
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular chirogenesis in zinc porphyrins by enantiopure hemicucurbit[n]urils (n = 6, 8)†
Lukas
Ustrnul
 a,
Sandra
Kaabel
a,
Tatsiana
Burankova
b,
Jevgenija
Martõnova
a,
Jasper
Adamson
c,
Nele
Konrad
a,
Peeter
Burk
d,
Victor
Borovkov
*ae and
Riina
Aav
*a
a,
Sandra
Kaabel
a,
Tatsiana
Burankova
b,
Jevgenija
Martõnova
a,
Jasper
Adamson
c,
Nele
Konrad
a,
Peeter
Burk
d,
Victor
Borovkov
*ae and
Riina
Aav
*a
aDepartment of Chemistry and Biotechnology, Tallinn University of Technology, Akadeemia tee 15, 12618 Tallinn, Estonia. E-mail: riina.aav@taltech.ee
bPaul Scherrer Institute, Laboratory for Neutron Scattering and Imaging, Forschungsstrasse 111, 5232 Villigen PSI, Switzerland
cNational Institute of Chemical Physics and Biophysics, Akadeemia tee 23, 12618 Tallinn, Estonia
dFaculty of Science and Technology, University of Tartu, Vanemuise 46-208, 51014 Tartu, Estonia
eCollege of Chemistry and Materials Science, South-Central University for Nationalities, 182# Minzu RD, Hongshan District, Wuhan, Hubei Province 430074, China. E-mail: victor.borovkov@scuec.edu.cn
First published on 16th November 2019
Abstract
Chiral cyclohexanohemicucurbit[n]urils (n = 6, 8) (cycHCs) are able to bind guests through multiple “outer surface interactions”, which in the case of planar zinc porphyrins leads to induction of chirality. Crystal structures of complexes of complementary sized hosts revealed social self-sorting, while in the solution phase one cycHC can accommodate up to three porphyrin molecules with log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Ktotal 9.
Ktotal 9.
Porphyrins are a special group of aromatic tetrapyrrolic compounds possessing unique spectroscopic properties. They are extensively used in a broad range of various applications, e.g., catalysis,1,2 building blocks for MOFs and artificial molecular machines,2–4 diagnostics and photodynamic therapy,5 chemical sensing,6 and recognition of chiral molecules.7–9 There are two basic strategies for their chiral applications: (1) employing covalently modified chiral porphyrins and (2) supramolecular chirogenesis, which is based on the asymmetry transfer from a chiral guest molecule to porphyrin(s) via noncovalent interactions.10 Such a chirogenic process leads to induced circular dichroism (ICD) in the region of porphyrin absorption. In general, the ICD intensity of porphyrin molecules is more prominent if several porphyrin units are involved7,8,11 and rather moderate for monomeric porphyrins either in organic12,13 or aqueous solutions.14–16 Nevertheless, porphyrin aggregates formed in aqueous media also exhibit intense ICD signals.17–24
Chiral macrocycles, cyclohexanohemicucurbit[n]urils (cycHC[6] and cycHC[8], see Fig. 1A), have complementary dimensions with porphyrins, with their height being close to a nanometer.25–27 They are easily accessible25,27 and have the ability to bind carboxylic acids enantioselectively.25,27 The main feature of the larger cycHC[8] is the selective binding of anions inside the cavity, governed by the size, shape, and charge distribution of the guest.28 While anion binding is widely studied for the whole family of hemicucurbiturils,29 the external binding to polar urea moieties has received much less attention. The study by Buschmann30 and some crystallographic evidence showed the coordination of thiophilic cations, Pd2+ and Hg2+, to the thiourea moiety of thiobambus[4]uril31 and Na+ cation interaction with the carbonyl oxygen of biotin[6]uril.32
