
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The role of neutral Rh(PONOP)H, free NMe2H, boronium and ammonium salts in the dehydrocoupling of dimethylamine-borane using the cationic pincer [Rh(PONOP)(η2-H2)]+ catalyst†‡
E. Anastasia K.
Spearing-Ewyn
a,
Nicholas A.
Beattie
b,
Annie L.
Colebatch
 a,
Antonio J.
Martinez-Martinez
a,
Andrew
Docker
a,
Timothy M.
Boyd
a,
Gregg
Baillie
b,
Rachel
Reed
b,
Stuart A.
Macgregor
*b and
Andrew S.
Weller
*a
a,
Antonio J.
Martinez-Martinez
a,
Andrew
Docker
a,
Timothy M.
Boyd
a,
Gregg
Baillie
b,
Rachel
Reed
b,
Stuart A.
Macgregor
*b and
Andrew S.
Weller
*a
aChemistry Research Laboratories, Department of Chemistry, University of Oxford, Oxford, OX1 3TA, UK. E-mail: andrew.weller@chem.ox.ac.uk
bInstitute of Chemical Sciences, Heriot Watt University, Edinburgh EH14 4AS, UK. E-mail: S.A.Macgregor@hw.ac.uk
First published on 20th September 2019
Abstract
The σ-amine-borane pincer complex [Rh(PONOP)(η1-H3B·NMe3)][BArF4] [2, PONOP = κ3-NC5H3-2,6-(OPtBu2)2] is prepared by addition of H3B·NMe3 to the dihydrogen precursor [Rh(PONOP)(η2-H2)][BArF4], 1. In a similar way the related H3B·NMe2H complex [Rh(PONOP)(η1-H3B·NMe2H)][BArF4], 3, can be made in situ, but this undergoes dehydrocoupling to reform 1 and give the aminoborane dimer [H2BNMe2]2. NMR studies on this system reveal an intermediate neutral hydride forms, Rh(PONOP)H, 4, that has been prepared independently. 1 is a competent catalyst (2 mol%, ∼30 min) for the dehydrocoupling of H3B·Me2H. Kinetic, mechanistic and computational studies point to the role of NMe2H in both forming the neutral hydride, via deprotonation of a σ-amine-borane complex and formation of aminoborane, and closing the catalytic cycle by reprotonation of the hydride by the thus-formed dimethyl ammonium [NMe2H2]+. Competitive processes involving the generation of boronium [H2B(NMe2H)2]+ are also discussed, but shown to be higher in energy. Off-cycle adducts between [NMe2H2]+ or [H2B(NMe2H)2]+ and amine-boranes are also discussed that act to modify the kinetics of dehydrocoupling.
Introduction
The dehydrocoupling of amine-boranes, H3B·NRR′H (R, R′ = H, alkyl), is an efficient way to produce new molecules and materials with B–N bonds, with hydrogen as the only by-product.1–5 Catalytic routes, mainly mediated by transition metal complexes, offer the possibility to influence kinetics and product distributions. This is especially important for the controlled dehydropolymerisation6–11 of primary amine-boranes, prototypically H3B·NMeH2, that form polymeric materials with main-chain B–N units, (BHNMe)n, that are isosteres of polyolefins and precursors to BN-containing materials.12–14 Challenges remain,15 in determining the precise mechanism of dehydropolymerisation which, at the current level of understanding, appears to be a complex and nuanced process. These are exemplified by: (i) the low catalyst loadings required to selectively produce polymers that hamper speciation studies, (ii) polymeric material that becomes insoluble at high molecular weight or with cross-linking, (iii) complex kinetics that often involve induction periods and modification by the hydrogen co-product, (iv) apparent changes in the precise mechanism dependent on the identity of the precatalyst, and (v) a general overarching process that requires two elementary transformations that need to work in concert: dehydrogenation of amine-borane to aminoborane and subsequent controlled polymerisation (Scheme 1).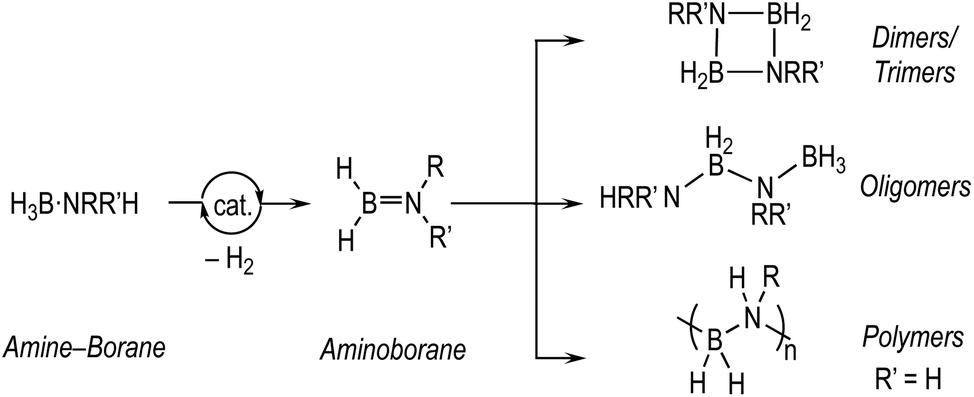 | ||
| Scheme 1 Generalised dehydrocoupling of methyl amine-boranes. R = H or Me. | ||
The dehydrocoupling of the secondary amine-borane H3B·NMe2H offers a more straightforward platform to study these processes as the product is a simple soluble dimer, [H2BNMe2]2 (Scheme 1), and selectivity for its formation is generally less strongly influenced by catalyst loading, meaning that catalyst speciation and kinetics are easier to study using techniques such as NMR spectroscopy. This provides opportunities to study, in closer detail, the elementary processes occurring in dehydrocoupling, with the caveat that the extra N-methyl group may influence both the kinetics and speciation of the catalyst when compared with H3B·NMeH2.16,17
Fundamental to any mechanism for dehydrocoupling is the initial dehydrogenation step to form an aminoborane via BH/NH activation and loss of H2. Depending on the catalyst system a number of different routes have been proposed to operate for this, that all invoke σ-amine-borane complexes18,19 as early intermediates (Scheme 2): (i) step-wise, or concerted, inner sphere;17,20–22 (ii) ligand-assisted cooperation;23–26 and (iii) hydride transfer to form a boronium27 cation that reprotonates the transiently formed hydride.28–31
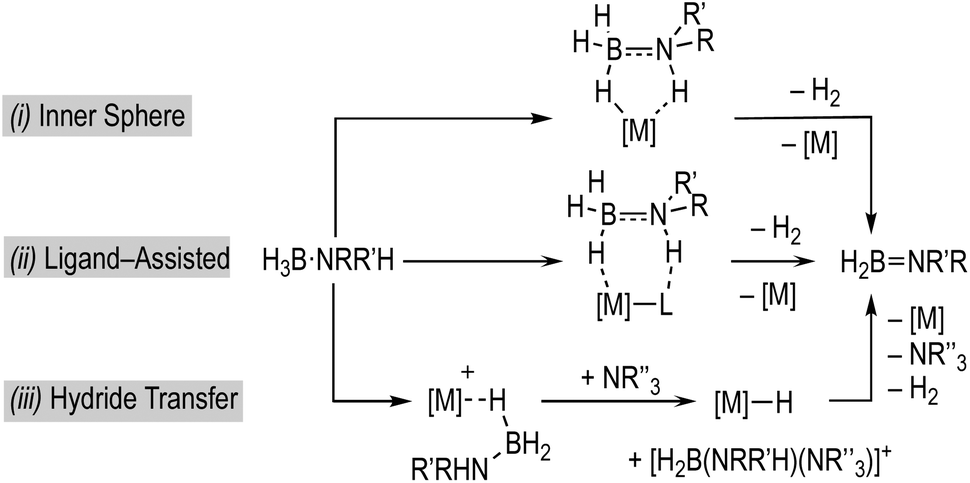 | ||
| Scheme 2 Generic amine-borane dehydrogenation pathways at a metal centre. The structures of the intermediates are illustrative and do not necessarily capture the order of elementary mechanistic steps for each isolated event. | ||
The initial reports of dehydropolymerisation of H3B·NMeH2 used the neutral pincer catalyst Ir(POCOP)H27,8,16 [A, POCOP = κ3-C6H3-2,6-(OPtBu2)2], Scheme 3. A number of closely related pincer-based systems have since been used to catalyse amine-borane dehydrocoupling.21,26,32–35 However to date no σ-amine-borane complexes have been reported with such systems, despite their key role in catalysis. Related off-cycle products have been characterised.36 Analogous POP ligand complexes (POP = e.g. Xantphos) have a richer coordination chemistry with amine-boranes, and η1 (e.g.B) and η2:η2-systems have been characterised that are also relevant to the dehydrocoupling of amine-boranes30,37,38 – although they may not actually lie on the catalytic cycle.11 For POCOP or PONOP-type systems the preparation, and deployment in catalysis, of a σ-amine-borane complex would provide valuable insight into the mechanism of dehydrocoupling.
