
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of organometallic pentalenide complexes
Stuart M.
Boyt
 a,
Niko A.
Jenek
a and
Ulrich
Hintermair
*ab
a,
Niko A.
Jenek
a and
Ulrich
Hintermair
*ab
aDepartment of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: u.hintermair@bath.ac.uk
bCentre for Sustainable Chemical Technologies, University of Bath, Claverton Down, Bath BA2 7AY, UK
First published on 13th March 2019
Abstract
While a number of reports have established the unique structures and electronic properties of mono- and dinuclear pentalenide complexes of s, p, d and f block elements, access to these intriguing compounds is restricted by synthetic challenges. Here we review various strategies for the synthesis, functionalisation and (trans)metalation of pentalenide complexes from a practical point of view, pointing out promising avenues for future research that may allow wider access to novel pentalenide complexes for application in many different areas.
 Stuart M. Boyt, Niko A. Jenek, Ulrich Hintermair | Stuart M. Boyt graduated from the University of Warwick with an MChem in Chemical Biology but developed a taste for organometallic chemistry after a masters thesis with Adrian Chaplin looking at NHC complexes of group 9 metals. In 2015 he moved to the University of Bath to pursue his PhD in the Hintermair lab. His project involves synthesizing new pentalenide ligands from readily available precursors and investigating their reactivity towards a range of s-, d- and p-block compounds. Niko A. Jenek obtained his BSc in Chemistry from the University of Stuttgart in 2013, after which he moved to the University of Freiburg to finish his master's studies with a thesis on the fluorination of silicon surfaces with Ingo Krossing in 2017. After further fluorination adventures at the Fraunhofer Institute for Solar Energy Systems he joined the Hintermair group at the University of Bath for his PhD on novel pentalenide complexes for catalysis. Ulrich Hintermair studied Chemistry and Chemical Engineering in Würzburg and Lyon with a stint at the University of St Andrews. After completing his PhD with Walter Leitner at Aachen he was a Humboldt postdoctoral fellow with Bob Crabtree at Yale University. In 2013 he started his independent academic career at the CSCT in Bath where he currently holds a Royal Society University Research Fellowship. Research in his group revolves around catalysis with metal complexes, including organometallic chemistry and real-time spectroscopy for mechanistic investigations. |
1. Introduction
Pentalene (Pn, C8H6) has long fascinated theoretical and synthetic organic chemists for its anti-aromatic 8π system.1 Unlike the flexible 8π cyclooctatetraene (COT, C8H8) Pn is planar due to its bicyclic ring structure, enforcing its anti-aromaticity. As a result, it readily dimerises above −196 °C to form two fulvene units isolated from each other by a cyclobutane linkage (Fig. 1).2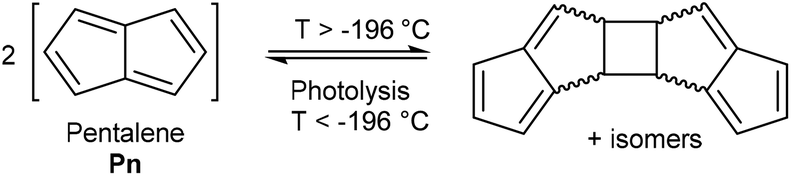 | ||
| Fig. 1 Reversible dimerisation of pentalene. | ||
Due to its inherent instability, Pn itself is of limited use for applications in synthetic chemistry.† However, similar to other 8π anti-aromatics like COT, double reduction of Pn to pentalenide (Pn2−, C8H62−) generates a stable 10π aromatic system that presents itself as a useful π ligand for organometallic chemistry.4 Its dianionic nature makes it a stronger donor for Lewis-acidic metals than neutral 10π hydrocarbons such as naphthalene, and the two five-membered ring systems enable symmetrical charge distribution for reversible hapticity shifts of the metals bound to it (Fig. 2).
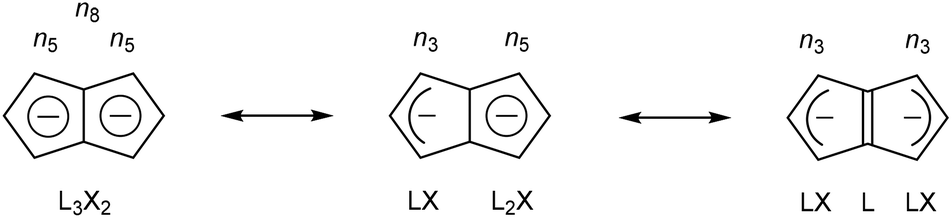 | ||
| Fig. 2 Charge distribution in the 10π aromatic pentalenide dianion. | ||
Due to this facile hapticity shift and a large degree of flexibility around the central C–C bridge, Pn2− ligands exhibit some uniquely adaptive coordination abilities that neither COT nor naphthalene derivatives display. Pn2− may wrap itself around, electron-deficient metal centres in η8 coordination mode, or form doubly η5/η3 coordinated homo- and hetero-bimetallic complexes with direct metal–metal interactions within syn-bimetallics and strong electronic coupling in anti-bimetallic systems.5 Given these intriguing properties it may appear surprising that organometallic Pn2− chemistry is far less explored and developed than that of its 6π congener cyclopentadienide (Cp−, C5H5−), arguably the most important and most widely used organometallic ligand to date.6 Whereas thousands of Cp− complexes including almost every metal in the periodic table are known7 and widely used in numerous applications including sensing, electrochemistry, magnetism, material synthesis, and of course catalysis, only about 150 examples of variously substituted Pn2− and HPn− complexes (including precursor salts) have been reported so far. This limitation is in no small part due to the practical challenges associated with accessing suitable synthons for Pn2− chemistry. Historically, only few dedicated organometallic labs have developed the expertise and equipment required for pentalenide chemistry, each following their own preferred synthetic method. However, a number of routes to suitable precursors have emerged in different parts of the chemical literature over the past 50 years. Here we summarise and compare various strategies for the synthesis and functionalisation of Pn2− complexes that promise to give more facile access to a wider range of precursors, and thus hopefully allow more widespread exploration of the intriguing properties of organometallic pentalenide complexes in different areas in the future.
2. Synthesis of pentalenides
In the context of discussing synthetic pentalenide chemistry, it is useful to distinguish the 8π antiaromatic pentalene (Pn, C8H6), the non-aromatic dihydropentalene (H2Pn, C8H8 existing as several double-bond isomers8), the 6π aromatic hydropentalenide (HPn−, C8H7−), and the 10π aromatic pentalenide (Pn2−, C8H62−). Their structures and interconversion pathways are laid out in Fig. 3. | ||
| Fig. 3 Nomenclature and interconversion of pentalene derivatives. | ||
2.1. Deprotonation of dihydropentalenes
Mono-anionic 6π HPn− can easily be generated by deprotonation of H2Pn using a weak base, as demonstrated by Katz and Mrowca.9 They showed that an isomeric mixture of H2Pn would react with KOH and Tl2SO4 in water to furnish TlPnH as a precipitate in moderate yields (Fig. 4). In contrast to the neutral, unsubstituted H2Pn, HPn− salts are thermally stable and can be isolated and stored at room temperature like the related MCp salts (M = Li, Na, K). Jones et al. later showed TlOEt in pentane to be equally effective in generating TlPnH from H2Pn.10 The first pKa of H2Pn must therefore be below 14, showing it to be slightly more acidic than HCp.11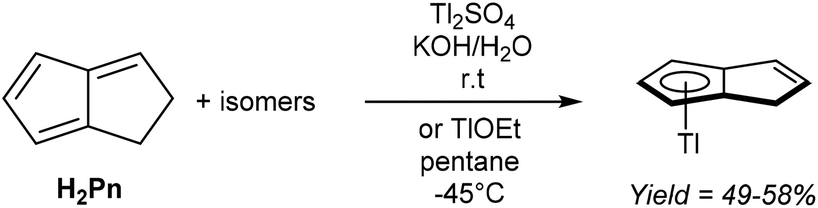 | ||
| Fig. 4 Synthesis of dilithium pentalenide by double deprotonation of dihydropentalenes. | ||
Presumably due to charge effects, deprotonation of the second ring in HPn− to furnish the fully 10π delocalised Pn2− requires a stronger base. Katz et al. showed that Li2Pn can be prepared by double deprotonation of H2Pn with an excess of nbutyllithium at −78 °C (Fig. 5).12 Although the second pKa value of H2Pn is not known with certainty, using two (or more) equivalents of bases with pKa values >25 usually leads to quantitative double deprotonation of all H2Pn double-bond isomers. While Katz's original preparation used heptane to precipitate Li2Pn, Stezowski et al. later used dimethoxyethane (DME) to obtain single crystals of η5[Li(DME)]2Pn that showed both metals to adopt anti configuration around the planar Pn2− in the solid state.13 A recent computational study confirmed this to be the preferred geometry for all alkali–metal Pn2− complexes in solution.14
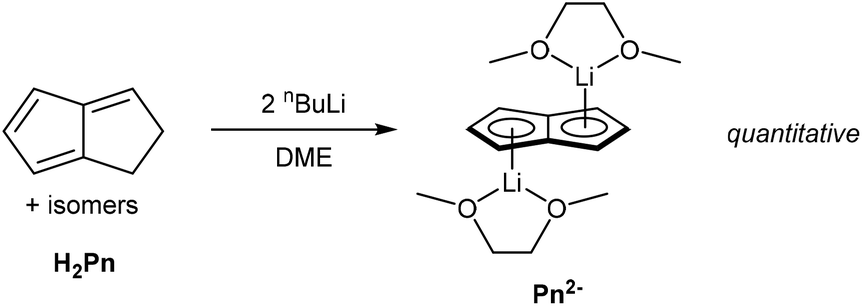 | ||
| Fig. 5 Synthesis of dilithium pentalenide by double deprotonation of dihydropentalenes. | ||
While these reactions look appealingly straightforward, and the utility of HPn− and Pn2− salts for transmetalation reactions is well documented (see section 3), the difficulty lies in generating the H2Pn starting material. In the following subsections we review the most prominent methods reported.
2.2. Pyrolytic routes to dihydropentalenes
H2Pn can be obtained from controlled, anaerobic pyrolysis of suitable precursors such as isodicyclopentadiene in Katz's method (Fig. 6).15 Isodicyclopentadiene was formed by treating dihydrodicyclopentadiene (obtained from partial hydrogenation of dicyclopentadiene) with acetic anhydride and selenium dioxide, followed by dehydration of the resulting alcohol by passing over alumina at 300 °C.16 Anaerobic pyrolysis of isodicyclopentadiene at 575 °C lead to the formation of 1,4-H2Pn (through release of ethylene) in ∼30% yield, to which hydroquinoline had to be added immediately to prevent polymerisation. However, Kazennova et al. later reported that passing the pyrolysis distillate through a plug of solid potassium hydroxide provided pure H2Pn samples that were stable to polymerisation at −78 °C.17 They also demonstrated an improved synthesis of isodicyclopentadiene, reporting a 40% overall yield of H2Pn starting from HCp.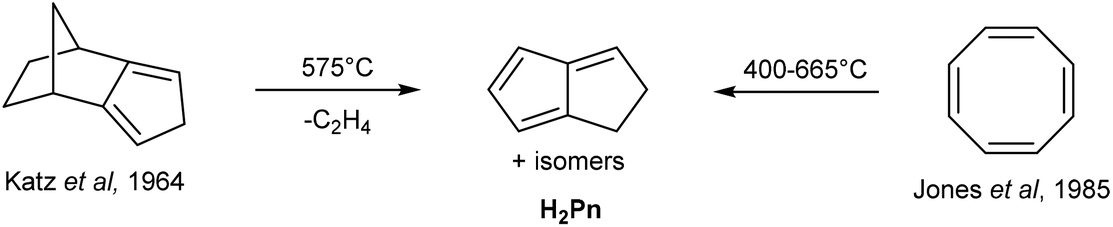 | ||
| Fig. 6 Synthesis of dihydropentalene by anaerobic flash vacuum pyrolysis. | ||
An alternative precursor was reported by Jones and Schwab, who showed that COT undergoes thermal rearrangement between 400–665 °C to give H2Pn (Fig. 6) in addition to acetylene, benzene and styrene.18 The selectivity to H2Pn was found to be highly temperature-dependent, with the optimum conversion occurring between 500–600 °C. An optimised procedure was later reported by Cloke et al., in which a 87% yield was achieved thanks to precise temperature and residence time control during the continuous-flow pyrolysis.19 In this set-up, the resulting H2Pn were condensed into a nbutyllithium solution in DME/hexane at −78 °C to be directly converted to the more stable [Li2(DME)x]Pn salts that precipitate to be further purified by washing with cold hexane.
While both routes are reasonably well established (with lower yields and more synthetic steps starting from inexpensive (HCp)2, or fewer steps and higher yields starting from the more valuable COT), they are limited to the synthesis of the parent (unsubstituted) H2Pn. Substituted iso-CpH or COT have not been explored as pyrolysis precursors for substituted H2Pn, likely due to the difficulty and expense of synthesising the starting materials and/or unwanted thermal rearrangement pathways. A slightly more versatile pyrolytic method has been reported by Griesbeck, who showed the use of two protected vinyl fulvenes, synthesised by Diels–Alder reaction of HCp and acroleins followed by pyrrolidine-facilitated Knoevenagel condensation with an additional equivalent of HCp (Fig. 7).20
 | ||
| Fig. 7 Flash vacuum pyrolysis of protected vinyl fulvenes derived from cyclopentadiene and acroleins. | ||
Anaerobic pyrolysis of these compounds at 520 °C furnished H2Pn (88% yield) or 2-Me-PnH2 (50% yield) through thermal cyclisation accompanied by loss of HCp. This methodology is appealing as the precursors are straightforward to synthesise from inexpensive starting materials, and potentially provide a route to other substituted H2Pn through use of substituted acroleins in the first step and/or substituted HCp in the second step. However, the final pyrolysis may require optimisation for each protected vinyl fulvene.
Another pyrolytic route to H2Pn and 2-Me-PnH2 has been reported by Stapersma et al., who showed that tetracyclo-oct-7-enes may be rearranged by anaerobic heating between 250 °C and 500 °C.21 These starting materials, derived from photolytic rearrangements of 7-carbomethoxynorbornadienes,22 are comparatively difficult to prepare, and their pyrolysis reactions gave relatively low H2Pn yields of 7–16% due to the formation of a large number of side products.