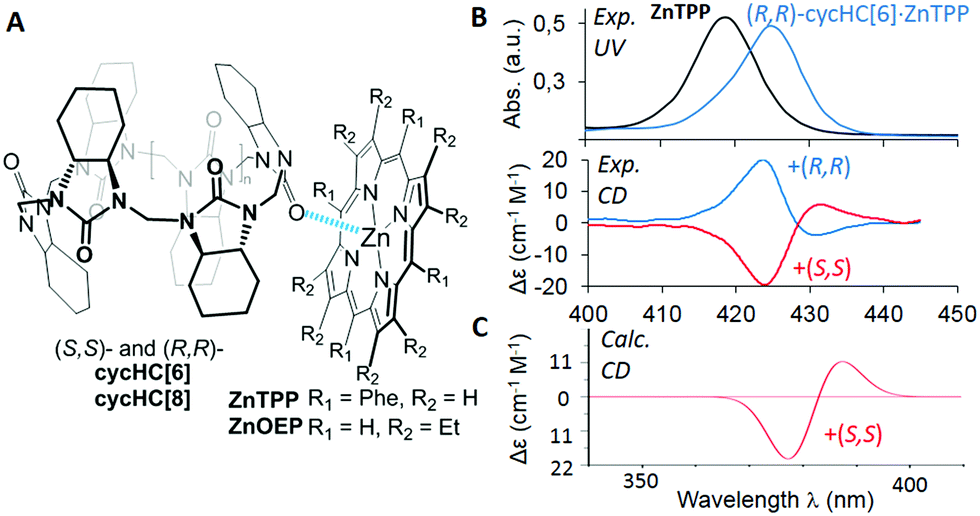 | ||
| Fig. 1 (A) Structures of studied complexes; (B) UV-vis and CD spectra of ZnTPP and its complexes with (S,S)-cycHC[6] and (R,R)-cycHC[6] in DCM; (C) TD-DFT simulated spectrum of ZnTPP·(S,S)-cycHC[6] complex. (See ESI† for details). | ||
Herein, we present the first example of chirality transfer from (R,R)- and (S,S)-enantiomers of cycHC[6] and cycHC[8] to achiral zinc octaethylporphyrin (ZnOEP) and zinc tetraphenylporphyrin (ZnTPP) (Fig. 1A) upon the supramolecular “outer surface interactions”, studied in solution and in the solid phase. The ability of cycHC[6] to imprint its chirality into achiral ZnTPP was investigated using circular dichroism in a nonpolar solvent and simulated computationally (Fig. 1B, C, and ESI†).
The addition of (S,S)-cycHC[6] to a dichloromethane (DCM) solution of ZnTPP resulted in the bathochromic shift of porphyrin absorption maxima up to 425 nm and the appearance of positive and negative Cotton effects in the region of the porphyrin Soret band at 431 and 424 nm, respectively (Fig. 1B).
The sole role of hemicucurbituril chirality in the ICD of ZnTPP was confirmed by employing antipodal cycHC[6]. Hence, (R,R)-cycHC[6] induced a perfect mirror image CD spectrum of ZnTPP. The CD spectra of other combinations of cycHCs and porphyrins; (S,S)/(R,R)-cycHC[6]·ZnOEP, (S,S)/(R,R)-cycHC[8]·ZnOEP, and (S,S)/(R,R)-cycHC[8]·ZnTPP gave similar results (Fig. S1–S4, ESI†), thus, confirming the generality of the chirogenic mechanism. The CD spectra were recorded in the presence of an excess of cycHCs, and therefore 1:1 complex formation was assumed. To probe the ICD ability of the monomeric unit of cycHCs, (R,R)-N,N′-dimethyl- and (R,R)-N,N′-diphenyl-cyclohexadiylurea (M1, M2) were used in a similar manner exhibiting no appreciable ICD in ZnTPP (Fig. S6, ESI†), pointing to differences in binding character of cycHCs compared with mono-ureas.
For unambiguous rationalization of the mechanism of chirality transfer and the mode of interaction of cycHCs with porphyrins, the corresponding time-dependent density functional theory (TD-DFT) simulation of cycHC[6]·ZnTPP complex was performed. The calculated CD spectrum of cycHC[6]·ZnTPP gave very good agreement with the experimental data (Fig. 1C), confirming the presence of 1:1 stoichiometry in the complex. The optimized geometry revealed the strongest interaction site between the porphyrin Zn cation and urea carbonyl of cycHC, with the Zn⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C distance being 2.203 Å. Localization of the highest occupied molecular orbital (HOMO) depicted in Fig. 2 shows distortion of porphyrin ring planarity that appears to be a prime cause of the chirality transfer mechanism. See ESI† for other orbitals involved in the electronic transitions.