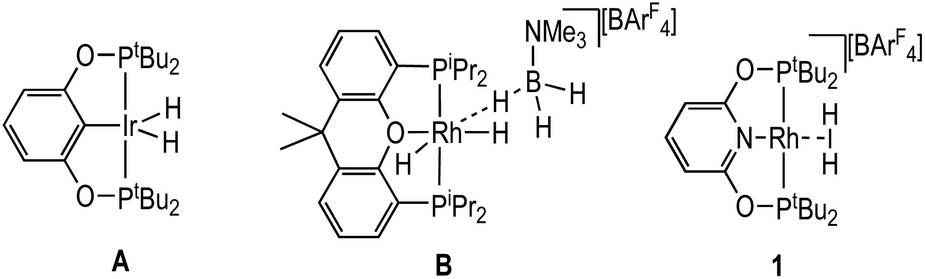 | ||
| Scheme 3 Examples of pincer complexes used in amine-borane dehydrocoupling and complex 1 used in this contribution. | ||
We now report that by use of the readily prepared cationic-precatalyst [Rh(PONOP)(η2-H2)][BArF4]39 [1, ArF = 3,5-(CF3)2C6H3, PONOP = κ3-NC5H3-2,6-(OPtBu2)2] a σ-amine-borane pincer complex can be prepared. 1 is also a competent catalyst for the dehydrocoupling of H3B·NMe2H, and detailed mechanistic studies probe the potential roles of amine (NMe2H), boronium ([H2B(NMe2H)2]+), ammonium ([NMe2H2]+) and a neutral Rh-hydride species in catalytic turnover. The importance of boronium and neutral hydride intermediates in the dehydrocoupling of H3B·NMe2H was first reported by Conejero and co-workers in cationic Pt-based systems.28 Hydride transfer from amine-boranes to cationic metal centres has been reported, for example, by Peruzzini40 and Jagirdar.41
Results and discussion
Synthesis, structures and reactivity of [Rh(PONOP)(H3B·NMe2R)][BArF4] R = Me, 2; H, 3
Dihydrogen σ-complexes offer convenient entry-points into amine-borane coordination chemistry, as the H2 ligand is readily displaced.42–45 The dihydrogen complex [Rh(PONOP)(η2-H2)][BArF4], 1, is prepared as a microcrystalline powder39 from addition of Na[BArF4]46 to Rh(PONOP)Cl47 under an atmosphere of H2 in CH2Cl2 solution followed by recrystallisation. Addition of one equivalent of H3B·NMe3 to 1 in 1,2-F2C6H4 solution resulted in displacement of H2 and the formation in quantitative yield by NMR spectroscopy of [Rh(PONOP)(η1-H3B·NMe3)][BArF4], 2 (Fig. 1). Recrystallisation of this solution from pentane afforded dark yellow crystals suitable for single crystal X-ray diffraction. The tertiary amine-borane H3B·NMe3 was used in these initial studies to stop onward dehydrocoupling by BH/NH activation.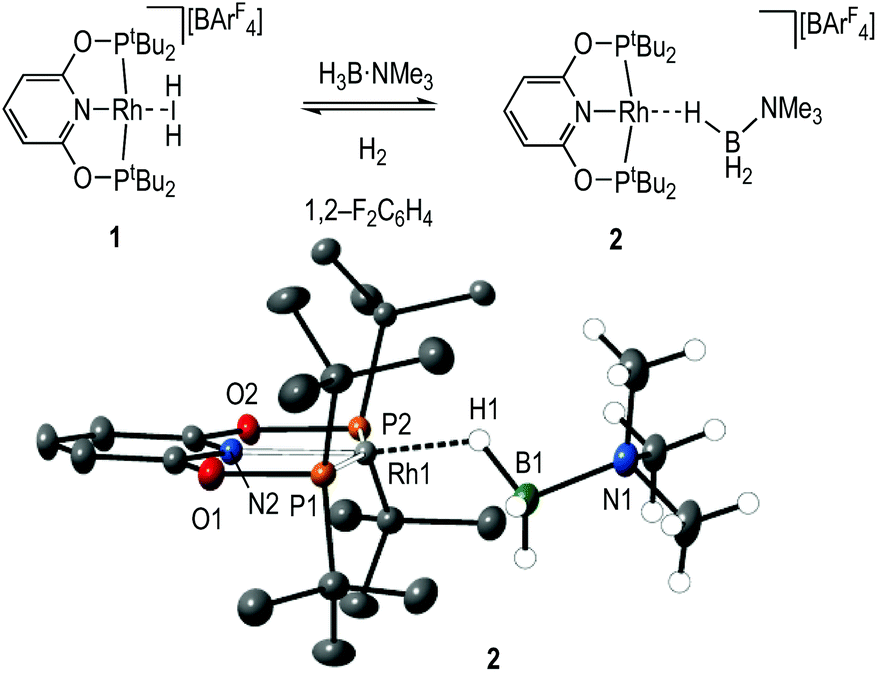 | ||
| Fig. 1 Synthesis and solid-state structure of complex 2. Displacement ellipsoids shown at the 30% level. [BArF4]− anion and most H-atoms are not shown for clarity. Selected bond distances (Å) and angles (°): Rh1–B1, 2.567(5); Rh1–P1, 2.2832(9); Rh1–P2, 2.2594(9); Rh1–N2, 2.026(3); B1–N1, 1.631(6); Rh1–H1, 1.72(5); P1–Rh1–P2, 161.63(3); N2–Rh1–H1, 172.6(17); Rh1–H1–B1, 121(4). | ||
The solid-state structure of the cationic portion of 2 is shown in Fig. 1. The hydrogen atoms associated with the boron were located and refined. The structure demonstrates an η1-bound H3B·NMe3 ligand, rather than bidentate η2:η2, as determined by a long Rh⋯B distance [2.567(5) Å] and a rather open Rh–H–B angle [121(4)°].48,49 There is one closer Rh–H distance [H1, 1.72(5) Å], with the other two considerably distant [2.8–2.9 Å]. In comparison, a pincer complex with a η2:η2 H3B·NMe3 bonding mode, [Rh(NNN)(H3B·NMe3)][BArF4] [NNN = 2,6-bis-[1-(2,6-diisopropylphenylimino)ethyl]pyridine] C, shows a closer Rh⋯B distance [2.305(5) Å] and more acute Rh–H–B angles [93(3)°], Scheme 4.38 Complex 2 is more closely related to [Ir(POCOP)(H)2(η1-HSiEt3)][B(C6F5)4], D,50 which has a far more pronounced η1 binding mode [Ir⋯Si 3.346(1) Å, Ir–H–Si 157°], as well as [Pt(ItBu’)(ItBu)(η1-H3B·pyridine)][BArF4], E [Pt⋯·B 2.8436(5) Å, Pt–H–B 147(4)°]51 (ItBu = 1,3-di-tert-butylimidazolylidene).
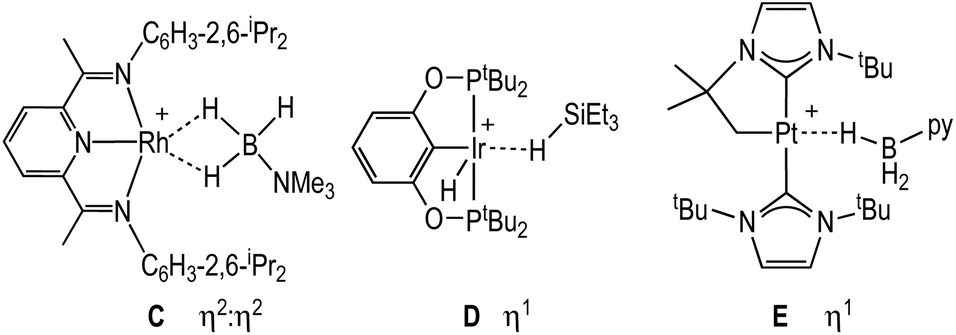 | ||
| Scheme 4 Comparison of η1 and η2:η2 amine-borane and silane complexes. | ||
Solution NMR spectroscopic data are in agreement with the η1-binding of the amine-borane being retained in CD2Cl2. In the 298 K 1H NMR spectrum a broad signal integrating to 3 H is observed at δ −2.55 that sharpens on decoupling 11B, that does not change on cooling to 173 K and no coupling to 103Rh is observed. This is indicative of a rapid exchange between terminal and bridging B–H positions.52,53 A single tBu environment was also observed. In the 11B NMR spectrum a broad signal at δ −15.2 that is assigned to the H3B·NMe3 group is shifted 7.9 ppm upfield from free H3B·NMe3 (δ −7.3). The 31P{1H} NMR spectrum displays a single doublet at δ 208.9 [J(RhP) = 142 Hz]. Electrospray-Ionisation Mass Spectrometry (ESI-MS) shows the correct isotope pattern for the cation (m/z = 575.26, calc. 575.26). Complex 2 is stable in 1,2-F2C6H4 solution for at least 24 hours.
H2 is a competitive ligand with H3B·NMe3 for coordination at the {Rh(PONOP)}+ fragment, and placing a sample of 2 (8.7 mM, 1,2-F2C6H4) under 4 atm H2 in a sealed NMR tube immediately results in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of 1:2 and free H3B·NMe3 – which is now observed as a sharp quartet in the 11B NMR spectrum. Degassing returns 2 as the major component showing that these two species are in equilibrium (Fig. 1). These experimental results are consistent with a computed ΔG from DFT calculations (see later) that is close to thermoneutral, being −1.2 kcal mol−1 in favour of 2. Under a D2 atmosphere (1 atm) H/D exchange at the borane also occurs, to afford the H2-, HD- and D2-isotopologues of 1, as identified by their distinctive isotopically-shifted 31P chemical shifts (ESI), free HxD3−xB·NMe3 – as shown by a loss of resolvable coupling in the 11B NMR spectrum – and dissolved H2/HD and D2. This H/D exchange likely involves reversible oxidative cleavage of B–H or D2 at the Rh(I)-centre followed by H/D exchange – via a σ-complex assisted metathesis (σ-CAM) mechanism.54 Similar H/D exchange has been noted in related Rh-dihydrogen pincer complexes.39,55
:1 ratio of 1:2 and free H3B·NMe3 – which is now observed as a sharp quartet in the 11B NMR spectrum. Degassing returns 2 as the major component showing that these two species are in equilibrium (Fig. 1). These experimental results are consistent with a computed ΔG from DFT calculations (see later) that is close to thermoneutral, being −1.2 kcal mol−1 in favour of 2. Under a D2 atmosphere (1 atm) H/D exchange at the borane also occurs, to afford the H2-, HD- and D2-isotopologues of 1, as identified by their distinctive isotopically-shifted 31P chemical shifts (ESI), free HxD3−xB·NMe3 – as shown by a loss of resolvable coupling in the 11B NMR spectrum – and dissolved H2/HD and D2. This H/D exchange likely involves reversible oxidative cleavage of B–H or D2 at the Rh(I)-centre followed by H/D exchange – via a σ-complex assisted metathesis (σ-CAM) mechanism.54 Similar H/D exchange has been noted in related Rh-dihydrogen pincer complexes.39,55
The reaction between 1 and one equivalent of H3B·NMe2H initially follows the same course as with H3B·NMe3 (Scheme 5). On time of mixing the complex [Rh(PONOP)(η1-H3B·NMe2H)][BArF4], 3, is observed to be the major organometallic product (greater than 95%), as identified by 1H [δ −1.98, 3 H], 11B [δ −21.3] and 31P{1H} [δ 212.9, J(RhP) = 138 Hz] NMR spectroscopies in comparison with complex 2. The remainder is complex 1. A triplet observed in the 11B NMR spectrum (∼10%) at δ −0.36 [J(BH) 110 Hz] is identified as H2B(NMe2)2H,56 and not boronium [H2B(NMe2H)2][BArF4] by comparison with an independently prepared sample of the latter in 1,2-F2C6H4 (δ −2.15, J(BH) 117 Hz).28 After 5 minutes aminoborane H2B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NMe2 [δ 37.8, t, J(BH) = 129 Hz]43,57 is observed, indicating dehydrogenation is proceeding that then eventually forms dimeric [H2BNMe2]2 [δ 5.7, J(BH) = 127 Hz]. After 35 minutes this solution has changed to return 1 as the sole organometallic complex, during which time the H3B·NMe2H has undergone dehydrocoupling to form [H2BNMe2]2 as the major product. The diamino-borane HB(NMe2)2 is the other, minor (2%), product [δ 28.7, d, J(BH) 130 Hz]. This formally comes from a hetero-dehydrocoupling of NMe2H and H3B·NMe2H.51,58
NMe2 [δ 37.8, t, J(BH) = 129 Hz]43,57 is observed, indicating dehydrogenation is proceeding that then eventually forms dimeric [H2BNMe2]2 [δ 5.7, J(BH) = 127 Hz]. After 35 minutes this solution has changed to return 1 as the sole organometallic complex, during which time the H3B·NMe2H has undergone dehydrocoupling to form [H2BNMe2]2 as the major product. The diamino-borane HB(NMe2)2 is the other, minor (2%), product [δ 28.7, d, J(BH) 130 Hz]. This formally comes from a hetero-dehydrocoupling of NMe2H and H3B·NMe2H.51,58
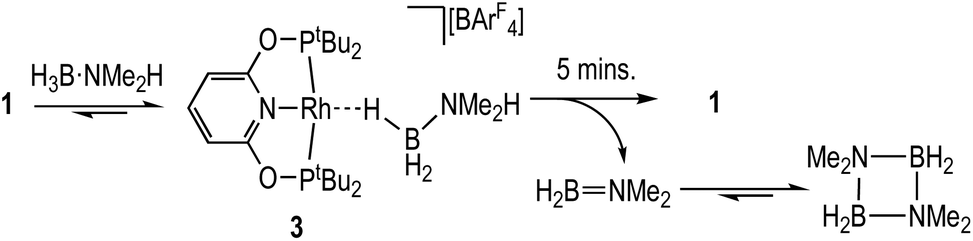 | ||
| Scheme 5 Dehydrogenation of H3B·NMe2H by complex 1. | ||
Catalysis and catalyst speciation: induction periods, change in resting state and a neutral hydride
These stoichiometric reactions demonstrate σ-complex formation, B–H activation, and competitive H2 binding, and thus set the scene for the catalytic studies on H3B·NMe2H dehydrocoupling. Initial studies using catalyst 1 in a sealed NMR tube [2 mol%, H3B·NMe2H 0.144 M, 298 K, 1,2-F2C6H4 solution] revealed, by 11B NMR spectroscopy (Fig. 2A), an induction period of approximately 300 seconds, followed by the consumption of H3B·NMe2H to finally give [H2BNMe2]2 after 2700 s. H2BNMe2 is observed to grow in and decay, with a temporal profile characteristic of an intermediate. Following the same reaction by interleaved 31P{1H} NMR spectroscopy (Fig. 2B) revealed the immediate formation of the σ-amine-borane complex 3, which reforms complex 1 as H3B·NMe2H is consumed and H2 builds up in the reaction head-space. A further complex is observed to grow in and then out again, which is characterised by a 31P{1H} resonance at δ 225.4 [J(RhP) = 170 Hz] and a broad, high field, signal in the 1H NMR spectrum at δ −10.13. This is identified by an independent synthesis as Rh(PONOP)H, 4. Notably, 4 appears at the early stages of reaction, post-induction period, and is then consumed as 1 grows in. A small amount (∼2%) of HB(NMe2)2 was also noted.