2.3. Thermal cyclisations of vinyl fulvenes
A mild thermal synthesis of H2Pn has been described by Gajewski and Cavender, who found that neat 6-vinyl fulvene will undergo thermal cyclisation at 110 °C (Fig. 8).23 This procedure gave exclusively the 1,5-PnH2 isomer due to facile 1,5-hydride shifts occurring.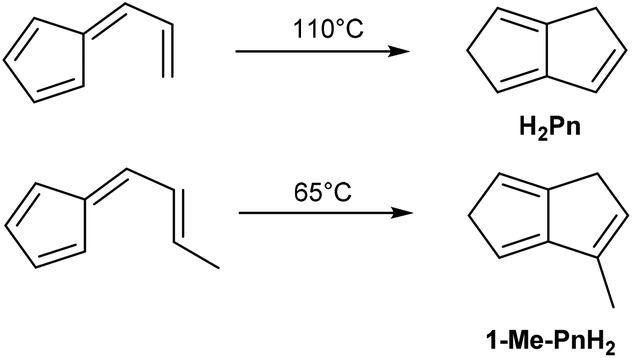 | ||
| Fig. 8 Thermal cyclisation of vinyl fulvenes to dihydropentalenes. | ||
6-Propenylfulvene was reported to undergo a thermal reaction at 65 °C, however, analysis of the products obtained was limited and no isolated yields were reported. The relatively low temperatures required to induce these rearrangements promise practical benefits over the higher temperature pyrolysis reactions discussed above. However, 6-vinyl fulvene is again not trivial to synthesise. Neuenschwander reported two preparations: the first reacting 1-hydroxymethyl-spiro-[2,4]-hepta-4,6-diene with HCl,24 the second reacting NaCp with 3-acetoxy-3-chloro-1-propene.25 Both routes give low yields (10% and 20%, respectively) due to significant side reactions occurring. Erden et al. describe an improved protocol, starting with condensation of HCp with 3-(methylthio)propanal, followed by oxidation with NaIO4 and sulfoxide elimination with DBU. The reported yield across the three steps was 37% including a purification after each step.26
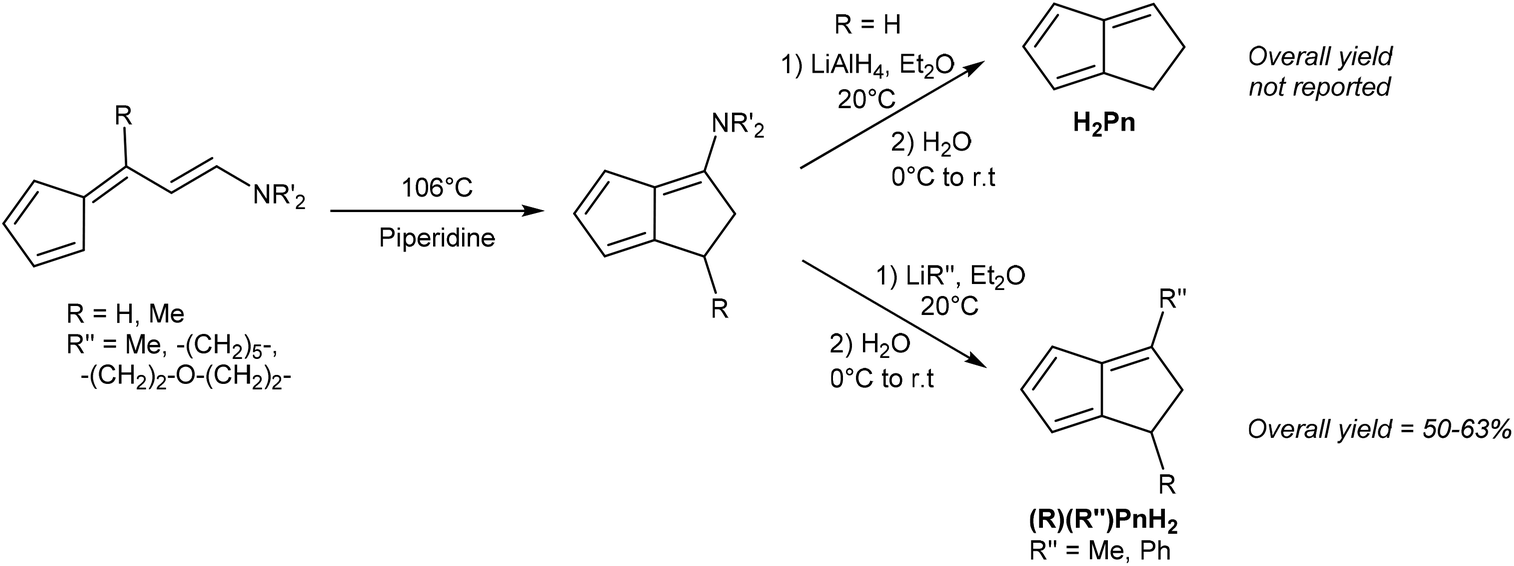
Kaiser and Hafner showed that 6-(2-aminovinyl)fulvenes would undergo a solution-phase thermal cyclisation and isomerisation in boiling piperidine to give 3-amino-PnH2 in high yields (Fig. 9).27 These species could be reduced with LiAlH4 to give an organoaluminium species that, upon hydrolysis, underwent amine elimination at room temperature to yield H2Pn. The syntheses of some mono- and di-substituted H2Pn were also described, using 6-substituted amino fulvenes in the first step, and using alkyl/aryl lithiums in the following reduction/elimination step. However, purification procedures for these compounds were not described.
 | ||
| Fig. 9 Thermal rearrangement of aminovinyl-fulvenes and subsequent reduction to dihydropentalenes. | ||
Similar to the above case of vinyl fulvenes, the amino-vinyl fulvene starting materials (derived from condensation of HCp with di(alkyl)-aminoacroleins) are difficult to handle due to their thermal instability.28 In theory, starting from substituted HCp could produce more stable amino-vinyl fulvenes and provide access to more highly substituted H2Pn. However, it is difficult to predict whether these species would follow the same thermal rearrangement pathways.
While some of the thermal routes reviewed above have been shown to be quite efficient in generating H2Pn precursors suitable for double deprotonation to Pn2−, the limitations of these rearrangement reactions are obvious: their precursors rely on multi-step syntheses and/or utilise expensive starting materials, they often require special apparatus, conditions need to be carefully controlled and optimised, and they are rather limited in scope.
2.4. Solution-phase rearrangement reactions
Several solution-phase syntheses of H2Pn that do not involve pyrolytic steps have been described in the literature. Jones et al. reported the synthesis of Li2Pn starting with the conversion of cycloheptatriene to 8,8-dibromobicyclo[5.1.0]octa-2,4-diene using bromoform and KOtBu (Fig. 10).10 Reacting this species with methyllithium triggered a carbene-induced rearrangement to give H2Pn which was trapped with nbutyllithium in DME/pentane to give [Li2(DME)x]Pn. Although the overall yields are quite low (<12%), this solution-phase method can be performed with standard laboratory equipment.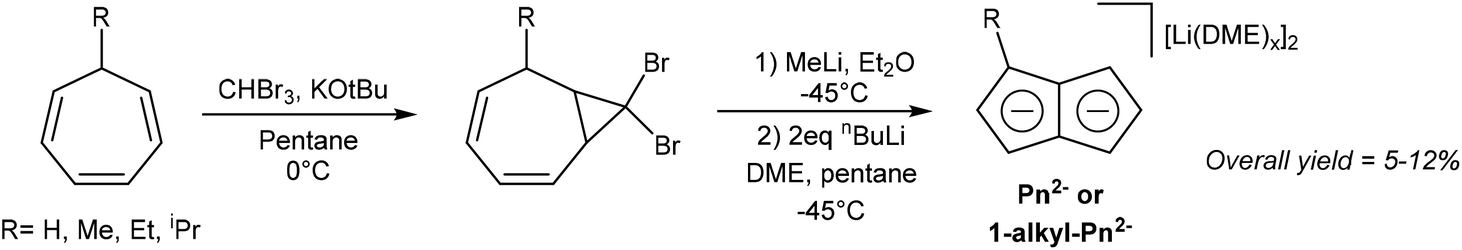 | ||
| Fig. 10 Synthesis of dilithium pentalenides from cycloheptatrienes. | ||
The Skattebøl rearrangement of the transient carbene generated from the geminal dihalo-cyclopropane unit allows for some variability in the substitution pattern. Cycloheptatriene can be converted to 7-alkylcycloheptatrienes through hydride abstraction, formation of the ethyl ether with EtOH and NaHCO3, then finally substitution of the ether with a Grignard reagent. Although 7-tert-butyl-cycloheptatriene was found not to react with CHBr3 and KOtBu, the methyl, ethyl and isopropyl variants did undergo this transformation (albeit with low yields of 9–15%). Treating the gem-dibromo cyclopropane unit in these species with methyllithium furnished 1-alkyl substituted H2Pn, which gave high yields of Li2[1-alkyl-Pn] upon double deprotonation with nbutyllithium (53–70%).10
Another synthesis of H2Pnvia carbene rearrangement has been reported by Brinker and Fleischhauer.29trans-1,2-Bis(2.2-dibromocyclopropyl)ethane, formed by twofold reaction of trans-1,3,5-hexatriene with CHBr3 and KOtBu, was found to rearrange to H2Pn when treated with methyllithium in diethyl ether. However, significant side products were formed, and attempts to separate the mixture using vapour-phase chromatography proved unsuccessful.
Ashley et al. developed a solution-phase synthesis of hexamethylpentalenide, Pn*2−, the pentalenide analogue of pentamethylcyclopentadienide (Cp*−).30 The starting material for this preparation is the so-called ‘Weiss-H4’ compound, obtained from reaction of dimethyl-1,3-acetonedicarboxylate with glyoxal.31 Reacting the ‘Weiss-H4’ compound with an excess of methyl iodide and K2CO3, followed by hydrolysis/decarboxylation with concentrated HCl yielded 2,4,6,8-tetramethylbicyclo[3.3.0]octane-3,7-dione (Fig. 11). The so-obtained diketone could be oxidised with bromine in MeOH, then reacted with ‘methylcerium dichloride’ (derived from methyllithium and cerium trichloride) at −78 °C in THF. The resulting alcohol is prone to polymerisation and thus extremely sensitive to acid. The final dehydration reaction therefore required an aprotic method, and it was found that LiCl in DMSO was effective in producing 1,3,4,5,6-pentamethyl-2-methylene-1,2-dihydropentalene, Me6-Pn or Pn*, an exocyclic isomer of hexamethylpentalene. This compound is stable due to its vinylic fulvene structure preventing cyclic conjugation of an otherwise anti-aromatic 8π system, and marks the only pentalenide synthon where the 8π Pn* is stable whereas the non-aromatic H2Pn* is unknown. As outlined in Fig. 3, to transform the Pn* into Pn*2− it must be doubly reduced. Direct reaction with alkali metals lead to degradation of the starting material through polymerisation, but reaction with a bulky trialkyl borohydride (LS-selectride) allowed for 1,4-addition of a hydride to yield LiHPn* (Fig. 12).32 Use of the less sterically demanding L-selectride resulted in a mixture of 1,2- and 1,4-addition products.
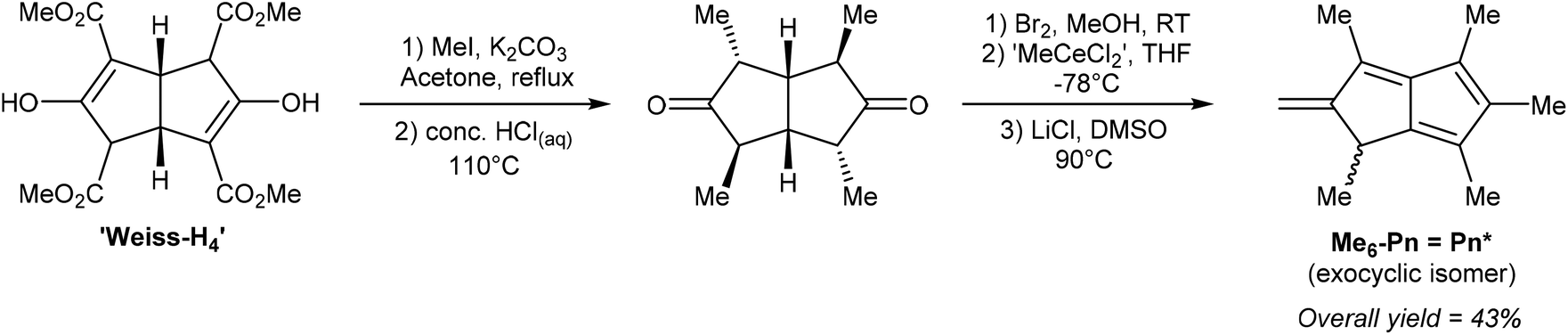 | ||
| Fig. 11 Synthesis of an exocyclic hexamethylpentalene isomer from ‘Weiss-H4’. | ||
 | ||
| Fig. 12 Reduction of hexamethylpentalene to lithium hexamethylhydropentalenide, and its subsequent deprotonation to dilithium hexamethylpentalenide. | ||
LiHPn* is reportedly insoluble in most organic solvents, requiring solubilisation with pyridine for NMR analysis. The final deprotonation of LiHPn* however was performed in a refluxing hexane slurry with nbutyllithium and TMEDA to afford the TMEDA adduct of Li2Pn*. In order to obtain single crystals suitable for XRD analysis [Li2(TMEDA)x]Pn* had to be converted into a bis-stannyl derivative using Me3SnCl (see Fig. 39, section 3.3), and transmetallated again using methyllithium in neat TMEDA.32
Despite requiring multiple steps involving several sensitive intermediates, the overall synthesis of [Li2(TMEDA)x]Pn* is well developed and gives access to a valuable synthon for organometallic pentalenide chemistry without the use of special apparatus. Using different electrophiles (for example, ethyl iodide or isopropyl bromide) in the first step could in principle allow for incorporation of different substituents into these four positions, but this change would likely require downstream modification of the overall synthesis. So far there is no report of such variants.