C distance being 2.203 Å. Localization of the highest occupied molecular orbital (HOMO) depicted in Fig. 2 shows distortion of porphyrin ring planarity that appears to be a prime cause of the chirality transfer mechanism. See ESI† for other orbitals involved in the electronic transitions.
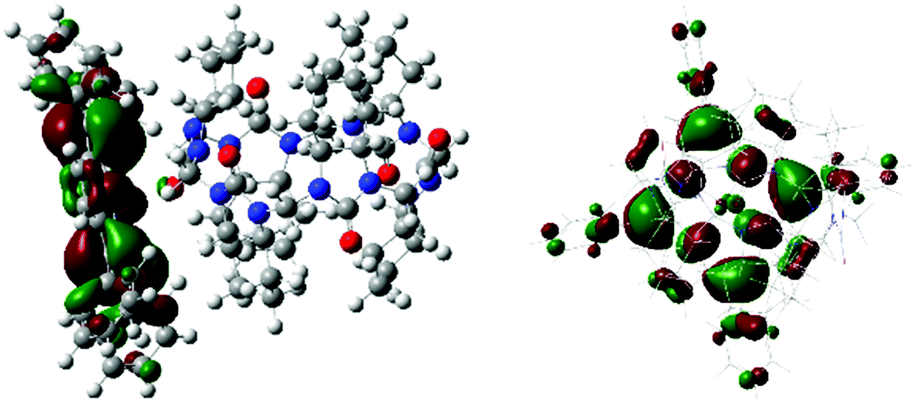 | ||
| Fig. 2 Visualization of the HOMO orbital of (S,S)-cycHC[6]·ZnTPP. | ||
We were fortunate to grow single crystals of the cycHC[6] and cycHC[8] complexes with ZnTPP (Fig. 3). Both the crystallographic structures showed distortion of the porphyrin plane in comparison to the planar conformation of noncomplexed porphyrin. The crystallographic structure of cycHC[6]·ZnTPP (Fig. 3A) revealed a rather unusual mode of binding with hexacoordinative zinc porphyrin forming a supramolecular linear chain with the cycHC[6] macrocycle and resulting in the overall 1:1 cycHC[6]·ZnTPP stoichiometry. Indeed, while zinc porphyrins are generally pentacoordinative in solution, several crystal structures of six-coordinated zinc porphyrins have been reported in the literature.33–37 Mostly, they are formed by self-association, however, there is one particular example of ZnTPP complexed with two tetrahydrofuran molecules.38 In addition, two examples of solution-phase studies of six-coordinated zinc porphyrin derivatives, utilizing chelate effect while coordinated to the Zn atom by heteroaromatic nitrogens, have been reported.39,40 In the case of cycHC[6]·ZnTPP complex, the average length of Zn⋯O is 2.39 Å, which is similar to other six-coordinated zinc complexes. Furthermore, the π-system of ZnTPP interacts with multiple C–H groups originating from the methylene bridges and cyclohexano moieties of cycHCs. The distances between the cycHC[6] hydrogens and the porphyrin mean plane are as follows: 2.39–2.66 Å for the methylene bridge and 2.64–3.10 Å for the cyclohexano group. Interestingly, the interacting protons of cyclohexano moieties are not of the urea subunit coordinated on the metal center but belong to the adjacent urea subunits. This observation suggests that the lack of these additional interactions with mono-urea derivative M1 allows free rotation around the coordination bond resulting in negligible ICD (see ESI†). At the same time, it emphasizes the appropriate preorganization of cycHCs’ shape allowing multiple-point interactions with porphyrins.