 | ||
| Fig. 2 Time course plots for dehydrogenation of H3B·NMe2H [0.144 M, 298 K, 1,2-F2C6H4 solvent] using complex 1 (2 mol%) as measured by in situ NMR spectroscopy in a sealed NMR tube. (A) 11B: △, H3B·NMe2H; ○, [H2BNMe2]2; □, H2BNMe2. (B) 31P{1H}: ◊, 3; ○, 4; □, 1. Dotted lines to guide the eye. | ||
Complex 4, Rh(PONOP)H, was independently synthesised by addition of the lithium amidoborane Li[NMe2BH3]59 to Rh(PONOP)Cl in pentane solution, and is formed alongside [H2BNMe2]2. Filtration and recrystallisation from methylcyclohexane afforded 4 as red crystals. Fig. 3 shows the solid-state structure of complex 4, which was refined successfully as a two-component twin. The structural solution shows a pseudo square-planar coordination geometry around Rh in which the hydride ligand was located. The associated structural metrics are unremarkable, and complex 4 adds to the relatively small number of structurally characterised planar pincer mono-hydrides.60–67 In the 1H NMR spectrum of the isolated product (C6D6), the hydride ligand is signalled by a doublet of triplets at δ −9.60 [J(RhH) = 19.5, J(PH) = 22.6 Hz] that collapses into a doublet in the 1H{31P} NMR spectrum. In the 31P{1H} NMR spectrum a doublet is observed at δ 226.2 [J(RhP) = 171 Hz]. While these data are very similar to those observed during catalysis in 1,2-F2C6H4 solution, the hydride chemical shift is different for pure material compared to that observed in situ during catalysis in this solvent: δ −9.92 and δ −10.13 respectively. This suggests the possibility of secondary, [Rh]–H⋯H–X, interactions68 that are discussed in the computational section.
 | ||
| Fig. 3 Independent synthesis, solid-state structure of complex 4, and possible intermediates. Displacement ellipsoids shown at the 30% level. Most H-atoms are not shown for clarity. Selected bond distances (Å) and angles (°): Rh1–H1, 1.54(5); Rh1–P1, 2.2179(17); Rh1–P2, 2.2020(17); Rh1–N1, 2.046(6); P1–Rh1–P2, 162.80(7). Free energies are DFT-calculated. | ||
The mechanism to form 4 from Rh(PONOP)Cl could operate via an amidoborane69–71 intermediate I (inset, Fig. 3) that undergoes β-elimination to generate 4 and H2BNMe2 (experimentally observed as the dimer). Arguing against this is that β-elimination processes in group-2 amidoboranes have been found to be rather high in energy,72 while the ethyl analogues M(PONOP)(CH2CH3) (M = Rh and Ir) have been reported to be unusually stable with regard to β-elimination and formation of the corresponding hydride.73,74 An alternative mechanism is that Li[NMe2BH3] acts as a simple hydride source,75,76 avoiding I and directly eliminating LiCl and H2BNMe2. DFT calculations suggest the latter scenario is more likely, with the σ-amidoborane adduct, Rh(PONOP)(H3BNMe2), II, computed to be 9.3 kcal mol−1 more stable than its N-bound isomer, Rh(PONOP)(NMe2BH3), I. Moreover, II exhibits a minimal barrier of 1.1 kcal mol−1 to B–H bond cleavage to form 4 and free H2BNMe2 (see ESI‡).
The role of boronium, [NMe2H2][BArF4], NMe2H and the neutral hydride in catalytic turnover
With the identity of the intermediate complex 4 determined as being a neutral hydride, we considered possible routes for its formation and consumption in the catalytic ensemble, taking into account the induction period that is observed, that also precedes significant productive turnover. We have recently reported11 that for cationic [Rh(DPEphos)]+-based dehydropolymerisation catalysts significant induction periods can be removed by adding amine, e.g. NMeH2, as this promotes the formation of the active catalyst. While the actual mechanism of the active catalyst being brought on cycle with the [Rh(DPEphos)]+-based catalysts is complex, one role of free amine (formed from B–N bond cleavage) is proposed to be attack at a cationic σ-amine-borane complex to afford a neutral hydride and boronium cation, e.g. [H2B(NMe2H)2]+.28,31 The role of boronium in productive turnover has also been probed in pincer-like [Rh(Xantphos-iPr)]+ systems, B,30 and Pt-based systems related to E,28Scheme 2, as well as others.29 We propose a similar set of fundamental processes operates here, in which amine-induced B–H bond cleavage gives a metal hydride that is then reprotonated. Two realistic scenarios presented themselves for this process: (a) the boronium route as described, or, (b) one that invoked the formation of ammonium (Scheme 6). This latter route is a deprotonation of σ-bound H3B·NMe2H to form an amidoborane, i.e.IIFig. 3, that then eliminates H2BNMe2. Experiments to probe these and other fundamental steps in catalysis are detailed below, and also explored in the computational section.
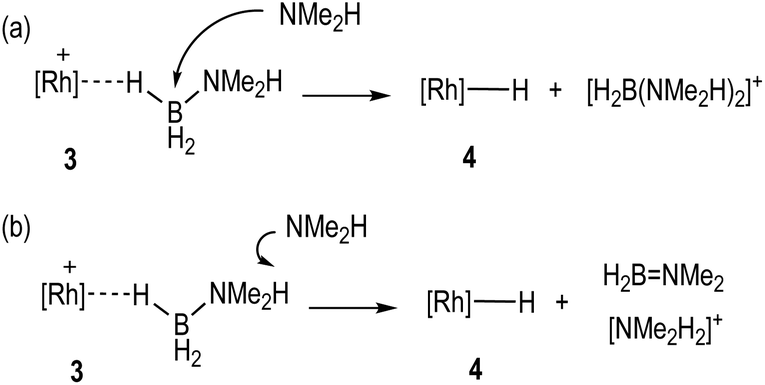 | ||
| Scheme 6 Possible mechanisms for the formation of hydride 4. | ||
(i) H2 evolution experiments (0.072 M H3B·NMe2H, 2 mol% 1, eudiometer, 298 K) confirm an induction period (∼450 s) also operates in an open system, before significant hydrogen production starts (TON = 50, max. rate (vmax) = 1.0 × 10−4 M s−1), Fig. 4, which is followed by an deceleration in rate consistent with substrate depletion. [H2BNMe2]2 is produced as the principal product (11B NMR), alongside a small amount of HB(NMe2)2. Periodic sampling demonstrates a similar profile for catalyst speciation as observed in a sealed NMR tube. There was no significant change in profile when Hg or sub-stoichiometric PPh3 (0.2 equivalents) was added – suggesting a homogeneous process.77 The post induction period reaction profile could not be reconciled with a simple kinetic model.
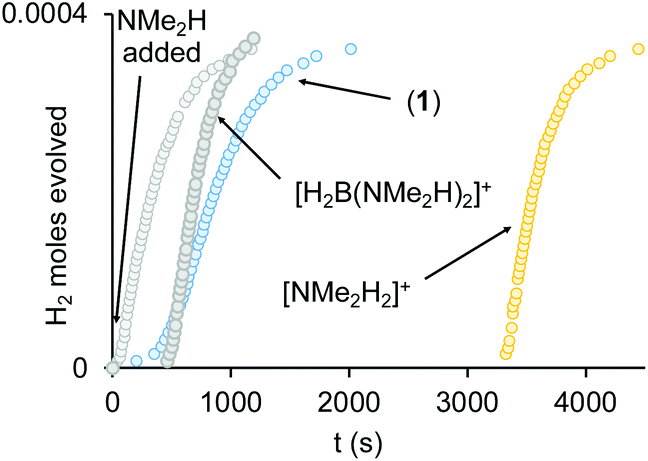 | ||
| Fig. 4 Time course plot for dehydrogenation of H3B·NMe2H [0.072 M, 298 K, 1,2-F2C6H4 solvent] using complex 1 (2 mol%), plus 1 equivalent NMe2H, [H2B(NMe2H)2][BArF4] or [NMe2H2][BArF4]. H2 as measured by H2 eudiometer. | ||
(ii) The induction period using catalyst 1 is removed by addition of one equivalent of NMe2H at the start of catalysis, that also promotes a slightly faster turnover (vmax = 1.6 × 10−4 M s−1), Fig. 4. Again, the temporal profile could not be reconciled with a simple kinetic model. Speciation experiments under these conditions (sealed NMR tube) indicate that 4 is now formed exclusively at the start of catalysis, with no 3 observed. The final resting state is 1. Amine thus promotes catalysis and moves the initial resting state to neutral 4.
(iii) Adding one equivalent of [H2B(NMe2H)2][BArF4] to catalyst 1 results in a much more pronounced induction profile (∼450 s), and once turnover starts catalysis is slightly faster (vmax = 1.8 × 10−4 M s−1), and decelerates slower. However, as boronium is also a source of free NMe2H on protonation of 4, we cannot discount that it is simply acts in this way to promote catalysis (vide infra).