2.5. Annulation reactions of cyclopentadienes
As already exemplified in some of the previous thermal rearrangement reactions, HCp provide a convenient starting material for the synthesis of pentalene derivatives. In the following section we review examples where selective 1,2 annulation reactions of HCp have been accomplished via mild solution-phase methods. An early example was provided by Hafner and Süss, who used Li[tBuCp] to synthesise tBu3Pn through its reaction with an iminium salt derived from 5-dimethylamino-2,2,6,6-tetramethyl-4-hepten-3-one and triethyloxonium tetrafluorborate (Fig. 13).33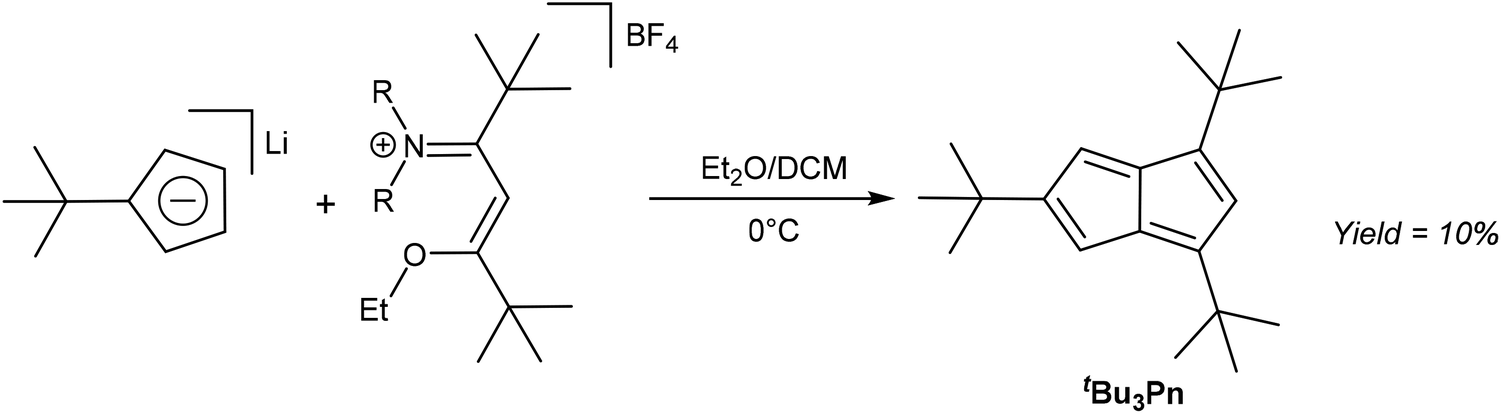 | ||
| Fig. 13 Synthesis of 1,3,5-tris(tbutyl)pentalene from lithium tbutylcyclopentadienide. | ||
The resulting deep blue species had to be purified by chromatography on alumina at −75 °C using pentane as the eluent. Kitschke and Lindner were later able to obtain crystals of tBu3Pn for XRD analysis by recrystallisation from hexane.34 Its stability is interesting in that although normally one would not approach the synthesis of an organometallic Pn2− ligand via its 8π Pn version, this might be a viable approach in the case of sterically demanding substitution patterns such as 1,3,5-tris(tert-butyl) that are effective in blocking dimerization pathways. Ashley et al. have shown that a permethylated Pn system may be reduced using a bulky borohydride to give HPn− and then Pn2− upon deprotonation (Fig. 11),32 so similar chemistry might be possible with other bulky Pn such as Hafner's tBu3Pn.
Due to their known stability and ease of deprotonation, routes that give access to H2Pn synthons are still preferable in most cases. Retrosynthetic analysis shows that Hafner's HCp annulation strategy to synthesise tBu3Pn is well suited to give access to H2Pn as well: 1,4-Michael addition by a Cp− to an electrophilic conjugated carbonyl species would yield a β-cyclopentadiene-functionalised ketone/aldehyde intermediate that is set up to undergo an intramolecular Knoevenagel condensation (Fig. 14). Compatible HCp merely require two adjacent unsubstituted carbons in order to accommodate the final ring-closing step. Thus, a wide range of mono-, 1,2 and 1,3 di-, as well as 1,2,3-trisubstituted HCp may be transformed into the corresponding H2Pn pro-ligands by reaction with simple enones (chalcones).
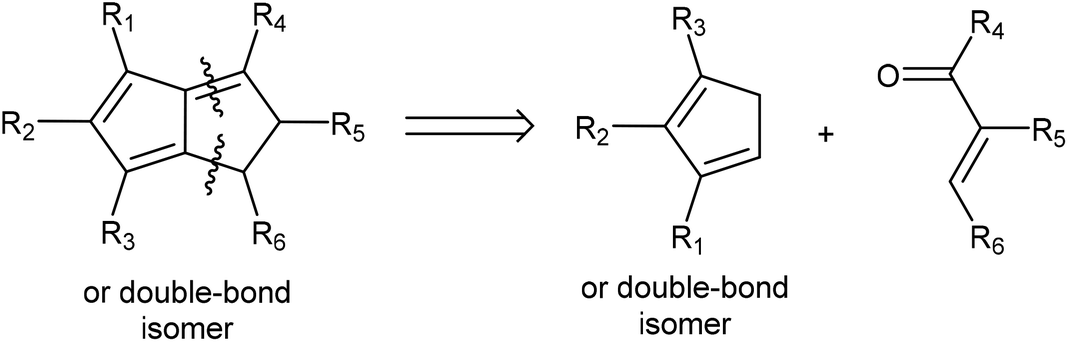 | ||
| Fig. 14 Retrosynthesis of substituted dihydropentalenes from cyclopentadienes and enones. | ||
The simplest unsubstituted H2Pn is currently not accessible by such a transformation, as HCp and acrolein are known to undergo a Diels–Alder reaction instead of the required Michael addition.35 However, Griesbeck showed that moving to slightly higher substituted reaction partners, and using pyrrolidine to activate the chalcone towards 1,4 nucleophilic attack via its enamine, indeed furnishes substituted H2Pn in good yields from a single step reaction under mild conditions (Fig. 15).36
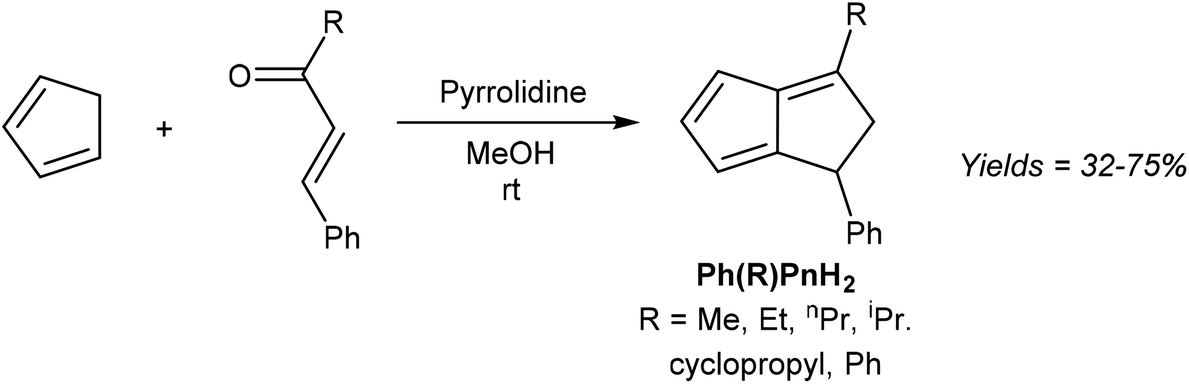 | ||
| Fig. 15 Synthesis of di-substituted dihydropentalenes by pyrrolidine-facilitated condensation of cyclopentadiene and substituted enones. | ||
The six different 1,3-disubstituted H2Pn thus prepared were air-stable compounds that did not polymerise and could be purified by high-vacuum distillation. The initially obtained 1,2 double-bond isomers were found to isomerise to the more stable 1,5 form when contacted with Brønsted acids (e.g. trifluoroacetic acid or activated alumina) or when heated above 500 °C. Curiously, none of the 1,3-disubstituted H2Pn reported in this study have been taken forward to the Pn2− form by double deprotonation or used as organometallic synthons in other reports so far.
Another, more highly substituted example of this strategy has been reported by Le Goff,37 who reported the fluoride-catalysed condensation of 1,2,3-Ph3CpH and 1,2,3-triphenylenone to yield Ph6PnH2 (Fig. 16). Oxidation with N-bromosuccinimide reportedly afforded the corresponding Ph6Pn as another example of an 8π pentalene stabilised by suitable substitution pattern, as inferred from its UV-vis spectrum.
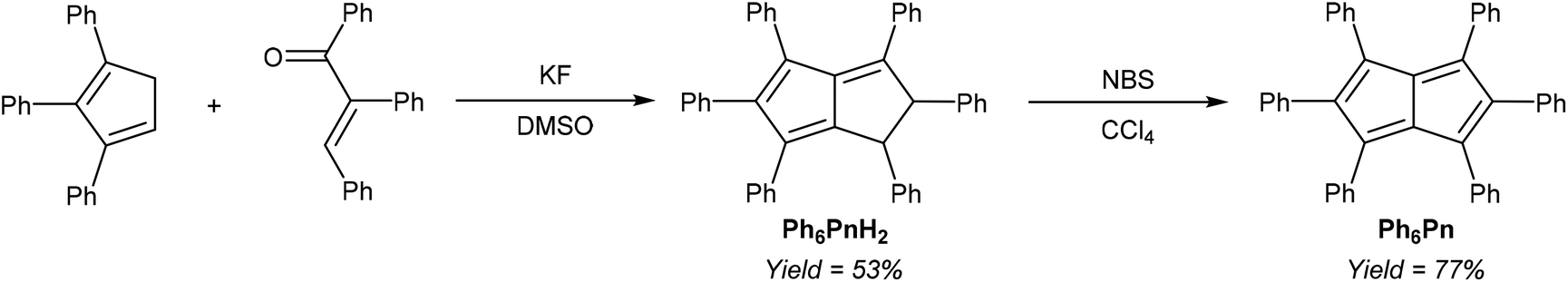 | ||
| Fig. 16 Synthesis of hexaphenyldihydropentalene, and its oxidation into hexaphenylpentalene. | ||
Its corresponding Ph6Pn2− would make an interesting organometallic ligand, however, this two-page single-author communication does not contain any synthetic or analytical details, and the synthesis has never been replicated in later literature.
2.6. Annulation reactions of fulvenes
In cases where the required chalcone is either difficult to access or does not display the desired reactivity, a step-wise approach to H2Pn synthesis viaHCp annulation is also possible. Condensation of a HCp with an aldehyde yields a 6-substituted fulvene that may be attacked by an enolate to ring-close via an intramolecular Knoevenagel condensation (Fig. 17). Although apparently introducing an additional reaction step, it eliminates the need for synthesising the enone in the first place. Formation of the fulvene intermediate from HCp and aldehyde follows a straightforward condensation reaction using pyrrolidine in MeOH as described by Stone and Little (Fig. 18).38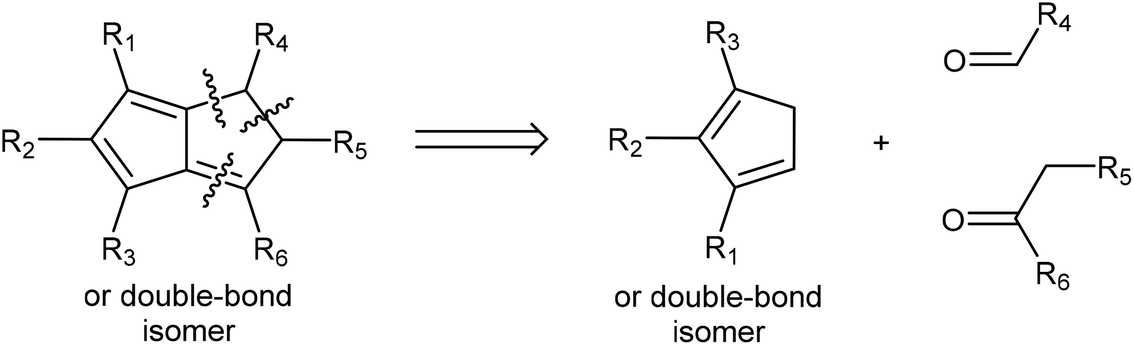 | ||
| Fig. 17 Retrosynthesis of substituted dihydropentalenes from cyclopentadienes, aldehydes and ketones. | ||
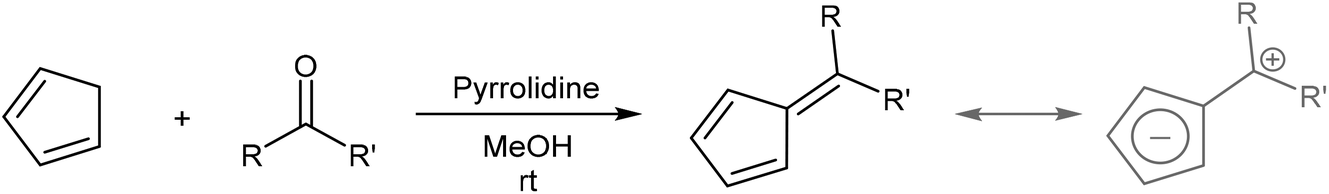 | ||
| Fig. 18 Synthesis of fulvenes from cyclopentadiene and carbonyl compounds and their resonance structure. | ||
6-Substituted fulvenes are stable enough to be isolated, however, with their exocyclic double bond being polarised to enable nucleophilic attack by a range of nucleophiles (Fig. 18).28 Coskun et al. have demonstrated the utility of this reactivity for H2Pn synthesis (Fig. 19).39 Reaction of HCp with a range of different aldehydes afforded 6-substituted fulvenes that cleanly condensed with acetone (used as the solvent for this transformation) at room temperature in the presence of pyrrolidine to give variously substituted 1,3-RMe-PnH2 in good yields. The air-stable products were obtained after evaporation of solvent and side products (mainly from cross-condensation of acetone) followed by flash chromatography on silica, which was reported not to induce any double bond isomerisation.
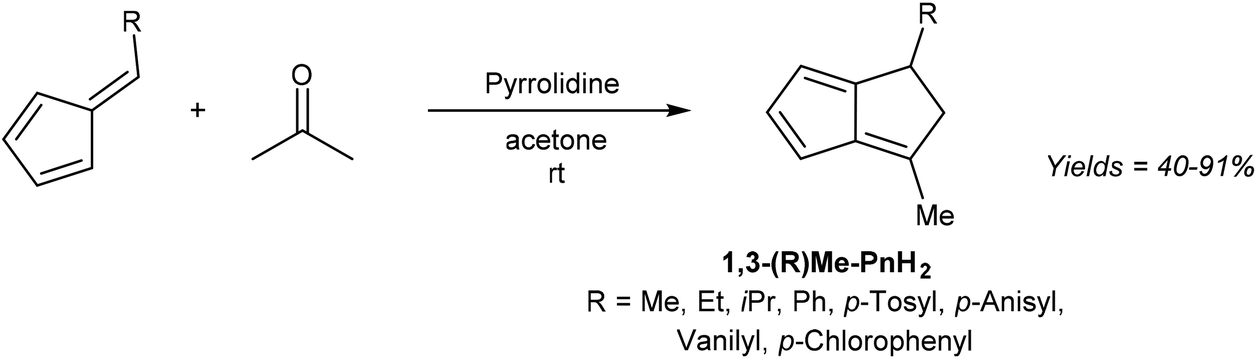 | ||
| Fig. 19 Synthesis of 1,3-disubstituted dihydropentalenes from 6-substituted fulvenes and acetone. | ||
Again, none of the H2Pn synthesised have been deprotonated to the corresponding Pn2− in this or other studies. Coskun et al. only describe the reactivity of 6-methyl fulvene, but it could be supposed that fulvenes derived from substituted HCp and various aldehydes could be reacted in an analogous manner to give a variety of more highly substituted H2Pn as outlined in Fig. 16. However, like the concerted double-condensation reaction using chalcones described above, the parent (unsubstituted) H2Pn is not likely to be accessible via this step-wise condensation method either due to the instability of its hypothetical reaction partners (unsubstituted fulvene and acetaldehyde).