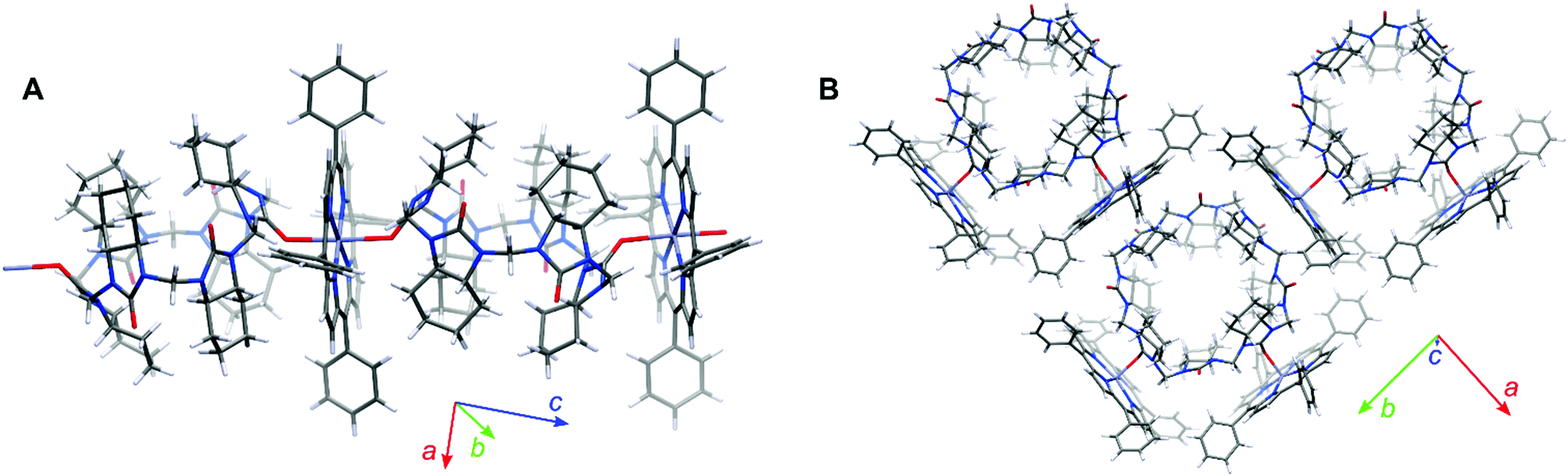 | ||
| Fig. 3 Single-crystal structure of (A) 1:1 1D polymer of (R,R)-cycHC[6]·ZnTPP, (B) 1:2 complex of (R,R)-cycHC[8]·ZnTPP. The solvent molecules have been omitted for clarity. Element colors: C gray, N blue, O red, H white, Zn light blue. | ||
In contrast, the crystal structure of cycHC[8]·ZnTPP shows the 1:2 complexation where one molecule of cycHC[8] is surrounded by four molecules of porphyrin with only two of them being complexed. Moreover, the two interacting porphyrins are bound pincer-like to the aligned urea subunits (Fig. 3B) separated by a single alternate monomer. However, the intermolecular interactions in crystals of cycHC[8]·ZnTPP are similar to those described for the cycHC[6] complexes.
The Zn⋯OC distance between the porphyrin and macrocycle moieties is 2.17 Å in cycHC[8] crystals, showing a larger overlap of van der Waals radii compared with the cycHC[6]·ZnTPP crystal. Besides, each porphyrin is also attached to cycHC[8]via a set of the C–H⋯π interactions with the strongest one found for the methylene bridge protons, where the distance from the porphyrin mean plane is 2.23–2.86 Å. In addition, weak interaction can be assumed for some of the protons of the cyclohexano group. These crystallographic structures revealed that the main attractive interaction occurs between the zinc cation of porphyrin and carbonyl oxygen of cycHCs, hence having six and eight potential binding sites in cycHC[6] and cycHC[8], respectively. However, it is sterically impossible for the porphyrins to be simultaneously attached to the neighboring monomeric units of cycHCs. Therefore, based on the crystallographic structures obtained, it is reasonable to assume that in the solution phase bulky cycHC[6] and cycHC[8] might be able to accommodate up to three and four porphyrin molecules, respectively.