(iv) Addition of one equivalent of [NMe2H2][BArF4] to catalyst 1 increases the induction period to ∼1 hour, but once turnover starts it is comparable to NMe2H and [H2B(NMe2H)2]+ doped systems (vmax = 2.0 × 10−4 M s−1).
(v) Complex 4 is a poor catalyst on its own (0.072 M H3BNMe2H, 2 mol% 1, eudiometer, 298 K), promoting slow turnover with only 25% conversion observed after ∼1 h (vmax = 0.5 × 10−4 M s−1).
(vi) When complex 4 is doped with one equivalent of [NMe2H2][BArF4] the induction period is removed and catalysis proceeds at a rate comparable to the NMe2H doped system (vmax = 1.6 × 10−4 M s−1). Doping with one equivalent of [H2B(NMe2H)2][BArF4] also removes the induction period, but turnover is slower (vmax = 0.5 × 10−4 M s−1).
(vii) Under stoichiometric conditions addition of [H2B(NMe2H)2][BArF4] or [NMe2H2][BArF4] to 4 recovers 1.78
These observations show the important role of NMe2H in catalysis, and that complex 1 can be regenerated by protonation of complex 4. However, the kinetic profiles are still complex, and in particular changes in induction periods on doping suggest off-cycle processes and more complex equilibria are operating, while the precise role of [NMe2H2][BArF4] and/or [H2B(NMe2H)2][BArF4] remain unclear. These points are explored in more detail next.
Kinetic analysis of H2 generation
Given non-trivial temporal profiles we have deployed a combination of maximum rates79 and Burés’ graphical approach of Variable Time Normalisation Analysis (VTNA) to interrogate the kinetics of catalysis.80,81 VTNA works by plotting reaction course (reactant or product) against t[cat]n for different [cat]TOTAL. By adjusting the power value until the various plots visually overlay the order in [cat]TOTAL can be determined independent of the complexity of the kinetic regime.82Fig. 5 shows this approach to determine the order in [Rh]TOTAL, that demonstrates an overall first order kinetic regime between 0.2 mol% and 4 mol% [Rh]TOTAL – thus excluding any dimer/monomer equilibria in catalysis and also that [Rh]TOTAL remains approximately constant throughout. The data was time-shifted (∼450 s) to remove the induction period that comes from the catalyst-independent B–N bond cleavage.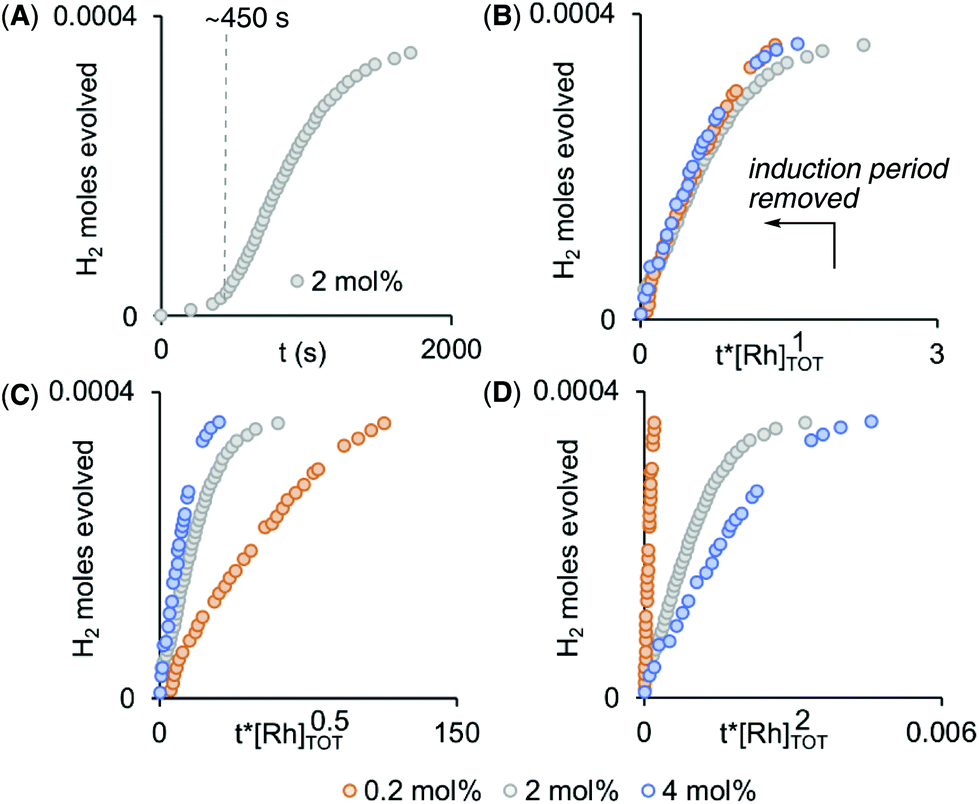 | ||
| Fig. 5 (A) Time course plot for dehydrogenation of H3B·NMe2H [0.072 M, 298 K, 1,2-F2C6H4 solvent] using complex 1 (2 mol%) as measured by H2 evolution. Variable Time Normalization Analysis80,81 for the order in [Rh]TOT (0.2 mol% – 4 mol%, induction periods removed): (B) [Rh]1, (C) [Rh]0.5, (D) [Rh]2. | ||
Given the complex kinetic profile, maximum rates (vmax) were used to determine the effect of isotopic substitution on the rate, and using H3B·NMe2D and D3B·NMe2H a kH/kD of 1.9 and 1.1 for NH and BH respectively was measured. This suggests that NH activation is involved in the rate-determining transition state, while BH activation is not.
The order in H3B·NMe2H was probed by determining the maximum rate measured as [H3B·NMe2H] was varied, while keeping [Rh]TOTAL fixed. Fig. 6 shows that this offers a profile that is initially positive order in amine-borane, but at higher concentrations of H3B·NMe2H the rate decelerates. This suggests that H3B·NMe2H is participating in an equilibrium that removes at least one of the reaction partners implicated in, or prior to, the turn-over limiting step. A scenario that explains these data is an off-cycle interaction between H3B·NMe2H and [H2B(NMe2H)2]+ or [NMe2H2]+ that would reduce the available concentration of H3B·NMe2H and [H2B(NMe2H)2]+ or [NMe2H2]+; and depending on the relative concentrations could act in an inhibitory manner. We discount a scenario where the product (i.e. [H2BNMe2]2) modifies the kinetics, as doping catalysis using 1 (2 mol%) with 50 equivalents of [H2BNMe2]2 leads to no change in the temporal profile (vmax = 1.0 × 10−4 M s−1).
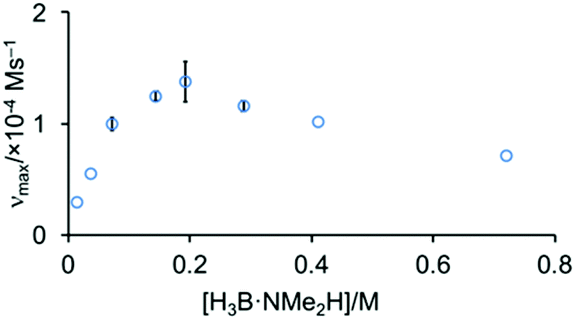 | ||
| Fig. 6 Maximum rate of catalysis versus [H3B·NMe2H] using complex 1 (0.00144 M) as measured by H2 evolution. | ||
Interaction between amine-boranes and [H2B(NMe2H)2][BArF4] or [NMe2H2][BArF4]: competing off-cycle equilibria
To probe the existence of off-cycle interactions, NMR titration experiments were carried out between [H2B(NMe2H)2][BArF4] and H3B·NMe3 or H3B·NMe2H. Monitoring the change in the chemical shift of the NH protons in [H2B(NMe2H)2]+ as a function of amine-borane concentration generated titration isotherms. WinEQNMR283 analysis of the H3B·NMe3 titration data determined a 1:1 stoichiometric association constant (Ka = 9.3(1) M−1), Fig. 7A. We suggest adducts such as III with non-classical dihydrogen bonds are formed.85 Related bisphosphine boronium adducts have been reported which show P–H⋯X hydrogen bonds.84 For H3B·NMe2H the situation is more complex. Although the titration data clearly demonstrate an interaction between the two species, 1:1, 2:1 or 1:2 binding models failed to provide satisfactory agreement with experimental data, implying higher stoichiometry and/or complex equilibria. Nevertheless, these experiments show that H3B·NMe2H and [H2B(NMe2H)2][BArF4] can form off-cycle adducts that attenuate the availability of both. Similar 1:1 equilibria are also operating with [NMe2H2][BArF4]/H3B·NMe3, Ka = 28(2) M−1, possibly via adducts such as IV, Fig. 7B. At high [H3B·NMe2H] the decrease in max rate suggests these interactions (with whichever partner) are significant enough to cause a relative reduction in rate. Similar adducts could also be involved in sequestering free amine-leading to the change in induction periods observed when [H2B(NMe2H)2][BArF4] or [NMe2H2][BArF4] are doped into catalysis; and productive turnover only occurs once sufficient NMe2H has been formed by B–N bond cleavage to overcome these off-cycle equilibria. Crystallographically characterized [R2NH2⋯NR2H]+ complexes are known.86
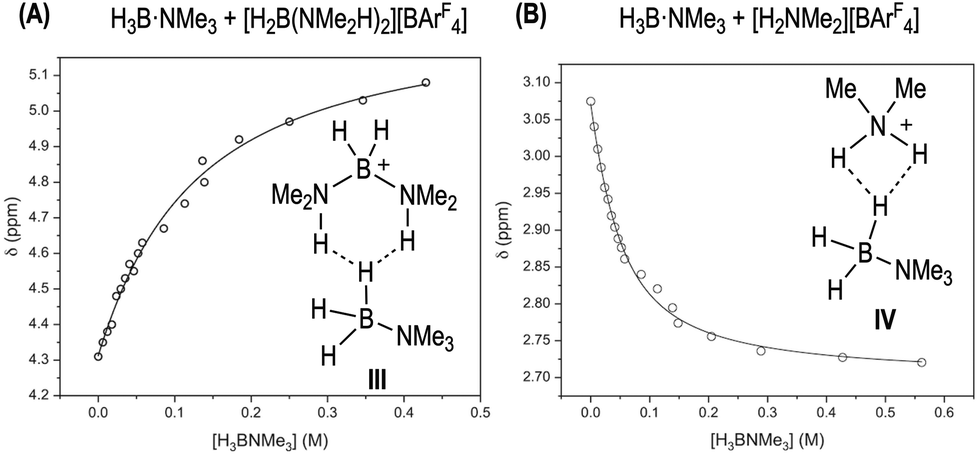 | ||
| Fig. 7 (A) Titration binding curve of [H2B(NMe2H)2][BArF4] (298 K, 30 mM, 1,2-F2C6H4) with H3B·NMe3. Fitted binding isotherm is indicated by line. Association Constant, Ka, 9.3(1) M−1, calculated using WinEQNMR283 monitoring the chemical shift data for the NH protons in [H2B(NMe2H)2][BArF4]. (B) Titration binding curve of [NMe2H2][BArF4] (298 K, 30 mM, 1,2-F2C6H4) with H3B·NMe3. Association Constant, Ka, 28(2) M−1 derived from monitoring the chemical shift data for the NMe protons in H3B·NMe3. | ||
Elementary steps of the mechanism
Pulling these observations together a catalytic cycle can be proposed, Scheme 7. ①: σ-Dihydrogen complex 1 reversibly reacts with H3B·NMe2H to form 3, that is observed to be the major species at the very start of catalysis. ②: NMe2H (formed from slow B–N bond cleavage of H3B·NMe2H) then rapidly reacts with 3 to form 4, and either [H2B(NMe2H)2]+ or [NMe2H2]+/H2BNMe2. ③: Protonation of 4 regenerates 1, free NMeH2 and in the case of boronium, H2BNMe2 that dimerises to give [H2BNMe2]2. The concentration of 1/4/3 follows temporal profiles (Fig. 2) that suggest they are closely matched in relative stabilities and their concentrations thus depend on how [H3B·NMe2H] and [H2] evolve throughout catalysis. Off-cycle equilibria between H3B·NMe2H and either [H2B(NMe2H)2]+ or [NMe2H2]+ operate to modify the available concentration of species involved in turnover. The measured KIE indicates NH activation is involved in the turnover limiting transition state. This could come from step ② being turnover limiting in the ammonium pathway (N–H cleavage of H3B·NMe2H), or step ③ in both boronium or ammonium pathways (N–H cleavage to reprotonate 4). The lack of a significant KIE measured for BH argues that B–H bond cleavage is not significant in the turnover limiting transition state. As the experimental data do not allow for discrimination between an ammonium or a boronium pathway we turned to computational studies to determine the relative energies of each pathway.