2.7. Acetylene oligomerisation
Akin to alkyne trimerization to give variously substituted 6π aromatics (Reppe chemistry),40 there are also some reports of alkyne oligomerisation reactions that produce pentalene scaffolds. Kuwabara et al.41 and Saito et al.42 reported the synthesis of bis-silylated dibenzopentalenides through reduction of phenylacetylenes with alkali metals (Fig. 20). When phenyl(trisipropylsilyl)acetylene was reacted with metallic lithium in Et2O only a small amount of Li2[Bn2Pn] was obtained. The major product of this reaction, a 1,4-dilithio-1,3-butadiene, could be reacted with Ba(HMDS)2 to give Ba[Bn2Pn], the hydrolysis of which yielded the silyl-substituted Bn2PnH2.43 Reduction of phenyl(trisipropylsilyl)acetylene with metallic potassium directly gave K2[Bn2Pn] in high yields, with other silyl-substituted phenylacetylenes reacting in the same manner (Fig. 20).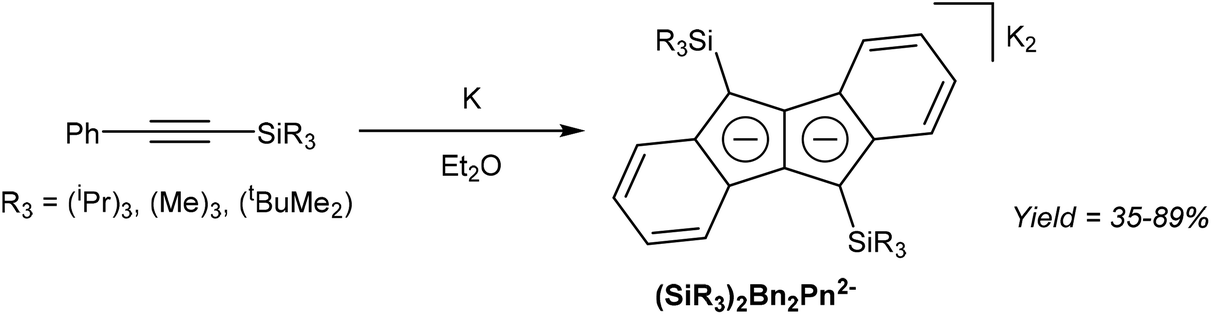 | ||
| Fig. 20 Formation of dibenzopentalenides by reaction of phenylsilylacetylenes with potassium. | ||
In another example, Bailey et al.44 described the PdCl2 catalysed cyclotetramerisation of phenylacetylene to Ph4PnH2 (Fig. 21). However, of the two isomeric products obtained from this reaction only one would be suitable for deprotonation into a Ph4Pn2−, and the report does not include methods of preparative work up and isolation.
 | ||
| Fig. 21 Palladium-mediated cyclotetramerization of phenylacetylene to tetraphenyldihydropentalenes. | ||
Although interesting examples of an alternative, potentially elegant synthetic approach of accessing H2Pn from simple alkyne starting materials, the generality and versatility of these methods remain to be explored more systematically.
2.8. Functionalisation of pentalenides
Although most solution-phase syntheses of H2Pn are easier for more highly substituted starting materials, the opposite is true for pyrolytic pathways. However, post-synthetic functionalisation of the parent (unsubstituted) H2Pn obtained from e.g. FVP is also possible to some degree. Cloke et al. for instance demonstrated the functionalisation of Li2Pn with of trialkylsilyl-groups (Fig. 22), which in addition to modifying their electronic and steric parameters imparts the complexes with enhanced solubility in hydrocarbon solvents.19 | ||
| Fig. 22 Reaction of dilithium pentalenide with silyl-electrophiles, and deprotonation of the resulting disilyl-dihydropentalenes. | ||
[Li(DME)]2Pn was shown to react regio-selectively with two equivalents of TMS-Cl in THF to give 1,4-TMS2PnH2 as a mixture of syn and anti isomers. These could be doubly deprotonated again with nbutyllithium in DME/pentane to give the corresponding [Li(DME)]2[1,4-TMS2Pn] for subsequent transmetalation reactions. When introduction of additional substituents was attempted at this stage, by further reaction of 1,4-TMS2Pn2− with another two equivalents of TMS-Cl, 1,1,4,4-TMS4PnH2 was obtained exclusively. Due to the two bis-silylated sp3 carbons in the 1 and 4 positions this compound cannot be converted into a Pn2− by way of deprotonation any more, limiting how much functionality may be introduced through this strategy. [Li(DME)]2Pn failed to react analogously with tris(ipropyl)silyl chloride, but did react with the more activated tris(ipropyl)silyl triflate to give the corresponding 1,4-TIPS2PnH2, forming the anti isomer only, presumably due to steric reasons in this case. Reacting this compound with KNH2 in Et2O gave high yields of K2[1,4-TIPS2Pn], a useful ligand for use in f-block chemistry where lithium salts tend to form ‘ate’ complexes (see section 3.3).19
The regioselectivity of the reaction of Pn2− with hard electrophiles is a consequence of its charge distribution. In addition to the fully delocalised (aromatic) η8 form and the partially delocalised (allylic) η3 form that govern reactivity with soft Lewis acids, its ground state η1 resonance structure conjugates both charges to the 1 and 4 positions (Fig. 23). Thus, post-synthetic functionalisation of Pn2− is often limited to these two positions.
 | ||
| Fig. 23 Resonance structures of the pentalenide dianion. | ||
3. Synthesis of pentalenide complexes of p-, d- and f-block metals
As mentioned in section 2.1 (Fig. 4 and 5), deprotonative metalation of H2Pn with suitably strong bases gives straightforward access to stable HPn− and Pn2− salts that can be used for transmetalation on to p-, d-, and f-block metals in the desired oxidation state. Before we review selected cases of these in section 3.3, there are also some examples of direct metalation reactions of neutral precursors via redox reactions (as in related HCp chemistry) that deserve mentioning. At present these appear less generally applicable than transmetalation from HPn− and Pn2− salts, but serve to illustrate the rich coordination chemistry of pentalenide ligands.3.1. Pentalenide and hydropentalenide complexes through direct metalation of neutral precursors
An early example of a direct redox metalation of Pn with a transition metal has been provided Hafner and Weidemüller,45 who reported that the reaction of Pn2 with an iron(0) precursor produced a small amount of (CO)5Fe2Pn (Fig. 24). According to the contemporary ionic electron counting model, and in line with the thermodynamic driving force of forming a 10π aromatic, this reaction implies a double reduction of Pn to Pn2− accompanied by cleavage of the dimer and oxidation of two Fe0 centres to FeI. Heating Pn2 to 50 °C in methylcyclohexane in the presence of Fe2(CO)9 resulted in the formation of a yellow crystalline solid, supposed to be the syn-diiron pentalenide complex shown based on its NMR spectroscopic and mass spectrometric signatures. However, no XRD analysis is available to confirm the geometry, and work up details were not provided in the original paper. A mixture of isomers of [1,3-Me2Pn]2 was reacted in a similar fashion to give an unidentified green diiron complex in 21% reported yield.45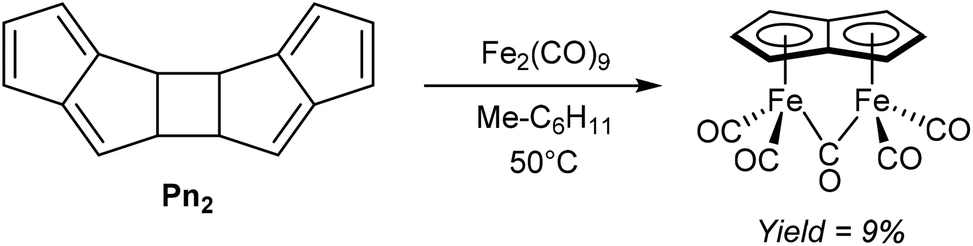 | ||
| Fig. 24 Reaction of pentalene dimer with diiron nonacarbonyl. | ||
In related examples of directly metalating H2Pn precursors, Hunt and Russel46 reported that reacting 3-Me2N-PnH2 or 3-Ph-PnH2 with Fe(CO)5 would produce analogous syn bimetallic (CO)5Fe2Pn complexes in similar yields (Fig. 25). However, none of the geometries have been confirmed by XRD either.
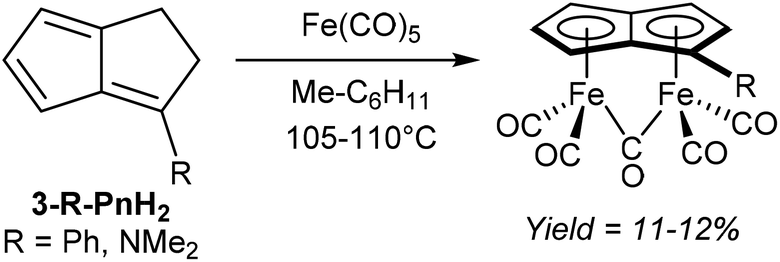 | ||
| Fig. 25 Reaction of mono-substituted dihydropentalenes with iron pentacarbonyl. | ||
Similar to direct metalation reactions of HCp with zero-valent metal precursors to give M(I)Cp complexes, this conversion of a H2Pn into a bimetallic Pn2− complex implies a double deprotonation and proton reduction to H2 to give two FeI centres in the resulting complex.
Another redox metalation of a Pn has been reported by Ashley et al.47 Refluxing Pn* (Fig. 11) with an excess of Fe2(CO)9 in toluene (added in portions) furnished a brown solid upon filtration and washing. Recrystallisation from toluene/hexane produced the η5(CO)5Fe2Pn* complex shown in Fig. 26 in good yields. In this case the syn geometry of the two metal centres has been established by XRD analysis. The same procedure could be applied to synthesise a related dicobalt complex using a stoichiometric amount of Co2(CO)8 (Fig. 26). Notably, both compounds could be synthesised on a multigram scale.
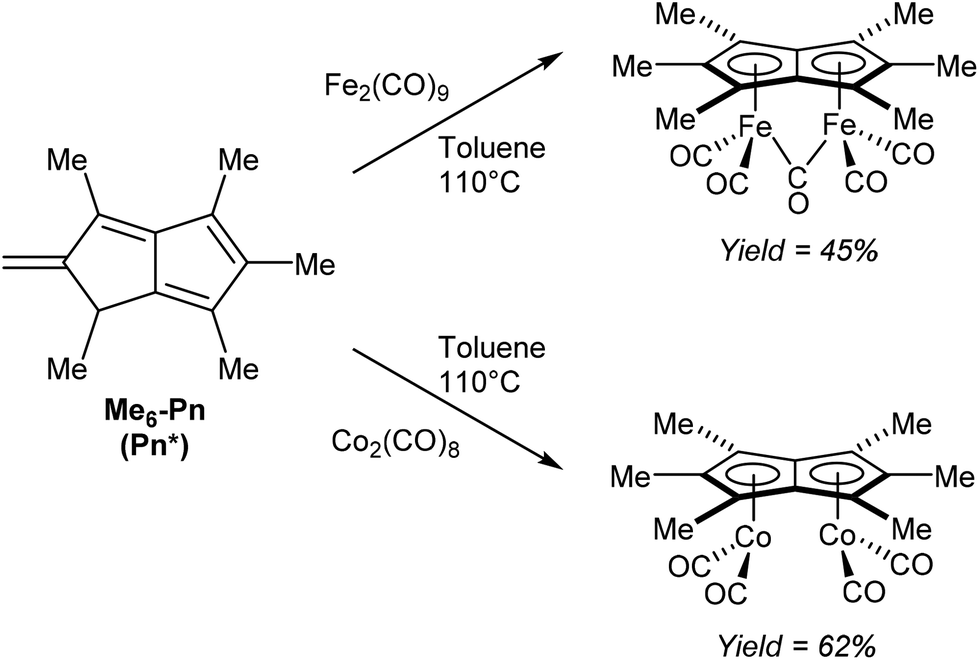 | ||
| Fig. 26 Reaction of hexamethylpentalene with diiron nonacarbonyl and dicobalt octacarbonyl. | ||
Both reactions involve a formal double reduction of the Pn* scaffold by the metal(0) precursors, accompanied by a two-bond hydrogen shift to the exocyclic methylene group. The similar reactivity but increased stability and enhanced crystallinity of these Pn*2− compounds compared to the parent Pn2− complexes are promising and provide motivation for further exploration of other substitution patterns in organometallic pentalenide chemistry.
Redox metallations of COT accompanied by skeletal rearrangement to Pn2− are also known for some d-block metal complexes. Brookes et al.48 reported the reaction of cis-[Ru(CO)4(GeMe3)2] with COT in refluxing heptane or octane (Fig. 27). Of the several compounds produced from this reaction, a small amount of the pale yellow syn bimetallic Ru2Pn complex shown in Fig. 27 could be isolated and analysed by XRD.
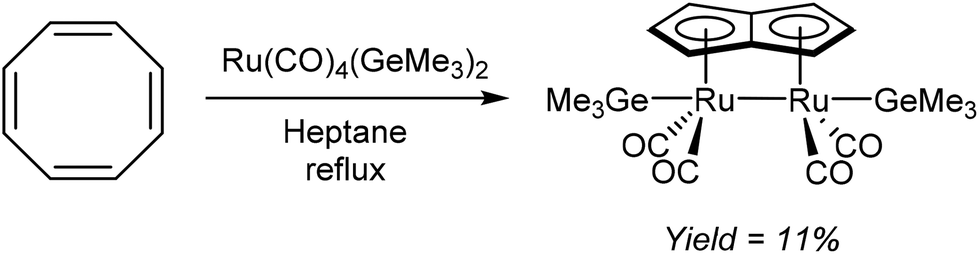 | ||
| Fig. 27 Rearrangement redox reaction of cyclooctatetraene with Ru(CO)4(GeMe3)2. | ||
Similarly, Howard et al.49 reported that COT would react with Ru3(CO)10 in refluxing heptane to give a trinuclear RuPn complex, albeit in low yields and in a mixture with various other RuCOT complexes. Silyl-substituted COT have been reported to react in an analogous manner, giving the correspondingly substituted trinuclear RuPn complexes.50 All of these reactions were very low yielding and no work up procedures are available, so the synthetic utility of these transformations appears limited.
