
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydroxynitrile lyases covalently immobilized in continuous flow microreactors†
Michelle P.
van der Helm‡
ab,
Paula
Bracco‡
a,
Hanna
Busch
a,
Katarzyna
Szymańska
*b,
Andrzej B.
Jarzębski
bc and
Ulf
Hanefeld
 *a
*a
aBiokatalyse, Afdeling Biotechnologie, Technische Universiteit Delft, Van der Maasweg 9, 2629HZ Delft, The Netherlands. E-mail: U.Hanefeld@tudelft.nl; Fax: +31(0)15 2781415; Tel: +31(0)15 2789304
bDepartment of Chemical Engineering and Process Design, Silesian University of Technology, Ks. M. Strzody 7, 44-100 Gliwice, Poland. E-mail: Katarzyna.Szymanska@polsl.pl; Fax: +48322371461; Tel: +48322371266
cInstitute of Chemical Engineering, Polish Academy of Sciences, Baltycka 5, 44-100 Gliwice, Poland
First published on 11th February 2019
Abstract
Enzymes are supreme catalysts when it comes to high enantiopurities and their immobilization will pave the way for continuous operation. In this context, we show the covalent immobilization of hydroxynitrile lyases HbHNL (from Hevea brasiliensis) and MeHNL (from Manihot esculenta) in a siliceous monolithic microreactor for continuous operation. A thorough characterization of the immobilized HNLs on mesoporous silicates indicated the conditions essential for a successful immobilization. Their application in a continuous flow system enabled a remarkably fast (3.2 min) production of chiral cyanohydrins with high conversion (97%) and high ee (98%) using minimal enzyme loading (STY = 71 g L−1 h−1 mgprotein−1). MeHNL showed increased operational stability, possibly due to a structural difference. The continuous flow microreactor outperformed batch systems, demonstrating the advantage of the mesoporous/macroporous environment for the expression of enzyme activity and the favorable characteristics of the microreactor. Overall, the system shows great potential for future industrial application of biocatalytic asymmetric syntheses.
Introduction
For large-scale production it is essential to operate continuously, both from an economical and sustainable perspective, and the petrochemical and food industries cannot be imagined without continuous processing. Naturally, this also holds for industrial biotransformations. Successful biocatalytic examples on a million ton scale include the production of high fructose corn syrup catalyzed by glucose isomerase and the transesterification of oils with lipases.1 Here, the development of immobilized enzymes paved the way for continuous operation in packed-bed reactors or stirred tank reactors in series. Despite these advances, the production of chiral compounds for the pharmaceutical and fine chemical industry is still dominated by batch-wise operation.2 For the realization of high enantiopurities, enzymes have established themselves as key catalysts and provide the most atom efficient route towards chiral building blocks, as recognized by many Presidential Green Chemistry awards.3,4 A prime example of highly selective biocatalysts are hydroxynitrile lyases (HNLs). This group of versatile enzymes catalyzes the enantioselective addition of nucleophilic cyanide to prochiral carbonyl compounds, realizing the formation of industrially important chiral cyanohydrins.5–13 Arguing from the green chemistry principles perspective,14 flow chemistry yields significant waste reductions by decreasing reaction volumes (principle 1) and energy consumption (principle 6), in particular if minimal back pressure needs to be overcome.Given the great asset of continuous operation and the versatility of HNLs, the covalent immobilization of HNLs in a siliceous monolithic microreactor and its exploration in a continuous flow system to achieve maximum control in minimum timeframes is herein presented. Recently, the application of immobilized enzymes in continuous flow (micro)reactors is gaining momentum.15–22 Flow systems are advantageous compared to batch operation as no catalyst separation step is required, the catalyst can easily be reused, higher productivities can be achieved and catalyst instability problems due to high shear forces are precluded.23–25 Additionally, the microreactor technology provides a handle to tune reaction conditions, facile scale-out by parallelizing multiple units and process intensification or even automation. On top of that, process safety is improved by reduced reactor volumes and possibilities for in situ generation of hazardous substances.15,18,26–31 Compared with the traditional packed-bed-reactor, siliceous monolithic microreactors are superior. They exhibit reduced back-pressure drops and the 3D hierarchical/torturous porous structure of the microchannels and mesopores creates a high surface-to-volume ratio. This results in very intensive homogenization of reactants even in the laminar flow rate range, which reduced diffusional times and enhanced mass transfer, while simultaneously providing ample surface for enzyme immobilization.32,33 To visualize this: several aspects change when switching from a dissolved enzyme or an enzyme attached to a small particle to an enzyme immobilized on a monolith reactor surface. Both, the enzyme in solution or the enzyme on a particle will move together with the reaction mixture and thus move with a similar motion as the reaction mixture. An enzyme attached to a surface will not move, the difference in motion between the enzyme and the reaction mixture will thus be large. With laminar flow mixing will however still not be optimal (Fig. 1a). In the twisting microchannels rapid mixing and a large difference in motion between the stationary enzyme and the reaction mixture enables the above described rate enhancements (Fig. 1b).
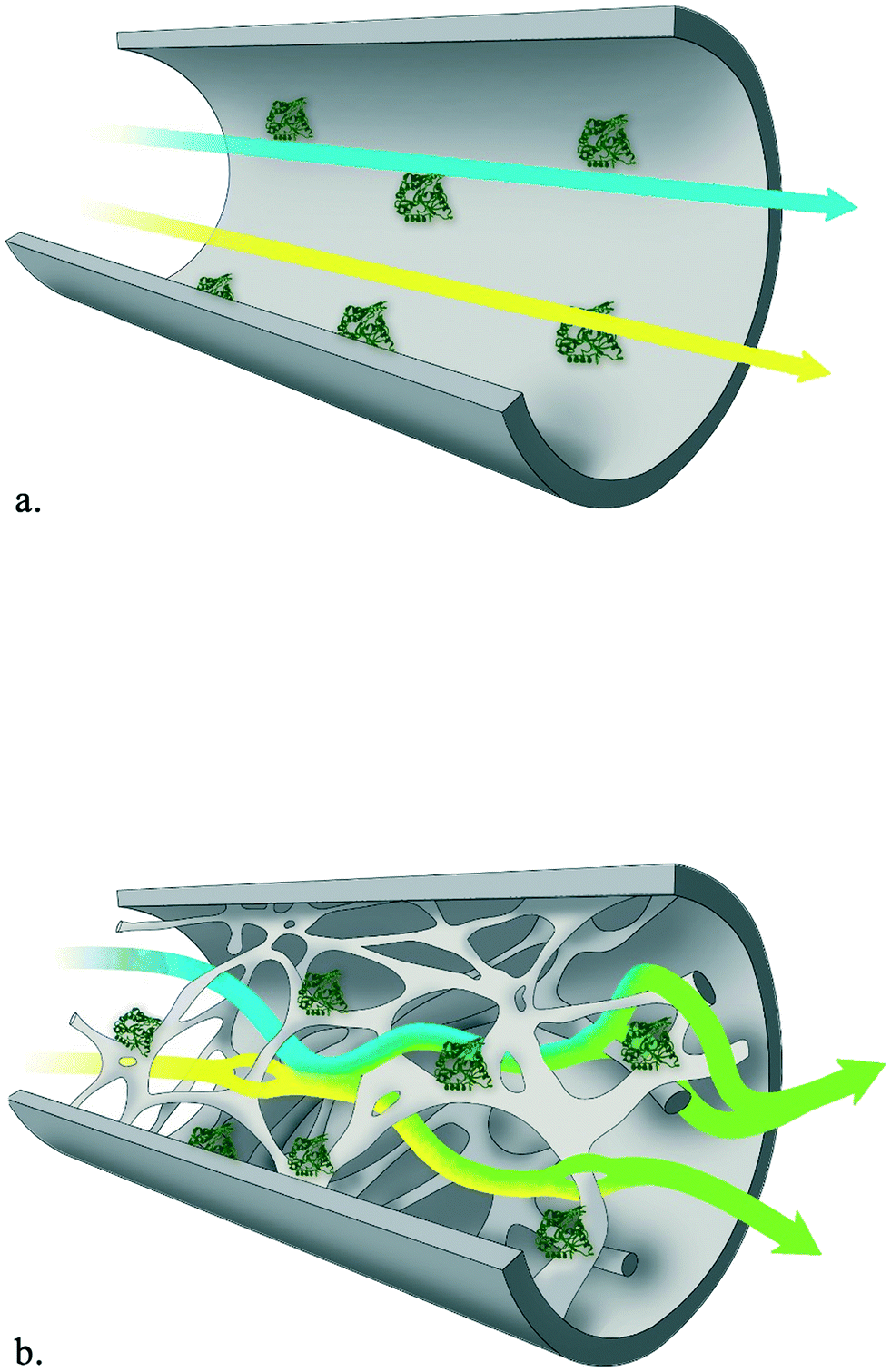 | ||
| Fig. 1 Portrayal of mixing and homogenization intensity in laminar flow (a) and in (micro)structured monolithic reactor (b). | ||
Given this background, the covalent immobilization of two homologous (S)-selective HNLs, HbHNL (from Hevea brasiliensis) and MeHNL (from Manihot esculenta), on modified silica supports and finally the design of an enzymatic monolithic microreactor is reported. As both enzymes are very similar, possible differences in the immobilization will indicate whether immobilization conditions are general or separate conditions are necessary for each enzyme. First, a thorough characterization of the immobilized HNLs on amino and epoxy grafted siliceous mesostructured cellular foam powders (MCF-APT and MCF-GPT respectively) is presented. This provides the foundation for the subsequent enzyme immobilization in the siliceous monolithic microreactor. The immobilized HNLs are explored in batch and continuous reaction systems. The first ever reported chiral synthesis, the enantioselective formation of chiral mandelonitrile, is employed as test case (Scheme 1).34