The stoichiometry of the cycHC[6]·ZnTPP complex in DCM solution was explored using a 1H NMR Job plot (Fig. S35, ESI†). Although this method is only indicative as was shown by Jurczak et al.,41 the results showed the existence of the complex with a stoichiometry higher than cycHC[6]:ZnTPP 1:2. Moreover, it is reasonable to suggest that the stability of subsequent complexations is decreased with increasing the stoichiometry (K1 ≥ K2 ≥ K3 ≥ K4) as a result of lowering the number of accessible binding sites. To characterize the binding and to evaluate the association constants of cycHC–porphyrin complexes in the solution phase, UV-vis and 1H NMR titration experiments were carried out. Because there is no commonly available tool for the calculation of 1:3 and 1:4 binding equilibria, we developed our own script for the analysis of this specific system. While a 1:4 binding model for the cycHC[8]-based systems appeared to be too overparameterized to deliver stable results, a simpler 1:3 model was used for all systems studied. We suppose that this simplification has a minor impact on the obtained values because the K4 value is expected to be small in comparison to K1, K2, and K3 and the abundance of 1:4 complex is negligible. We focused on the cycHC[8]·ZnTPP complex as a representative example because it can be additionally utilized for anion binding.28 Thus, data from four UV-vis and three NMR titrations, covering the 0.6–5000 μM concentration range of ZnTPP were collected. Independent fitting of these data resulted in similar stepwise decreasing association constants; K1 > K2 > K3 (Table 1, see ESI† for fitting methodology). In addition, two isothermal titration calorimetry (ITC) experiments have been carried out and analyzed in terms of the sequential binding model, while the initial values for fitting were the association constants obtained from the UV-vis and NMR titrations. Thermodynamic parameters obtained using ITC indicated the independence of the binding sites, with very close values of binding enthalpies for each individual binding step (Table 1). In addition, these experiments unambiguously confirmed the initial assumption that K1 ≥ K2 ≥ K3. It must be noted that fairly strong total external binding constants can be reached due to the multiple site binding (Table 1).
:3 binding modela of cycHC[8]·ZnTPP obtained in DCM solutions by NMR, UV-vis, and ITC methods and their thermodynamic parameters ΔH and TΔS obtained from ITC at 293 K
| No. | K 1 (M−1) | K 2 (M−1) | K 3 (M−1) | log Ktotalb |
|
|---|---|---|---|---|---|
a
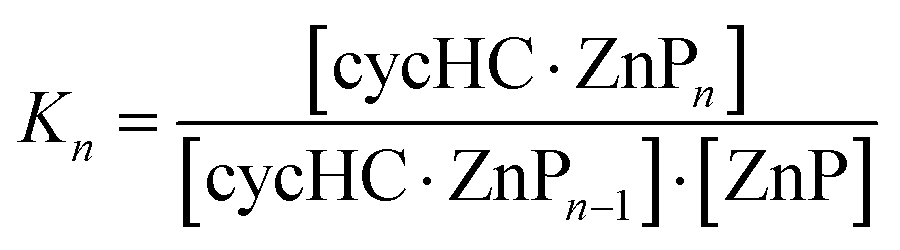 , where ZnP is Zn–porphyrin and n is stoichiometry.
b
K
total = K1·K2·K3. , where ZnP is Zn–porphyrin and n is stoichiometry.
b
K
total = K1·K2·K3.