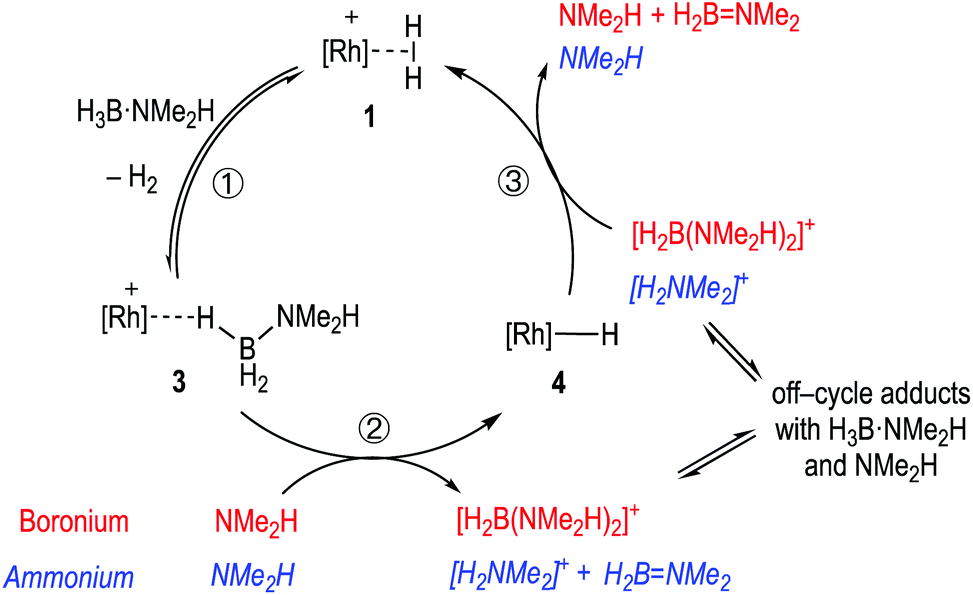 | ||
| Scheme 7 Suggested simplified catalytic cycle with boronium (red) ammonium (blue, italics) and pathways. | ||
Computational studies
The details of the dehydrogenation and protonation steps (3 → 4 → 1) were also probed with DFT calculations. The model used incorporated the full PONOP ligand with geometries optimised with the BP86 functional. Free energies were then corrected for solvation, dispersion (BJD3) and basis set effects (def2TZVP, see ESI‡ for full details). 2-Hexanone (ε = 14.14) was employed as solvent as a model for 1,2-difluorobenzene (ε = 13.81) for which parameters are not currently available. Two pathways were considered for H3B·NMe2H dehydrogenation from 3 and free NMe2H to form 1 (see Scheme 8): initial nucleophilic attack at B to form boronium (following the proposal of Conejero and co-workers28) and N–H deprotonation of the bound H3B·NMe2H ligand, i.e. the ammonium route.87 Both processes initially form 4 which is then reprotonated by either [H2B(NMe2H)2]+ or [NMe2H2]+ to form 1. Alternative processes based on B–H and/or N–H activation from 3 without amine or boronium involvement were also considered and shown to be significantly higher in energy (see Fig. S1–S3, ESI‡).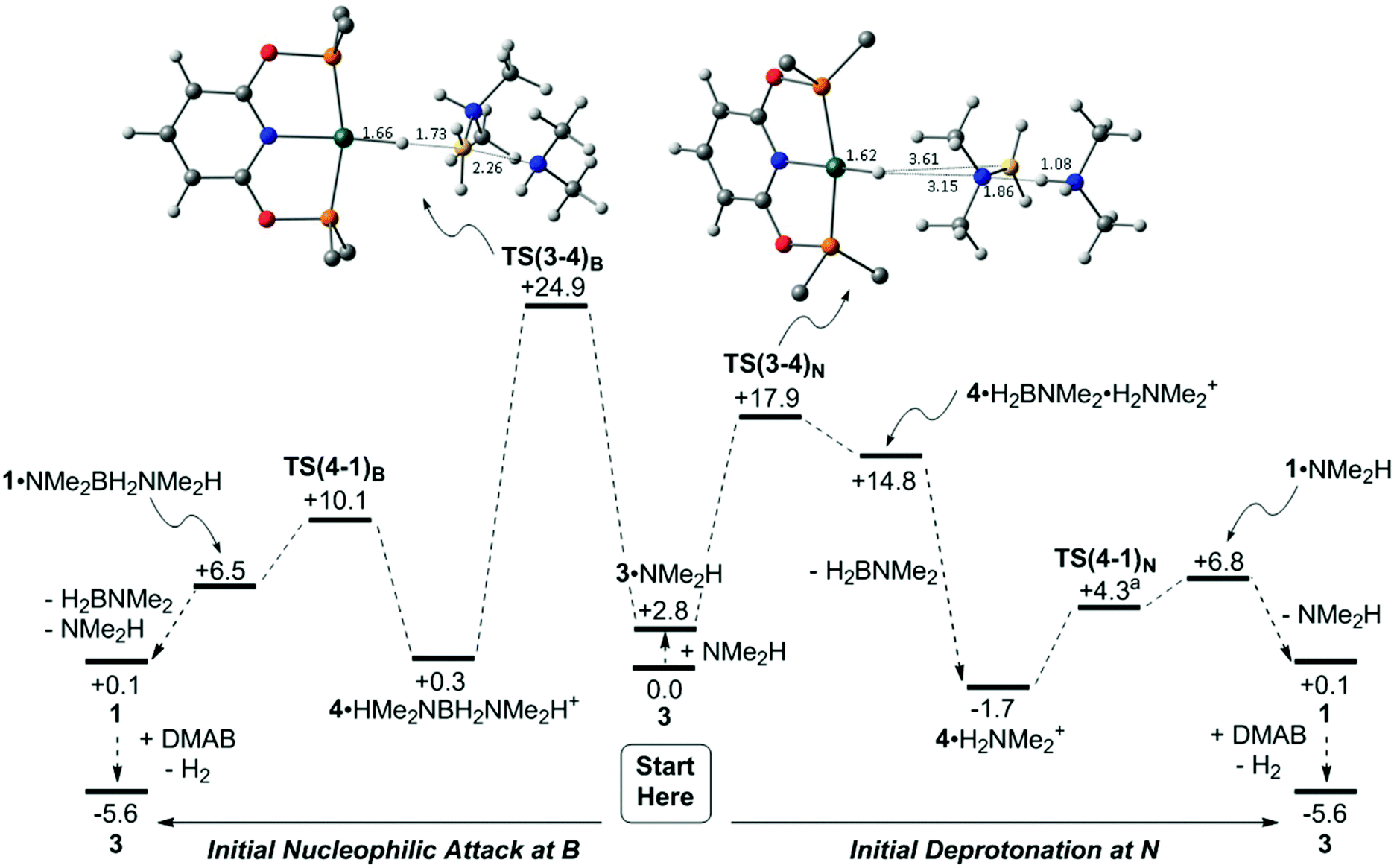 | ||
| Scheme 8 Computed free energy profiles (kcal mol−1) for dehydrogenation of H3B·NMe2H from H-bonded adduct 3 (centre) via initial nucleophilic attack at B (left) or initial deprotonation at N (right). [Rh] = (PONOP)Rh. Energies are quoted relative to 3 + free H3B·NMe2H and NMe2H set to 0.0 kcal mol−1. Ball and stick representations of TS(3-4)B and TS(3-4)N have the tBu Me groups removed for clarity and all distances are given in Å. Atom colouring scheme: Rh (teal); C (grey); H (while); O (red); N (blue); P (orange); B (yellow). aTS(4-1)N is a true stationary point on the electronic energy surface but differential zero-point energy effects cause the free energy of this species to fall below 1·NMe2H. | ||
Starting from 3 (Scheme 8 centre), a H-bonded adduct, 3·NMe2H (+2.8 kcal mol−1), is formed that features a short NH⋯N distance of 1.76 Å. Nucleophilic attack at B then passes through TS(3-4)B at +24.9 kcal mol−1 to form 4·HMe2NBH2NMe2H+ with a strong Rh–Hδ−⋯Hδ+–N dihydrogen interaction (1.75 Å). This is thus set up for facile proton transfer viaTS(4-1)B (+10.1 kcal mol−1) to give [(PONOP)Rh(η2-H2)]+, 1, initially as an H-bonded adduct with the Me2NBH2NMe2H moiety. Release of H2BNMe2 and NMe2H along with substitution of H2 in 1 by H3B·NMe2H (see Scheme 9) then reforms 3 and completes the catalytic cycle. The overall process is exergonic by 5.6 kcal mol−1 and has an energy span of 24.9 kcal mol−1. The alternative N–H deprotonation of the H3B·NMe2H ligand in 3·NMe2H proceeds through TS(3-4)N at +17.9 kcal mol−1 and occurs with concomitant B–H bond cleavage to give neutral hydride 4, as a weakly bound adduct with H2BNMe2 and [H2NMe2]+. Removal of H2BNMe2 allows formation of 4·H2NMe2+ (−1.7 kcal mol−1). This again features a strong Rh–Hδ−⋯Hδ+–N interaction (1.55 Å) and allows proton transfer to give 1·NMe2H at +6.8 kcal mol−1. Loss of NMe2H and H2/H3B·NMe2H substitution again completes the cycle. The overall barrier for this N-deprotonation mechanism is 17.9 kcal mol−1 and is therefore predicted to be favoured over nucleophilic attack at B.