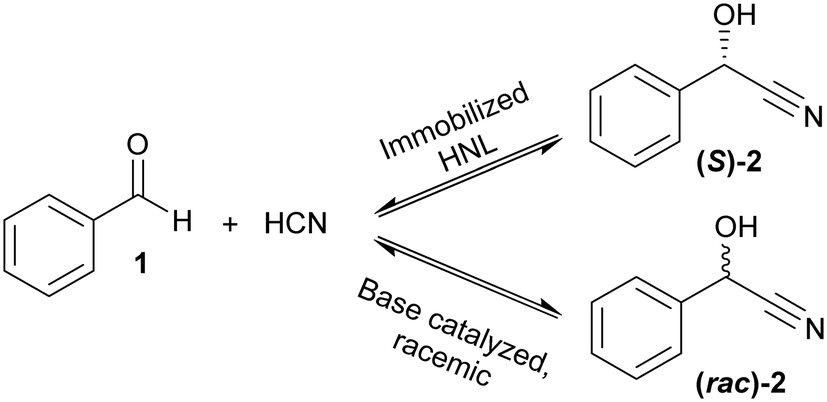 | ||
| Scheme 1 Enantioselective synthesis of (S)-mandelonitrile ((S)-2) from benzaldehyde (1) and hydrogen cyanide catalyzed by immobilized HNL (top) and base catalyzed racemic mandelonitrile ((rac)-2) synthesis (chemical background reaction) (bottom). | ||
Results and discussion
Enzyme immobilization and characterization
The (S)-selective HbHNL and MeHNL were recombinantly expressed and purified from bacterial hosts. Utilizing the PDB structures of HbHNL (3C6X)35 and MeHNL (1DWP)36 the relevant enzyme parameters for the immobilization were determined with PyMOL Molecular Graphics System.37,38 For both biocatalysts, the homodimer structures measure less than 10 nm in their longest extension (Fig. 2a and b). With the aim of a covalent immobilization, the number of surface lysine residues was determined as 36 lysines per homodimer (in magenta in Fig. 2c and d).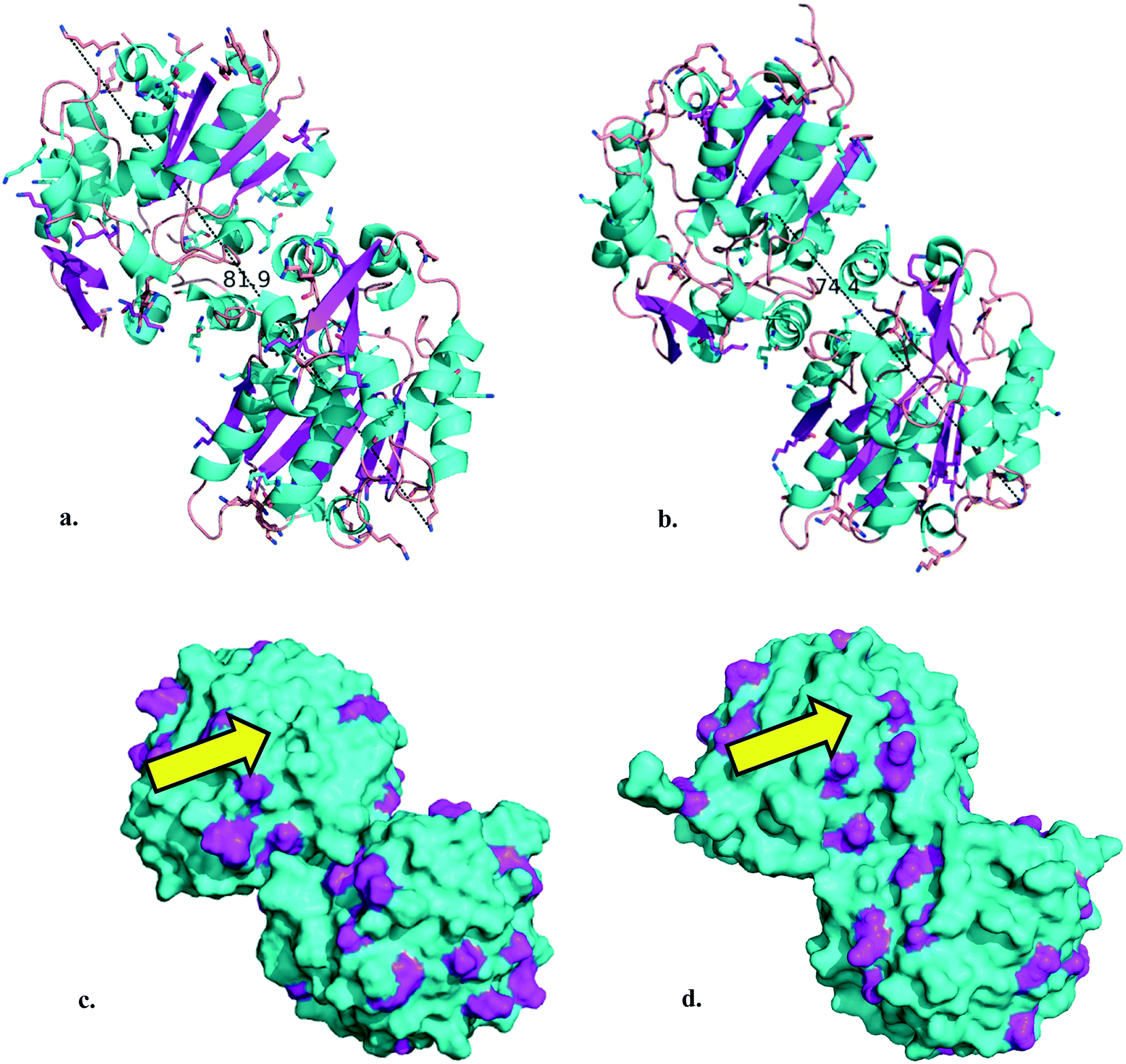 | ||
| Fig. 2 a. HbHNL homodimer cartoon (longest extension: 81.9 Å). b. MeHNL homodimer cartoon (longest extension: 74.4 Å). Lysine residues are shown as sticks, α-helices are highlighted in cyan, β-sheets in magenta, nitrogen atoms in blue and hydrogen atoms are hidden in the image. c. HbHNL homodimer surface representation. d. MeHNL homodimer surface representation. Surface lysine residues are highlighted in magenta. The yellow arrow indicates the active site entrance with catalytic triad residues (Ser 80, His 235/236 and Asp 207/208).39 Structural data 3C6X35 and 1DWP36 were obtained from the PDB38 and the images were created using the PyMOL Molecular Graphics System.37N.B. the HbHNL homodimer images were constructed by duplication of the asymmetric unit structural data with the ‘symexp’ command. | ||
Both enzymes are discussed as homodimers in neutral aqueous solution here to ease comparison. For HbHNL rigorous proof for this was provided40 while for MeHNL this is supported only by crystallography.12 In solution a tetrameric structure is more likely but not proven.41 The consequences of this tetrameric structure will also be discussed below. The amino acid arrangements show that the HNLs possess a more hydrophilic surface, as can be expected for enzymes that display their natural activity in aqueous surroundings (Fig. S1a and b†). A Poisson–Boltzmann electrostatic surface potential calculation indicates that they are overall negatively charged in neutral aqueous solution (Fig. S1c and d†). It is precisely for those reasons that a hydrophobic carrier should not be deployed for the HNL immobilization, as it can induce adverse folding effects and lead to unfavorable non-specific interactions with enzymes and/or products.42 Hence, the HNLs were covalently immobilized on hydrophilic mesoporous silicates, i.e.: mesostructured cellular foam functionalized with 3-aminopropyltrimethoxysilane (MCF-APT) and 3-glycidyloxypropyltrimethoxysilane (MCF-GPT). With a collection of transmission electron microscopy (TEM) images the pore size diameters (dp) of the MCF carriers were estimated to be 26 nm (MCF-APT) and 27 nm (MCF-GPT) (Fig. S2†) and thus even in the most unfavorable orientation both HNLs will fit in the mesopores. Moreover, this translates to an internal single pore volume of 9200 nm3 (MCF-APT) and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 nm3 (MCF-GPT), which is more than 100 times the volume of the enzyme homodimer. Ideally, the covalent immobilization can be accomplished by reacting the surface lysines of the enzyme with either reactive epoxides or aldehydes (obtained after glutaraldehyde activation of amino functionalized carriers43) of the MCF carrier, creating either an amine or diimine (Scheme S1†). The number of accessible surface lysines (Fig. 2c and d) and the amount of grafted organic content on the MCF carriers, determined as 9 wt% for MCF-APT and 7 wt% for MCF-GPT by thermogravimetric analysis (TGA) (Fig. S3†), were used for the calculation of the minimum enzyme:support immobilization ratio. For the covalent immobilization experiments at least a 1:1 enzyme versus MCF-APT w/w ratio and a 1:2 w/w ratio enzyme versus MCF-GPT has to be deployed to ensure that all enzyme units will be covalently immobilized, which overall gives a 2–3 times molar excess of functional carrier groups (Table 1). With this excess of carrier functionalities a covalent attachment can be guaranteed (Table 1).44
300 nm3 (MCF-GPT), which is more than 100 times the volume of the enzyme homodimer. Ideally, the covalent immobilization can be accomplished by reacting the surface lysines of the enzyme with either reactive epoxides or aldehydes (obtained after glutaraldehyde activation of amino functionalized carriers43) of the MCF carrier, creating either an amine or diimine (Scheme S1†). The number of accessible surface lysines (Fig. 2c and d) and the amount of grafted organic content on the MCF carriers, determined as 9 wt% for MCF-APT and 7 wt% for MCF-GPT by thermogravimetric analysis (TGA) (Fig. S3†), were used for the calculation of the minimum enzyme:support immobilization ratio. For the covalent immobilization experiments at least a 1:1 enzyme versus MCF-APT w/w ratio and a 1:2 w/w ratio enzyme versus MCF-GPT has to be deployed to ensure that all enzyme units will be covalently immobilized, which overall gives a 2–3 times molar excess of functional carrier groups (Table 1). With this excess of carrier functionalities a covalent attachment can be guaranteed (Table 1).44
| Entry | Structurea | Molecular weight | mmol amino groups per g |
|---|---|---|---|
| a X-ray structures 3C6X35 and 1DWP36 from the PDB.38 b Molecular weight of homodimer according to ref. 45. c Considering 36 surface lysine residues for HbHNL and MeHNL homodimers. d Epoxy functionalities (glycidyl ether). | |||
| HbHNL |

|
58.4 (kDa)b | 0.62c |
| MeHNL |

|
60.0 (kDa)b | 0.60c |
| MCF-APT |
|
58 (g mol−1) | 1.6 |
| MCF-GPT |
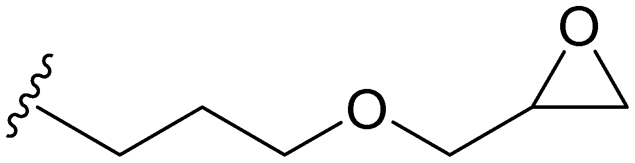
|
115 (g mol−1) | 0.64d |
However, in order to minimize the biocatalyst load higher enzyme:support w/w ratios were tested (Table 2), 1:10, 1:20 and 1:40 respectively. The data in Table 2 disclose that HNLs immobilized on the activated MCF-APT carriers gave the highest protein loadings and were most active in the cleavage of (rac)-mandelonitrile ((rac)-2) (cyanolysis assay).