|
|||||
| 1 | NMR | 1890 ± 60 | 1100 ± 80 | 50 ± 25 | 8.02 |
| 2 | UV-vis | 2070 ± 20 | 900 ± 20 | 430 ± 120 | 8.90 |
| 3 | ITC | 2280 ± 150 | 770 ± 20 | 400 ± 20 | 8.84 |
| 4 | ΔH (kJ mol−1) | −24.0 ± 0.15 | −26.8 ± 0.5 | −28.0 ± 0.4 | |
| 5 | TΔS (kJ mol−1) | −5.1 ± 0.6 | −10.5 ± 0.5 | −13.4 ± 0.4 | |
Because the binding mechanism for all supramolecular systems studied is essentially the same, just a comparison of the corresponding K1 values is sufficient to evaluate the complex stability. Therefore, other cycHCs and Zn–porphyrin complexes were studied to an extent necessary to obtain the K1 values (Table 2). In addition, the K1 values for mono-urea M1 and porphyrins were determined using the 1:1 binding model42,43 to reveal approximately 100 times weaker interaction in comparison to cycHCs. Bulkier M2 provided negligible binding (Fig. S22, ESI†). Analogous observations were made earlier by us in the study of external binding of a Brønsted acid to cycHC[6]s and M1.44 The significantly lower external binding between M1 and Zn–porphyrins is assumed to be caused by the lack of multiple-point interaction. A general trend in the stability of complexes is as follows: cycHC[6]·ZnTPP > cycHC[6]·ZnOEP > cycHC[8]·ZnTPP > cycHC[8]·ZnOEP ≫ M1·ZnTPP > M1·ZnOEP. The higher stability of ZnTPP over ZnOEP complexes clearly reflects the dependence of binding upon the electron-accepting ability of the Zn ion as a result of the presence of Ph groups in ZnTPP. The stability of cycHC[6] complexes over the cycHC[8] is not so evident and needs further investigations. Apparently, the clear differences in binding six and eight membered cycHC homologues in solution and solid phase reflect the strong influence of geometry on their interaction mode.
:3 binding model for cycHCs complexes and according to 1:1 binding model for M1 in DCM solutions
| No. | Complex | Titration method | |
|---|---|---|---|
| NMR | UV-vis | ||
|
a From three titrations obtained also K2 = 1900 ± 200 M−1, K3 = 190 ± 16 M−1.
b In addition, K2 = 950 ± 40 M−1 obtained.
c For all Kn see Table 1.
d Results evaluated using the 1:1 model from http://supramolecular.org.
|
|||
| 1 | cycHC[6]·ZnTPP | 5000 ± 200a | 5340 ± 60 |
| 2 | cycHC[6]·ZnOEP | 4200 ± 130b | 3070 ± 30 |
| 3 | cycHC[8]·ZnTPP | 1890 ± 60 | 2070 ± 20 |
| 4 | cycHC[8]·ZnOEP | 705 ± 18 | 880 ± 20 |
| 5 | M1·ZnTPP | 50.5 ± 0.8 | 57.4 ± 1.1 |
| 6 | M1·ZnOEP | — | 12.4 ± 0.3 |
In conclusion, for the first time, we demonstrated that zinc porphyrins can be effectively used to sense hemicucurbituril chirality. The used macrocycles have large association constants with porphyrins thanks to their suitable preorganization. The main interaction point is coordination between the electron-deficient porphyrin zinc ion and electron-rich carbonyl oxygen on the outer part of cycHCs. The collected data fully support the 1:3 cycHC:porphyrin stoichiometry as the major complexation process in solution. Chiroptical studies indicated an efficient chirality transfer process from chiral cycHCs to achiral zinc porphyrins via a chiral ring distortion mechanism. The observed chirogenic phenomenon opens further prospects to utilize the cycHCs's cavity available for another guest molecule to modulate induced chirality by formation of more complexed supramolecular systems and other molecular networks for sensing and/or catalytic applications.
The authors acknowledge Mohammed Hasan, Kamini Mishra, Karina Barsunova, Aleksandra Gluškova, Tatsiana Shalima, and Paul Kerner for experimental assistance and Pall Thordarson for useful discussions. We also thank the Estonian MER for financial support through Grants PUT692, PRG399, IUT 19-9; the ERDF through the CoE 3.2.0101.08-0017, CoE 2014- 2020.4.01.15-0013, CoE TK134; the H2020-FETOPEN, 828779 (INITIO); and the start-up research grant YZZ16005 from South-Central University for Nationalities; as well as the High-Performance Computing Center of the University of Tartu.