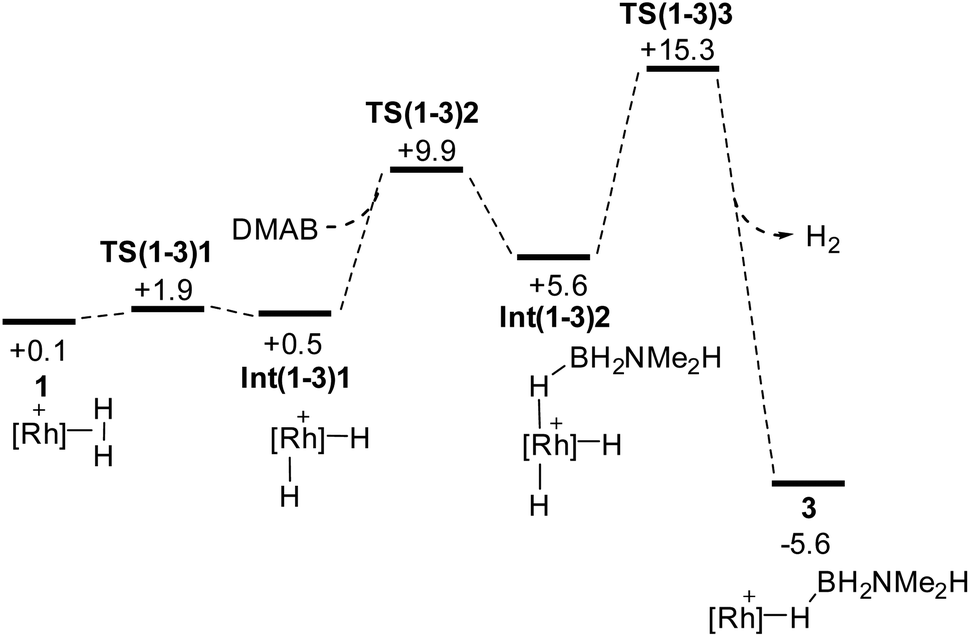 | ||
| Scheme 9 Computed free energy profiles (kcal mol−1) for H2/H3B·NMe2H substitution in 1 to reform 3; [Rh]+ = [Rh(PONOP)]+. Energies are quoted relative to 3 + free H3B·NMe2H and NMe2H set to 0.0 kcal mol−1. | ||
Scheme 9 shows the details of the H2/H3B·NMe2H substitution to reform 3 from 1. An associative transition state could not be located, but instead initial, very facile oxidative cleavage of the η2-H2 ligand permits formation of an η1-H3B·NMe2H Rh(III) adduct, Int(1-3)2, at +5.6 kcal mol−1. Reductive elimination of H2 then proceeds viaTS(1-3)3 at +15.3 kcal mol−1 to form 3.88
Returning to Scheme 8, the computed geometries of the key transition states show TS(3-4)B is similar to that located by Conejero and co-workers in their study,28 and features significant B–H stretching corresponding to hydride transfer onto the Rh centre. In contrast TS(3-4)N exhibits a much later geometry in which hydride transfer to Rh is complete and significant H+ transfer to form the [H2NMe2]+ cation is evident. As a result TS(3-4)N displays a very large dipole moment (24.7 D) making its energy sensitive to solvation effects.89TS(3-4)N is favoured on energetic grounds and would be expected to show a small N–H/N–D KIE (and no B–H/B–D KIE). In contrast the significant B–H bond stretching in TS(3-4)B would imply a significant B–H/B–D KIE that is not seen experimentally.
The computed energies of 1, 3 and 4 in Scheme 8 bear comparison with the time course plots derived from experiment in Fig. 2. 1 and 3 are computed to be most stable as isolated species, however 4 is present as the dihydrogen-bonded adduct 4·H2NMe2+. This formulation is also consistent with the small shift in 1H chemical shift associated with this species under catalytic conditions. Overall these three species are all within 1.8 kcal mol−1 of each other. The barriers for the formation of 4 (as 4·H2NMe2+, ΔG‡ = 17.9 kcal mol−1viaTS(3-4)N) and its onward reaction to reform 3 (ΔG‡ = 17.0 kcal mol−1, i.e. from 4·H2NMe2+ at −1.7 kcal mol−1 to TS(1-3)3 at +15.3 kcal mol−1) are also finely balanced. The computed energetics therefore reflect the observation of 1, 3 and 4 during catalysis.
Finally, H3B·NMe2H dehydrogenation at the neutral hydride 4 was also assessed and a novel concerted transition state, TS(4-4), involving B–H transfer onto the Rh–H ligand, with concomitant N–H transfer onto the Rh centre was characterised (Fig. 8). This releases not only H2BNMe2 but also H2 in a single step, regenerating 4 with a computed barrier of 26.2 kcal mol−1. This barrier is significantly higher than that for the amine-assisted pathway in Scheme 8 and so is consistent with the poor performance of isolated 4 as a dehydrocoupling catalyst.90 We have recently reported a similar amine-borane dehydrogenation transition state at an Fe–H species.91
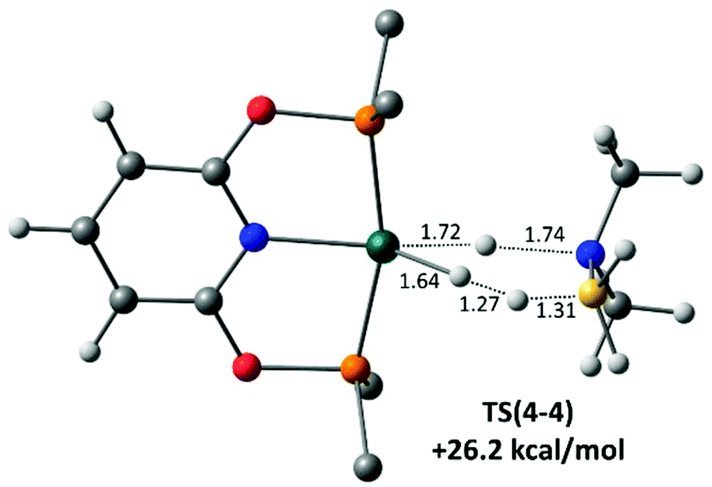 | ||
| Fig. 8 Computed geometry of TS(4-4) for dehydrogenation of H3B·NMe2H at 4, with selected distances in Å. Energies is quoted relative to 4 + free H3B·NMe2H set to 0.0 kcal mol−1. Rh (teal); C (grey); H (while); O (red); N (blue); P (orange); B (yellow). | ||
Conclusions
By using the [Rh(PONOP)(η2-H2)][BArF4] precatalyst, which has a labile dihydrogen ligand, we have been able to map out the catalytic dehydrocoupling of H3B·NMe2H using a pincer complex. As is becoming increasingly apparent,28,30,31 for cationic systems a hydride transfer/reprotonation route from a σ-bound amine-borane is a viable pathway for dehydrogenation when using cationic catalysts. While this can occur via a nucleophilic attack on B, via a boronium (i.e.Scheme 2), we show here that an alternative pathway of deprotonation of the σ-bound amine-borane to form an intermediate ammonium salt is also a viable route (Scheme 10). Central to both these processes is the generation of free amine from B–N bond cleavage to act as a nucleophile or base respectively. We,11 and others,24 have recently commented on the role of amine in promoting catalytic turnover in a variety of dehydrocoupling systems; and there is a very recent complementary report of the stoichiometric role of ammonium/amine in the protonation of borohydride complexes to eventually form σ-bound aminoboranes.92 In the system under discussion here the amine-assisted formation of the neutral hydride, i.e.4, and [NMe2H2]+ is rate limiting in catalysis, although this may not necessarily be the case for every system in a more general sense. Whatever the precise barriers of each step, the off-cycle equilibria involving [NMe2H2]+ (or [H2B(NMe2H)2]+ in a boronium route) will likely have an additional influence on the overall kinetics. A more detailed understanding of the role of initial B–N bond cleavage, to form free amine and the resulting co-catalysts, in dehydrocoupling – and especially dehydropolymerisation – could well be important in building an overarching general mechanism for such processes.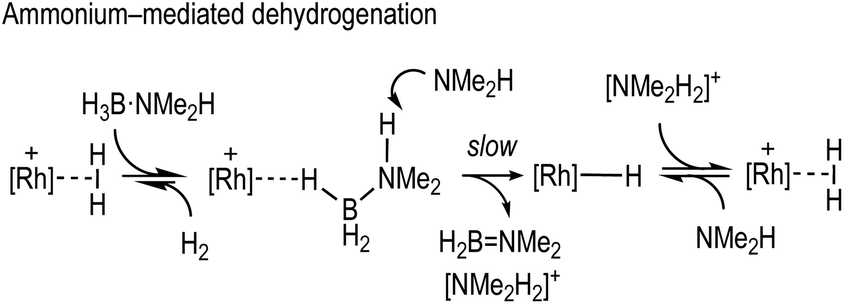 | ||
| Scheme 10 Hydride transfer and reprotonation via ammonium. | ||
Experimental
All manipulations, unless otherwise stated, were performed under an argon atmosphere using standard Schlenk line and glovebox techniques. Glassware was oven dried at 130 °C overnight and flame dried under vacuum prior to use. Pentane, hexane, toluene, Et2O and CH2Cl2 were dried using a Grubbs-type solvent purification system (MBraun SPS-800) and degassed by three successive freeze–pump–thaw cycles.93 THF was dried over Na/benzophenone, vacuum distilled, degassed by three successive freeze–pump–thaw cycles and stored over 3.0 Å molecular sieves. 1,2-F2C6H4 (pre-treated with alumina) and CD2Cl2 were dried over CaH2, vacuum distilled, degassed by three successive freeze–pump–thaw cycles and stored over 3.0 Å molecular sieves. H3B·NMe3 and H3B·NMe2H were purchased from Sigma-Aldrich and sublimed prior to use (5.0 × 10−2 mbar, 298 K and 303 K respectively). Hg (99.9995%) was purchased from Sigma-Aldrich, washed with 1,2-F2C6H4 and dried in vacuo prior to use. PPh3 and n-butyllithium (2.5 M in hexanes) were purchased from Sigma-Aldrich and used as received. BH3·THF (1.0 M in THF) and NMe2H (2.0 M in THF) were purchased from Fisher Scientific and used as received to form solutions in 1,2-F2C6H4 of the desired concentrations. Na[BArF4] (ArF = 3,5-(CF3)2C6H3),46 [BH2(NMe2H)2][BArF4],28,94 D3B·NMe3,95 H3B·NMe2D,96 Rh(PONOP)C47 and 139,97 were prepared by literature methods.
NMR spectra were recorded on a Bruker AVIIIHD 500 or Bruker AVIIIHD 400 nanobay spectrometer at room temperature, unless otherwise stated. Residual protio solvent was used as a reference for 1H NMR spectra in deuterated solvent samples. For 1,2-F2C6H4 solvent the NMR spectrometer was pre-locked to a sample of C6D6 (25%) and 1,2-F2C6H4 (75%) and referenced to the centre of the downfield solvent multiplet, δ = 7.07. 31P and 11B NMR spectra were referenced externally against 85% H3PO4 and BF3·OEt2 respectively. Chemical shifts (δ) are quoted in ppm and coupling constants (J) in Hz. Electrospray ionization mass spectrometry (ESI-MS) of organometallic complexes were recorded using a Bruker MicrOTOF instrument directly connected to a modified Innovative Technology glovebox.98 Samples were diluted in 1,2-F2C6H4 to a concentration of approximately 1.0 × 10−6 M before analysis. Elemental microanalyses were performed by Stephen Boyer at London Metropolitan University.