| Enzyme:support w/w ratio |
Immobilization yield (%) | Protein loada (mg per g carrier) | Measured activitya,b,c (U per mg carrier) | |
|---|---|---|---|---|
| a Weight of carrier is final total preparation, including protein weight. b Corrected for the blank reaction with added (activated) MCF carrier. c U (units) stands for μmol benzaldehyde per min. Reported error values are standard deviations based on triplicate (n = 3) measurements of the Bradford and cyanolysis assays (see Experimental section). | ||||
| HbHNL–MCF-APT | 1:10 |
92 | 86.5 ± 1.8 | 6.8 ± 0.3 |
| HbHNL–MCF-APT | 1:20 |
88 | 42.3 ± 1.2 | 3.1 ± 0.2 |
| HbHNL–MCF-APT | 1:40 |
94 | 23.0 ± 0.6 | 1.6 ± 0.1 |
| MeHNL–MCF-APT | 1:10 |
88 | 81.4 ± 4.3 | 1.8 ± 0.3 |
| HbHNL–MCF-GPT | 1:20 |
36 | 18.4 ± 1.3 | 0.2 ± 0.1 |
| MeHNL–MCF-GPT | 1:20 |
52 | 25.6 ± 2.3 | 1.1 ± 0.1 |
In the case of MCF-GPT carrier most protein was washed off during the buffer washing steps. These results were in accordance with earlier research, which proved the glutaraldehyde crosslinked systems as superior.46,47 This can be ascribed to the creation of a favorable microenvironment for the expression of enzyme activity, which is cooperatively generated by the pores of the MCF material with the glutaraldehyde tether.46,48 The glutaraldehyde linkage forms a dimeric or trimeric structure and creates a longer spacer between the carrier and the enzyme surface. The microenvironment with favorable substrate and product concentrations and increased enzyme activity was not observed in systems without linkage. A reasonable explanation is that a close contact between enzyme and carrier surface could negatively influence the enzyme's superstructure and consequently its activity.42,46,49 Furthermore, the successful immobilization of HbHNL and MeHNL on the amino and epoxy functionalized carriers was proven by Fourier transform infrared (FTIR) spectroscopy (Fig. S4†), TGA (Fig. S5†), TEM (Fig. S6†) and scanning electron microscopy (SEM) (Fig. S7a and b†). In the FTIR spectra of the free and immobilized enzyme samples the amide I stretching (∼1650 cm−1) and the amide II bending vibration mode (∼1540 cm−1) of protein secondary amides (black dashed box on the right in Fig. S4†) were identified and TGA confirmed a twofold increase in organic content for HbHNL-MCF-APT (20 wt%), MeHNL-MCF-APT (19 wt%) and MCF-APT-GA (glutaraldehyde activated carrier) (19 wt%) in Fig. S5.† The latter finding is in line with the fact that unreacted glutaraldehyde crosslinks were washed off during the buffer washing after immobilization. This idea is further supported by the high immobilization yields for MCF-APT based on the analysis of the buffer washing steps (Table 2). For HbHNL-MCF-GPT and MeHNL-MCF-GPT on the other hand the organic material content did not increase significantly, which is in conjunction with the results in Table 2, indicating that most protein was washed off. Moreover, TEM images after immobilization showed denser silica network arrays than before (Fig. S6†). Yet, with SEM, no morphological differences were observed for the immobilized enzymes compared to the functionalized carriers (Fig. S7a and b†), which retained the characteristic MCF coral-type morphology.50 Having demonstrated the effective HNL immobilization on the powdered MCF carriers with MCF-APT having more favorable properties, the amino functionalized (and glutaraldehyde activated) siliceous monolithic microreactor (6 × 40 mm, 0.96 mL, 260 mg silica) was tested in continuous reactions. Structural details of the silica monolith are included in Fig. S7c and d.† As with the powdered MCF-APT, the monolith immobilization led to high protein loadings with 89% (11.3 mg total protein; 1120 U per monolith) and 72% (17.4 mg total protein; 1310 U per monolith) immobilization yields for HbHNL and MeHNL respectively, indicating the overall versatility of the covalent immobilization method.
Batch and continuous synthesis of (S)-mandelonitrile ((S)-2)
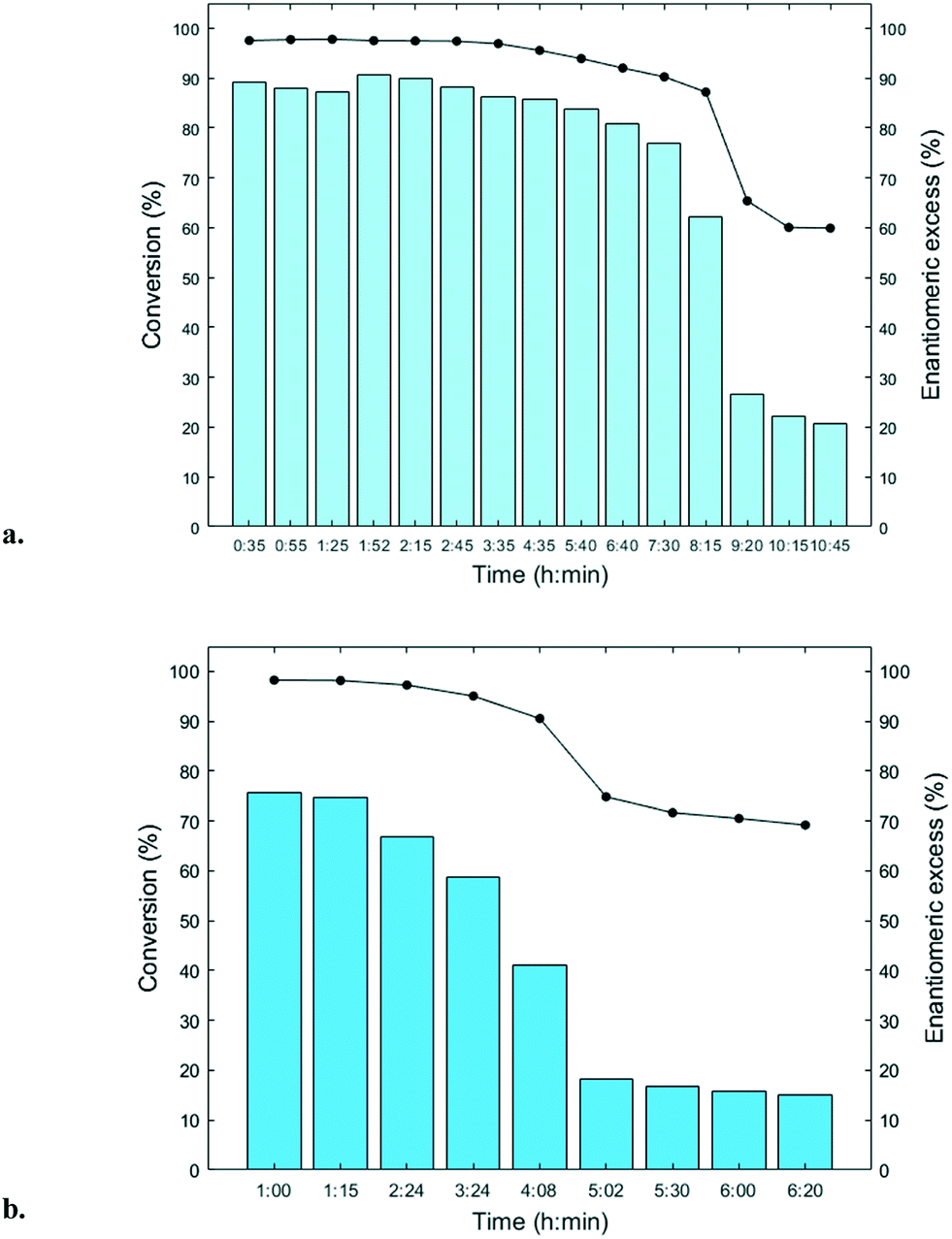
The immobilized HNLs were deployed as catalysts in the enantioselective synthesis of (S)-mandelonitrile ((S)-2) (Scheme 1) in batch and continuous flow systems in monophasic buffer saturated (citrate/phosphate pH 5.0, 50 mM) MTBE. The advantage offered by the organic phase is typically the realization of high substrate concentrations and increased enzyme stability.5,51 Additionally, the organic medium also enables the efficient suppression of the undesired non-selective base catalyzed chemical background reaction (Scheme 1).5 The batch system resulted in a final ee of 87% (S)-2 at a reasonable conversion (38%) with HbHNL-MCF-APT (2.5 mg total protein; 150 U) after 3 h (Fig. 3a and S8†). Moreover, the pH of the reaction played a key role and the silica support catalyzed the racemic background reaction (Fig. S9 and S10† for blank reaction profiles with and without carrier), hence limiting the final ee of the product. This observation has also been reported for other silica carriers, such as Celite.52–54 Due to this non-selective background reaction, the enantiomeric purity of the product can only reach high values at the beginning of the reaction. However, almost full conversion (94%) of benzaldehyde (1) was only observed after ∼30 h, leading to a poor final product ee of 42% (S). Fascinatingly, switching to a continuous microreactor flow system with HbHNL (11.3 mg total protein; 1120 U) immobilized on the amino functionalized (and glutaraldehyde activated) monolithic microreactor (Scheme 2) led to major improvement of the catalytic performance. Full