Akin to the alkali metal induced and palladium catalysed alkyne oligomerisation reactions that produce substituted H2Pn (section 2.7), the direct formation of HPn− complexes from cyclotetramerization of acetylenes has also been reported. For example, Coffield et al.51 showed that heating Mn2(CO)10 in THF under a pressure of acetylene resulted in the formation of η5(CO)3MnPnH as a viscous yellow oil that was purified by distillation (Fig. 28). The same transformation could also be accomplished using MeMn(CO)5 but with lower yields of 27%. Further deprotonation of the bound HPn− was not attempted.
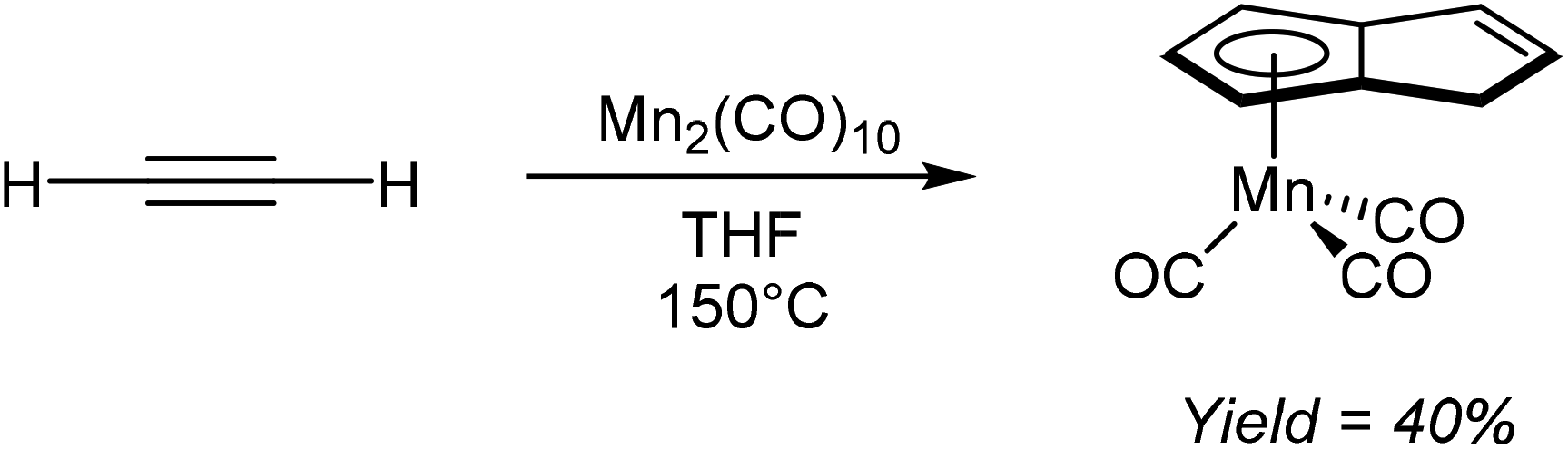 | ||
| Fig. 28 Reaction of acetylene with dimanganese decacarbonyl. | ||
Komatsu et al. reported the formation of η5RhPnH complexes through RhI mediated tetramerisation of bulky mono-substituted alkynes (Fig. 29). The reaction of tbutylacetylene with [Rh(COD)Cl]2 and NEt3 in cyclohexane furnished η5(COD)Rh[tbu4PnH] in 13% yield after purification by chromatography on alumina followed by recrystallization from EtOH.52
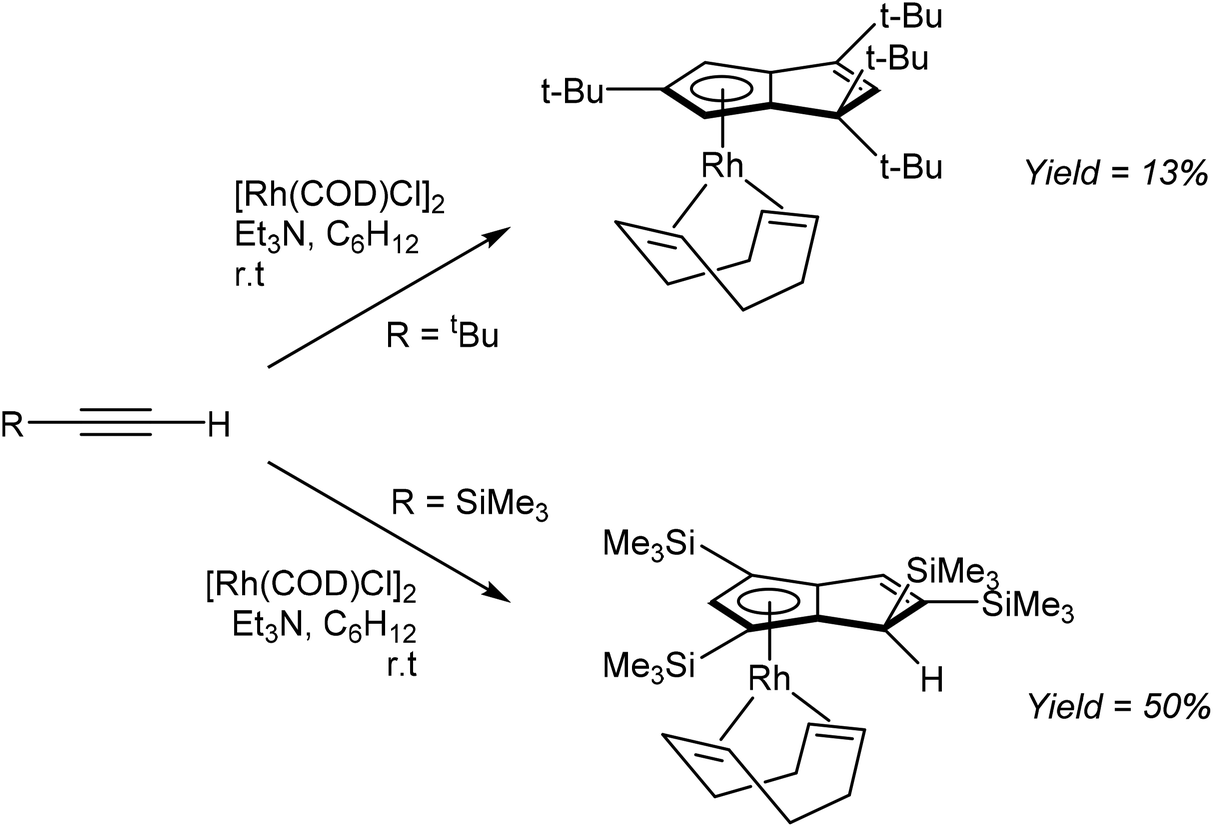 | ||
| Fig. 29 Reaction of substituted acetylenes with [Rh(COD)Cl]2. | ||
The analogous reaction of TMS-acetylene with [Rh(COD)Cl]2 gave the corresponding η5(COD)Rh[TMS4PnH] complex in 50% isolated yield. Interestingly, whereas tetramerization of tbutylacetylene produced 1,1,3,5-R4PnH−, TMS-acetylene yielded the 1,2,4,6-R4PnH− regio-isomer. While not possible with the former, the latter could in principle allow for a second deprotonation to a 1,2,4,6-R4Pn2−. However, this has not been reported yet.
3.2. Hydropentalenide complexes through transmetalation of hydropentalenides
As shown in Fig. 3 and section 2.4, and illustrated in some of the examples in section 3.1, the monoanionic, 6π aromatic HPn− synthon may be obtained from either hydride addition to a stable Pn or mono-deprotonation of H2Pn. Katz and Rosenberger provided an early demonstration of its utility as a transmetalation agent: in situ generation from H2Pn with 1.1 equivalents of nbutyllithium in THF/hexane followed by addition of iron(II) precursors yielded the ferrocene analogue η5Fe[PnH]2 as a mixture of two isomers (Fig. 30).53 The stable compound was obtained in 54% yield after aqueous work up followed by sublimation and recrystallisation from hexane.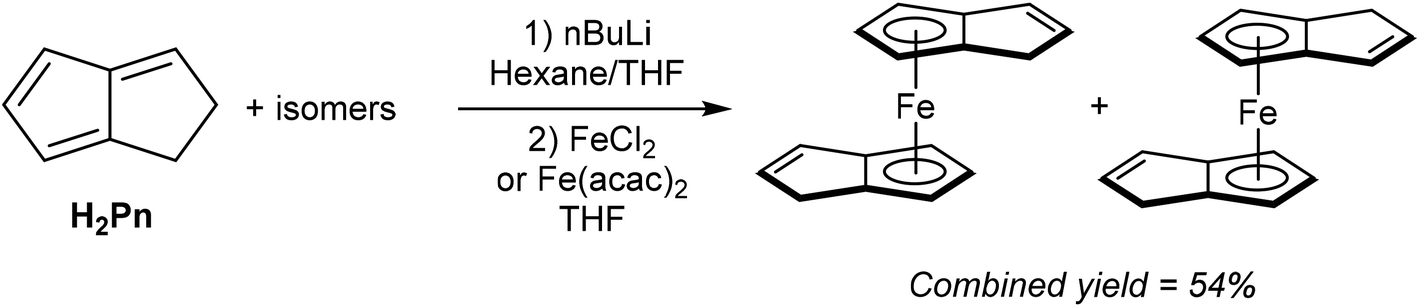 | ||
| Fig. 30 Synthesis of di(hydropentalenide)iron from dihydropentalene. | ||
TlCp is known to be a useful transmetalation agent in instances where alkali metal Cp− salts are too reducing.54 In the same manner, TlPnH (Fig. 4) has been shown to act as an efficient, mild transmetalating agent with d-block metal precursors: reaction with FeCl2 in THF was shown to give the ferrocene derivatives shown in Fig. 30 in 47% isolated yield.9 Similalry, reaction of TlPnH with [Rh(COD)Cl]2 in THF smoothly yielded η5(COD)Rh[PnH] (Fig. 31).9 The TlCl by-product was easily removed by filtration, and sublimation of the product gave the pure RhI complex in high yield. The same study also demonstrated the successful reaction of TlPnH with Me3PtI, but the Me3Pt[PnH] complex produced required gas-phase chromatography in order to furnish pure product.
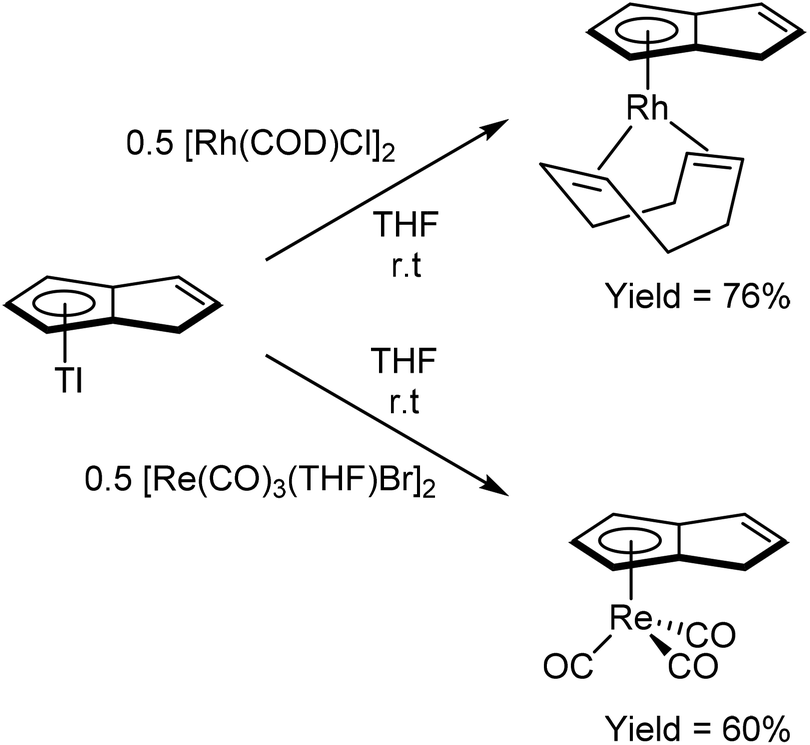 | ||
| Fig. 31 Transmetalation reactions of thallium hydropentalenide. | ||
Reaction of TlPnH with a suspension of [Re(CO)3(THF)Br]2 in THF in the absence of light gave η5(CO)3Re[PnH] as a colourless solid after purification by chromatography on silica (Fig. 31).10 Recrystallisation from Et2O yielded crystals suitable for XRD analysis. The analogous manganese complex could also be prepared through the reaction of [Mn(CO)3(py)2Br] with TlPnH in THF to give the same product of the cyclotetramerisation of acetylene with Mn2(CO)10 (Fig. 28).5a Reacting a mixture of Tl[MePnH] isomers with [Re(CO)3(THF)Br]2 in THF yielded the corresponding η5(CO)3Re[MePnH] complexes as a mixture of double bond isomers.10 Although good combined yields were obtained after purification on silica, the isomers (distinguishable by NMR) could not be separated by chromatography or fractional crystallisation.
Direct metalation reactions of H2Pn with other heavy p-block metal bases has also been achieved. Ustynyuk et al. reported the synthesis of mono- and bis-Me3Sn derivatives of H2Pn using tin amides.55 The reaction of H2Pn with one equivalent of Me3Sn(NEt2) in boiling hexane produced, after distillation, a mixture of 1- and 2-η1-(Me3Sn)PnH (Fig. 32). Higher yields were obtained by stirring the reagents in heptane at room temperature for 48 hours. According to Fig. 23, the 1-isomer would be the primary reaction product from formal addition of a Me3Sn+ moiety to the HPn− intermediate, and NMR analysis indeed showed rapid suprafacial 1–2–3 shifts of the TMT group within (Me3Sn)PnH (as also observed in related η1-TMT Cp and Ind complexes56) to be responsible for formation of the 2-isomer.
 | ||
| Fig. 32 Reactions of dihydropentalene with trimethyltin-diethylamide. | ||
The same reaction with two equivalents of Me3Sn(NEt2) produced 1,4-(Me3Sn)2Pn in high yield without purification. A 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of anti- and syn-isomers was obtained which did not interconvert; although VT 13C NMR analysis showed suprafacial Me3Sn shifts to be possible within each (Me3Sn)2Pn isomer there was no evidence for superfacial migration or intermolecular exchange. A pure sample of the anti-isomer could be obtained after separation by fractional distillation and crystallisation from pentane, enabling structural analysis by XRD. The authors also showed how the anti- and syn-isomers may be distinguished by their 119Sn chemical shift values and assigned via characteristic 119Sn–117Sn coupling constants within a mixture in solution.
