outdoor O3 mixing ratios and morbidity and mortality.1,2 Since North Americans spend the majority (∼85%) of their time indoors,3 most O3 exposure is expected to occur in indoor environments.4 Ozone can be generated indoors by appliances such as photocopiers, printers, and air cleaning devices, but in most buildings the predominant source of indoor O3 is transport from outdoors through ventilation.5,6 Whether originating indoors or outdoors, O3 in indoor environments can undergo homogeneous and heterogeneous chemistry. Secondary organic aerosols and other potentially irritating species, such as aldehydes, ketones, acids, and epoxides can be generated and cause additional adverse health effects.7–11 Indoor exposure to O3 is accompanied by exposure to the products of O3-initiated indoor chemistry.Ozone-initiated chemistry in indoor environments has received significant attention.12,13 As an oxidant, O3 reacts in ventilation systems and on interior materials, including carpets and indoor furnishing surfaces.7,14,15 It also initiates gas-phase reactions with common indoor volatile organic compounds (VOCs) and those from tobacco smoke, cleaning products, and air fresheners.16,17 Recently, the human envelope has received increasing attention as skin oils on human surfaces and soiled clothing are an important O3 sink.9,15,18 In addition to reducing O3 levels, O3-initiated reactions are a source of two other important indoor oxidants: hydroxyl and nitrate radicals. Ozone/alkene reactions are considered an important source of hydroxyl radicals (OH) indoors,19 while the reaction of O3 with nitrogen dioxide (NO2) is the major source of nitrate radicals indoors.20,21 With these recent scientific efforts, our knowledge of O3 reactivity indoors has expanded greatly. However, the listed studies have been conducted in either mechanically ventilated buildings, under laboratory settings, or in simulated office environments; all are non-residential environments.
A large number of studies focused on understanding O3 levels in commercial buildings, museums, and classrooms have been undertaken in the past four decades.13,22–27 The air exchange rate (AER) has been shown to affect the indoor concentration of O3, with a higher AER yielding higher indoor to outdoor ratios (I/O), which often range from 0.3–0.8.13,24 Far fewer measurements of O3 mixing ratios have been made in residences, where people spend most of their time. Mean O3 mixing ratios in homes range from sub-ppbv levels to 60 ppbv and I/O span a large range between <0.1 and 0.7, depending on ventilation rates.13,23,24,28–32 Most measurements conducted in residences were made with passive O3 samplers that were exposed to the indoor environment for extended periods of time (1 to 14 days). The few studies that report time-resolved O3 in American homes were conducted prior to the implementation of the 8 hour O3 National Ambient Air Quality Standard (NAAQS) in 1997.32 For measurements made in US homes subsequent to the NAAQS implementation, indoor O3 levels were generally lower than 2 ppbv.31,43–45 However, model studies investigating O3 chemistry and predicting OH and hydroperoxy (HO2) radicals in indoor environment use O3 levels of up to 50 ppbv.15,19,33 Many laboratory experiments studying O3-initiated reactions in residential settings use O3 levels ranging from 30 to 250 ppbv.8,34,35 There is a lack of real-time characterization of the mixing ratio of O3 in residences and investigation of O3-initiated chemistry under realistic residential conditions.
To help fill this knowledge gap, we deployed the Mobile Indoor Laboratory for Oxidative Species (MILOS), a custom-built non-invasive mobile laboratory that provides time-resolved measurements of a suite of oxidants and oxidant precursors (collectively referred to as oxidant*), in three different indoor environments including an occupied residence. The mixing ratios, sources, and sinks of O3, nitric oxide (NO), NO2, and nitrous acid (HONO) were characterized in the residence and compared to those in a chemistry lab and an office. Secondary gas-phase and heterogeneous reactions involving these species were also investigated.
2 Methods
2.1 Experimental design
Continuous, time-resolved measurements were conducted in three indoor environments in Syracuse, New York, including an occupied single-family residence, a chemistry laboratory, and an academic office using MILOS. A detailed description of the instrumental setup, including limits of detection (LOD) for the various analyzers, can be found elsewhere.28 In brief, air was drawn through a perfluoroalkoxy (PFA) tubing inlet placed at head height followed by ¼ inch PFA tubing before entering a UV photometric analyser for O3 measurements (Ecotech Serinus 10, UV photometric) and a chemiluminescence NOx analyzer (Ecotech Serinus 40) that nominally measures NO, NOx (NO + NO2), and NO2. The NOx channel of this instrument responds quantitatively to HONO and nitric acid (HNO3) in addition to NO2 and NO, and has an unquantified response to alkyl nitrates. Interference from alkyl nitrates is assumed to be small based on the current knowledge about oxidation chemistry indoors,36 and measured indoor HNO3 levels have been reported to be very low relative to that of HONO.37 The nominal NO2 (NO2* = NO2 + HONO) was measured throughout the field study. Distinct separate measurements of NO2 and HONO were made occasionally following the method described by Zhou et al.28 CO2, relative humidity (RH), and temperature indoors were monitored with a TSI IAQ7545 air quality monitor. Data were acquired at 30 s intervals for all species except speciated HONO, for which data were acquired every 5 minutes.Air sampling was carried out sequentially between three indoor environments. Each location was sampled in fall/winter and in spring, with the purpose of capturing seasonal differences. The fall/winter campaign lasted for more than 2 months from September 27 to December 9, 2017, and the spring campaign spanned 5 weeks from March 23 to May 2, 2018. Outdoor mixing ratios were measured intermittently by moving the inlet to an outdoor location. Average I/O was calculated for individual species.