conversion and excellent ee (99% (S)-2) were achieved within minutes with minimal enzyme loading (Fig. 3b and S11†). HbHNL showed the highest conversion (98%) with excellent ee (99% (S)-2) at 0.075 mL min−1 flow rate (residence time of 12.8 min). To directly compare the rate of the enzyme under the different conditions rather than overall conversions the specific rates were calculated. These are normalized for the amount of enzyme immobilized and can thus be compared directly. In terms of specific reaction rate (r) – mmol of product formed per minute per g enzyme (explanation of calculation given in ESI† section N)20,55 – at a similar conversion degree (∼43%), the HbHNL continuous flow system is 8 times faster than the batch analogue with rflow of 5.6 mmol min−1 gprotein−1vs. rbatch of 0.70 mmol min−1 gprotein−1. The continuous flow microreactor system can effectively suppress the racemic background reaction at pH 5.0, especially when reaching higher flow rates (shorter residence times) (Fig. S13† for blank reaction). The background reaction (monolith activated with glutaraldehyde – without enzyme) showed a maximum of 15% conversion when a low flow was applied (0.045 mL min−1), whereas 8–10% conversion was reached with flows between 0.1 and 0.2 mL min−1. This finding is in agreement with the batch reaction results for which longer reaction times also resulted in a competing racemic background reaction. The enzyme-catalyzed enantioselective reaction is significantly faster when boosted by operating at higher flow rates, due to greatly enhanced mixing of liquid reagents as well as the intensified mass transport to the liquid-catalyst/enzyme interface (Fig. 1b). These outcomes are in line with the results reported for acyltransferase from Mycobacterium smegmatis (MsAcT) immobilized on silica monoliths.56 The excellent performance of the biocatalyst and control of product formation might be ascribed to the favorable expression of enzyme activity in a distinct, mesoporous/macroporous environment with increased surface-to-volume ratio. However, this should not differ between a batch and continuous reaction given similar surface to volume ratios. For the monolithic reactor the latter value is about 60 m2 mL−1 (6 × 107 m2 m−3), i.e. three orders of magnitude larger than for the batch slurry system. Another distinct difference between the two approaches is the intrinsic property of the microreactor with its tortuous pore structure (SEM images in Fig. S7c and d†). This induces very intensive lateral mixing and enhanced mass transport. Consequently, diffusional limitation of the reaction rate is greatly relaxed and a very strong effect on the overall catalytic activity and productivity is observed (Fig. 1b).56 In effect, protein loading of the monolith may potently be much larger, and this would translate into enhanced activity and thus reactor productivity. The application of hydrophilic mesoporous/macroporous monoliths with meandering/twisting flow-through channels facilitates a chaotic movement of reactants and results in intensive mixing and enhanced mass transport to the enzyme and decreased diffusional times.26,57 Typically, characteristic reaction times (inverse of kcat) of 0.01 to 1 s are found for enzyme catalyzed reactions, and they are perfectly in the range of (micro)mixing times observed in stirred tank (batch) reactors.58 Thus, under homogenous conditions (one-phase systems, but also a slurry system with fine particles) the apparent (observed) reaction rate is seriously limited, in fact controlled, by the intensity of mass transfer. Overall the substrates as well as the (immobilized) enzymes all move into the same general direction. However, in our system transport has to take place from the fluid (MTBE with HCN and benzaldehyde) to the solid stagnant phase/microreactor with immobilized biocatalyst. Here, intensification of mass transfer and mixing appeared to have a very significant impact on the apparent reaction rate and hence overall productivity. Similar effects have been shown in previous studies with heterogeneously catalyzed reactions and liquid–liquid biphasic solvent system.15,16,59 In particular, when comparing the batch and flow systems it is evident that the batch system is hampered by the competing racemic background reaction and mass transfer to the active sites of the enzyme whereas in the continuous flow system these problems can be effectively overcome by the chaotic movement induced by the tortuous microreactor structure.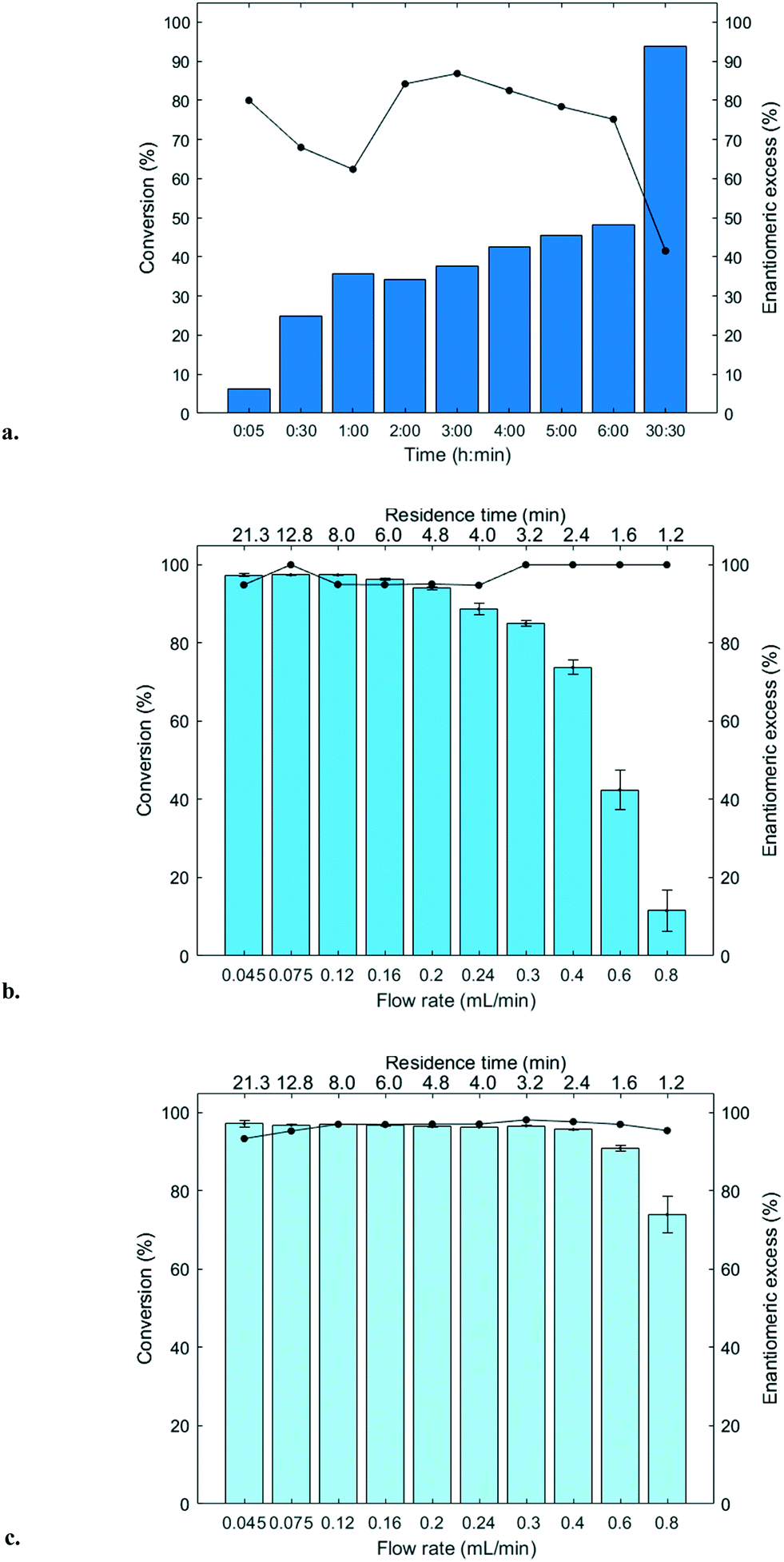 | ||
| Fig. 3 (S)-Mandelonitrile ((S)-2) synthesis reaction: a. HbHNL-MCF-APT (50 mg; 2.5 mg total protein; 150 U), 2 mL HCN in MTBE (1.5–2 M) saturated with citrate phosphate buffer (pH 5.0, 50 mM), benzaldehyde (1) 100 μL (1 mmol), ISTD 1 (1,3,5-triisopropylbenzene) 27.5 μL (0.1 mmol), shaken at 1000 rpm at room temperature (18–22 °C) under nitrogen atmosphere, conversion (bars)/enantiomeric excess (black line) vs. time b. HbHNL-silica monolith (11.3 mg total protein; 1120 U per monolith) conversion (bars)/enantiomeric excess (black line) (%) vs. flow rate (mL min−1)/residence time (min). c. MeHNL-silica monolith (17.4 mg total protein; 1310 U per monolith) conversion (bars)/enantiomeric excess (black line) vs. flow rate (mL min−1)/residence time (min). Flow rate is total of two pumps. Pump 1 pumps benzaldehyde (1) in MTBE (0.50 M, containing 0.066 mM ISTD 1) and pump 2 pumps HCN in MTBE (1.5–2.0 M, containing 0.066 mM ISTD 2 saturated with citrate phosphate buffer (pH 5.0, 50 mM)). Error bars indicate the standard deviation based on triplicate (n = 3) HPLC samples (see Experimental section). | ||