Synthesis of [Rh(PONOP)(H3B·NMe3)][BArF4] (2)
1 (50 mg, 0.036 mmol) and H3B·NMe3 (2.62 mg, 0.036 mmol) were dissolved in 1,2-F2C6H4 (2 mL) and the reaction stirred at room temperature for 24 h. The solution was then concentrated in vacuo to ca. 1 mL, cooled to 0 °C and pentane (5 mL) was added to give a precipitate. The solid was isolated by filtration and washed with pentane (2 mL × 2) before being dried under vacuum to give the product as a yellow powder. An isolated yield 32.8 mg (63%) was obtained. Layering a 1,2-F2C6H4 solution of complex 2 with pentane and storing at 5 °C overnight yielded dark yellow crystals suitable for single crystal X-ray diffraction.Spectroscopic data for [Rh(PONOP)(H3B·NMe2H)][BArF4] (3)
Synthesis of Li[H3B·NMe2]59
Hexane (20 mL) was added to H3B·NMe2H (400 mg, 6.79 mmol) to give a white suspension, which was then cooled to −78 °C. n-Butyllithium (2.5 M, 3 mL, 7.5 mmol) was added dropwise by syringe over 15 minutes. The reaction was stirred at −78 °C for 1 hour and then allowed to come to room temperature and stirred for an additional hour. The solution was then filtered, leaving a white solid which was washed with hexane. The solid was dried cold in vacuo for 2 hours to yield a white powder (412.9 mg, 94% yield).Synthesis of Rh(PONOP)H (4)
Rh(PONOP)Cl (150 mg, 0.279 mmol) and Li[H3B·NMe2] (36.2 mg, 0.558 mmol) were dissolved in pentane (5 mL) to give an orange/red solution, which was stirred at room temperature for 24 hours. The solution was then filtered and the solvent removed in vacuo and dried under vacuum overnight to yield a red powder. An isolated yield of 113.3 mg (81% yield) was obtained. A concentrated solution of 4 in methylcyclohexane was stored at 5 °C for 1 hour and then moved to −20 °C and after 3 days, red crystals suitable for single crystal X-ray diffraction were formed.Synthesis of D3B·NMe2H
D3B·NMe2H was prepared by a modification of the literature procedure for the synthesis of D3B·NMeH2.95 D3B·NMe3 (200 mg, 2.6 mmol, 99% D) was added to a J. Young flask and cooled to −78 °C. NMe2H (2.0 M in THF, 7 mL, 132.0 mmol) was added to the D3B·NMe3, the solution allowed to warm to room temperature and was stirred for 72 hours. The volatiles were removed in vacuo at 0 °C and the resulting solid purified by sublimation (5.0 × 10−2 mbar, 303 K) to give a white solid (130 mg, 80% yield). NMR data as reported previously in the literature.96General procedure for kinetic measurements of the catalytic dehydrogenation of H3B·NMe2H
Conflicts of interest
There are no conflicts of interest.Acknowledgements
University of Oxford, Heriot-Watt University, the EPSRC (DTP and EP/M024210/1) and SCG Chemicals Co. Ltd for funding. The ToC for this article was prepared using Catacycle.99 David Ryan for stimulating and insightful discussions.Notes and references
- A. Staubitz, A. P. M. Robertson, M. E. Sloan and I. Manners, Chem. Rev., 2010, 110, 4023–4078 CrossRef CAS PubMed.
- R. Waterman, Chem. Soc. Rev., 2013, 42, 5629–5641 RSC.
- H. C. Johnson, T. N. Hooper and A. S. Weller, Top. Organomet. Chem., 2015, 49, 153–220 CrossRef CAS.
- A. Rossin and M. Peruzzini, Chem. Rev., 2016, 116, 8848–8872 CrossRef CAS PubMed.
- S. Bhunya, T. Malakar, G. Ganguly and A. Paul, ACS Catal., 2016, 6, 7907–7934 CrossRef CAS.
- E. M. Leitao, T. Jurca and I. Manners, Nat. Chem., 2013, 5, 817–829 CrossRef CAS PubMed.
- A. Staubitz, A. Presa Soto and I. Manners, Angew. Chem., Int. Ed., 2008, 47, 6212–6215 CrossRef CAS PubMed.
- A. Staubitz, M. E. Sloan, A. P. M. Robertson, A. Friedrich, S. Schneider, P. J. Gates, J. Schmedt auf der Günne and I. Manners, J. Am. Chem. Soc., 2010, 132, 13332–13345 CrossRef CAS PubMed.
- D. Han, F. Anke, M. Trose and T. Beweries, Coord. Chem. Rev., 2019, 380, 260–286 CrossRef CAS.
- Smart Inorganic Polymers: Synthesis, Properties, and Emerging Applications in Materials and Life Sciences, ed. E. Hey-Hawkins and M. Hissler, Wiley-VCH, Weinhiem, 2019 Search PubMed.
- G. M. Adams, D. E. Ryan, N. A. Beattie, A. I. McKay, G. C. Lloyd-Jones and A. S. Weller, ACS Catal., 2019, 9, 3657–3666 CrossRef CAS PubMed.
- V. Du, G. Whittell and I. Manners, Dalton Trans., 2016, 45, 1055–1062 RSC.
- D. A. Resendiz-Lara, N. E. Stubbs, M. I. Arz, N. E. Pridmore, H. A. Sparkes and I. Manners, Chem. Commun., 2017, 53, 11701–11704 RSC.
- X. Wang, T. N. Hooper, A. Kumar, I. K. Priest, Y. Sheng, T. O. M. Samuels, S. Wang, A. W. Robertson, M. Pacios, H. Bhaskaran, A. S. Weller and J. H. Warner, CrystEngComm, 2017, 19, 285–294 RSC.
- A. L. Colebatch and A. S. Weller, Chem. – Eur. J., 2019, 25, 1379–1390 CrossRef CAS PubMed.
- B. L. Dietrich, K. I. Goldberg, D. M. Heinekey, T. Autrey and J. C. Linehan, Inorg. Chem., 2008, 47, 8583–8585 CrossRef CAS PubMed.
- A. Kumar, H. C. Johnson, T. N. Hooper, A. S. Weller, A. G. Algarra and S. A. Macgregor, Chem. Sci., 2014, 5, 2546–2553 RSC.
- M. Shimoi, S.-i. Nagai, M. Ichikawa, Y. Kawano, K. Katoh, M. Uruichi and H. Ogino, J. Am. Chem. Soc., 1999, 121, 11704–11712 CrossRef CAS.
- M. A. Esteruelas, A. M. López and M. Oliván, Chem. Rev., 2016, 116, 8770–8847 CrossRef CAS PubMed.
- A. Paul and C. B. Musgrave, Angew. Chem., Int. Ed., 2007, 46, 8153–8156 CrossRef CAS PubMed.
- E. M. Titova, E. S. Osipova, A. A. Pavlov, O. A. Filippov, S. V. Safronov, E. S. Shubina and N. V. Belkova, ACS Catal., 2017, 7, 2325–2333 CrossRef CAS.
- M. A. Esteruelas, P. Nolis, M. Oliván, E. Oñate, A. Vallribera and A. Vélez, Inorg. Chem., 2016, 55, 7176–7181 CrossRef CAS PubMed.
- A. N. Marziale, A. Friedrich, I. Klopsch, M. Drees, V. R. Celinski, J. Schmedt auf der Günne and S. Schneider, J. Am. Chem. Soc., 2013, 135, 13342–13355 CrossRef CAS PubMed.
- A. Glüer, M. Förster, V. R. Celinski, J. Schmedt auf der Günne, M. C. Holthausen and S. Schneider, ACS Catal., 2015, 5, 7214–7217 CrossRef.
- X. Zhang, L. Kam, R. Trerise and T. J. Williams, Acc. Chem. Res., 2017, 50, 86–95 CrossRef CAS PubMed.
- P. Bhattacharya, J. A. Krause and H. Guan, J. Am. Chem. Soc., 2014, 136, 11153–11161 CrossRef CAS PubMed.
- W. E. Piers, S. C. Bourke and K. D. Conroy, Angew. Chem., Int. Ed., 2005, 44, 5016–5036 CrossRef CAS PubMed.
- M. Roselló-Merino, J. López-Serrano and S. Conejero, J. Am. Chem. Soc., 2013, 135, 10910–10913 CrossRef PubMed.
- S. Pal, S. Kusumoto and K. Nozaki, Organometallics, 2018, 37, 906–914 CrossRef CAS.
- G. M. Adams, A. L. Colebatch, J. T. Skornia, A. I. McKay, H. C. Johnson, G. C. Lloyd-Jones, S. A. Macgregor, N. A. Beattie and A. S. Weller, J. Am. Chem. Soc., 2018, 140, 1481–1495 CrossRef CAS PubMed.
- A. Kumar, N. A. Beattie, S. D. Pike, S. A. Macgregor and A. S. Weller, Angew. Chem., Int. Ed., 2016, 55, 6651–6656 CrossRef CAS PubMed.
- A. St John, K. I. Goldberg and D. M. Heinekey, in Organometallic Pincer Chemistry, ed. G. van Koten and D. Milstein, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, pp. 271–287 Search PubMed.
- T.-P. Lin and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 15310–15313 CrossRef CAS PubMed.
- A. Rossin, G. Bottari, A. M. Lozano-Vila, M. Paneque, M. Peruzzini, A. Rossi and F. Zanobini, Dalton Trans., 2013, 42, 3533–3541 RSC.
- D. Han, M. Joksch, M. Klahn, A. Spannenberg, H. J. Drexler, W. Baumann, H. Jiao, R. Knitsch, M. R. Hansen, H. Eckert and T. Beweries, Dalton Trans., 2016, 45, 17697–17704 RSC.
- T. J. Hebden, M. C. Denney, V. Pons, P. M. B. Piccoli, T. F. Koetzle, A. J. Schultz, W. Kaminsky, K. I. Goldberg and D. M. Heinekey, J. Am. Chem. Soc., 2008, 130, 10812–10820 CrossRef CAS PubMed.
- H. C. Johnson, C. L. McMullin, S. D. Pike, S. A. Macgregor and A. S. Weller, Angew. Chem., Int. Ed., 2013, 52, 9776–9780 CrossRef CAS PubMed.
- A. Johnson, A. J. Martínez-Martínez, S. A. Macgregor and A. S. Weller, Dalton Trans., 2019, 48, 9776–9781 RSC.
- M. Findlater, K. M. Schultz, W. H. Bernskoetter, A. Cartwright-Sykes, D. M. Heinekey and M. Brookhart, Inorg. Chem., 2012, 51, 4672–4678 CrossRef CAS PubMed.
- A. Rossin, M. Caporali, L. Gonsalvi, A. Guerri, A. Lledós, M. Peruzzini and F. Zanobini, Eur. J. Inorg. Chem., 2009, 2009, 3055–3059 CrossRef.
- R. Kumar and B. R. Jagirdar, Inorg. Chem., 2013, 52, 28–36 CrossRef CAS PubMed.