2.2 Site description
The single-family residence is a 4-bedroom 1.5-storey house in an urban residential area. The main floor (where measurements were made) has an open floor plan with doors and windows on all four sides. Gas appliances include a stove (5 burners and an oven), water heater, fireplace, and furnace for space heating. The sampling inlet was placed in the kitchen (approximately the center of the main floor) or occasionally in the living room at head height (∼1.5 m above the floor). The chemistry lab (basement, no window) and office (2nd floor, 1 window) are in the same building in the Syracuse University campus, ∼1.8 km away from the residence. All appliances in the university use electricity. The estimated air volume is 250, 90, and 24 m3 in the residence, lab, and office, respectively. The residence utilized natural ventilation (sometimes opening patio doors) and a forced-air system (heating or air conditioning) during the sampling periods, whereas the lab and office used mechanical ventilation that exchanged room air with outdoor air. During the sampling period, the residence was occupied by two non-smoking adults, one child, and a dog. The lab was occupied by five to seven non-smoking adults and the office was occupied by one non-smoking adult. Occupants performed their usual daily activities throughout the field campaign. Activities such as cooking using the stove and opening doors and windows were logged by the occupants.2.3 In-house environmental chamber
To investigate the indoor oxidizing environment under higher O3 levels in the residence, O3-perturbation experiments were conducted in the residence during both measurement campaigns. A 5 L Pyrex glass environmental chamber (60 cm in length × 10 cm in diameter) was placed in the shade and connected to the sampling line between the sampling inlet and MILOS. MILOS continuously sampled room air through the chamber. A known amount of O3 was then injected into the environmental chamber, where it mixed with ambient indoor air. Air composition in the chamber was monitored for approximately one hour during O3 injection, followed by a ∼30 minute injection of zero air as a negative control. In all perturbation experiments, the chamber air flow was 1.3 L min−1, yielding a residence time of 4.5 min.2.4 Air exchange rate
The air exchange rate (AER) was estimated using a decay method of the indoor CO2 concentration.28 Only periods with a dominant indoor source (e.g., cooking in the residence, occupants or dry ice in the lab and office) which led to CO2 concentrations significantly higher than the background values, followed by a non-source period, were considered for these calculations. Assuming constant removal rates and background mixing ratios of CO2 during the decay period and an even distribution of the indoor CO2 concentration the AER can be calculated.| Ct = e−ktCo + Cb | (1) |
3 Results and discussion
3.1 Oxidant levels in the three indoor environments
The statistics of the air exchange rate and oxidant* levels in the residence, chemistry lab, and office over the entire sampling period are shown in Fig. 1. The residence had a distinctly different oxidizing environment than the academic building. The average (±SD) AER in the residence was 0.69 (±0.30) h−1, substantially lower than that in the lab (3.80 (±0.45) h−1) and the office (5.02 (±0.24) h−1). This is consistent with the reported low AER for US residences and the higher recommended AER for commercial buildings (4–15 h−1).38–40 Indoor O3 levels followed the same trends as the AER. The lowest average O3 was observed in the residence, 0.62 (±1.77) ppbv, which is on average 30 times lower than outdoor O3 levels measured over the same time period (20.5 (±9.6) ppbv), yielding an average I/OO3 of 0.03. Mean O3 levels measured in US homes prior to the implementation of the 8 hour O3 National Ambient Air Quality Standards in 1997 were 13–60 ppbv.32,41,42 Studies subsequent to the implementation, on the other hand, reported substantially lower O3 levels that ranged from 0.027 to 1.9 ppbv,31,43–45 similar to our observations. In these studies, I/OO3 generally spanned from 0.1 to 0.3, but increased substantially to 0.6–0.7 when windows were open.13,32,45 Ozone levels in the lab and office were comparable (average of 21.5 (±8.7) vs. 22.5 (±8.6) ppbv; Fig. 1); mean values were approximately 30 times higher than those in the residence. Outdoor measurements were made during the office sampling campaign in spring, but not during office sampling in fall/winter, or during either of the laboratory sampling periods. The average I/OO3 in the office was 0.67 based on the spring data, well within the range of I/O in buildings with negligible indoor O3 sources (<0.1–0.8).13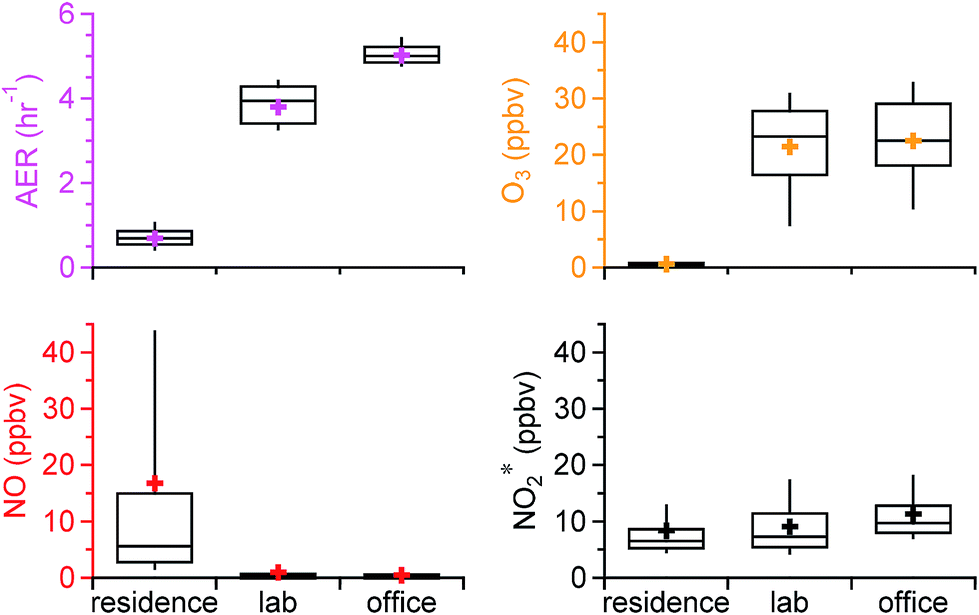 | ||
| Fig. 1 Statistics of the air exchange rate (AER) and mixing ratios of O3, NO, and the sum of NO2 and HONO (NO2*) observed in three indoor environments in Syracuse, NY, over the entire field study period. The box and whisker plots show the median (line), mean (marker), upper and lower quartiles (box), and the 10th and 90th percentiles (whiskers). | ||
Lower O3 levels and I/OO3 in the residence than in the academic building may be due to the low AER. Without significant indoor sources, O3 indoors is transported from outside through air change. The literature has shown that homes and offices tend to have comparable O3 loss rates to indoor surfaces, ranging from 2.5 to 5.4 h−1.13 Surface loss rates in this and other US residences are generally much larger than the AER, whereas surface loss rates in the academic building is comparable to the AER. As a result, O3 levels and I/OO3 in the residence are lower. This is consistent with reported observations that an increased outdoor ventilation rate increases indoor O3 concentrations and I/OO3, as less O3 will be lost (e.g., between walls) as it makes its way into the indoor space.4,22,24
NO showed an opposing trend compared to O3. Its mixing ratios were substantially higher in the residence (16.8 (±31.0) ppbv) than in the lab (1.0 (±3.7) ppbv) and the office (0.5 (±1.5) ppbv). The average I/ONO in the residence was 11.7 over the entire campaign and 0.13 in the office based on the spring data. The higher NO levels in the residence likely resulted from the combined effect of direct indoor sources and lower loss to O3. Cooking was an important source of NO in the residence, with peak NO levels of ∼350 ppbv. The low AER and low O3 levels in the house resulted in high NO mixing ratios after cooking which were sustained for hours, as discussed in detail by Zhou et al.28 Low NO mixing ratios in the lab and office were likely explained by a lack of indoor sources and by titration by O3. NO2* followed a similar trend as O3, with lower mixing ratios in the residence (8.4 (±6.8) ppbv) followed by the lab (9.1 (±5.3) ppbv) and the office (11.3 (±5.5) ppbv), although the differences between the residential and non-residential locations were much less pronounced than for O3 or NO. The average I/ONO2 was 1.26 for the residence (all data) and 1.70 for the office (spring data only). Outdoor-to-indoor transport has been shown to be important for NOx.46–48 It could be a major source of NO2* in the office and lab as a result of a high AER in the academic building. Secondary production from reactions between NO and O3 may also be important. The residence had negligible influence from outdoor NOx emissions due to a low AER;28 cooking was often the dominant source of NO2*, and – as discussed in Sections 3.3 and 3.4 – the reaction between NO and O3 was also an important NO2* source under some conditions.