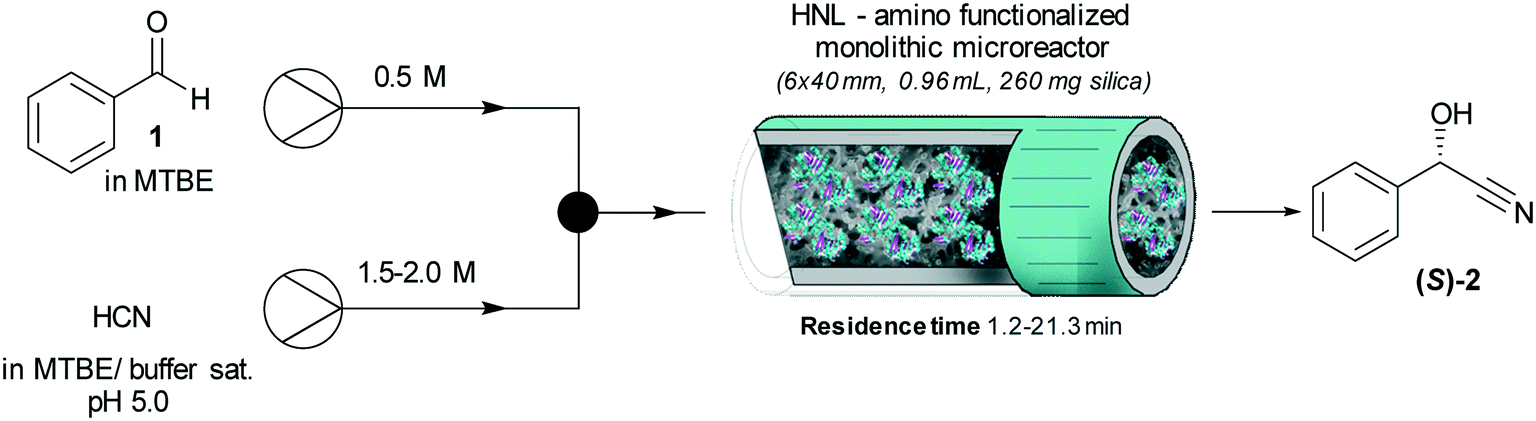 | ||
| Scheme 2 Synthesis of (S)-mandelonitrile ((S)-2) in buffer saturated MTBE (citrate/phosphate (pH 5.0, 50 mM)) continuous flow reaction system with HNL immobilized on amino functionalized (and glutaraldehyde activated) siliceous monolithic microreactor. | ||
Next, the MeHNL amino functionalized (and glutaraldehyde activated) monolithic microreactor (17.4 mg total protein; 1310 U) was tested in continuous flow reactions and even showed 97% conversion and 98% ee (S)-2 with a flow of 0.3 mL min−1 and residence time of only 3.2 min (Fig. 3c and S12†). The increased stability of MeHNL at higher flow rates compared to HbHNL is most likely attributed to the immobilization of MeHNL tetrameric structures inside the pores (Fig. 4).60,61 The MeHNL tetramer with a longest extension of 9.8 nm still fits in the mesopores and harbors more surface lysines for immobilization. This way, the tetramer can create more anchor points with the carrier surface and forms a rigid structure, which is more resistant against high flow. Subsequently, the stability of the MeHNL-silica monolith was tested for prolonged reaction times at two different flow rates (Fig. 5). At a flow rate of 0.1 mL min−1MeHNL was stable for about 8 h on stream, while at 0.2 mL min−1 flow this was reduced to 4 h. Clearly, higher flow rates have a negative impact on the enzyme stability, which leads to a trade-off between minimization of the racemic background reaction (at higher flows) and increased enzyme stability (at lower flows). Placing this into context, the native MeHNL is reported to be stable at 4 °C for up to one year, while HbHNL has a storage stability of a few months.12 Likewise, the immobilized HNLs on the powdered MCF carriers in this work did not lose their activity even after five weeks of storage in potassium phosphate buffer (pH 7.0, 100 mM) at 4 °C. On that account, the most plausible cause for the limited enzyme stability in the flow system (Fig. 5) and subsequent enzyme deactivation is the exposure to high shear forces, rather than the intrinsic stability of the (immobilized) enzymes.
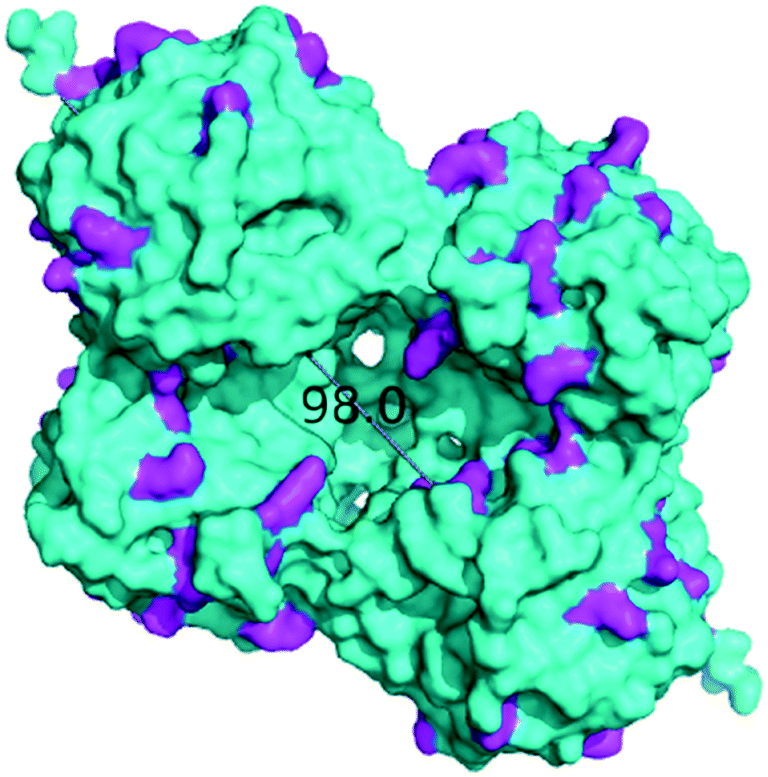 | ||
| Fig. 4 MeHNL tetramer surface representation (longest extension: 98.0 Å).60 Surface lysine residues are highlighted in magenta. X-ray structure 1DWP36 was obtained from the PDB38 and the image was created using the PyMOL Molecular Graphics System.37 | ||
 | ||
| Fig. 5 Stability of MeHNL-silica monolith for (S)-mandelonitrile ((S)-2) synthesis reaction in continuous flow system: a. 2 cm piece of MeHNL-silica monolith (3.3 mg total protein; 185 U per monolith) conversion (bars)/enantiomeric excess (black line) vs. time (min) on stream with flow rate of 0.1 mL min−1. b. 2 cm piece of MeHNL-silica monolith (3.3 mg total protein; 185 U per monolith) conversion (bars)/enantiomeric excess (black line) vs. time (min) on stream with flow rate of 0.2 mL min−1. Flow rate is total of two pumps. Pump 1 pumps benzaldehyde (1) in MTBE (0.50 M, containing 0.066 mM ISTD 1) and pump 2 pumps HCN in MTBE (1.5–2.0 M, containing 0.066 mM ISTD 2 saturated with citrate phosphate buffer (pH 5.0, 50 mM)). | ||
Ultimately, the productivity of the enzymatic microreactor can be expressed in terms of space time yield (STY) and in this case MeHNL shows a remarkable estimated STY of 1229 g L−1 h−1, whereas HbHNL shows STYs up to 613 g L−1 h−1. However, by considering the amount of total protein immobilized, the STYs read 54 and 71 g L−1 h−1 mgprotein−1 for HbHNL and MeHNL respectively, thus showing excellent productivities in the same order of magnitude. Moreover, the volume of the reaction is reduced by switching from batch (2.2 ml) to flow (1 ml) reactor volume, realizing the desired waste reduction.
Overall, the combination of HNLs in a siliceous monolithic microreactor created a very active catalyst, which as of now has never been reported for HNL-based microreactors or even for enzyme-based microreactors in asymmetric synthesis. Recently a continuous flow cascade with hydroxynitrile lyase (AtHNL from Arabidopsis thaliana) was described. In this system a packed-bed reactor (microbore column 3 × 50 mm) filled with celite-AtHNL was used for the enantioselective production of (R)-mandelonitrile ((R)-2). The highest conversion (85%) and ee of 96% (R) were achieved with 100 mg celite-AtHNL (25 mg enzyme loading) at 0.04 mL min−1 flow rate (i.e. residence time of 35.3 min).62 Yet, with our HNL monolithic microreactor we show (almost) full conversion and very good ee (99% (S)) with remarkable residence times ranging between 3–22 minutes (flow rates 0.045–0.3 mL min−1) with minimal enzyme loading (11.3 mg and 17.4 mg total protein for HbHNL and MeHNL). Another very recent example of a continuous flow system in biocatalytic asymmetric synthesis is the esterification of 2-phenylpropionic acid by CALB (Novozyme 435, supported lipase B from Candida antarctica).63 A packed-bed reactor (HPLC column 4.6 × 150 mm) was equipped with 700 mg of the well-known, highly stable CALB and was operated for three weeks, converting 15 g of the acid and thus giving STYs of 19 g L−1 h−1 and 0.03 g L−1 h−1 mgprotein−1. Evidently, the reported STYs values here for the HbHNL and MeHNL-silica monoliths are a significant step forward. We believe that the specific covalent immobilization method and the intrinsic properties of the microreactor are more suited for achieving maximum control in short timeframes than the traditional packed-bed reactors.