Conflicts of interest
There are no conflicts to declare.Notes and references
- H. Lu and X. P. Zhang, Chem. Soc. Rev., 2011, 40, 1899–1909 RSC.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed.
- W.-Y. Gao, M. Chrzanowski and S. Ma, Chem. Soc. Rev., 2014, 43, 5841–5866 RSC.
- S. Singh, A. Aggarwal, N. V. S. D. K. Bhupathiraju, G. Arianna, K. Tiwari and C. M. Drain, Chem. Rev., 2015, 115, 10261–10306 CrossRef CAS PubMed.
- R. Paolesse, S. Nardis, D. Monti, M. Stefanelli and C. Di Natale, Chem. Rev., 2017, 117, 2517–2583 CrossRef CAS PubMed.
- H. Lu and N. Kobayashi, Chem. Rev., 2016, 116, 6184–6261 CrossRef CAS PubMed.
- V. Borovkov, Symmetry, 2014, 6, 256–294 CrossRef CAS.
- G. A. Hembury, V. V. Borovkov and Y. Inoue, Chem. Rev., 2008, 108, 1–73 CrossRef CAS PubMed.
- V. V. Borovkov and Y. Inoue, in Supramolecular Chirality, ed. M. Crego-Calama and D. N. Reinhoudt, Springer Berlin Heidelberg, Berlin, Heidelberg, 2006, pp. 89–146 Search PubMed.
- P. Liu, P. Neuhaus, D. V. Kondratuk, T. S. Balaban and H. L. Anderson, Angew. Chem., Int. Ed., 2014, 53, 7770–7773 CrossRef CAS PubMed.
- T. Mizutani, T. Ema, T. Yoshida, Y. Kuroda and H. Ogoshi, Inorg. Chem., 1993, 32, 2072–2077 CrossRef CAS.
- H. Ogoshi and T. Mizutani, Acc. Chem. Res., 1998, 31, 81–89 CrossRef CAS.
- M. Balaz, M. D. Napoli, A. E. Holmes, A. Mammana, K. Nakanishi, N. Berova and R. Purrello, Angew. Chem., Int. Ed., 2005, 44, 4006–4009 CrossRef CAS PubMed.
- A. D’Urso, P. F. Nicotra, G. Centonze, M. E. Fragalà, G. Gattuso, A. Notti, A. Pappalardo, S. Pappalardo, M. F. Parisi and R. Purrello, Chem. Commun., 2012, 48, 4046–4048 RSC.
- A. D’Urso, N. Marino, M. Gaeta, M. S. Rizzo, D. A. Cristaldi, M. E. Fragalà, S. Pappalardo, G. Gattuso, A. Notti, M. F. Parisi, I. Pisagatti and R. Purrello, New J. Chem., 2017, 41, 8078–8083 RSC.
- E. Bellacchio, R. Lauceri, S. Gurrieri, L. M. Scolaro, A. Romeo and R. Purrello, J. Am. Chem. Soc., 1998, 120, 12353–12354 CrossRef CAS.
- R. Lauceri, A. Raudino, L. M. Scolaro, N. Micali and R. Purrello, J. Am. Chem. Soc., 2002, 124, 894–895 CrossRef CAS PubMed.
- R. Lauceri, G. F. Fasciglione, A. D’Urso, S. Marini, R. Purrello and M. Coletta, J. Am. Chem. Soc., 2008, 130, 10476–10477 CrossRef CAS PubMed.
- G. D. Luca, A. Romeo, L. M. Scolaro and R. F. Pasternack, Chem. Commun., 2010, 46, 389–391 RSC.
- I. Occhiuto, G. D. Luca, V. Villari, A. Romeo, N. Micali, R. F. Pasternack and L. M. Scolaro, Chem. Commun., 2011, 47, 6045–6047 RSC.
- I. G. Occhiuto, M. Samperi, M. Trapani, G. De Luca, A. Romeo, R. F. Pasternack and L. M. Scolaro, J. Inorg. Biochem., 2015, 153, 361–366 CrossRef CAS PubMed.
- M. Gaeta, D. Raciti, R. Randazzo, C. M. A. Gangemi, A. Raudino, A. D’Urso, M. E. Fragalà and R. Purrello, Angew. Chem., Int. Ed., 2018, 57, 10656–10660 CrossRef CAS PubMed.
- R. Randazzo, A. Mammana, A. D’Urso, R. Lauceri and R. Purrello, Angew. Chem., Int. Ed., 2008, 47, 9879–9882 CrossRef CAS PubMed.
- R. Aav, E. Shmatova, I. Reile, M. Borissova, F. Topić and K. Rissanen, Org. Lett., 2013, 15, 3786–3789 CrossRef CAS PubMed.
- M. Öeren, E. Shmatova, T. Tamm and R. Aav, Phys. Chem. Chem. Phys., 2014, 16, 19198–19205 RSC.
- E. Prigorchenko, M. Öeren, S. Kaabel, M. Fomitšenko, I. Reile, I. Järving, T. Tamm, F. Topić, K. Rissanen and R. Aav, Chem. Commun., 2015, 51, 10921–10924 RSC.
- S. Kaabel, J. Adamson, F. Topić, A. Kiesilä, E. Kalenius, M. Öeren, M. Reimund, E. Prigorchenko, A. Lõokene, H. J. Reich, K. Rissanen and R. Aav, Chem. Sci., 2017, 8, 2184–2190 RSC.
- N. N. Andersen, M. Lisbjerg, K. Eriksen and M. Pittelkow, Isr. J. Chem., 2018, 58, 435–448 CrossRef CAS.
- H.-J. Buschmann, A. Zielesny and E. Schollmeyer, J. Inclusion Phenom. Macrocyclic Chem., 2006, 54, 181–185 CrossRef CAS.
- M. Singh, E. Solel, E. Keinan and O. Reany, Chem. – Eur. J., 2015, 21, 536–540 CrossRef CAS PubMed.
- M. Lisbjerg, B. M. Jessen, B. Rasmussen, B. E. Nielsen, A. Ø. Madsen and M. Pittelkow, Chem. Sci., 2014, 5, 2647–2650 RSC.
- T.-L. Teo, M. Vetrichelvan and Y.-H. Lai, Org. Lett., 2003, 5, 4207–4210 CrossRef CAS PubMed.
- B. P. Borah and J. Bhuyan, J. Chem. Sci., 2018, 130, 117 CrossRef.
- Y. Gao, X. Zhang, C. Ma, X. Li and J. Jiang, J. Am. Chem. Soc., 2008, 130, 17044–17052 CrossRef CAS PubMed.
- B. M. J. M. Suijkerbuijk, D. M. Tooke, A. L. Spek, G. van Koten and R. J. M. Klein Gebbink, Chem. – Asian J., 2007, 2, 889–903 CrossRef CAS PubMed.
- J. Bhuyan and S. Sarkar, Cryst. Growth Des., 2011, 11, 5410–5414 CrossRef CAS.
- C. K. Schauer, O. P. Anderson, S. S. Eaton and G. R. Eaton, Inorg. Chem., 1985, 24, 4082–4086 CrossRef CAS.
- M. Schmittel and S. K. Samanta, J. Org. Chem., 2010, 75, 5911–5919 CrossRef CAS PubMed.
- L. Favereau, A. Cnossen, J. B. Kelber, J. Q. Gong, R. M. Oetterli, J. Cremers, L. M. Herz and H. L. Anderson, J. Am. Chem. Soc., 2015, 137, 14256–14259 CrossRef CAS PubMed.
- F. Ulatowski, K. Dąbrowa, T. Bałakier and J. Jurczak, J. Org. Chem., 2016, 81, 1746–1756 CrossRef CAS PubMed.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- D. B. Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792–12805 RSC.
- E. Prigorchenko, S. Kaabel, T. Narva, A. Baškir, M. Fomitšenko, J. Adamson, I. Järving, K. Rissanen, T. Tamm and R. Aav, Chem. Commun., 2019, 55, 9307–9310 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1949778 and 1949779. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc07150d |
| This journal is © The Royal Society of Chemistry 2019 |