3.2 Influence of season and occupant activity on oxidant levels in the residence
Background periods without human perturbations in the residence (i.e., patio doors closed and no cooking events) were selected for seasonal comparisons of indoor oxidant* levels. The AER in the residence in spring was ∼15% higher than that in fall/winter (0.74 (±0.24) vs. 0.65 (±0.36) h−1; Fig. 2), consistent with previous studies that have demonstrated a higher AER in spring and summer compared to winter.29,30,49 Daytime (8 am to 4 pm) outdoor O3 mixing ratios were also higher in spring than in fall/winter, by an average of 10.3 ppbv. Although indoor background O3 mixing ratios were low and noisy in both seasons, only 10% of fall/winter background O3 data was above the limit of detection (LOD; 0.6 ppbv), while 36% of the spring data was above the LOD. Since there was a negligible indoor source of O3 in the residence, the higher indoor O3 levels in spring were likely a result of the higher AER and outdoor O3 during warmer months. This subsequently affected background NO and NO2* levels. As seen in Fig. 2, the background NO in spring was 1.7 ppbv lower than that in fall/winter, while the oxidation products (NO2 and HONO, reported as NO2*) were ∼2 ppbv higher in spring. This is consistent with O3 reacting with NO indoors to form NO2 – and subsequently HONO – in spring. Some small seasonal differences in the AER and oxidant* levels during background periods (closed window and no cooking) were observed in the lab and office (Fig. S1 in the ESI†), but no firm conclusions could be drawn.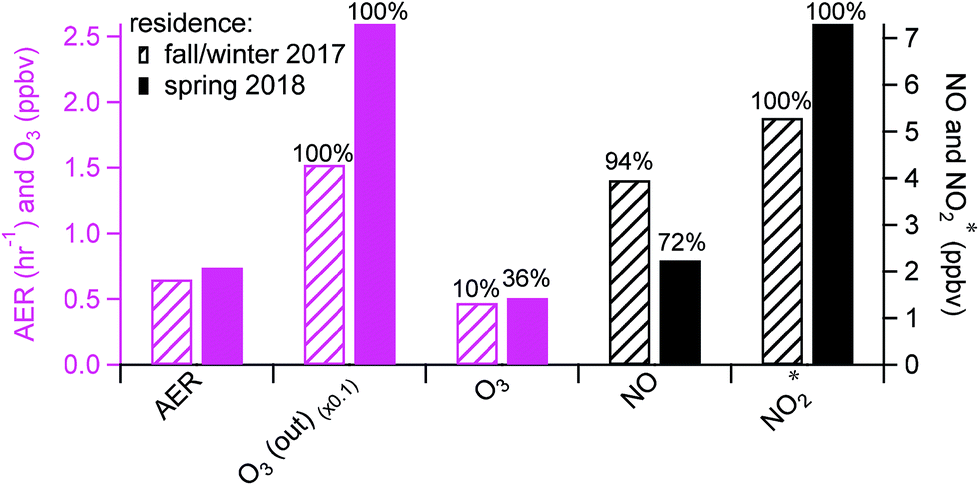 | ||
| Fig. 2 Average AER, ambient outdoor O3 levels, and mixing ratios of O3 and nitrogen oxides in the residence under background conditions in two seasons. Data below the detection limit were included to calculate averages. Text above the bars indicates the percentage of data above the detection limit. | ||
Human activities greatly influenced oxidant* mixing ratios in the residence. As seen in Fig. 3a, the average mixing ratios of O3, NO, and NO2* under background conditions during the residential sampling periods averaged 0.47, 3.96, and 5.29 ppbv, respectively. While cooking did not affect O3 levels, it led to substantially higher levels of nitrogen oxides. Peak NO and NO2* mixing ratios during cooking events (N = 98) were on average about 30 and 6 times greater, respectively, than the background levels (Fig. 3a). A detailed discussion of the influence of cooking from the fall data in this residence has been previously reported.28
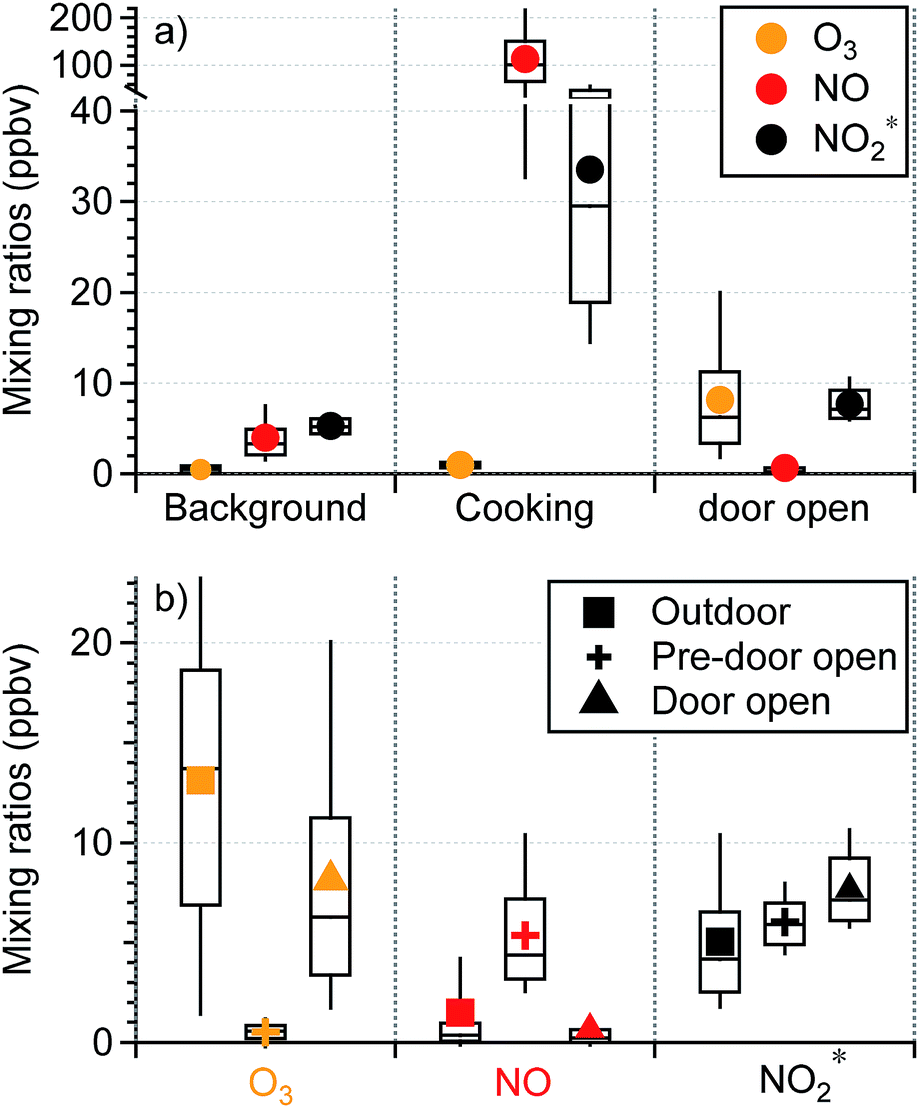 | ||
| Fig. 3 Statistics of oxidant* observed (a) in the residence during background, cooking peak (5 min), and patio door open periods; (b) outside the residence (outdoor), indoors during undisturbed background conditions over a period of 1–2 hours prior to opening patio doors (pre-door open), and indoors with open patio doors (door open). The box and whisker plots show the median (line), mean (marker), upper and lower quartiles (box), and the 10th and 90th percentiles (whiskers). | ||
During the fall/winter 2017 campaign, the patio doors were sometimes opened (1–3 hours; N = 14) for increased ventilation. Although the AER was not measured during these door-open events, the AER was likely much larger during these periods than when doors were closed. Opening windows has been shown to increase the AER by up to 3 h−1.49,50 The increase of the AER is associated with several factors including the width of the opening and the elevation of windows/doors, number of windows/doors opened, indoor–outdoor temperature difference, and wind speed and direction. During the door-open events, large (4.8 m2) patio doors were fully opened; differences between indoor and outdoor temperature ranged from ∼3 °C to 18.2 °C and outdoor wind speed reached 11.8 m s−1 (based on meteorology data at Syracuse Hancock International Airport,51 7.3 km north of the residence). Given this combination of factors, it is possible that the AERs during the door open periods in our campaign exceeded those reported in other studies, which had smaller area of openings and smaller indoor–outdoor temperature differences. As shown in Fig. 3b, outdoor O3 mixing ratios averaged 13.1 (±7.9) ppbv. Indoor O3 levels under background conditions measured prior to opening the patio doors were substantially lower with an average of 0.53 (±0.60) ppbv, consistent with the typical background O3 levels in this residence. When the patio doors were open, indoor O3 mixing ratios increased substantially from the background sub-ppbv level to an average of 8.1 (±6.5) ppbv, approximately 62% of the level outdoors. This is due to a combination of increased ventilation rates and decreased filtration by the building envelope with open doors.