Conclusion
HbHNL and MeHNL were successfully immobilized on porous silica supports with high yields and deployed as catalysts in batch and continuous synthesis of (S)-mandelonitrile ((S)-2). With the design of a continuous flow microreactor containing minimal HNL loadings, a drastic enhancement of the catalytic performance as compared to the batch system was achieved, leading to full conversion and high ee (99% (S)-2) within minutes. Thus the process intensification resulting in exceptional STYs, solvent reduction and lower energy consumption is achieved. Moreover, no catalysts removal step is necessary in the continuous reaction, improving the step economy. MeHNL displayed increased operational stability compared to HbHNL, reasonably due to immobilization of tetrameric structures. Our studies clearly highlight the importance of carefully selecting a reaction system for a specific biotransformation, show that enzyme-catalyzed reactions can benefit greatly from reaction engineering and further the assembly of continuous flow microreactor cascades. The successful application of a monolithic microreactor as enzyme carrier for the first ever described enantioselective synthesis bodes well for future industrial continuous production of chiral building blocks.Experimental section
Caution: Potassium/sodium cyanide and hydrogen cyanide (HCN) are poisonous. Experiments were performed in a well-ventilated fume hood with a calibrated HCN detector. HCN liquid and solid wastes were neutralized with commercial bleach and disposed.64General
Chemicals were purchased in the highest available purity from Sigma-Aldrich, VWR, Acros Organics or Avantor Poland and used without further purification unless reported otherwise. Petroleum ether (PE) was distilled prior to use. (rac)-Mandelonitrile ((rac)-2) was freshly purified by column chromatography (gradient PE/EtOAc 9:1/3:7) with the flash chromatography system (Reveleris X2 Flash instrument) using a pre-packed silica column (40 μm, 12 mg, GRACE) and stored at −20 °C protected from light and under nitrogen atmosphere. Benzaldehyde (1) was distilled and stored at 4 °C under nitrogen atmosphere protected from light. Analytical grade salts and Millipore water were used for the preparation of aqueous buffers. Hydrogen cyanide (1.5–2.0 M) in methyl tert-butyl ether (MTBE) was prepared according to ref. 64 and determination of the concentration was carried out as in ref. 65. The final HCN solution was kept over citrate/phosphate buffer (pH 5.0, 50 mM) in a 1:1 ratio and stored in a dark bottle at 4 °C. FTIR spectroscopy was performed with a Nicolet™ 6700 FT-IR spectrometer from Thermo Electron Corporation equipped with OMNIC Software using the potassium bromide (KBr) method. Spectra were recorded at a wavenumber range from 4000–400 cm−1 with a 4 cm−1 resolution. TGA was carried out with the TGA 2 SF/1100 STARe System (Mettler-Toledo). The selected method was 30–800 °C (heating rate of 10 °C min−1) with 100 mL min−1 air flow rate. SEM was performed on a JEOL JSM-6010LA analytical microscope equipped with JEOL JSM 6010 software. The field-emission gun operated at 5.0 kV acceleration voltage. TEM was carried out on a JEOL JEM-1400 PLUS with electron tomography software operating at 120 kV (LaB6 filament). For the TEM sample preparation, the material was finely ground with ethanol in an agate mortar and a droplet thereof was placed on the TEM grid (carbon–copper). UV spectroscopy was carried out with a Cary 60 UV-vis spectrophotometer (Agilent Technologies) connected to a Cary single cell Peltier accessory (Agilent Technologies). HPLC analyses were performed on a chiral HPLC system (Waters or Agilent Technologies 1200 series) with differential refractometer and UV/VIS detector, column thermostat, autosampler, HPLC pump and equipped with a chiralpak AD-H column (Daicel, 4.6 × 250 mm, 5 μm). NMR spectra were recorded on an Agilent-400 MR DD2 and are internally referenced to residual solvent signals. Data for 1H NMR are reported as follows: chemical shift (δ ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet) and integration. Data for 13C NMR are given in terms of chemical shift. Room temperature in the laboratory was measured as 18–22 °C.
Synthesis of MCF carriers
In a typical procedure Pluronic P123 (poly(ethylene oxide)-block-poly(propylene oxide)-block-poly(ethylene oxide); EO20-PO70-EO20) (2 g) was dissolved in 1.6 M HCl (75 mL) at room temperature.66 Then, the organic swellings agents, 1,3,5-trimethylbenzene (5.8 mL) and NH4F (0.023 g) were added under vigorous stirring and the mixture was heated to 40 °C. After 1 h stirring, tetraethyl orthosilicate (TEOS) (silica and ethanol source) was added (4.7 mL), whereupon the mixture was stirred for 1 h, then stored at 40 °C for 20 h and finally at 100 °C for 24 h. After cooling to room temperature, the white precipitate was filtered, dried at room temperature for 4 days and calcined at 500 °C for 8 h, finally obtaining the siliceous MCF material.Post-functionalization of MCF carriers
MCFs were grafted with aminopropyl (amino functionality) and glycidyloxypropyl (epoxy functionality) groups using the respective organosilane precursors. Before functionalisation, MCFs were brought into contact with water vapour for 5 h and then dried at 200 °C for 2 h. Functional groups were grafted onto the MCFs surface by reacting the surface silanol groups of silica with the suitable organosilanes (3-aminopropyltrimethoxysilane (APTS) and 3-glycidyloxypropyltrimethoxysilane (GPTS)) dissolved in toluene under reflux (24 h, 85 °C), eventually obtaining a load of the functional moiety of about 1.5 mmol per g of silica. In particular, 20 mL of the solution containing APTS or GPTS were stirred under reflux with 1 g of MCF for 24 h. After filtration, the solvent was evaporated at 60 °C.Synthesis of silica monolith
The general procedure was as follows:32,66 first polyethylene glycol 35000 (PEG, 9.09 g) was dissolved in 1 M HNO3 (104.6 mL), after which tetraethoxysilane (TEOS, 87.1 mL) was added slowly under stirring (500 rpm) to the PEG solution in an ice bath followed by the addition of cetyltrimethylammonium bromide (CTAB, 4.016 g). The solution was mixed and then left to gel in polypropylene tubes at 40 °C and aged for 10 days. Next the alcogels obtained were impregnated with 1 M ammonia solution for 9 h at 90 °C. The samples were washed with water, dried at room temperature and then calcined at 550 °C for 8 h (ramp of 1 °C min−1) to obtain silica rods 50 mm in length and 6 mm in diameter. The monoliths were functionalized and clad with polymer resin (L285MGS-H285MGS type) to obtain single-rod continuous-flow microreactors with a reactor volume of 0.96 mL (cylinder volume corrected with porosity factor of 85%).
Functionalization of silica monolith-amino functionalized carrier
Pristine silica monoliths were modified to attach the enzyme non-specifically by means of amino glutaraldehyde linkage. In short, oven-dried (overnight, 150 °C) silica monoliths (1 g) were gently stirred and refluxed with 3-aminopropyltrimethoxysilane (0.27 mL) in 35 mL of dry toluene for 72 h. After taking out the monoliths, they were washed with toluene and dried.Cloning, overexpression and purification of HbHNL and MeHNL
Nucleotide and protein sequences of HbHNL and MeHNL are given in ESI† section O. The synthetic gene of HbHNL (accession number U40402.1) was subcloned using NcoI and HindIII restriction enzymes and ligated into an equality treated pSE420 vector (Invitrogen). The resulting construct was named pSE420hbhnl. The gene of MeHNL (without His-tag, as confirmed by sequencing) was amplified by PCR from vector pBADmehnl using primers 5′-CATGCCATGGTGACCGCACATTTTGTTCTGATTC-3′ (fwd, including NcoI) and 5′-CGTTTCACTTCTGAGTTCGGCATGGG-3′ (rev, including BsmFI). Later, digestion using NcoI and BsmFI restriction enzymes was carried out. Using the same strategy, the linearised pSE420 vector was obtained from the construct pSE420hbhnl, and ligated to mehnl resulting in vector named pSE420mehnl. Constructs pSE420hbhnl and pSE420mehnl were transformed into electrocompetent E. coli TOP10 cells via standard procedure.Stocked (−80 °C) E. coli TOP10 pSE420mehnl or E. coli TOP10 pSE420hbhnl cells were spread in a LB agar plate containing ampicillin (100 μg mL−1 final concentration) and grown overnight at 37 °C. A single colony was used to inoculate a 200 mL LB media supplemented with ampicillin (100 μg mL−1). After 16 h at 180 rpm and 37 °C (New Brunswick Scientific Incubator Shaker Innova 44 Series), a 15 L fermenter vessel containing 10 L LB and ampicillin (100 μg mL−1) was inoculated with 1.5% v/v of preculture. Expression was carried out at 37 °C, 400–450 rpm, pH 7.00 (acid solution: HCl 2 M, basic solution: KOH 2 M), air flow 1.6 standard liter per minute. When the optical density (OD600) reached 0.8 the system was induced with isopropyl β-D-1-thiogalactopyranoside (IPTG, 0.1 mM final concentration) and the temperature reduced to 18 °C. After 24 h, the cells were harvested and washed with sodium acetate buffer (25 mM, pH 5.8, buffer A) and stored at −20 °C.