- G. Alcaraz, A. B. Chaplin, C. J. Stevens, E. Clot, L. Vendier, A. S. Weller and S. Sabo-Etienne, Organometallics, 2010, 29, 5591–5595 CrossRef CAS.
- C. J. Stevens, R. Dallanegra, A. B. Chaplin, A. S. Weller, S. A. Macgregor, B. Ward, D. McKay, G. Alcaraz and S. Sabo-Etienne, Chem. – Eur. J., 2011, 17, 3011–3020 CrossRef CAS PubMed.
- Y. Gloaguen, G. Bénac-Lestrille, L. Vendier, U. Helmstedt, E. Clot, G. Alcaraz and S. Sabo-Etienne, Organometallics, 2013, 32, 4868–4877 CrossRef CAS.
- T. M. Douglas, A. B. Chaplin, A. S. Weller, X. Yang and M. B. Hall, J. Am. Chem. Soc., 2009, 131, 15440–15456 CrossRef CAS PubMed.
- A. J. Martínez-Martínez and A. S. Weller, Dalton Trans., 2019, 48, 3551–3554 RSC.
- W. H. Bernskoetter, C. K. Schauer, K. I. Goldberg and M. Brookhart, Science, 2009, 326, 553 CrossRef CAS PubMed.
- M. L. H. Green and G. Parkin, in The Chemical Bond III: 100 years old and getting stronger, ed. D. M. P. Mingos, Springer International Publishing, Cham, 2017, pp. 79–139 Search PubMed.
- A. J. Martínez-Martínez, B. E. Tegner, A. I. McKay, A. J. Bukvic, N. H. Rees, G. J. Tizzard, S. J. Coles, M. R. Warren, S. A. Macgregor and A. S. Weller, J. Am. Chem. Soc., 2018, 140, 14958–14970 CrossRef PubMed.
- J. Yang, P. S. White, C. K. Schauer and M. Brookhart, Angew. Chem., Int. Ed., 2008, 47, 4141–4143 CrossRef CAS PubMed.
- M. Roselló-Merino, R. J. Rama, J. Díez and S. Conejero, Chem. Commun., 2016, 52, 8389–8392 RSC.
- Y. Kawano, T. Kakizawa, K. Yamaguchi and M. Shimoi, Chem. Lett., 2006, 35, 568–569 CrossRef CAS.
- A. G. Algarra, L. J. Sewell, H. C. Johnson, S. A. Macgregor and A. S. Weller, Dalton Trans., 2014, 43, 11118–11128 RSC.
- R. N. Perutz and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2007, 46, 2578–2592 CrossRef CAS PubMed.
- A. B. Chaplin and A. S. Weller, Organometallics, 2011, 30, 4466–4469 CrossRef CAS.
- L. K. Krannich, C. L. Watkins, D. K. Srivastava and R. K. Kanjolia, Coord. Chem. Rev., 1992, 112, 117–129 CrossRef CAS.
- H. Nöth and H. Vahrenkamp, Chem. Ber., 1967, 100, 3353–3362 CrossRef.
- P. Bellham, M. S. Hill, G. Kociok-Köhn and D. J. Liptrot, Chem. Commun., 2013, 49, 1960–1962 RSC.
- Adapted from: G. B. Fisher, J. C. Fuller, J. Harrison, S. G. Alvarez, E. R. Burkhardt, C. T. Goralski and B. Singaram, J. Org. Chem., 1994, 59, 6378–6385 CrossRef CAS.
- S. P. Semproni, C. Milsmann and P. J. Chirik, J. Am. Chem. Soc., 2014, 136, 9211–9224 CrossRef CAS PubMed.
- L. M. Guard, T. J. Hebden, D. E. Linn and D. M. Heinekey, Organometallics, 2017, 36, 3104–3109 CrossRef CAS.
- M. Feller, U. Gellrich, A. Anaby, Y. Diskin-Posner and D. Milstein, J. Am. Chem. Soc., 2016, 138, 6445–6454 CrossRef CAS PubMed.
- W. D. Bailey, W. Kaminsky, R. A. Kemp and K. I. Goldberg, Organometallics, 2014, 33, 2503–2509 CrossRef CAS.
- M. C. Haibach, D. Y. Wang, T. J. Emge, K. Krogh-Jespersen and A. S. Goldman, Chem. Sci., 2013, 4, 3683–3692 RSC.
- M. A. Esteruelas, M. Oliván and A. Vélez, Inorg. Chem., 2013, 52, 5339–5349 CrossRef CAS PubMed.
- L. M. Martínez-Prieto, C. Melero, D. del Río, P. Palma, J. Cámpora and E. Álvarez, Organometallics, 2012, 31, 1425–1438 CrossRef.
- R. Johansson and O. F. Wendt, Organometallics, 2007, 26, 2426–2430 CrossRef CAS.
- N. V. Belkova, L. M. Epstein, O. A. Filippov and E. S. Shubina, Chem. Rev., 2016, 116, 8545–8587 CrossRef CAS PubMed.
- T. D. Forster, H. M. Tuononen, M. Parvez and R. Roesler, J. Am. Chem. Soc., 2009, 131, 6689–6691 CrossRef CAS PubMed.
- H. Helten, B. Dutta, J. R. Vance, M. E. Sloan, M. F. Haddow, S. Sproules, D. Collison, G. R. Whittell, G. C. Lloyd-Jones and I. Manners, Angew. Chem., Int. Ed., 2013, 52, 437–440 CrossRef CAS PubMed.
- J. Spielmann, D. Piesik, B. Wittkamp, G. Jansen and S. Harder, Chem. Commun., 2009, 3455–3456 RSC.
- P. Bellham, M. D. Anker, M. S. Hill, G. Kociok-Köhn and M. F. Mahon, Dalton Trans., 2016, 45, 13969–13978 RSC.
- M. D. Walter, P. S. White, C. K. Schauer and M. Brookhart, J. Am. Chem. Soc., 2013, 135, 15933–15947 CrossRef CAS PubMed.
- The 31P{1H} NMR chemical shift of Ir(PONOP)H has been reported in ESI in ref. 73, the synthesis of which comes from β-elimination from Ir(PONOP)Et. While Rh(PONOP)Et was also structurally characterised its stability towards β-elimination was not reported.
- C. L. Bailey, A. Y. Joh, Z. Q. Hurley, C. L. Anderson and B. Singaram, J. Org. Chem., 2016, 81, 3619–3628 CrossRef CAS PubMed.
- G. Barozzino Consiglio, P. Queval, A. Harrison-Marchand, A. Mordini, J.-F. Lohier, O. Delacroix, A.-C. Gaumont, H. Gérard, J. Maddaluno and H. Oulyadi, J. Am. Chem. Soc., 2011, 133, 6472–6480 CrossRef CAS PubMed.
- C. A. Jaska and I. Manners, J. Am. Chem. Soc., 2004, 126, 9776–9785 CrossRef CAS PubMed.
- Under these conditions a small amount (∼20%) of a new species identified as [Rh(PONOP)(NMe2H)][BArF4] was also observed, δ 198.2 [J(RhP) = 151 Hz]. This does not react with H3B·NMe3 and we have not observed it in any other experiments – suggesting that it does not play a role in catalysis.
- A. A. Mikhailine, M. I. Maishan, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2012, 134, 12266–12280 CrossRef CAS PubMed.
- J. Burés, Angew. Chem., Int. Ed., 2016, 55, 2028–2031 CrossRef PubMed.
- J. Burés, Angew. Chem., Int. Ed., 2016, 55, 16084–16087 CrossRef PubMed.
- V. Vasilenko, C. K. Blasius and L. H. Gade, J. Am. Chem. Soc., 2018, 140, 9244–9254 CrossRef CAS PubMed.
- M. J. Hynes, J. Chem. Soc., Dalton Trans., 1993, 311–312 RSC.
- T. A. Shuttleworth, M. A. Huertos, I. Pernik, R. D. Young and A. S. Weller, Dalton Trans., 2013, 42, 12917–12925 RSC.
- X. Chen, J.-C. Zhao and S. G. Shore, Acc. Chem. Res., 2013, 46, 2666–2675 CrossRef CAS PubMed.
- M. A. Fox, A. E. Goeta, A. K. Hughes and A. L. Johnson, J. Chem. Soc., Dalton Trans., 2002, 2132–2141 RSC.
- Mechanisms involving B–H and N–H activation at the Rh centre entailed significantly higher barriers – see ESI.‡.
- The energy of 3-coordinate [Rh(PONOP)]+ (+17.5 kcal mol−1) suggests a purely dissociative mechanism is less favourable.
- In comparison the computed dipole for TS(3-4)B is 9.1 D and for 3·NMe2H 6.3 D. TS(3-4)N remains more stable in the absence of the solvent correction, but only by 0.5 kcal mol−1.
- An alternative pathway involving stepwise B–H and N–H activation was also characterised but this has a higher barrier of 34.2 kcal mol−1.
- M. Espinal-Viguri, S. E. Neale, N. T. Coles, S. A. Macgregor and R. L. Webster, J. Am. Chem. Soc., 2019, 141, 572–582 CrossRef CAS PubMed.
- K. Kirchner, N. Gorgas, B. Stöger and L. F. Veiros, Angew. Chem., Int. Ed., 2019 DOI:10.1002/anie.201906971.
- A. B. Pangborn, M. A. Giardello, R. H. Grubbs, R. K. Rosen and F. J. Timmers, Organometallics, 1996, 15, 1518–1520 CrossRef CAS.
- O. J. Metters, A. M. Chapman, A. P. M. Robertson, C. H. Woodall, P. J. Gates, D. F. Wass and I. Manners, Chem. Commun., 2014, 50, 12146–12149 RSC.
- A. L. Colebatch, B. W. Hawkey Gilder, G. R. Whittell, N. L. Oldroyd, I. Manners and A. S. Weller, Chem. – Eur. J., 2018, 24, 5450–5455 CrossRef CAS PubMed.
- M. E. Sloan, A. Staubitz, T. J. Clark, C. A. Russell, G. C. Lloyd-Jones and I. Manners, J. Am. Chem. Soc., 2010, 132, 3831–3841 CrossRef CAS PubMed.
- G. M. Adams, F. M. Chadwick, S. D. Pike and A. S. Weller, Dalton Trans., 2015, 44, 6340–6342 RSC.
- A. T. Lubben, J. S. McIndoe and A. S. Weller, Organometallics, 2008, 27, 3303–3306 CrossRef CAS.
- ToC graphic prepared using: J. McFarlane, B. Henderson, S. Donnecke and J. S. McIndoe, An Information-Rich Graphical Representation of Catalytic Cycles, ChemRxiv, 2019 DOI:10.26434/chemrxiv.8206637.v1 . Preprint.
Footnotes |
| † Dedication: In recognition of Professor Robin N. Perutz's outstanding scientific contributions to physical organometallic chemistry, and also the mentoring and friendship RNP has generously shared with SAM and ASW over their careers. |
| ‡ Electronic supplementary information (ESI) available: Synthesis, characterisation data, structures of 2 and 4, computational details. CCDC 1917326 and 1917160. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt03358k |
| This journal is © The Royal Society of Chemistry 2019 |