:1 mixture of anti- and syn-isomers was obtained which did not interconvert; although VT 13C NMR analysis showed suprafacial Me3Sn shifts to be possible within each (Me3Sn)2Pn isomer there was no evidence for superfacial migration or intermolecular exchange. A pure sample of the anti-isomer could be obtained after separation by fractional distillation and crystallisation from pentane, enabling structural analysis by XRD. The authors also showed how the anti- and syn-isomers may be distinguished by their 119Sn chemical shift values and assigned via characteristic 119Sn–117Sn coupling constants within a mixture in solution.
Similar to the η5 thallium reagents, η1 stannylated Pn act as mild transmetalation agents in cases where alkali metal Pn2− salts are too reducing (see below). This reactivity is not seen with the structurally related but non-fluxional η1 silylated Pn derivatives discussed in section 2.8 due to the strength of the covalent Si–C bond, preventing the trisalkylsily substituent from acting as a leaving group.
Turner et al. demonstrated the use of mono-stannylated HPn*− as a ligand transfer agent for synthesising hydropentalenide complexes of group 4 metals (Fig. 33).57 The reaction of LiHPn* with SnMe3Cl in pentane gave η1(Me3Sn)HPn* as a 1:1 mixture of syn and anti isomers in near quantitative yields after isolation by filtration.
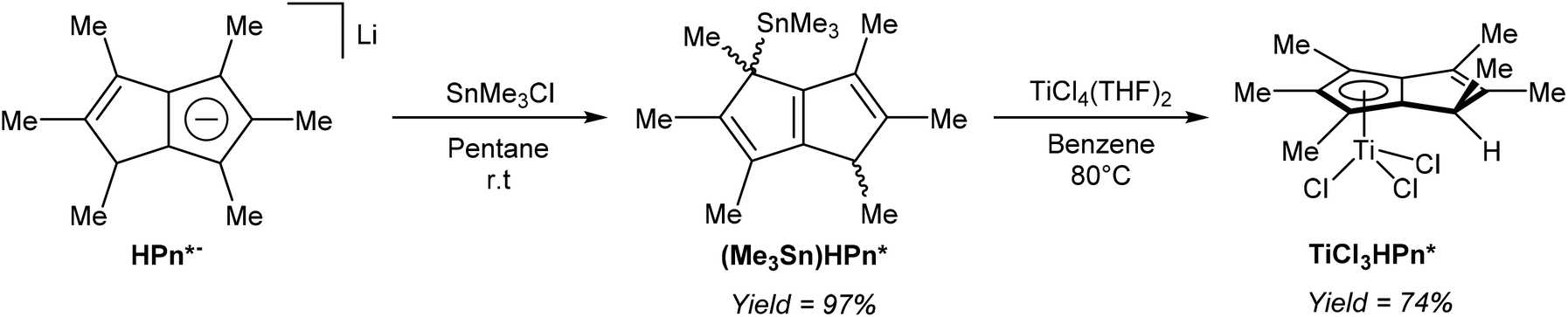 | ||
| Fig. 33 Synthesis of a stannylated derivative of HPn*− and its reaction with a titanium(IV) precursor. | ||
Reacting (Me3Sn)HPn* with TiCl4(THF)2 in refluxing benzene gave a single isomer of η5TiCl3[HPn*] in high yields, and single crystals for XRD analysis could be grown from diethyl ether. Chlorine-bridged, mono-nuclear zirconium and hafnium HPn* dimer complexes could also be prepared from (Me3Sn)HPn*.57 Although deprotonation of the bound HPn* ligand in the Ti complex was not attempted, the reverse reaction (mono-protonation of a titanium Pn*2− complex) was later reported by the same group (see Fig. 40).
3.3. Pentalenide complexes through transmetalation of pentalenides
Due to their stability and ease of preparation, alkali metal Pn2− salts are the most prevalent reagents for synthesising organometallic pentalenide complexes. Again similar to MCp chemistry, Jonas et al. reported that [Li2(DME)x]Pn would react cleanly with Cp2VCl by substitution of one Cp− ligand and chloride.58 The resulting η5CpVη8Pn complex (Fig. 34) is air sensitive but could be sublimed under vacuum to give pure product in high yields. η5Cp*Vη8Pn and η5IndVη8Pn could be prepared in the same manner, and recrystallisation from hexane at low temperatures gave samples suitable for XRD analysis.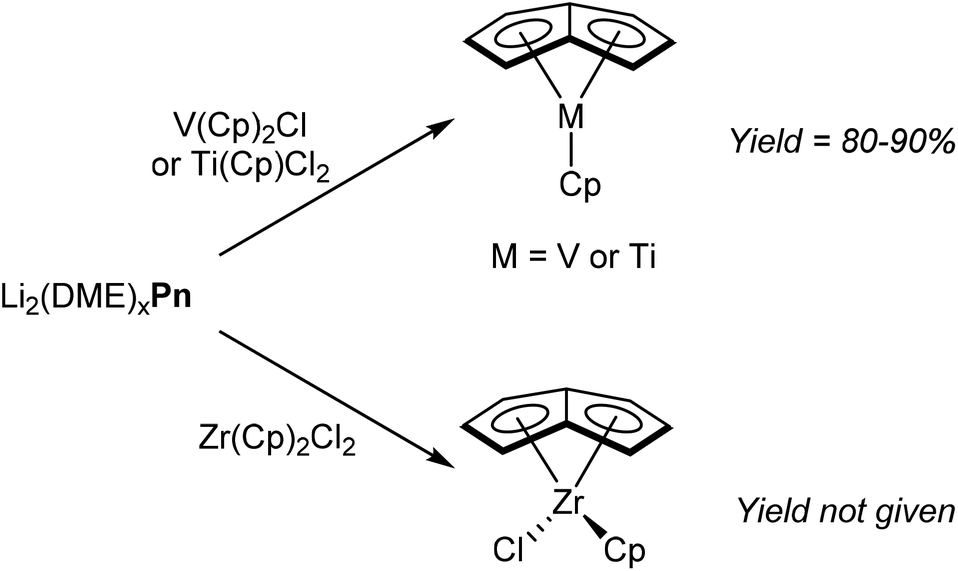 | ||
| Fig. 34 Transmetalation reactions of dilithium pentalenide with early d-block metals. | ||
The reaction of CpTiCl2 with [Li2(DME)x]Pn yielded η5CpTiη8Pn in high yield, and the analogous reaction of Cp2ZrCl2 gave η5CpClZrη8Pn (Fig. 34).59 However, details of solvents for these transformations is missing from these reports.
Jones et al. managed to access several new bimetallic group 7 carbonyl Pn2− complexes by way of transmetalation.60 The reaction of [Li2(DME)x]Pn with 2 equivalents of [Mn(CO)3(py)2Br] in THF gave exclusively the anti-isomer of η5[(CO)3Mn]2Pn (Fig. 35), which after extraction and filtration in air was purified by chromatography on silica. When the analogous reaction was performed with one equivalent of [Re(CO)3(THF)Br]2, a mixture of syn- and anti η5[(CO)3Re]2Pn was obtained, with the ratio being dependent on the reaction temperature. The two isomers could be separated through fractional crystallisation.
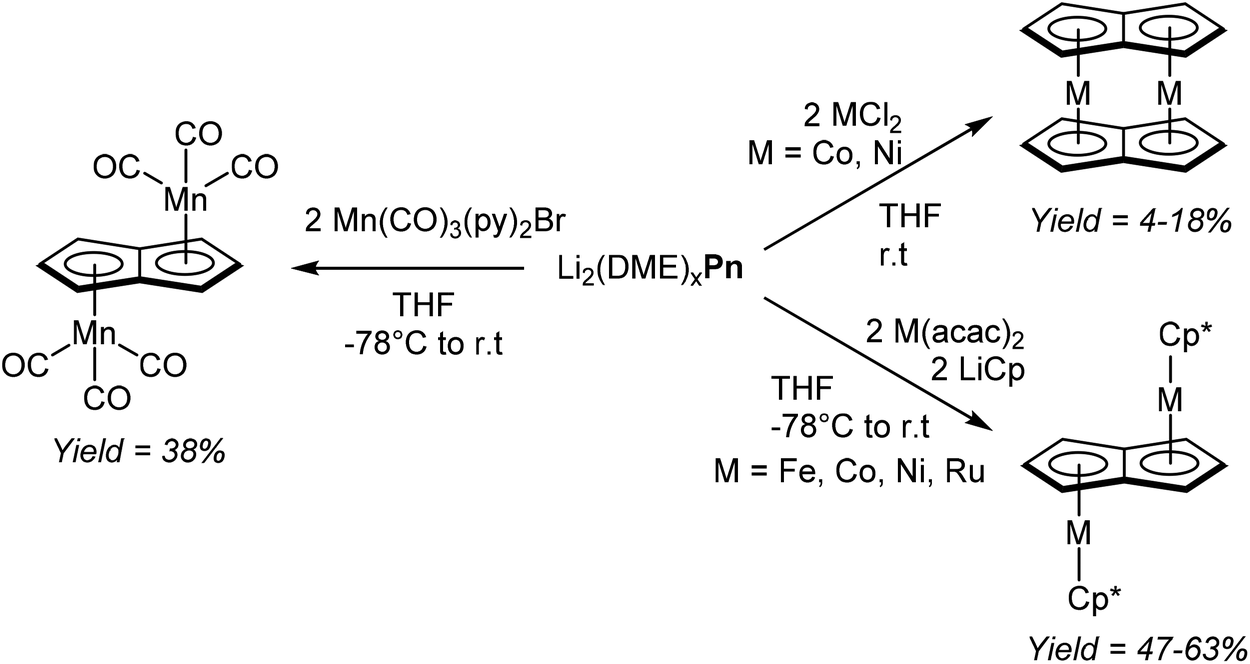 | ||
| Fig. 35 Transmetalation reactions of dilithium pentalenide with late d-block metals. | ||
Katz et al. showed that dinuclear Pn2− sandwich complexes of cobalt and nickel could be synthesised by reacting their respective dihalide salts with [Li2(DME)x]Pn in THF (Fig. 35).61 Both compounds were obtained in low yields after purification by sublimation. The analogous reaction with FeCl2 however did not produce the analogous dinuclear sandwich complex (“double ferrocene”),61b but a mononuclear sandwich complex with two bridging HPn− ligands instead.62 Binding et al. were later able to synthesise the dinuclear sandwich complex [FePn*]2 by reacting [Li2(TMEDA)x]Pn* with Fe(acac)2 in THF at room temperature,63 providing yet another example of the utility of more highly substituted pentalenide scaffolds.
Manriquez et al. showed that Cp*Fe(acac), generated in situ from LiCp* and Fe(acac)2 in THF, would react smoothly with [Li2(DME)x]Pn to produce anti η5[Cp*Fe]η8Pn (Fig. 35).64 The complex was obtained in high yields after recrystallization from toluene; analogous cobalt, nickel and ruthenium complexes could be prepared in the same manner.
Exploring Pn2− complexes of f-block metals, Cloke at al successfully synthesised thorium and uranium complexes of 1,4-(TIPS)2Pn2− by way of transmetallation.65 The reaction of ThCl4 and K2[1,4-(TIPS)2Pn] suspended in THF yielded a bright orange solid containing a mixture of staggered and eclipsed isomers of η8[1,4-(TIPS)2Pn]2Th (Fig. 36). The product could be purified by sublimation, and crystals for XRD analysis were grown from pentane. The same reaction with UCl4 produced a dark green product as the analogous isomeric mixture of η8[1,4-(TIPS)2Pn]2U again in high yields.
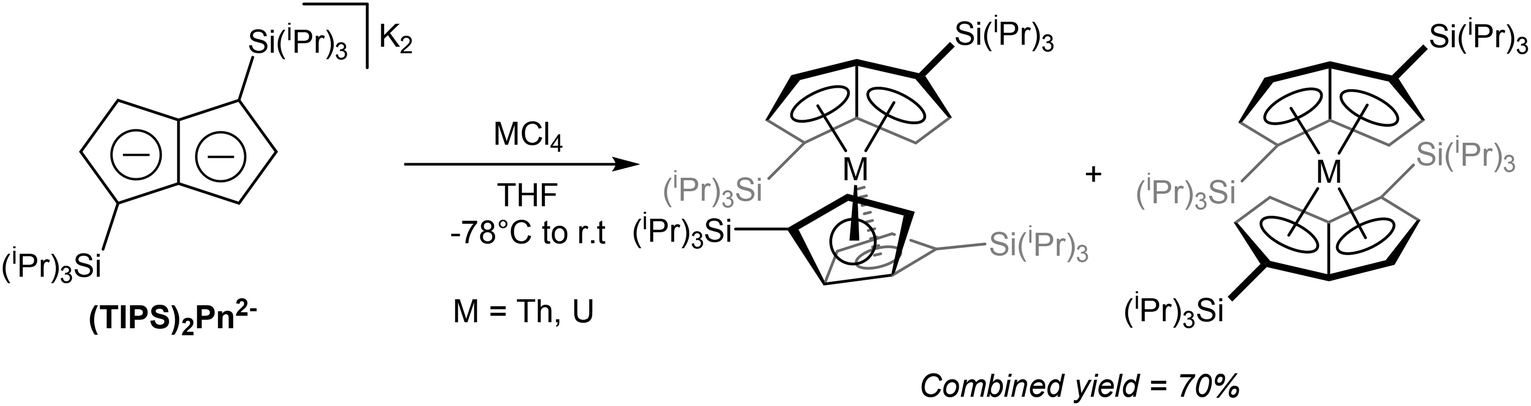 | ||
| Fig. 36 Transmetalation of dipotassium bis(trimethylsilyl) pentalenide with f-block metal halides. | ||
Balazs et al. synthesised the anionic cerium(III) sandwich complex K[Ce(1,4-TIPS2Pn)2] by reacting K2[1,4-TIPS2Pn] with CeCl3 in THF at room temperature.66 This sand-coloured species was purified by filtration and washing with pentane, and characterised crystallographically by complexing the potassium ion with 18-crown-6 and recrystallising the resulting solid from toluene. The oxidation of K[Ce(1,4-TIPS2Pn)2] was shown using a slurry of AgBPh4 in THF, and the resulting cerium(IV) sandwich complex [Ce(1,4-TIPS2Pn)2] was purified by extraction with pentane and crystallisation. Ashley et al. showed the analogous synthesis of CePn*2 by reacting [Li2(TMEDA)x]Pn* and CeCl3 in THF at room temperature, followed by oxidation of the intermediate Li[CePn*2] using an excess of dichloroethane.67
In another example of a substituted Pn2− transmetallation reaction, Cooper et al. described the reactivity of Pn*2− with group 4 metal halides.68 ZrCl4 and HfCl4 mixed with [Li2(TMEDA)x]Pn* in benzene gave, after filtration and recrystallization, halide-bridged dinuclear η8 complexes of zirconium and hafnium as LiCl adducts (Fig. 37). Attempts to remove coordinated THF via high vacuum lead to decomposition.
 | ||
| Fig. 37 Reactions of dilithium hexamethylpentalenide with group IV metal halides. | ||
When the analogous reaction was performed with titanium(IV) precursors, no titanium complex could be obtained but ligand degradation was observed instead. Reaction of [Li2(TMEDA)x]Pn* with TiCl3 and subsequent oxidation with PbCl2 did give a dinuclear TiIV complex, however, only in 8% yield.
In order to access TiIV2Pn* complexes, less reducing transmetalation agents were required. [Li2(TMEDA)x]Pn* was thus reacted with two equivalents of Me3SnCl to give η1(Me3Sn)2Pn* derivatives (Fig. 38), analogous to those reported for the parent H2Pn (section 3.2, Fig. 32), in an extension to the η1(Me3Sn)HPn* shown in Fig. 33.68
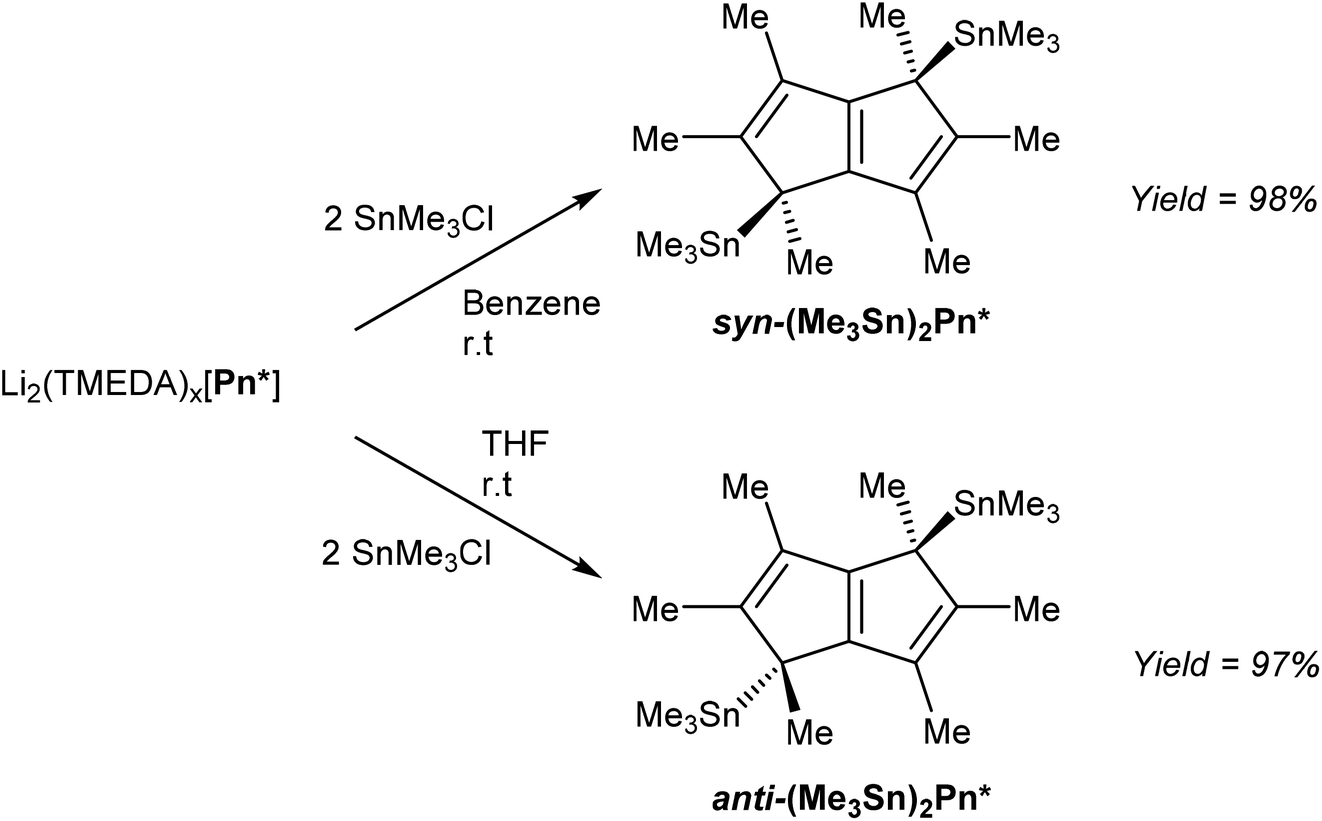 | ||
| Fig. 38 Synthesis of bis-stannylated hexamethylpentalenides. | ||
Interestingly, the use of a non-polar solvent such as benzene lead to formation of the syn product, whereas the use of THF exclusively gave the anti-isomer. Both could be isolated in very high yields after filtration and recrystallisation from toluene or pentane. It was proposed that the second lithiation step in the synthesis of [Li2(TMEDA)x]Pn* initially precipitated the syn dilithio complex as the kinetic product, which when reacted further in a benzene slurry produced syn TMT derivatives, whereas the use THF allowed interconversion to the thermodynamic anti dilithio configuration during the dissolution, producing anti TMT derivatives. This represents an interesting opportunity to steer the face selectivity of M2Pn reagents in transmetalation reactions in general, but remains to be explored for other pentalenide reagents than [Li2(TMEDA)x]Pn*.
In terms of transmetalation ability, reaction of syn η1(Me3Sn)2Pn* with TiCl4 in toluene indeed smoothly produced η8[TiCl2Pn*]2 in high yields after recrystallization (Fig. 39).68 The analogous reactions with ZrCl4 and HfCl4 failed to produce the desired complexes, suggesting the presence of LiCl is required to stabilise the Zr/Hf Pn* complexes shown in Fig. 37.
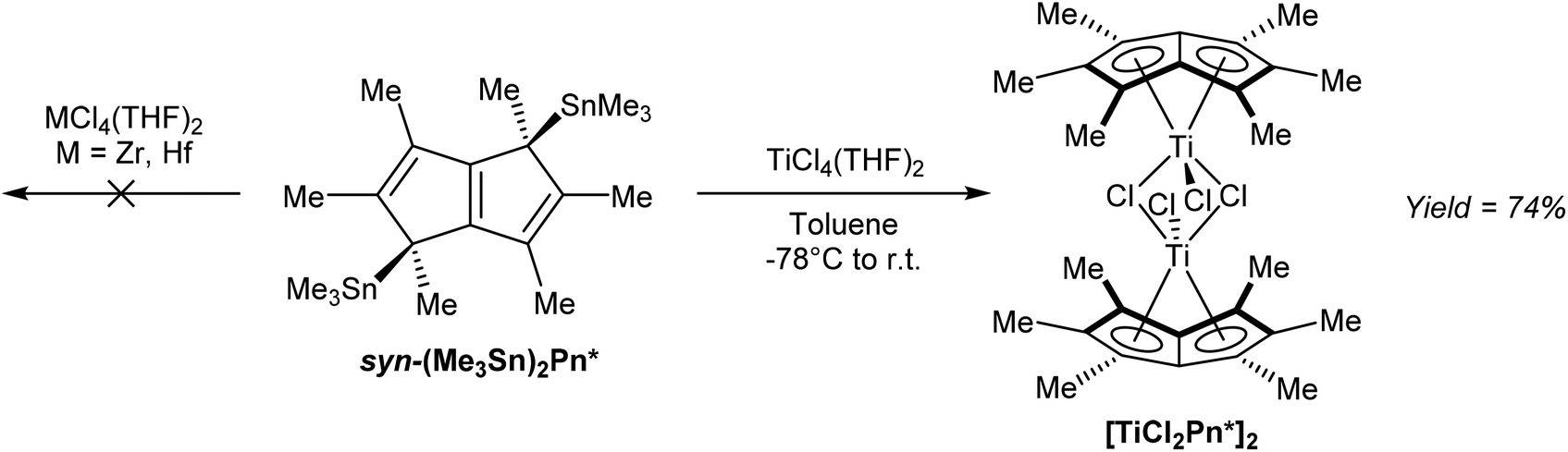 | ||
| Fig. 39 Reactions of bis(trimethylstannyl)-hexamethylpentalenide with group IV metal halides. | ||
In a controlled hydrolysis experiment, Clement et al. showed that reacting [TiCl2Pn*]2 with one equivalent of 2,6-xylenol (pKa ∼10) in toluene resulted in clean mono-protonation of the bound Pn* ligand and cleavage of the dimer by coordination of the alkoxide (Fig. 40), giving a single isomer of the mono-nuclear hydropentalenide complex Ti(OR)Cl2[HPn*] in 68% yield.69 This transformation represents an interesting example of accessing a HPn− complex via protonation of a Pn2− complex, but on its own does not allow commenting on the reversibility of the reaction.
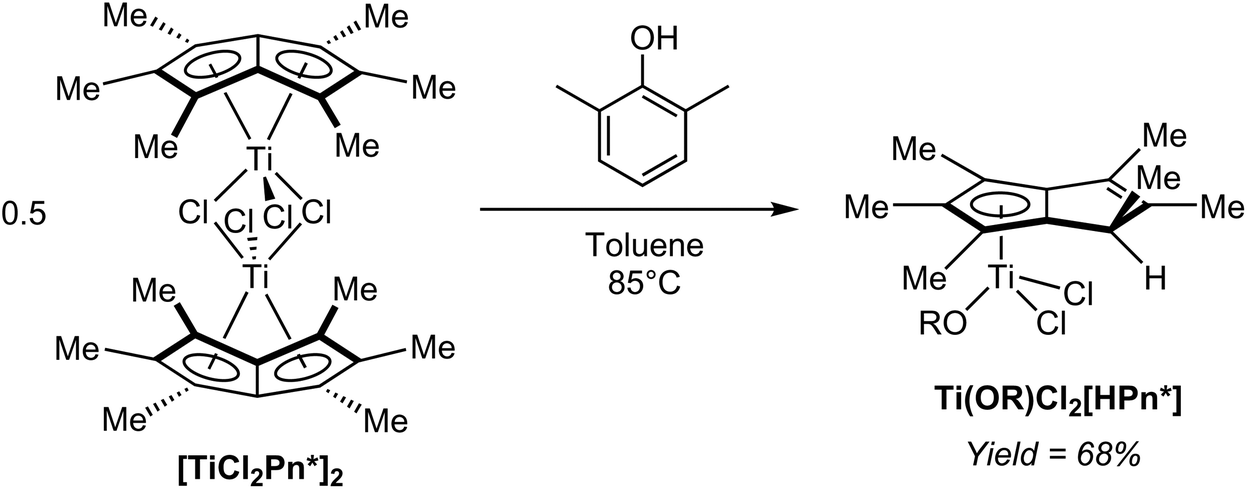 | ||
| Fig. 40 Partial hydrolysis of a titanium(IV) Pn* complex with 2,6-xylenol. | ||
In a further demonstration of the usefulness of (Me3Sn)2Pn* as mild transmetalation agent, Chadwick et al. successfully synthesised dinuclear Pn*2− complexes of easily reducible rhodium(I) and iridium(I) precursors.70 Reactions of [M(CO)2Cl]2 (M = Rh, Ir) with syn η1(Me3Sn)2Pn* (generated in situ) selectively produced the corresponding syn η5[(CO)2M]2Pn* complexes (Fig. 41). Both the rhodium and iridium complexes have been analysed by XRD, with suitable crystal samples grown from hexane at low temperatures.
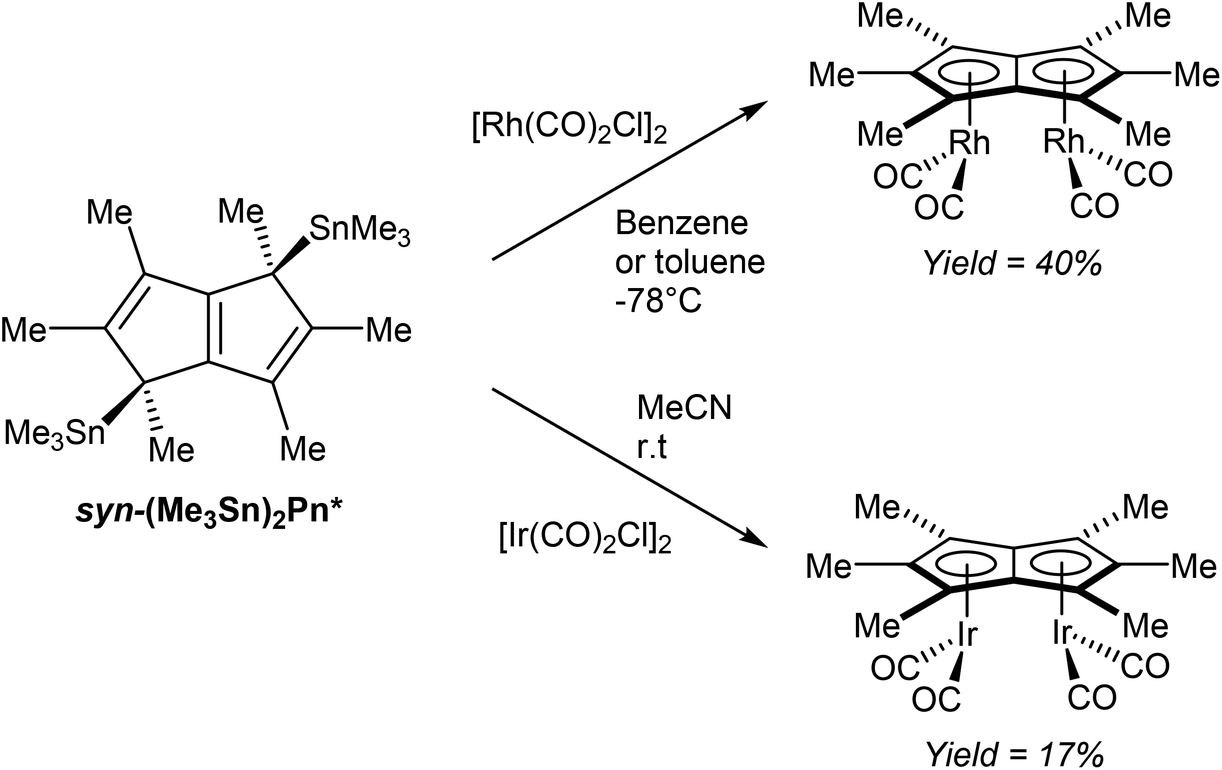 | ||
| Fig. 41 Reactions of bis(trimethylstannyl)hexamethylpentalenide with group IX metal carbonyl dimers. | ||
Whether the observed syn selectivity of the transmetalation was a result of using syn(Me3Sn)2Pn* or due to the dimeric nature of the metal precursor is not clear, however.
4. Conclusion
From the selected examples reviewed it is clear that a number of different synthetic routes to pentalenide complexes exist. Unsubstituted H2Pn is mostly synthesised by controlled anaerobic pyrolysis of various starting materials, whereas several relatively straightforward solution-phase methods for substituted H2Pn exist. It is interesting that not all of these, mostly developed in the realm of organic synthesis, have been put to use in organometallic pentalenide chemistry yet. Symmetrically substituted H2Pn appear most desirable to reduce the number of possible isomers, which may be hard to separate or transform selectively from a mixture. Given that their reactivity is mostly governed by their core π electronic system, it is not unreasonable to expect that most new substituted H2Pn would be amenable to the same deprotonation, functionalisation and transmetalation methods developed for the two archetypical synthons, unsubstituted H2Pn and permethylated Pn*. This would allow a wider range of novel Pn2− complexes to be synthesised for a more systematic study of Pn2− ligand effects on their electronic structure and reactivity. Thallium salts and Me3Sn derivatives have proven to be mild and effective transmetalation agents in addition to the alkali metal salts, but the development of less toxic alternatives would be desirable for practical reasons. The prospect of controlling the face selectivity of Pn2− double metalation via either kinetically controlled deprotonation or installation of syn-configured leaving groups represents an exciting opportunity for selectively synthesising syn-dinuclear half-sandwich complexes with interesting properties for binding and activating various ligands across the two metal sites. The stability of mono-deprotonated HPn− and its metal complexes may even allow consecutive installation of different metals to synthesise heterodinuclear compounds in a controlled manner. In general, the flexible and adaptive nature of Pn2− to bind a range of different metals together with the ability of bringing two metals together in close proximity with electronic coupling hold great potential for future applications in sensing, electrochemistry, and homogeneous catalysis.Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
We thank the Royal Society (awards UF160458 and EA180091 to UH) and the University of Bath (studentships to SMB and NAJ) for funding, Prof. F. G. N. Cloke and Dr A. F. R. Kilpatrick for advice, support and assistance in pentalenide chemistry, and Dr E. Suturina for help with access to Russian chemical literature.References
- H. J. Lindner, in Houben-Weyl Methods of Organic Chemistry, Vol. 5: Carbocyclic p-Electron Systems, Georg Thieme Verlag, Stuttgart, 4th edn, 1985, pp. 103–122 Search PubMed.
- T. Bally, S. Chai, M. Neuenschwander and Z. Zhu, J. Am. Chem. Soc., 1997, 119, 1869–1875 CrossRef CAS.
- H. Hopf, Angew. Chem., Int. Ed., 2013, 52, 12224–12226 CrossRef CAS PubMed.
- S. A. R. Knox and F. G. A. Stone, Acc. Chem. Res., 1974, 7, 321–328 CrossRef CAS.
- (a) O. T. Summerscales and F. G. N. Cloke, Coord. Chem. Rev., 2006, 250, 1122–1140 CrossRef CAS; (b) F. G. N. Cloke, J. C. Green, A. F. R. Kilpatrick and D. O'Hare, Coord. Chem. Rev., 2017, 344, 238–262 CrossRef CAS.
- R. H. Crabtree, The Organometallic Chemistry of the Transition Metals, John Wiley & Sons, 6th edn, 2014 Search PubMed.
- J. F. Hartwig, Organotransition Metal Chemistry: From Bonding To Catalysis, University Science Books, Sausalito, California, 2010 Search PubMed.
- A. Pauli, H. Kolshorn and H. Meier, Chem. Ber., 1987, 120, 1611–1616 CrossRef CAS.
- T. J. Katz and J. J. Mrowca, J. Am. Chem. Soc., 1967, 89, 1105–1111 CrossRef CAS.
- S. C. Jones, P. Roussel, T. Hascall and D. O'Hare, Organometallics, 2006, 25, 221–229 CrossRef CAS.
- A. V. Bandura and S. N. Lvov, J. Phys. Chem. Ref. Data, 2006, 35, 15–30 CrossRef CAS.
- T. J. Katz and M. Rosenberg, J. Am. Chem. Soc., 1962, 84, 865–866 CrossRef CAS.
- J. J. Stezowski, H. Hoier, D. Wilhelm, T. Clark and P. V. Schleyer, J. Chem. Soc., Chem. Commun., 1985, 1263–1264 RSC.
- J. Barroso, S. Mondal, J. L. Cabellos, E. Osorio, S. Pan and G. Merino, Organometallics, 2017, 36, 310–317 CrossRef CAS.
- T. J. Katz, R. K. Ohara and M. Rosenberg, J. Am. Chem. Soc., 1964, 86, 249–252 CrossRef CAS.
- K. Alder, F. H. Flock and P. Janssen, Chem. Ber., 1956, 89, 2689–2697 CrossRef CAS.
- N. B. Kazennova, A. K. Shestakova, V. A. Chertkov, N. A. Ustynyuk and Y. A. Ustynyuk, Zh. Org. Khim., 1986, 22, 2108–2111 CAS.
- M. Jones and L. O. Schwab, J. Am. Chem. Soc., 1968, 90, 6549–6550 CrossRef CAS.
- F. G. N. Cloke, M. C. Kuchta, R. M. Harker, P. B. Hitchcock and J. S. Parry, Organometallics, 2000, 19, 5795–5798 CrossRef CAS.
- A. G. Griesbeck, Synthesis, 1990, 144–147 CrossRef CAS.
- J. Stapersma, I. D. C. Rood and G. W. Klumpp, Tetrahedron, 1982, 38, 2201–2211 CrossRef CAS.
- J. Stapersma, I. D. C. Rood and G. W. Klumpp, Tetrahedron, 1982, 38, 191–199 CrossRef CAS.
- J. J. Gajewski and C. J. Cavender, Tetrahedron Lett., 1971, 1057 CrossRef CAS.
- M. Neuenschwander, H. Schaltegger and D. Meuche, Helv. Chim. Acta, 1963, 46, 1760–1765 CrossRef CAS.
- R. Kyburz, H. Schaltegger and M. Neuenschwander, Helv. Chim. Acta, 1971, 54, 1037–1046 CrossRef CAS.
- I. Erden, J. Sabol, A. Gubeladze and A. Ruiz, Turk. J. Chem., 2013, 37, 519–524 CAS.
- R. Kaiser and K. Hafner, Angew. Chem., Int. Ed. Engl., 1970, 9, 892 CrossRef CAS.
- K. Hafner, K. H. Häfner, C. König, M. Kreuder, G. Ploss, G. Schulz, E. Sturm and K. H. Vöpel, Angew. Chem., Int. Ed. Engl., 1963, 2, 123–134 CrossRef.
- U. H. Brinker and I. Fleischhauer, Tetrahedron, 1981, 37, 4495–4502 CrossRef CAS.
- A. E. Ashley, A. R. Cowley and D. O'Hare, Eur. J. Org. Chem., 2007, 2239–2242 CrossRef CAS.
- S. H. Bertz, J. M. Cook, A. Gawish and U. Weiss, Org. Synth., 1986, 64, 27–38 CrossRef CAS.
- A. E. Ashley, A. R. Cowley and D. O'Hare, Chem. Commun., 2007, 15, 1512–1514 RSC.
- K. Hafner and H. U. Süss, Angew. Chem., Int. Ed. Engl., 1973, 12, 575–577 CrossRef.
- B. Kitschke and H. J. Lindner, Tetrahedron Lett., 1977, 18, 2511–2514 CrossRef.
- A. G. Griesbeck, J. Org. Chem., 1989, 54, 4981–4982 CrossRef CAS.
- A. G. Griesbeck, Chem. Ber., 1991, 124, 403–405 CrossRef CAS.
- E. Le Goff, J. Am. Chem. Soc., 1962, 84, 3975–3976 CrossRef CAS.
- K. J. Stone and R. D. Little, J. Org. Chem., 1984, 49, 1849–1853 CrossRef CAS.
- N. Coskun, J. X. Ma, S. Azimi, C. Gartner and I. Erden, Org. Lett., 2011, 13, 5952–5955 CrossRef CAS PubMed.
- N. E. Schore, Chem. Rev., 1988, 88, 1081–1119 CrossRef CAS.
- T. Kuwabara, K. Ishimura, T. Sasamori, N. Tokitoh and M. Saito, Chem. – Eur. J., 2014, 20, 7571–7575 CrossRef CAS PubMed.
- M. Saito, M. Nakamura, T. Tajima and M. Yoshioka, Angew. Chem., Int. Ed., 2007, 46, 1504–1507 CrossRef CAS PubMed.
- H. Li, B. Wei, L. Xu, W.-X. Zhang and Z. Xi, Angew. Chem., Int. Ed., 2013, 52, 10822–10825 CrossRef CAS PubMed.
- P. M. Bailey, B. E. Mann, I. D. Brown and P. M. Maitlis, J. Chem. Soc., Chem. Commun., 1976, 238–239 RSC.
- W. Weidemüller and K. Hafner, Angew. Chem., Int. Ed. Engl., 1973, 12, 925–925 CrossRef.
- (a) D. F. Hunt and J. W. Russell, J. Am. Chem. Soc., 1972, 94, 7198–7199 CrossRef CAS; (b) D. F. Hunt and J. W. Russell, J. Organomet. Chem., 1972, 46, C22–C24 CrossRef CAS.
- A. E. Ashley, G. Balazs, A. R. Cowley, J. C. Green and D. O'Hare, Organometallics, 2007, 26, 5517–5521 CrossRef CAS.
- A. Brookes, F. Gordon, J. Howard, S. A. R. Knox and P. Woodward, J. Chem. Soc., Chem. Commun., 1973, 587–589 RSC.
- J. A. K. Howard, S. A. R. Knox, V. Riera, F. G. A. Stone and P. Woodward, J. Chem. Soc., Chem. Commun., 1974, 452–453 RSC.
- J. A. K. Howard, S. A. R. Knox, F. G. A. Stone, A. C. Szary and P. Woodward, J. Chem. Soc., Chem. Commun., 1974, 788–789 RSC.
- T. H. Coffield, K. G. Ihrman and W. Burns, J. Am. Chem. Soc., 1960, 82, 4209–4210 CrossRef CAS.
- H. Komatsu, Y. Suzuki and H. Yamazaki, Chem. Lett., 2001, 16, 998–999 CrossRef.
- T. J. Katz and M. Rosenberger, J. Am. Chem. Soc., 1963, 85, 2030–2031 CrossRef CAS.
- C. Janiak, Coord. Chem. Rev., 1997, 163, 107–216 CrossRef CAS.
- Y. A. Ustynyuk, A. K. Shestakova, V. A. Chertkov, N. N. Zemlyansky, I. V. Borisova, A. I. Gusev, E. B. Tchuklanova and E. A. Chernyshev, J. Organomet. Chem., 1987, 335, 43–57 CrossRef CAS.
- N. M. Sergeyev, Prog. Nucl. Magn. Reson. Spectrosc., 1973, 9, 71–144 CrossRef.
- Z. R. Turner, J.-C. Buffet and D. O'Hare, Organometallics, 2014, 33, 3891–3903 CrossRef CAS.
- K. Jonas, B. Gabor, R. Mynott, K. Angermund, O. Heinemann and C. Kruger, Angew. Chem., Int. Ed. Engl., 1997, 36, 1712–1714 CrossRef CAS.
- K. Jonas, P. Korb, G. Kollbach, B. Gabor, R. Mynott, K. Angermund, O. Heinemann and C. Kruger, Angew. Chem., Int. Ed. Engl., 1997, 36, 1714–1718 CrossRef CAS.
- S. C. Jones, T. Hascall, S. Barlow and D. O'Hare, J. Am. Chem. Soc., 2002, 124, 11610–11611 CrossRef CAS PubMed.
- (a) T. J. Katz and N. Acton, J. Am. Chem. Soc., 1972, 94, 3281–3283 CrossRef CAS; (b) T. J. Katz, N. Acton and J. Mcginnis, J. Am. Chem. Soc., 1972, 94, 6205–6206 CrossRef CAS.
- M. R. Churchill and K. K. G. Lin, Inorg. Chem., 1973, 12, 2274–2279 CrossRef CAS.
- S. C. Binding, J. C. Green, W. K. Myers and D. O'Hare, Inorg. Chem., 2015, 54, 11935–11940 CrossRef CAS PubMed.
- J. M. Manriquez, M. D. Ward, W. M. Reiff, J. C. Calabrese, N. L. Jones, P. J. Carroll, E. E. Bunel and J. S. Miller, J. Am. Chem. Soc., 1995, 117, 6182–6193 CrossRef CAS.
- (a) F. G. N. Cloke, J. C. Green and C. N. Jardine, Organometallics, 1999, 18, 1080–1086 CrossRef CAS; (b) F. G. N. Cloke and P. B. Hitchcock, J. Am. Chem. Soc., 1997, 119, 7899–7900 CrossRef.
- G. Balazs, F. G. N. Cloke, J. C. Green, R. M. Harker, A. Harrison, P. B. Hitchcock, C. N. Jardine and R. Walton, Organometallics, 2007, 26, 3111–3119 CrossRef CAS.
- A. Ashley, G. Balazs, A. Cowley, J. Green, C. H. Booth and D. O'Hare, Chem. Commun., 2007, 15, 1515–1517 RSC.
- R. T. Cooper, F. M. Chadwick, A. E. Ashley and D. O'Hare, Organometallics, 2013, 32, 2228–2233 CrossRef CAS.
- D. D. Clement, S. C. Binding, T. A. Q. Arnold, F. M. Chadwick, I. J. Casely, Z. R. Turner, J.-C. Buffet and D. O'Hare, Polyhedron, 2019, 157, 146–151 CrossRef CAS.
- F. M. Chadwick, A. E. Ashley, R. T. Cooper, L. A. Bennett, J. C. Green and D. M. O'Hare, Dalton Trans., 2015, 44, 20147–20153 RSC.
Footnote |
| † Electronically stabilized benzopentalenes have proven useful compounds for organic materials chemistry (see ref. 3 for a recent overview) but are beyond the scope of this review. |
| This journal is © The Royal Society of Chemistry 2019 |