Overexpressed cells (12 grams) carrying MeHNL or HbHNL were resuspended in 100 mL of buffer A and lysed with a French press (Multi Shot, Constant Cell Disruption Systems LA Biosystems). After centrifugation the supernatant was filtered with Whatman filters (GE Healthcare, pore size 0.45 μm) and loaded into a previously equilibrated Fast Flow Q-Sepharose column (61 mL) with buffer A in a NGC™ 10 Medium-Pressure Chromatography Systems (Bio-Rad). After washing, the target proteins eluted in a gradient from 10% to 100% sodium acetate buffer (25 mM, pH 5.8 + 0.5 M NaCl) (12 CV, 7 mL min−1) and monitored via UV detection at 280 nm. Fractions containing the desired protein (as analysed by SDS-PAGE) were collected, slowly salted with 1.5 M (NH4)2SO4, filtered and loaded into a previously equilibrated Phenyl Sepharose Fast Flow column (18 mL) with 50 mM potassium phosphate buffer (50 mM, pH 7.0 + 1.5 M (NH4)2SO4). After washing, the elution step started with a gradient (5 CV, 6 mL min−1) from 0% to 50% of elution buffer potassium phosphate (50 mM, pH 7.0). Then an isocratic flow of 50% of elution buffer was performed (5 CV, 6 mL min−1). Finally, a slower gradient (15 CV, 6 mL min−1) from 50% to 100% of elution was applied. Fractions containing the desired protein (as analyzed by SDS-PAGE) were collected, concentrated by ultrafiltration using Amicon filters (10 kDa cutoff, Merck) and desalted using PD10 (GE healthcare), 100 mM potassium phosphate pH 7.0 was used as storage buffer. As a result, 18 mg and 24 mg of total protein were isolated from 12 g of cells with a purity of 88% and 55% for Hb and MeHNL respectively. From the above described protocols 120 mg of each HNL can be isolated from respective 10 L expression. Purified samples were stored at −80 °C until immobilization. SDS PAGE scans of purified HNLs are provided in Fig. S14.†
Protein quantification
Quantification of the average total protein concentration was performed by the Bradford Assay67 or the direct detect FTIR method based on the infrared absorption of the peptide bond. A bovine serum albumin (BSA) calibration curve was used as a standard.Cyanolysis assay
HNL activities were quantified by means of a cyanolysis assay, in which the cleavage of (rac)-mandelonitrile ((rac)-2) to benzaldehyde (1) and hydrogen cyanide (cyanolysis) is followed by measuring the increase in absorbance at wavelength (λ) of 280 nm at 25 °C. The procedure was according to ref. 64. Measured specific activity values for HbHNL and MeHNL were 99 U mg−1 and 75 U mg−1 of total protein respectively. Unit is defined as μmol of benzaldehyde formed per minute.SDS polyacrylamide gel electrophoresis (PAGE) procedure
20 μL of sample were mixed with 20 μL loading dye and heated to 95 °C for 5–10 min. The sample pockets of a Criterion™ Precast XT gel (Bio-Rad 12% Bis-Tris) were filled with 15 μL sample and 10 μL marker (Bio-Rad Precision Plus Protein™ Unstained Standards (MW size range 10–250 kDa)). SDS-PAGE was conducted at 200 V for 45–50 min with 3-morpholinopropane-1-sulfonic acid (MOPS) running buffer (Bio-Rad). Afterwards, gels were stained for 1–2 h in SimplyBlue™ SafeStain and destained with Millipore water overnight.Enzyme immobilization MCF carriers
Enzyme immobilization on MCF carriers and MCF-APT glutaraldehyde activation was performed as previously reported.68–71 In short, MCF-APT was activated with 2.5 vol% buffered glutaraldehyde solution (potassium phosphate, 100 mM, pH 7.0). The mixture was gently stirred for 1 h. Glutaraldehyde activated MCF-APT or MCF-GPT carrier were suspended in 10 mL protein solution in potassium phosphate buffer (100 mM, pH 7.0), amounts and concentrations adopted to obtain a 1:10/1:20 or 1:40 enzyme:carrier w/w ratio. The mixture was stored for 24 h at 4 °C and the excess protein was washed off with 20 mL potassium phosphate buffer (100 mM, pH 7.0), potassium phosphate buffer (100 mM, pH 7.0 + 0.5 M NaCl), sodium acetate buffer (100 mM, pH 5.0) and distilled water. Filtered preparations were suspended in 10 mL Tris-HCl buffer (500 mM, pH 8.0) for 24 h at 4 °C. After washing with 20 mL potassium phosphate buffer (100 mM, pH 7.0), the immobilized enzyme preparations were stored resuspended in potassium phosphate buffer (100 mM, pH 7.0). Eluates from the immobilization steps were collected and analyzed for the presence of protein (Bradford assay) and the remaining HNL activity of the immobilized enzymes was determined by the cyanolysis assay.
Enzyme immobilization silica monolith
Monolith (6 × 40 mm, 0.96 mL, 260 mg silica) immobilization was carried out as in ref. 56. In short, amino groups were activated with 2.5 vol% buffered glutaraldehyde solution (potassium phosphate, 100 mM, pH 7.0) for 60 min (0.5 mL min−1). Excess glutaraldehyde was washed off with distilled water for 60 min (0.5 mL min−1) and potassium phosphate buffer (pH 7.0, 100 mM) for additional 40 min (0.5 mL min−1). The protein solution in potassium phosphate buffer (100 mM, pH 7.0) was circulated through the reactor under recycling conditions for 2.5 h (0.5 mL min−1) at room temperature and suspended overnight at 4 °C. The protein concentration was adopted in order to establish a 1:10/1:20/1:40 enzyme:support w/w ratio. The excess of protein was washed off with potassium phosphate buffer (100 mM, pH 7.0) for 15 min (0.1 mL min−1) and additional 20 min (0.5 mL min−1), potassium phosphate buffer (100 mM, pH 7.0 + 0.5 M NaCl) for 40 min (0.5 mL min−1), sodium acetate buffer (100 mM, pH 4.5) for 35 min (0.5 mL min−1) and finally distilled water for 40 min (0.5 mL min−1). The monolith was rinsed with Tris-HCl buffer (500 mM, pH 8.0) for 15 min (0.5 mL min−1) and suspended in Tris-HCl buffer overnight at 4 °C. Finally, the monolith with immobilized enzyme was washed and stored resuspended in potassium phosphate buffer (100 mM, pH 7.0). Eluates from the immobilization steps were collected and analyzed for the presence of protein using the direct detect FTIR method based on the infrared absorption of the peptide bond.
Synthesis reactions of (S)-mandelonitrile ((S)-2) batch procedure.
50 mg HbHNL-MCF-APT (2.5 mg total protein; 150 U) (rinsed with citrate phosphate buffer (pH 5.0, 50 mM)) was added to a 4 mL reaction vial, followed by benzaldehyde (1) (100 μl, ±106 mg, 1 mmol) and 1,3,5-triisopropylbenzene (ISTD 1, 27.5 μL, ±25 mg, 0.1 mmol) under nitrogen. The reaction was started by addition of 2 mL HCN solution in MTBE (1.5–2 M, saturated with citrate/phosphate buffer 50 mM, pH 5.0). Reaction mixture was shaken (1000 rpm) at room temperature (18–22 °C). A small sample (10–25 μL) was taken at regular intervals and immediately analyzed by chiral HPLC (reaction set-up in Fig. S15†).Synthesis reactions of (S)-mandelonitrile ((S)-2) continuous flow procedure
Prior to the reaction, the monolith (6 × 40 mm, 0.96 mL, 260 mg silica) was washed with citrate phosphate buffer (pH 5.0, 50 mM) for 30 min (0.5 mL min−1). Two high pressure pumps (Azura, KNAUER) connected to a mixing unit (with a T-piece assembly) were deployed (Fig. S16†). Pump 1 was used for the benzaldehyde (1) solution in MTBE (0.50 M with 0.066 mM 1,3,5-triisopropylbenzene ISTD 1) and pump 2 for the HCN solution in MTBE (1.5–2 M, saturated with citrate/phosphate buffer 50 mM pH 5.0 with 0.066 mM mesitylene ISTD 2). A standard T-mixer (see Fig. S16†) in which the two streams collided head-on was connected to the microreactor via 15 cm Peek tubing, inner diameter 0.76 mm with and an overall dead volume of max. 0.068 ml. The combined flows were passed through the microreactor (reactor volume 0.96 mL), containing immobilized HbHNL (11.3 mg total protein; 1120 U) or MeHNL (17.4 mg total protein; 1310 U) on the silica monolith. For every single flow rate three samples were collected in defined time and every single sample was weighted to check the flow rate (flow rate = (weight of sample/density)/time). Concentrations of substrate and product at different flow rates (i.e. residence times) were determined with chiral HPLC.Stability studies
For the stability testing a 2 cm piece of MeHNL-silica monolith (3.3 mg total protein; 185 U) was left for prolonged time on stream at 0.1 mL min−1 or 0.2 mL min−1 flow rate. Samples of the reactor outflow were taken at regular intervals and analyzed by chiral HPLC.HPLC method and sampling
Samples (10–25 μL) were added to 1 mL heptane/isopropanol (95:5), dried over anhydrous MgSO4 and centrifuged. 10 μL of the sample was injected into the HPLC system equipped with chiralpak AD-H column (Daicel, 4.6 × 250 mm, 5 μm). The following HPLC method settings were applied: mobile phase: heptane:isopropanol 95:5 (0.1% v/v TFA), UV detection wavelength: λ = 216 nm, oven temperature: 40 °C and flow rate: 1 mL min−1. The specific compound retention times are: ∼2.9 min 1,3,5-triisopropylbenzene (ISTD 1), ∼3.0 min mesitylene (ISTD 2), ∼5.0 min benzaldehyde (1), ∼7.1 min benzoic acid, ∼11.5 min (S)-mandelonitrile ((S)-2) and ∼13.0 min (R)-mandelonitrile ((R)-2).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to express our gratitude to Guzman Torrelo, Jianfeng Jin, Isabel Nieuwenhuijse and Mattia Lazzarotto from Delft University of Technology for their help with the cloning, protein expression and purification. Professor Anna Chrobok from the Silesian University of Technology is acknowledged for her advice with chemical purification procedures and professor Martina Pohl from Forschungszentrum Jülich GmbH for sharing the tetrameric PyMOL model of MeHNL. Generous funding to H. B. (STW; grant number 14170) is acknowledged. We gratefully acknowledge Monika Kostka for the design of the cartoon displaying micromixing in the (micro)structured monolithic reactor.References
- R. DiCosimo, J. McAuliffe, A. J. Poulose and G. Bohlmann, Chem. Soc. Rev., 2013, 42, 6437–6474 RSC.
- C. Wiles and P. Watts, Green Chem., 2012, 14, 38–54 RSC.
- G. Torrelo, U. Hanefeld and F. Hollmann, Catal. Lett., 2015, 145, 309–345 CrossRef CAS.
- In Catalysis: An Integrated Textbook for Students, ed. U. Hanefeld and L. Lefferts, Wiley-VCH, Weinheim, 2017, vol. 4, p. 128 Search PubMed.
- P. Bracco, H. Busch, J. van Langermann and U. Hanefeld, Org. Biomol. Chem., 2016, 14, 6375–6389 RSC.
- J. Holt and U. Hanefeld, Curr. Org. Synth., 2009, 6, 15–37 CrossRef CAS.
- M. Breuer, K. Ditrich, T. Habicher, B. Hauer, M. Kesseler, R. Stürmer and T. Zelinski, Angew. Chem., Int. Ed., 2004, 43, 788–824 CrossRef CAS PubMed.
- T. Purkarthofer, W. Skranc, C. Schuster and H. Griengl, Appl. Microbiol. Biotechnol., 2007, 76, 309–320 CrossRef CAS PubMed.
- M. North, Tetrahedron: Asymmetry, 2003, 14, 147–176 CrossRef CAS.
- M. North, Tetrahedron, 2004, 60, 10383–10384 CrossRef CAS.
- M. Gruber-Khadjawi, M. H. Fechter and H. Griengl, in Enzyme Catalysis in Organic Synthesis, ed. K. Drauz, H. Gröger and O. May, Wiley-VCH Verlag GmbH & Co. KGaA, 2012, pp. 947–990 Search PubMed.
- M. Dadashipour and Y. Asano, ACS Catal., 2011, 1, 1121–1149 CrossRef CAS.