While the majority of O3 loss during transport from outdoors can be attributed to reaction with building materials and surface deposition, gas phase chemistry may have also contributed. The average mixing ratios of outdoor NO and NO2* were 1.5 (±3.7) and 5.0 (±3.4) ppbv, respectively, and those for the pre-door-open periods were 5.4 (±3.1) and 6.0 (±1.4) ppbv, respectively. If only physical mixing occurred during the door-open periods, NO and NO2* levels would lie between the background and outdoor values, because the house volume would contain a mixture of the original indoor air and infiltrated outdoor air. However, NO decreased to an average of 0.67 (±1.7) ppbv during the door-open periods, which was even lower than outdoor ambient values. This indicates an extra loss mechanism in addition to dilution. On the other hand, NO2* increased by ∼1.7 ppbv during the door-open periods, resulting in levels higher than those outdoors. This is suggestive of secondary production. These observations together suggest that O3 introduced indoors via door opening reacted with NO indoors to yield secondary oxidation products (specifically NO2). Although windows in this residence were often closed in winter, they can be open for more than half the time in warmer months.52 This highlights the importance of considering door/window open conditions in investigations of indoor chemistry.
3.3 Ozone–NOx chemistry implied from temporal trends
Temporal trends of oxidant* in the residence and the lab on a typical day during the sampling periods are shown in Fig. 4. Distinct features of the indoor oxidant* can be seen. In the residence, O3 was stable and often below the LOD, whereas NO and NO2* had stable background levels of approximately 5 and 8 ppbv, respectively (Fig. 3a and 4a). Sharp increases of NO and NO2* (up to 230 and 50 ppbv) were observed during meal times due to cooking, as reported previously.28 In the lab, O3 levels were generally between 10 and 30 ppbv. Mixing ratios of NO in the lab were very low and remained below the LOD except during periods consistent with morning rush-hour traffic or night-time traffic possibly associated with evening events at the university, during which high NO2* levels were also observed (Fig. 4b). Similar temporal profiles were observed in the office and the lab; both showed clear diurnal patterns with an O3 trough and NO and NO2* peaks during morning rush-hour (Fig. S2†). The fact that O3 and NO were inversely related in these indoor environments suggests that one species was generally titrated by the other. In addition, O3 showed a clear anti-correlation with NO2* in the lab and office (Fig. 4b and S2†), which further suggests the importance of chemical reactions between O3 and NOx.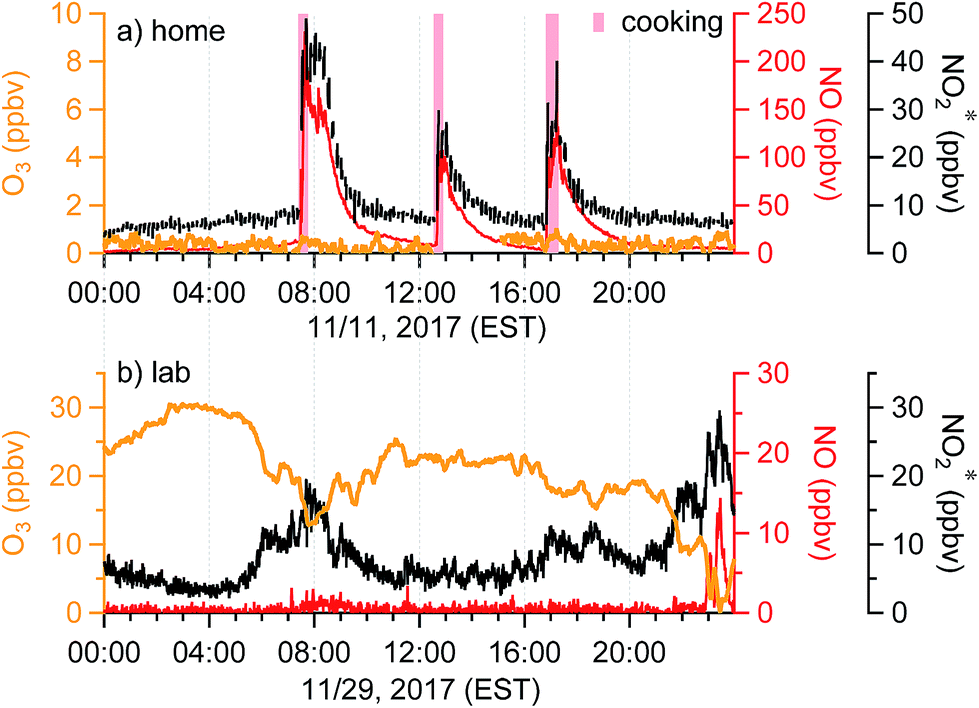 | ||
| Fig. 4 Time series of oxidant* on a typical day in (a) the residence in Syracuse, NY, and (b) a chemistry lab in Syracuse University. Windows and doors were closed in both scenarios. | ||
When doors were closed, NO and NO2* mixing ratios displayed a smooth exponential decay after cooking (Fig. 4a), with mean decay rate constants of 0.92 (±0.37) and 1.5 (±0.54) h−1, respectively.28 These levels remained elevated above background mixing ratios often for more than four hours. Opening the patio doors changed the profiles dramatically. For example, after cooking lunch on October 7, 2017, NO and NO2* levels in the residence rose rapidly and then decayed to ∼70 and 11 ppbv, respectively (Fig. 5). Indoor O3 remained low (∼0.6 ppbv) throughout this time period. When the patio doors were opened O3 levels rose rapidly to 20–30 ppbv and NO quickly dropped below the LOD. The behavior of O3 and NO could be explained by physical mixing of outdoor and indoor air masses. However, NO2* showed a rapid initial increase to 20 ppbv when the doors were opened and then remained elevated higher than the mixing ratios observed prior to opening doors until the doors were closed. This suggests chemical production of NO2* in the residence when doors were open, consistent with previous discussions (Section 3.2). We note that NO2 photolysis has been suggested to be a source of O3 indoors.53 Given the NO2 mixing ratios measured in this campaign, as well as photon fluxes measured previously,28 we predict that NO2 photolysis could increase O3 levels by up to 5× in sunlit regions in this residence. Low O3 levels, small illuminated volumes, and rapid mixing of air inside the house prevented us from testing this hypothesis.
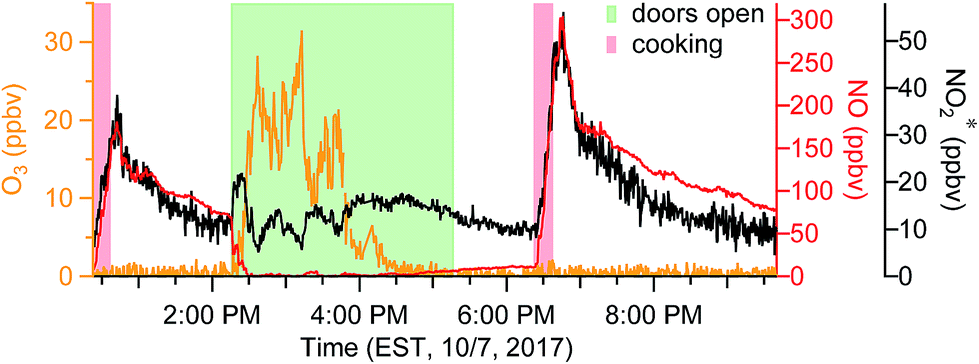 | ||
| Fig. 5 Time series of oxidant* on a typical day in the residence during periods when patio doors were open. | ||
3.4 Chamber experiments (O3-perturbation experiments)
To validate the hypothesis that NO reacts with O3 and generates NO2* under door-open conditions in the residence, O3-perturbation experiments were performed in the residence. One was performed under background conditions and six were performed after cooking. In these experiments approximately 35 ppbv of O3 was added into the environmental chamber (after accounting for dilution) while MILOS continuously pulled room air through it. Fig. 6 shows an example of a typical experiment performed after cooking. Prior to perturbation the combustion products NO, NO2, and HONO decayed exponentially with rate constants of 0.7, 1.1, and 1.0 h−1, respectively. With an addition of 35 ppbv O3, a sharp drop in NO occurred concomitantly with an increase of NO2 and HONO. This provides direct evidence that O3 reacts with NO and forms NO2* in indoor air. A negative control replacing O3 with zero air (ZA) following O3 perturbation resulted in the same dilution factor yet removed the oxidant from the chemical reactions. This resulted in a slight increase in NO (∼8 ppbv), while both NO2 and HONO (as well as NO2*) dropped rapidly. This behaviour was reproducible, as shown in Fig. S3–S5 in the ESI.† This supports the hypothesis that O3–NO chemistry occurs in the indoor atmospheric environment.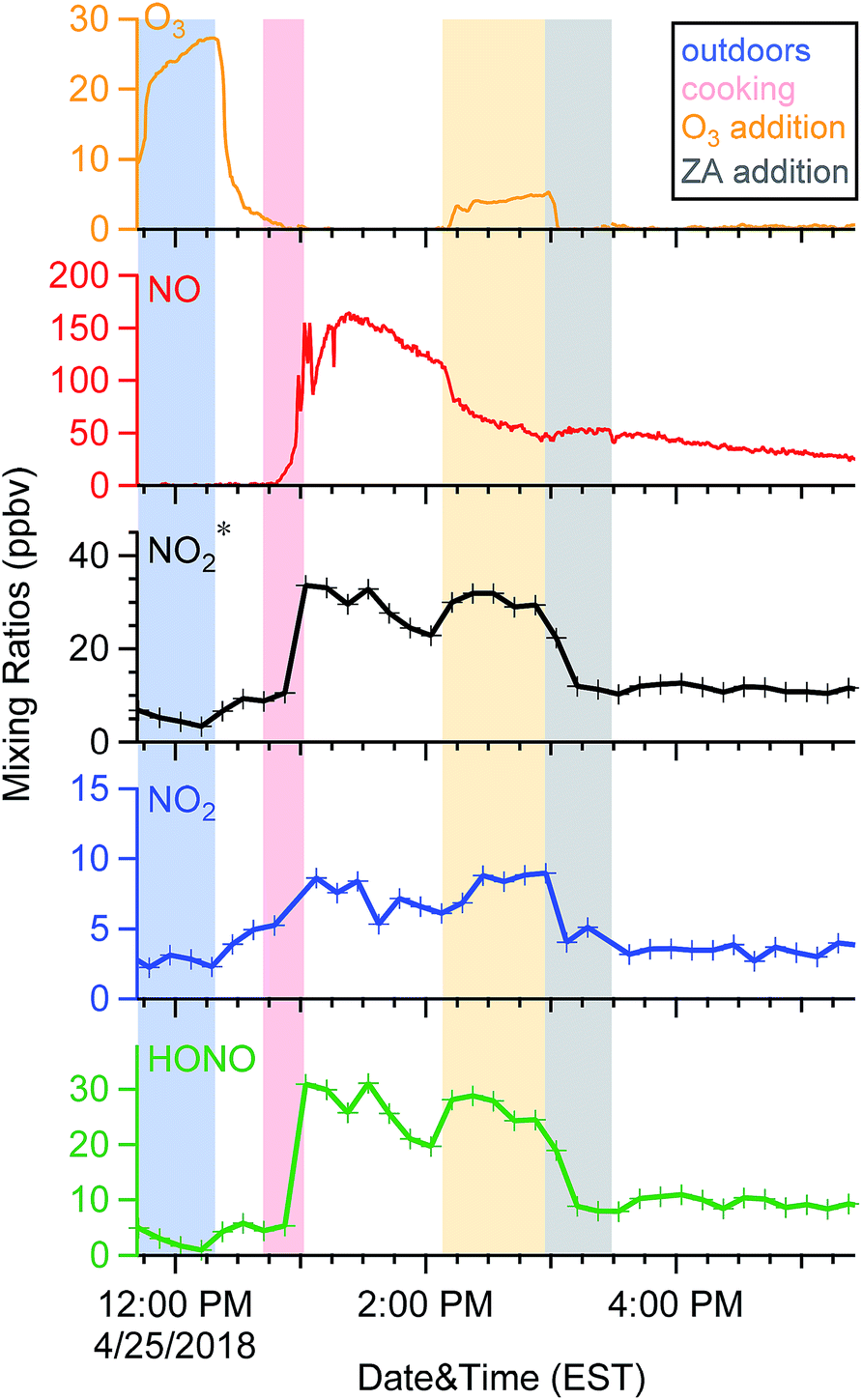 | ||
| Fig. 6 Time series of oxidant* observed in the residence sampled through the environmental chamber during an O3-perturbation experiment performed after cooking. Shaded areas indicate different conditions noted in the legend of the graph. | ||
Similar changes of oxidant* levels were observed in the other five experiments performed after cooking (Fig. S3–S5†). In the post-cooking experiments, the O3 mixing ratio decreased by an average of 29.1 (±4.7) ppbv (after accounting for dilution). After accounting for dilution in the chamber, NO mixing ratios decreased by 21.1 (±4.1) ppbv following O3 injection. Concurrent increases in NO2* mixing ratios (17.4 (±4.2) ppbv) were observed. For individual experiments, the observed increase in NO2* was 64–108% of the observed NO loss (average 84 (±18)%), suggesting that the majority of NO loss was due to reaction with O3. Speciated NO2 and HONO were detected during these experiments. Ratios of NO2 to HONO were variable, with NO2 contributing 22–66% to the total increase in NO2* (with an average of 42%). Thin films of water have been observed to form on indoor surfaces at a relative humidity (RH) as low as 20%.54 Heterogeneous “wet” chemistry of NO2 on indoor surfaces (RH > 20%) has been shown to generate HONO.55,56 Comparing elevated surface area to volume (S/V) ratios in indoor environments to those outdoors, heterogeneous reactions of NO2 have been projected to be important indoors.57 The NO2-to-HONO conversion is greatly enhanced in the presence of light (λ < 400 nm).58,59 However, as the chamber was in the dark (i.e., shade), heterogeneous photochemistry likely did not contribute to HONO formation in these experiments. Since the RH during the perturbation experiments was between 30% and 40%, our results are consistent with gas-phase NO2 (formed from reactions between O3 and NO) reacting on “wet” glass surfaces to form HONO, as would happen on indoor surfaces with the intrusion of O3-containing outdoor air.
We performed control experiments (N = 3) injecting O3 in ZA only (no room air) into the environmental chamber to investigate the contribution of uptake to glass on O3 mixing ratios in the chamber. In these experiments, the O3 mixing ratio immediately dropped by 5–17 ppbv in the chamber and recovered exponentially with an average rate constant of 5.5 (±0.35) h−1. Over the course of the experiments, which ranged from 41 to 60 minutes, an average of 3.8 (±1.2) ppbv O3 was consumed. No clear trends in ozone uptake were observed with successive experiments. The shape of the recovery curve is consistent with reactive uptake of O3 onto the glass chamber surface. Clean glass has very low O3 uptake rates. However, thin organic films have been observed to form on impervious indoor glass surfaces.60 Since the chamber was exposed to room air during the entire field campaign, and it was preconditioned (room air pulled through the chamber before instrumentation) prior to most perturbation experiments, we expect the surface composition of the chamber walls to reflect that of indoor surfaces in the residence. Reactive uptake of O3 by adsorbed surface materials could be responsible for the observed O3 depletion. This will be relevant to many indoor locations due to high S/V ratios.
Reaction with NO and uptake to the chamber surface accounted for, on average, 86.0 (±8.9)% of the observed ozone loss. The remaining O3 sink (∼4.2 ppbv) is likely unsaturated hydrocarbons in the residential air. These were scrubbed in the ZA but would be components in the sampled residential air. They can be emitted from building materials, carpets, paints, and indoor plants, and tend to be present at higher levels in residences than outdoors.17,61 Using speciated alkene mixing ratios from Weschler and Shields62 and associated reaction rate constants, we calculated a loss of approximately 1.3 ppbv O3 due to reaction with alkenes in the environmental chamber. This is smaller than the average unattributed O3 loss of 4.2 ppbv, but it does, when combined with NO and surface uptake, account for between 93 and 100% of the observed O3 loss in four of the six post-cooking experiments; an additional 7–9 ppbv O3 remains unattributed in the other two experiments. Given the variability of indoor alkene mixing ratios, the three processes considered (reaction with NO, uptake to surfaces, and reaction with alkenes) appear to account for the majority of O3 loss in the environmental chamber and in the residence.
While only one perturbation experiment was performed under background conditions (Fig. S6†), it is worth discussing briefly. Initial NO levels were much lower prior to O3 injection compared to those in the post-cooking experiments (∼4.3 vs. 77.3 ppbv). Accordingly, the decrease in the O3 mixing ratio was smaller in this experiment (13.8 ppbv vs. ∼29 ppbv in the post-cooking experiments). After accounting for dilution, NO decreased by 3.3 ppbv and NO2* increased by 1.7 ppbv following injection of O3. Approximately 6.7 ppbv of consumed O3 remains unattributed after accounting for uptake to the chamber walls. This is within the range determined for the post-cooking experiments (1.3–8.6 ppbv) and is likely due to reaction with alkenes. Fig. 7 shows relative strengths of the O3 sinks considered (NO, alkenes, and surface uptake) under post-cooking and background conditions. It is clear that different O3 loss processes dominate under different conditions. While NO dominates O3 loss after cooking (73%), alkene reactions are the most important single sink under background conditions (accounting for 48% of O3 loss). Uptake to the chamber surface accounted for less than 14% of O3 loss in the post-cooking experiments but was as large a sink as NO under background conditions (27%). While conditions in the environmental chamber (especially the surface composition and S/V ratio) do not fully reflect those in most residences, the conclusion that human activities such as cooking and opening doors will affect oxidant* mixing ratios and fates is likely generalizable to many indoor environments.
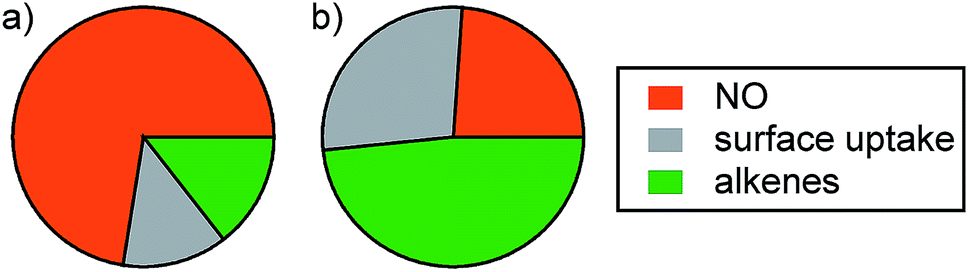 | ||
| Fig. 7 Relative contribution of the three sinks to O3 loss in the environmental chamber under (a) post-cooking and (b) background conditions. The contribution of alkenes is determined based on the O3 loss that is not accounted for by the other two processes. | ||
4 Conclusions and implications
We made continuous time-resolved measurements of O3 and nitrogen oxides in three indoor environments (home, laboratory, and office) in Syracuse, New York, in two seasons. Oxidant* mixing ratios were very different in different indoor environments. Ozone, which is typically considered a major (or the only) indoor oxidant, was present at very low levels (<1 ppbv) in the residence, whereas NO and NO2* levels were higher compared to those in the laboratory and office environments with higher AERs. As NO concentrations have been reported to be quite high in North American residences (mean values ranging from 30 to 77 ppbv (ref. 28 and references therein)), O3 will likely often be fully titrated by NO. This suggests that the observation of low O3 levels in such residences may be generalizable. This will have implications for the fate of other species indoors. For example, alkene oxidation will be slow, and OH steady state mixing ratios – in the absence of other OH sources – will be lower than those often predicted based on higher (∼20 ppbv) indoor O3 mixing ratios.Our work highlights the effects of perturbations caused by human activity on the relative levels of oxidants* in the residence compared to those in the office and laboratory environments. Cooking increased NO and NO2*, while opening patio doors for natural ventilation increased O3 and reduced NO. In situ chamber experiments further confirmed that chemistry was responsible for the observed NO2* increase during natural ventilation periods – specifically the formation of NO2 from reactions of O3 with NO in the gas phase and the heterogeneous production of HONO on indoor surfaces. These results suggest that when predicting chemistry in indoor environments, the type of building (residence vs. commercial) and perturbations (e.g., cooking and opening windows/doors) need to be taken into account.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding for this work was provided by the Alfred P. Sloan Foundation CIE program (G-2018-11062). The authors acknowledge participating homeowners and Mr Corey Kroptavich for assistance with the experimental setup, and thank Dr C. J. Weschler for valuable discussions.References
- B. J. Hubbell, A. Hallberg, D. R. McCubbin and E. Post, Health-Related Benefits of Attaining the 8-Hr Ozone Standard, Environ. Health Perspect., 2005, 113(1), 73–82 CrossRef CAS PubMed.
- M. L. Bell, F. Dominici and J. M. Samet, A meta-analysis of time-series studies of ozone and mortality with comparison to the national morbidity, mortality, and air pollution study, Epidemiology, 2005, 16(4), 436–445 CrossRef PubMed.
- N. E. Klepeis, W. C. Nelson, W. R. Ott, J. P. Robinson, A. M. Tsang, P. Switzer, J. V. Behar, S. C. Hern and W. H. Engelmann, The National Human Activity Pattern Survey (NHAPS): a resource for assessing exposure to environmental pollutants, J. Exposure Anal. Environ. Epidemiol., 2001, 11, 231 CrossRef CAS PubMed.
- C. Chen, B. Zhao and C. J. Weschler, Assessing the Influence of Indoor Exposure to “Outdoor Ozone” on the Relationship between Ozone and Short-term Mortality in U. S. Communities, Environ. Health Perspect., 2012, 120(2), 235–240 CrossRef CAS PubMed.
- Q. Zhang and P. L. Jenkins, Evaluation of ozone emissions and exposures from consumer products and home appliances, Indoor Air, 2017, 27(2), 386–397 CrossRef CAS PubMed.
- K. J. Boelter and J. H. Davidson, Ozone generation by indoor, electrostatic air cleaners, Aerosol Sci. Technol., 1997, 27(6), 689–708 CrossRef CAS.
- H. Wang and G. C. Morrison, Ozone-Initiated Secondary Emission Rates of Aldehydes from Indoor Surfaces in Four Homes, Environ. Sci. Technol., 2006, 40(17), 5263–5268 CrossRef CAS PubMed.
- B. K. Coleman, M. M. Lunden, H. Destaillats and W. W. Nazaroff, Secondary organic aerosol from ozone-initiated reactions with terpene-rich household products, Atmos. Environ., 2008, 42(35), 8234–8245 CrossRef CAS.
- A. Wisthaler and C. J. Weschler, Reactions of ozone with human skin lipids: sources of carbonyls, dicarbonyls, and hydroxycarbonyls in indoor air, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(15), 6568–6575 CrossRef CAS PubMed.
- S. Zhou, L. W. Y. Yeung, M. W. Forbes, S. Mabury and J. P. D. Abbatt, Epoxide formation from heterogeneous oxidation of benzo[a]pyrene with gas-phase ozone and indoor air, Environ. Sci.: Processes Impacts, 2017, 19(10), 1292–1299 RSC.
- C. J. Weschler, Ozone's Impact on Public Health: Contributions from Indoor Exposures to Ozone and Products of Ozone-Initiated Chemistry, Environ. Health Perspect., 2006, 114(10), 1489–1496 CrossRef CAS PubMed.
- C. J. Weschler, Chemistry in indoor environments: 20 years of research, Indoor Air, 2011, 21(3), 205–218 CrossRef CAS PubMed.
- C. J. Weschler, Ozone in indoor environments: concentration and chemistry, Indoor Air, 2000, 10(4), 269–288 CrossRef CAS PubMed.
- D. Rim, E. T. Gall, R. L. Maddalena and W. W. Nazaroff, Ozone reaction with interior building materials: influence of diurnal ozone variation, temperature and humidity, Atmos. Environ., 2016, 125, 15–23 CrossRef CAS.
- M. Kruza, A. C. Lewis, G. C. Morrison and N. Carslaw, Impact of surface ozone interactions on indoor air chemistry: a modeling study, Indoor Air, 2017, 27(5), 1001–1011 CrossRef CAS PubMed.
- W. W. Nazaroff and C. J. Weschler, Cleaning products and air fresheners: exposure to primary and secondary air pollutants, Atmos. Environ., 2004, 38(18), 2841–2865 CrossRef CAS.
- Z. H. Fan, P. Lioy, C. Weschler, N. Fiedler, H. Kipen and J. F. Zhang, Ozone-initiated reactions with mixtures of volatile organic compounds under simulated indoor conditions, Environ. Sci. Technol., 2003, 37(9), 1811–1821 CrossRef CAS PubMed.
- S. Liu, R. Li, R. J. Wild, C. Warneke, J. A. de Gouw, S. S. Brown, S. L. Miller, J. C. Luongo, J. L. Jimenez and P. J. Ziemann, Contribution of human-related sources to indoor volatile organic compounds in a university classroom, Indoor Air, 2016, 26(6), 925–938 CrossRef CAS PubMed.
- N. Carslaw, L. Fletcher, D. Heard, T. Ingham and H. Walker, Significant OH production under surface cleaning and air cleaning conditions: impact on indoor air quality, Indoor Air, 2017, 27(6), 1091–1100 CrossRef CAS PubMed.
- C. Arata, K. J. Zarzana, P. K. Misztal, Y. Liu, S. S. Brown, W. W. Nazaroff and A. H. Goldstein, Measurement of NO3 and N2O5 in a Residential Kitchen, Environ. Sci. Technol. Lett., 2018, 5(10), 595–599 CrossRef CAS.
- G. Drakou, C. Zerefos, I. Ziomas and M. Voyatzaki, Measurements and numerical simulations of indoor O3 and NOx in two different cases, Atmos. Environ., 1998, 32(4), 595–610 CrossRef CAS.
- M. O. Fadeyi, K. Alkhaja, M. B. Sulayem and B. Abu-Hijleh, Evaluation of indoor environmental quality conditions in elementary schools' classrooms in the United Arab Emirates, Frontiers of Architectural Research, 2014, 3(2), 166–177 CrossRef.
- I. Romieu, M. C. Lugo, S. Colome, A. M. Garcia, M. H. Avila, A. Geyh, S. R. Velasco and E. P. Rendon, Evaluation of indoor ozone concentration and predictors of indoor-outdoor ratio in Mexico City, J. Air Waste Manage. Assoc., 1998, 48(4), 327–335 CrossRef CAS.
- M. O. Fadeyi, Ozone in indoor environments: research progress in the past 15 years, Sustainable Cities and Society, 2015, 18, 78–94 CrossRef.
- S. Bhangar, S. C. Cowlin, B. C. Singer, R. G. Sextro and W. W. Nazaroff, Ozone Levels in Passenger Cabins of Commercial Aircraft on North American and Transoceanic Routes, Environ. Sci. Technol., 2008, 42(11), 3938–3943 CrossRef CAS PubMed.
- J. Xiang, C. J. Weschler, J. Mo, D. Day, J. Zhang and Y. Zhang, Ozone, Electrostatic Precipitators, and Particle Number Concentrations: Correlations Observed in a Real Office during Working Hours, Environ. Sci. Technol., 2016, 50(18), 10236–10244 CrossRef CAS PubMed.
- D. Saraga, S. Pateraki, A. Papadopoulos, C. Vasilakos and T. Maggos, Studying the indoor air quality in three non-residential environments of different use: a museum, a printery industry and an office, Build. Environ., 2011, 46(11), 2333–2341 CrossRef.
- S. Zhou, C. J. Young, T. C. VandenBoer, S. F. Kowal and T. F. Kahan, Time-Resolved Measurements of Nitric Oxide, Nitrogen Dioxide, and Nitrous Acid in an Occupied New York Home, Environ. Sci. Technol., 2018, 52(15), 8355–8364 CrossRef CAS PubMed.
- S. Langer, G. Bekö, E. Bloom, A. Widheden and L. Ekberg, Indoor air quality in passive and conventional new houses in Sweden, Build. Environ., 2015, 93, 92–100 CrossRef.
- S. Uchiyama, T. Tomizawa, A. Tokoro, M. Aoki, M. Hishiki, T. Yamada, R. Tanaka, H. Sakamoto, T. Yoshida, K. Bekki, Y. Inaba, H. Nakagome and N. Kunugita, Gaseous chemical compounds in indoor and outdoor air of 602 houses throughout Japan in winter and summer, Environ. Res., 2015, 137, 364–372 CrossRef CAS PubMed.
- G. B. Diette, N. N. Hansel, T. J. Buckley, J. Curtin-Brosnan, P. A. Eggleston, E. C. Matsui, M. C. McCormack, D. L. Williams and P. N. Breysse, Home indoor pollutant exposures among inner-city children with and without asthma, Environ. Health Perspect., 2007, 115(11), 1665–1669 CrossRef PubMed.
- J. F. Zhang and P. J. Lioy, Ozone In Residential Air – Concentrations, I/O Ratios, Indoor Chemistry, And Exposures, Indoor Air, 1994, 4(2), 95–105 CrossRef CAS.
- N. Carslaw, A mechanistic study of limonene oxidation products and pathways following cleaning activities, Atmos. Environ., 2013, 80, 507–513 CrossRef CAS.
- M. O. Fadeyi, C. J. Weschler, K. W. Tham, W. Y. Wu and Z. M. Sultan, Impact of Human Presence on Secondary Organic Aerosols Derived from Ozone-Initiated Chemistry in a Simulated Office Environment, Environ. Sci. Technol., 2013, 47(8), 3933–3941 CrossRef CAS PubMed.
- H. Destaillats, M. M. Lunden, B. C. Singer, B. K. Coleman, A. T. Hodgson, C. J. Weschler and W. W. Nazaroff, Indoor secondary pollutants from household product emissions in the presence of ozone: a bench-scale chamber study, Environ. Sci. Technol., 2006, 40(14), 4421–4428 CrossRef CAS PubMed.
- C. J. Young, S. Zhou, J. A. Siegel and T. F. Kahan, Illuminating the dark side of indoor oxidants, Environ. Sci.: Processes Impacts, 2019 10.1039/c9em00111e.
- M. L. Fischer, D. Littlejohn, M. M. Lunden and N. J. Brown, Automated Measurements of Ammonia and Nitric Acid in Indoor and Outdoor Air, Environ. Sci. Technol., 2003, 37(10), 2114–2119 CrossRef CAS PubMed.
- A. K. Persily, Field measurement of ventilation rates, Indoor Air, 2016, 26(1), 97–111 CrossRef CAS PubMed.
- R. Reichman, E. Shirazi, D. G. Colliver and K. G. Pennell, US residential building air exchange rates: new perspectives to improve decision making at vapor intrusion sites, Environ. Sci.: Processes Impacts, 2017, 19(2), 87–100 RSC.
- ASHRAE, ANSI/ASHRAE Standard 62.1-2010, Ventilation for acceptable indoor air quality, ed. American Society of Heating, R., and Air-Conditioning Engineers, Inc., Atlanta, GA, 2010 Search PubMed.
- E. L. Avol, W. C. Navidi and S. D. Colome, Modeling Ozone Levels in and around Southern California Homes, Environ. Sci. Technol., 1998, 32(4), 463–468 CrossRef CAS.
- K. Lee, J. Xue, A. S. Geyh, H. Ozkaynak, B. P. Leaderer, C. J. Weschler and J. D. Spengler, Nitrous acid, nitrogen dioxide, and ozone concentrations in residential environments, Environ. Health Perspect., 2002, 110(2), 145–150 CrossRef CAS PubMed.
- E. Simons, J. Curtin-Brosnan, T. Buckley, P. Breysse and P. A. Eggleston, Indoor environmental differences between inner city and suburban homes of children with asthma, Journal of Urban Health, 2007, 84(4), 577–590 CrossRef PubMed.
- P. N. Breysse, T. J. Buckley, D. Williams, C. M. Beck, S. J. Jo, B. Merriman, S. Kanchanaraksa, L. J. Swartz, K. A. Callahan, A. M. Butz, C. S. Rand, G. B. Diette, J. A. Krishnan, A. M. Moseley, J. Curtin-Brosnan, N. B. Durkin and P. A. Eggleston, Indoor exposures to air pollutants and allergens in the homes of asthmatic children in inner-city Baltimore, Environ. Res., 2005, 98(2), 167–176 CrossRef CAS PubMed.
- M.-E. Héroux, N. Clark, K. Van Ryswyk, R. Mallick, N. L. Gilbert, I. Harrison, K. Rispler, D. Wang, A. Anastassopoulos, M. Guay, M. MacNeill and A. J. Wheeler, Predictors of Indoor Air Concentrations in Smoking and Non-Smoking Residences, Int. J. Environ. Res. Public Health, 2010, 7(8), 3080–3099 CrossRef PubMed.
- N. Carslaw, A new detailed chemical model for indoor air pollution, Atmos. Environ., 2007, 41(6), 1164–1179 CrossRef CAS.
- M. S. Waring and J. R. Wells, Volatile organic compound conversion by ozone, hydroxyl radicals, and nitrate radicals in residential indoor air: magnitudes and impacts of oxidant sources, Atmos. Environ., 2015, 106, 382–391 CrossRef CAS PubMed.
- C. J. Weschler, H. C. Shields and D. V. Nalk, Indoor chemistry involving O3, NO, and NO2 as evidenced by 14 months of measurements at a site in Southern California, Environ. Sci. Technol., 1994, 28(12), 2120–2132 CrossRef CAS PubMed.
- Y. Liu, P. K. Misztal, J. Xiong, Y. Tian, C. Arata, W. W. Nazaroff and A. H. Goldstein, Detailed investigation of ventilation rates and airflow patterns in a northern California residence, Indoor Air, 2018, 28(4), 572–584 CrossRef CAS PubMed.
- C. Howard-Reed, L. A. Wallace and W. R. Ott, The effect of opening windows on air change rates in two homes, J. Air Waste Manage. Assoc., 2002, 52(2), 147–159 CrossRef CAS.
- National Oceanic and Atmospheric Administration NOAA National Centers for Environmental Information Intergrated Surface Database, https://www.ncdc.noaa.gov/isd.
- L. A. Wallace, S. J. Emmerich and C. Howard-Reed, Continuous measurements of air change rates in an occupied house for 1 year: the effect of temperature, wind, fans, and windows, J. Exposure Anal. Environ. Epidemiol., 2002, 12, 296 CrossRef CAS PubMed.
- S. F. Kowal, S. R. Allen and T. F. Kahan, Wavelength-Resolved Photon Fluxes of Indoor Light Sources: Implications for HOx Production, Environ. Sci. Technol., 2017, 51(18), 10423–10430 CrossRef CAS PubMed.
- A. L. Sumner, E. J. Menke, Y. Dubowski, J. T. Newberg, R. M. Penner, J. C. Hemminger, L. M. Wingen, T. Brauers and B. J. Finlayson-Pitts, The nature of water on surfaces of laboratory systems and implications for heterogeneous chemistry in the troposphere, Phys. Chem. Chem. Phys., 2004, 6(3), 604–613 RSC.
- B. J. Finlayson-Pitts, L. M. Wingen, A. L. Sumner, D. Syomin and K. A. Ramazan, The heterogeneous hydrolysis of NO2 in laboratory systems and in outdoor and indoor atmospheres: an integrated mechanism, Phys. Chem. Chem. Phys., 2003, 5(2), 223–242 RSC.
- D. B. Collins, R. F. Hems, S. Zhou, C. Wang, E. Grignon, M. Alavy, J. A. Siegel and J. P. D. Abbatt, Evidence for Gas-Surface Equilibrium Control of Indoor Nitrous Acid, Environ. Sci. Technol., 2018, 52(21), 12419–12427 CrossRef CAS PubMed.
- S. Gligorovski, Nitrous acid (HONO): an emerging indoor pollutant, J. Photochem. Photobiol., A, 2016, 314, 1–5 CrossRef CAS.
- V. Bartolomei, M. Sorgel, S. Gligorovski, E. Gomez Alvarez, A. Gandolfo, R. Strekowski, E. Quivet, A. Held, C. Zetzsch and H. Wortham, Formation of indoor nitrous acid (HONO) by light-induced NO2 heterogeneous reactions with white wall paint, Environ. Sci. Pollut. Res., 2014, 21(15), 9259–9269 CrossRef CAS PubMed.
- E. Gomez Alvarez, M. Sorgel, S. Gligorovski, S. Bassil, V. Bartolomei, B. Coulomb, C. Zetzsch and H. Wortham, Light-induced nitrous acid (HONO) production from NO2 heterogeneous reactions on household chemicals, Atmos. Environ., 2014, 95, 391–399 CrossRef CAS.
- V. W. Or, M. R. Alves, M. Wade, S. Schwab, R. L. Corsi and V. H. Grassian, Crystal Clear? Microspectroscopic Imaging and Physicochemical Characterization of Indoor Depositions on Window Glass, Environ. Sci. Technol. Lett., 2018, 5(8), 514–519 CrossRef CAS.
- S. K. Brown, M. R. Sim, M. J. Abramson and C. N. Gray, Concentrations of Volatile Organic Compounds in Indoor Air – A Review, Indoor Air, 1994, 4(2), 123–134 CrossRef CAS.
- C. J. Weschler and H. C. Shields, Production of the Hydroxyl Radical in Indoor Air, Environ. Sci. Technol., 1996, 30(11), 3250–3258 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9em00129h |
| This journal is © The Royal Society of Chemistry 2019 |