- K. Steiner, A. Glieder and M. Gruber-Khadjawi, in Science of Synthesis: Biocatalysis in Organic Synthesis, ed. K. Faber, W. D. Fessner and N. Turner, Georg Thieme Verlag, 2015, vol. 2, pp. 1–30 Search PubMed.
- R. A. Sheldon and J. M. Woodley, Chem. Rev., 2017, 118, 801–838 CrossRef PubMed.
- J. M. Bolivar and B. Nidetzky, Chim. Oggi, 2013, 31, 50–54 CAS.
- J. M. Bolivar, J. Wiesbauer and B. Nidetzky, Trends Biotechnol., 2011, 29, 333–342 CrossRef CAS PubMed.
- E. Laurenti and A. dos Santos Vianna Jr, Biocatalysis, 2015, 1, 148–165 Search PubMed.
- N. Miložič, M. Lubej, M. Lakner, P. Žnidaršič-Plazl and I. Plazl, Chem. Eng. J., 2017, 313, 374–381 CrossRef.
- R. Wohlgemuth, I. Plazl, P. Žnidaršič-Plazl, K. V. Gernaey and J. M. Woodley, Trends Biotechnol., 2015, 33, 302–314 CrossRef CAS PubMed.
- L. Tamborini, P. Fernandes, F. Paradisi and F. Molinari, Trends Biotechnol., 2018, 36, 73–88 CrossRef CAS PubMed.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- (a) J. Britton, S. Majumdar and G. A. Weiss, Chem. Soc. Rev., 2018, 47, 5891–5918 RSC; (b) J. Britton and C. L. Raston, Chem. Soc. Rev., 2017, 46, 1250–1271 RSC; (c) J. C. Pastre, D. L. Browne and S. V. Ley, Chem. Soc. Rev., 2013, 42, 8849–8869 RSC.
- (a) R. Munirathinam, J. Huskens and W. Verboom, Adv. Synth. Catal., 2015, 357, 1093–1123 CrossRef CAS; (b) F. Costantini, E. M. Benetti, D. N. Reinhoudt, J. Huskens, G. J. Vancso and W. Verboom, Lab Chip, 2010, 10, 3407–3412 RSC.
- N. N. Rao, S. Lütz, K. Würges and D. Minör, Org. Process Res. Dev., 2009, 13, 607–616 CrossRef CAS.
- K. G. Hugentobler, M. Rasparini, L. A. Thompson, K. E. Jolley, A. J. Blacker and N. J. Turner, Org. Process Res. Dev., 2017, 21, 195–199 CrossRef CAS.
- Y. Asano, S. Togashi, H. Tsudome and S. Murakami, Pharmaceutical Engineering, 2010, 30, 32 Search PubMed.
- J. M. Bolivar, M. A. Tribulato, Z. Petrasek and B. Nidetzky, Biotechnol. Bioeng., 2016, 113, 2342–2349 CrossRef CAS PubMed.
- R. L. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502–7519 CrossRef CAS PubMed.
- R. H. Ringborg and J. Woodley, React. Chem. Eng., 2016, 1, 10–22 RSC.
- R. Singh, H.-J. Lee, A. K. Singh and D.-P. Kim, Korean J. Chem. Eng., 2016, 33, 2253–2267 CrossRef CAS.
- B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS.
- K. Szymańska, M. Pietrowska, J. Kocurek, K. Maresz, A. Koreniuk, J. Mrowiec-Białoń, P. Widłak, E. Magner and A. Jarzębski, Chem. Eng. J., 2016, 287, 148–154 CrossRef.
- K. Szymańska, K. Odrozek, A. Zniszczoł, W. Pudło and A. B. Jarzębski, Chem. Eng. J., 2017, 315, 18–24 CrossRef.
- L. Rosenthaler, Biochem. Z., 1908, 14, 238–253 CAS.
- A. Schmidt, K. Gruber, C. Kratky and V. S. Lamzin, J. Biol. Chem., 2008, 283, 21827–21836 CrossRef CAS PubMed.
- H. Lauble, S. Förster, B. Miehlich, H. Wajant and F. Effenberger, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2001, 57, 194–200 CrossRef CAS.
- PyMOL Molecular Graphics System, Version 1.8, Schrödinger, LLC, 2015 Search PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- K. Gruber, G. Gartler, B. Krammer, H. Schwab and C. Kratky, J. Biol. Chem., 2004, 279, 20501–20510 CrossRef CAS PubMed.
- U. Hanefeld, G. Stranzl, A. J. J. Straathof, J. J. Heijnen, A. Bergmann, R. Mittelbach, O. Glatter and C. Kratky, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2001, 1544, 133–142 CrossRef CAS.
- J.-K. Guterl, J. N. Andexer, T. Sehl, J. von Langermann, I. Frindi-Wosch, T. Rosenkranz, J. Fitter, K. Gruber, U. Kragl and T. Eggert, J. Biotechnol., 2009, 141, 166–173 CrossRef CAS PubMed.
- (a) U. Hanefeld, L. Gardossi and E. Magner, Chem. Soc. Rev., 2009, 38, 453–468 RSC; (b) U. Hanefeld, Chem. Soc. Rev., 2013, 42, 6308–6321 RSC.
- O. Barbosa, C. Ortiz, Á. Berenguer-Murcia, R. Torres, R. C. Rodrigues and R. Fernandez-Lafuente, RSC Adv., 2014, 4, 1583–1600 RSC.
- L. Gardossi, P. B. Poulsen, A. Ballesteros, K. Hult, V. K. Švedas, Đ. Vasić-Rački, G. Carrea, A. Magnusson, A. Schmid and R. Wohlgemuth, Trends Biotechnol., 2010, 28, 171–180 CrossRef CAS PubMed.
- F. Effenberger, S. Förster and H. Wajant, Curr. Opin. Chem. Biol., 2000, 11, 532–539 CrossRef CAS.
- K. Szymańska, J. Bryjak, J. Mrowiec-Białoń and A. B. Jarzębski, Microporous Mesoporous Mater., 2007, 99, 167–175 CrossRef.
- E. Wehtje, P. Adlercreutz and B. Mattiasson, Appl. Microbiol. Biotechnol., 1988, 29, 419–425 CrossRef CAS.
- C. Lei, Y. Shin, J. Liu and E. J. Ackerman, J. Am. Chem. Soc., 2002, 124, 11242–11243 CrossRef CAS.
- M. Hoarau, S. Badieyan and E. N. G. Marsh, Org. Biomol. Chem., 2017, 15, 9539–9551 RSC.
- P. Schmidt-Winkel, W. W. Lukens, P. Yang, D. I. Margolese, J. S. Lettow, J. Y. Ying and G. D. Stucky, Chem. Mater., 2000, 12, 686–696 CrossRef CAS.
- D. Costes, G. Rotčnkovs, E. Wehtje and P. Adlercreutz, Biocatal. Biotransform., 2001, 19, 119–130 CrossRef CAS.
- D. Costes, E. Wehtje and P. Adlercreutz, Enzyme Microb. Technol., 1999, 25, 384–391 CrossRef CAS.
- D. Okrob, M. Paravidino, R. V. Orru, W. Wiechert, U. Hanefeld and M. Pohl, Adv. Synth. Catal., 2011, 353, 2399–2408 CrossRef CAS.
- P. Bracco, G. Torrelo, S. Noordam, G. De Jong and U. Hanefeld, Catalysts, 2018, 8, 287 CrossRef.
- C. Csajagi, G. Szatzker, E. R. Tőke, L. Uerge, F. Darvas and L. Poppe, Tetrahedron: Asymmetry, 2008, 19, 237–246 CrossRef CAS.
- K. Szymańska, K. Odrozek, A. Zniszczoł, G. Torrelo, V. Resch, U. Hanefeld and A. B. Jarzębski, Catal. Sci. Technol., 2016, 6, 4882–4888 RSC.
- A. D. Stroock, S. K. Dertinger, A. Ajdari, I. Mezić, H. A. Stone and G. M. Whitesides, Science, 2002, 295, 647–651 CrossRef CAS PubMed.
- A. Nie, Z. Gao, L. Xue, Z. Cai, G. M. Evans and A. Eaglesham, Chem. Eng. Sci., 2018, 184, 14–24 CrossRef CAS.
- H. Griengl, N. Klempier, P. Pöchlauer, M. Schmidt, N. Shi and A. A. Zabelinskaja-Mackova, Tetrahedron, 1998, 54, 14477–14486 CrossRef CAS.
- D. Okrob, J. Metzner, W. Wiechert, K. Gruber and M. Pohl, ChemBioChem, 2012, 13, 797–802 CrossRef CAS PubMed.
- R. Fernandez-Lafuente, Enzyme Microb. Technol., 2009, 45, 405–418 CrossRef CAS.
- A. Brahma, B. Musio, U. Ismayilova, N. Nikbin, S. B. Kamptmann, P. Siegert, G. E. Jeromin, S. V. Ley and M. Pohl, Synlett, 2016, 27, 262–266 CAS.
- A. Iemhoff, J. Sherwood, C. R. McElroy and A. J. Hunt, Green Chem., 2018, 20, 136–140 RSC.
- G. Torrelo, N. van Midden, R. Stloukal and U. Hanefeld, ChemCatChem, 2014, 6, 1096–1102 CrossRef CAS.
- L. M. van Langen, F. van Rantwijk and R. A. Sheldon, Org. Process Res. Dev., 2003, 7, 828–831 CrossRef CAS.
- K. Zielińska, K. Szymańska, R. Mazurkiewicz and A. B. Jarzębski, Tetrahedron: Asymmetry, 2017, 28, 146–152 CrossRef.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- A. B. Jarzębski, K. Szymańska, J. Bryjak and J. Mrowiec-Białoń, Catal. Today, 2007, 124, 2–10 CrossRef.
- J. Bryjak, Biochem. Eng. J., 2003, 16, 347–355 CrossRef CAS.
- J. Bryjak, A. Trochimczuk and A. Noworyta, J. Chem. Technol. Biotechnol., 1993, 57, 73–78 CrossRef CAS.
- K. Szymańska, J. Bryjak and A. B. Jarzębski, Top. Catal., 2009, 52, 1030–1036 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cy02192a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |