
Exogenous-oxidant-free electrochemical oxidative C–H phosphonylation with hydrogen evolution†
Yong
Yuan‡
 ab,
Jin
Qiao‡
a,
Yangmin
Cao
a,
Jingmei
Tang
a,
Mengqin
Wang
a,
Guojuan
Ke
a,
Yichen
Lu
a,
Xue
Liu
a and
Aiwen
Lei
*ab
ab,
Jin
Qiao‡
a,
Yangmin
Cao
a,
Jingmei
Tang
a,
Mengqin
Wang
a,
Guojuan
Ke
a,
Yichen
Lu
a,
Xue
Liu
a and
Aiwen
Lei
*ab
aNational Research Center for Carbohydrate Synthesis, Jiangxi Normal University, Nanchang 330022, P. R. China. E-mail: aiwenlei@whu.edu.cn
bCollege of Chemistry and Molecular Sciences, Institute for Advanced Studies (IAS), Wuhan University, Wuhan 430072, P. R. China
First published on 15th March 2019
Abstract
We herein report a versatile and environmentally friendly electrochemical oxidative C–H phosphonylation protocol. This protocol features a broad substrate scope; not only C(sp2)–H phosphonylation, but also C(sp3)–H phosphonylation is tolerated well under exogenous-oxidant-free and metal catalyst-free electrochemical oxidation conditions.
Phosphorus containing compounds are of much importance because they are widely found in a vast number of natural products, biological compounds, and agricultural chemicals,1 as well as represent outstanding synthetic intermediates for the preparation of complex organic molecules.2 As such, methods for the preparation of these valuable molecules are very important synthetic technologies. Over the past decades, considerable works have been conducted on the synthesis of significant phosphorus containing compounds.3 Compared with conventional synthetic methods that employed pre-functionalized aryl halides or arylboronic acids as the starting materials,4 direct C–H phosphonylation represents a much more attractive strategy for C–P bond formation.5 Although this strategy avoided the use of pre-functionalized starting materials and has achieved considerable development, most of the reported reactions still suffer from some drawbacks, such as the need for metal catalysts or stoichiometric oxidants. Therefore, developing a much more elegant method for producing phosphorus containing compounds under metal catalyst-free and exogenous-oxidant-free reaction conditions is urgently needed.
As an ideal alternative to chemical oxidants, electrochemical anodic oxidation provides an efficient and environmentally benign synthetic method for C–H functionalization.6 In this context, considerable progress has been made, such as in electrochemical oxidative C–H amination,7 oxygenation,8 thiolation,9 alkylation,10etc.11 Nevertheless, electrochemical oxidative C–H phosphonylation is still underexplored.12 As part of our recent research interest in the area of electrochemical oxidative C–H functionalization, we herein report an elegant and versatile electrochemical oxidative C–H phosphonylation protocol. Overall, the notable features of this reaction include the following: (1) neither a metal catalyst nor an exogenous-oxidant/additive is required; and (2) this protocol features a broad substrate scope – not only C(sp2)–H phosphonylation, but also C(sp3)–H phosphonylation is tolerated well under electrochemical conditions.
We initiated our studies by investigating the reaction of 2-phenylimidazo[1,2-a]pyridine (1a) with triethyl phosphite (2a) because the imidazopyridine unit is a widely existing substructure in many commercially available drugs and the resultant C–H phosphonylation products may also have a potential utility in pharmaceutical chemistry (Table 1).13 To our delight, when the reaction was performed in an undivided cell employing a carbon rod as the anode and a platinum plate as the cathode as well as using nBu4NBF4 as electrolyte, the reaction worked smoothly in acetonitrile under a constant current of 4 mA, providing the desired C–H phosphonylation product 3a in 70% NMR yield (Table 1, entry 1). At the same time, H2 was also detected by GC-TCD. Further explorations showed that nBu4NPF6 was a better choice for electrolyte, affording 3a in 77% NMR yield and 70% isolated yield (Table 1, entries 2 and 3). In comparison, conducting the reaction with a nickel plate cathode or a carbon cloth anode showed decreased reaction efficiency (Table 1, entries 4 and 5). In addition, a control experiment showed that the constant current was crucial for the reaction (Table 1, entry 6) (for more details about optimization of reaction conditions, see the ESI†). Note that for some conditions with low yields, 1a and its homo-coupling product were often observed.
|
|
||
|---|---|---|
| Entry | Electrolysis conditions | Yieldb (%) |
| a Reaction conditions: carbon rod anode, platinum plate cathode, undivided cell, 1a (0.3 mmol), 2a (0.6 mmol), nBu4NBF4 (0.1 mmol), MeCN (10 mL), 50 °C, 4 mA, 6 h (3 F mol−1). b Yields were determined by 31P NMR using PPh3 as an internal standard. c Isolated yield. | ||
| 1 | Carbon rod anode, platinum plate cathode, nBu4NBF4 | 70 |
| 2 | Entry 1 but nBu4NCIO4 as electrolyte | 54 |
| 3 | Entry 1 but nBu4NPF6 as electrolyte | 77, 70c |
| 4 | Entry 3 but nickel plate cathode | 61 |
| 5 | Entry 3 but carbon cloth anode | 49 |
| 6 | Entry 3 but no electric current | 0 |
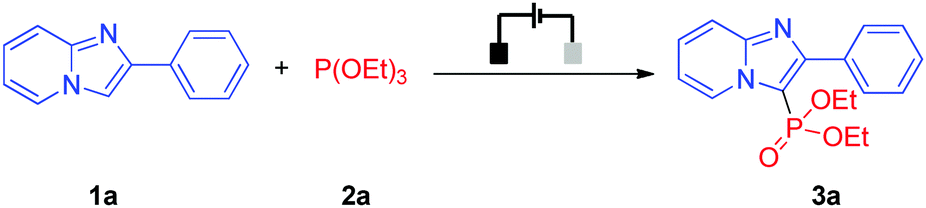
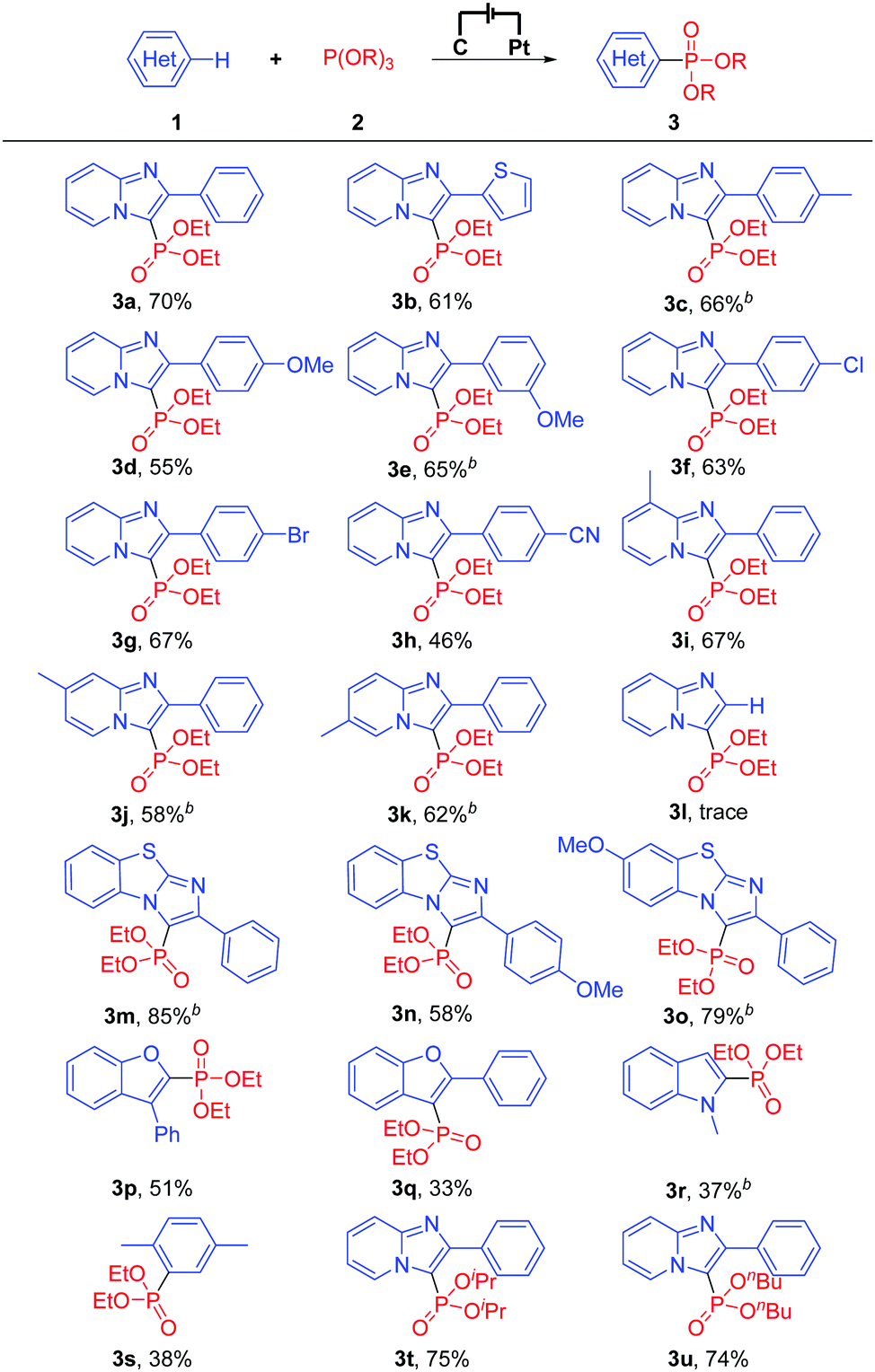
With the optimized reaction conditions in hand, the scope of the exogenous-oxidant-free electrochemical oxidative C(sp2)–H phosphonylation was subsequently explored. Initially, different heteroarenes were investigated. As shown in Table 2, 2-phenylimidazo-[1,2-a]pyridine and 2-thiophenylimidazo-[1,2-a]pyridine could be transformed into the C–H phosphonylation products in 70% and 61% yields, respectively (Table 2, 3a and 3b). 2-Phenylimidazo-[1,2-a]pyridines bearing both electron-donating groups and weak electron-withdrawing groups at the 2-phenyl moiety afforded the products in moderate to good yields (Table 2, 3c–3g). In comparison, 2-phenylimidazo-[1,2-a]pyridine with a strong electron-withdrawing group at the 2-phenyl moiety delivered the C(sp2)–H phosphonylation product in moderate yield (Table 2, 3h). When 8-methyl, 7-methyl, and 6-methyl substituted imidazopyridines were treated with triethyl phosphite (2a), the corresponding products could still be obtained in moderate to good yields (Table 2, 3i–3k). However, when using imidazo[1,2-a]pyridine as the substrate, only a trace amount of the C(sp2)–H phosphonylation product (Table 2, 3l) was obtained; the reason for this may be that imidazo[1,2-a]pyridine has a relatively high oxidation potential and is not easily oxidized. It is worth noting that other heteroarenes such as important benzo[d]-imidazo[2,1-b]thiazole derivatives, 3-phenylbenzofuran, 2-phenylbenzofuran, N-methylindole, and p-xylene were also suitable for this reaction, affording the respective products in 33% to 85% yields (Table 2, 3m–3s). Other phosphonylation reagents for this electrochemical oxidative C(sp2)–H phosphonylation protocol were investigated as well. Delightfully, despite the bulky steric hindrance or a long alkyl chain, P(OiPr)3 and P(OnBu)3 were well suitable for the reaction and afforded the C(sp2)–H phosphonylation products in 75% and 74% yields, respectively (Table 2, 3t and 3u). Note that for some examples with longer durations of electrolysis (Table 2, 3c, 3e, 3j, 3k, 3m, 3o, and 3r), the reason for prolonging the duration of electrolysis is mainly to increase the conversion rates, thereby further improving the yields.
| a Reaction conditions: carbon rod anode, platinum plate cathode, undivided cell, 4 (0.3 mmol), 2 (0.6 mmol), nBu4NPF6 (0.1 mmol), MeCN (10 mL), 70 °C, 4 mA, 6 h (3 F mol−1), isolated yield. b 50 °C. c Yields were determined by 31P NMR using PPh3 as an internal standard. d 4 (0.6 mmol) and 2 (0.3 mmol). |
|---|
|
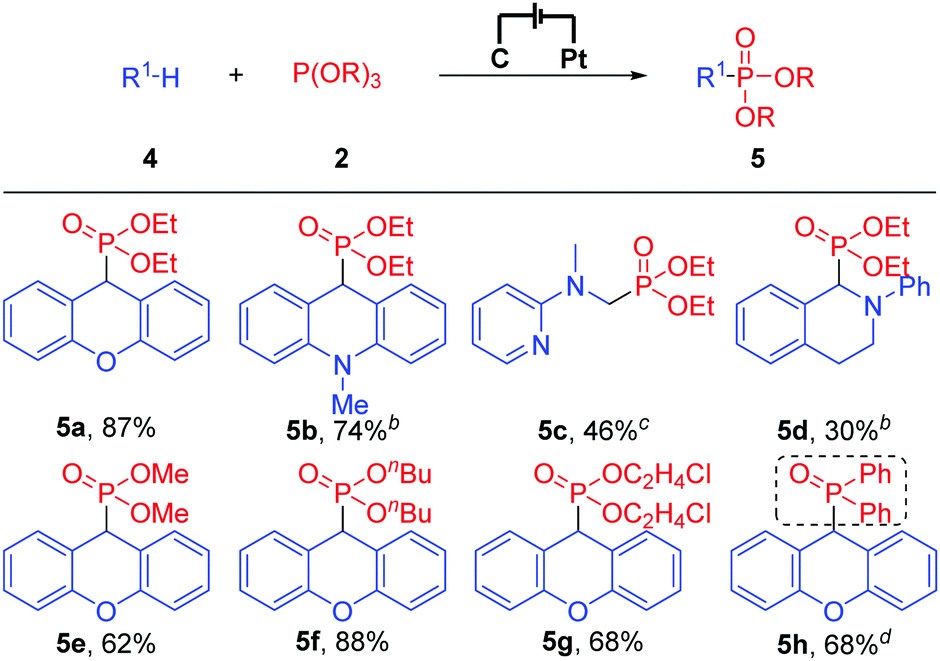
Subsequently, we turned our attention to the more challenging C(sp3)–H phosphonylation because no examples have been reported in which C(sp2)–H phosphonylation and C(sp3)–H phosphonylation were accomplished concurrently under similar or the same reaction conditions. Gratefully, under the electrochemical conditions, C(sp3)–H phosphonylation was also accomplished. As shown in Table 3, when xanthene and N-methyl-9,10-dihydroacridine were employed as substrates, the reaction worked smoothly and afforded the corresponding C(sp3)–H phosphonylation products in 87% and 74% yields, respectively (Table 3, 5a and 5b). Besides, an N,N-dimethylaniline derivative and N-phenyl tetrahydroisoquinoline were also compatible with the reaction conditions (Table 3, 5c and 5d). Notably, other phosphonylation reagents such as P(OMe)3, P(OnBu)3, P(OC2H4Cl)3, and even diphenylphosphine oxide all were suitable substrates and afforded the C(sp3)–H phosphonylation products in good to high yields (Table 3, 5e–5h). Unfortunately, only a trace amount of the product was detected when P(OiPr)3 was applied as a substrate, possibly because of its bulky steric hindrance.
To demonstrate the practicality of this electrochemical oxidative C–H phosphonylation, reactions of P(OEt)3 with 1a and 4a on a 6.0 mmol scale were performed. The reactions worked smoothly and afforded the corresponding C–H phosphonylation products in 55% and 50% yields (for more details about gram scale synthesis, see the ESI†), respectively.
To shed light on this exogenous-oxidant-free electrochemical oxidative C–H phosphonylation mechanism, cyclic voltammetry (CV) experiments on 1a, 2a, and 4a were performed, respectively. The oxidation peaks of 1a and 4a were observed at 1.68 V and 2.28 V, respectively, while no obvious oxidation peak of 2a was observed at 0–2.5 V (for more details about cyclic voltammetry (CV) experiments, see the ESI†). This result suggests that our electrochemical oxidative C–H phosphonylation might begin with the oxidation of 1a or 4a at the anode. To further gain some insights into the mechanism, the reactions of 1a and 4a with HPO(OEt)2 or PO(OEt)3 were performed (Scheme 1), respectively. Besides, the reaction of 4a with HPO(OEt)2 afforded the desired product in 12% yield, and no desired products were detected in any other reactions. These results indicated that neither HPO(OEt)2 nor PO(OEt)3 is the reaction intermediate, and once again indicated that our electrochemical oxidative C–H phosphonylation really begins with the oxidation of 1a or 4a instead of 2a.
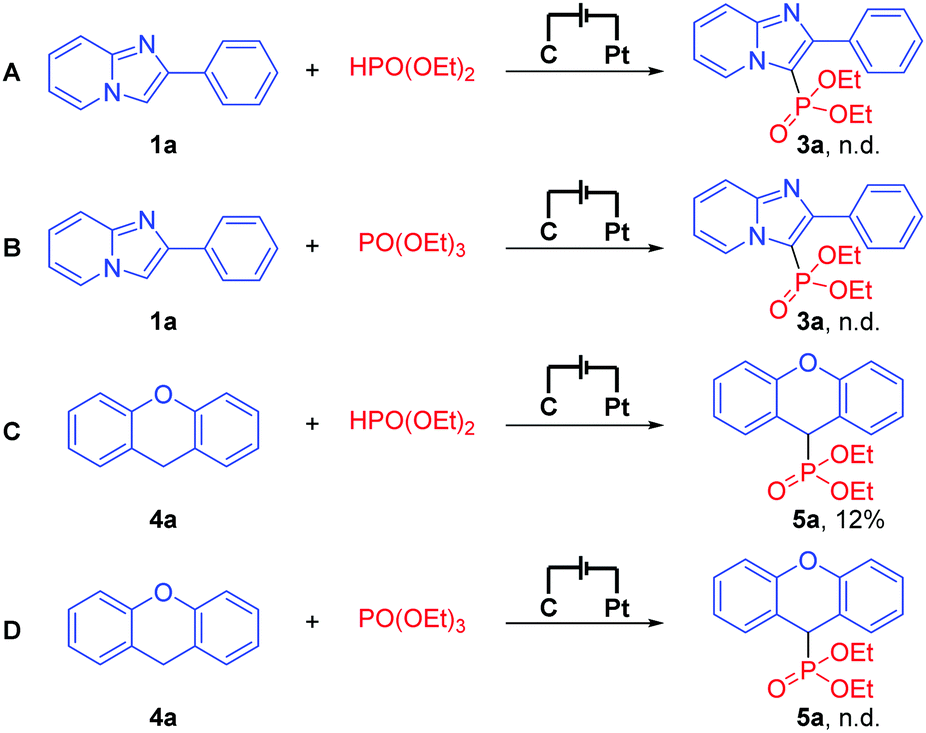 | ||
| Scheme 1 Control experiments. | ||
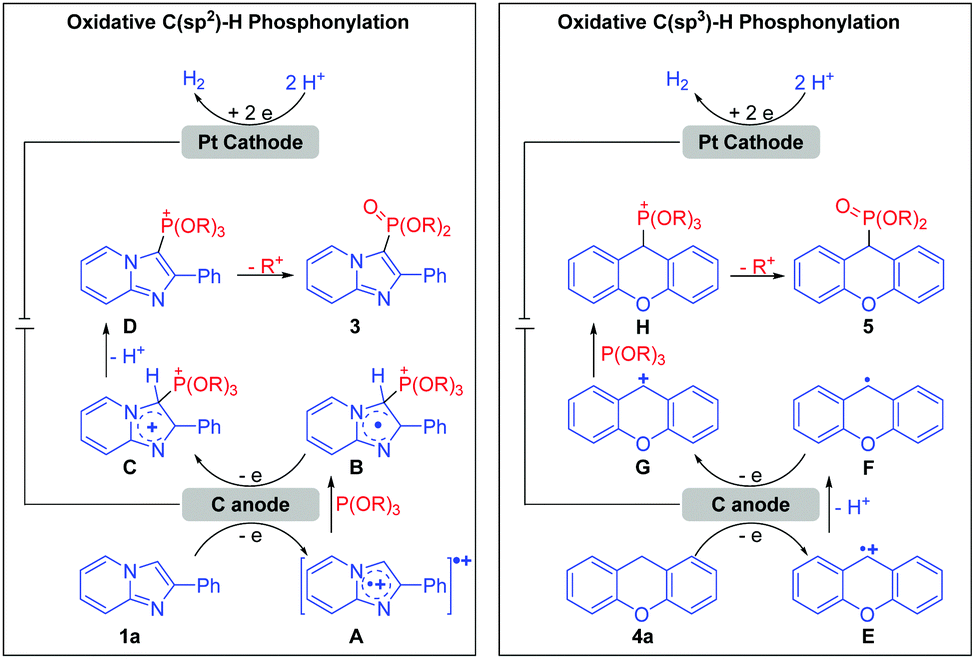
Based on the studies above and previous reports,3b,5b a plausible mechanism for this electrochemical oxidative C–H phosphonylation is proposed in Scheme 2. For C(sp2)–H phosphonylation, 1a is first oxidized at the anode to form the radical cation intermediate A, which is then captured by P(OR)3 (2) to deliver adduct B. B undergoes further anodic oxidation and dehydrogenation, resulting in the formation of phosphorus cation intermediate D. Finally, the ensuing dealkylation of D forms the C(sp2)–H phosphonylation product 3. At the same time, protons were reduced at the cathode to form H2. With respect to C(sp3)–H phosphonylation, the generated intermediate E does not directly react with P(OR)3 (2), but sequentially loses one proton and one electron to afford intermediate G; G is then captured by P(OR)3 (2) to deliver the phosphorus cation intermediate H. Subsequently, as with the C(sp2)–H phosphonylation, the ensuing dealkylation of H leads to the C(sp3)–H phosphonylation product 5.
 | ||
| Scheme 2 Proposed mechanism. | ||
In summary, we have disclosed a versatile and environmentally friendly electrochemical oxidative C–H phosphonylation protocol. Under exogenous-oxidant-free and metal catalyst-free electrochemical oxidation conditions, a series of complex and significant phosphorus containing compounds were constructed in moderate to high yields accompanied by hydrogen evolution. Notably, during the reaction, neither an exogenous oxidant nor a metal catalyst is required. With respect to the substrate scope, not only C(sp2)–H phosphonylation, but also C(sp3)–H phosphonylation is tolerated well under the electrochemical conditions.
This work was supported by the National Natural Science Foundation of China (21520102003, 21702081, 21702150), the Hubei Province Natural Science Foundation of China (2017CFA010), and the Jiangxi Provincial Education Department Foundation (GJJ160325). The Program of Introducing Talents of Discipline to Universities of China (111 Program) is also acknowledged.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) M. Pramanik, N. Chatterjee, S. Das, K. D. Saha and A. Bhaumik, Chem. Commun., 2013, 49, 9461–9463 RSC; (b) X. Chen, D. J. Kopecky, J. Mihalic, S. Jeffries, X. Min, J. Heath, J. Deignan, S. Lai, Z. Fu, C. Guimaraes, S. Shen, S. Li, S. Johnstone, S. Thibault, H. Xu, M. Cardozo, W. Shen, N. Walker, F. Kayser and Z. Wang, J. Med. Chem., 2012, 55, 3837–3851 CrossRef CAS PubMed; (c) E. A. Wydysh, S. M. Medghalchi, A. Vadlamudi and C. A. Townsend, J. Med. Chem., 2009, 52, 3317–3327 CrossRef CAS PubMed; (d) K. Moonen, I. Laureyn and C. V. Stevens, Chem. Rev., 2004, 104, 6177–6216 CrossRef CAS PubMed.
- (a) R. M. Yebeutchou, F. Tancini, N. Demitri, S. Geremia, R. Mendichi and E. Dalcanale, Angew. Chem., Int. Ed., 2008, 47, 4504–4508 CrossRef CAS PubMed; (b) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029–3070 CrossRef CAS PubMed; (c) M. R. Harnden, A. Parkin, M. J. Parratt and R. M. Perkins, J. Med. Chem., 1993, 36, 1343–1355 CrossRef CAS; (d) T. Minami and J. Motoyoshiya, Synthesis, 1992, 333–349 CrossRef CAS.
- (a) X. Ma, Q. Xu, H. Li, C. Su, L. Yu, X. Zhang, H. Cao and L.-B. Han, Green Chem., 2018, 20, 3408–3413 RSC; (b) L. Niu, J. Liu, H. Yi, S. Wang, X.-A. Liang, A. K. Singh, C.-W. Chiang and A. Lei, ACS Catal., 2017, 7, 7412–7416 CrossRef CAS; (c) R. E. Islas and J. J. García, ChemCatChem, 2017, 9, 4125–4131 CrossRef CAS; (d) S. Wang, D. Qiu, F. Mo, Y. Zhang and J. Wang, J. Org. Chem., 2016, 81, 11603–11611 CrossRef CAS PubMed; (e) S.-Y. Chen, R.-S. Zeng, J.-P. Zou and O. T. Asekun, J. Org. Chem., 2014, 79, 1449–1453 CrossRef CAS PubMed.
- (a) I. Ghosh, R. S. Shaikh and B. König, Angew. Chem., Int. Ed., 2017, 56, 8544–8549 CrossRef CAS PubMed; (b) R. S. Shaikh, S. J. S. Düsel and B. König, ACS Catal., 2016, 6, 8410–8414 CrossRef CAS; (c) R. A. Dhokale and S. B. Mhaske, Org. Lett., 2013, 15, 2218–2221 CrossRef CAS PubMed; (d) G. Yang, C. Shen, L. Zhang and W. Zhang, Tetrahedron Lett., 2011, 52, 5032–5035 CrossRef CAS; (e) C. S. Demmer, N. Krogsgaard-Larsen and L. Bunch, Chem. Rev., 2011, 111, 7981–8006 CrossRef CAS PubMed; (f) Y.-L. Zhao, Y. Li, S.-M. Li, Y.-G. Zhou, F.-Y. Sun, L.-X. Gao and F.-S. Han, Adv. Synth. Catal., 2011, 353, 1543–1550 CrossRef CAS; (g) R. Zhuang, J. Xu, Z. Cai, G. Tang, M. Fang and Y. Zhao, Org. Lett., 2011, 13, 2110–2113 CrossRef CAS PubMed.
- (a) J. Wang, J. Li, Y. Wei, J. Yang and C. Huo, Org. Chem. Front., 2018, 5, 3534–3537 RSC; (b) Q. Chen, X. Wang, G. Yu, C. Wen and Y. Huo, Org. Chem. Front., 2018, 5, 2652–2656 RSC; (c) R. S. Shaikh, I. Ghosh and B. König, Chem. – Eur. J., 2017, 23, 12120–12124 CrossRef CAS PubMed; (d) Q. Gui, L. Hu, X. Chen, J. Liu and Z. Tan, Chem. Commun., 2015, 51, 13922–13924 RSC; (e) M. Yadav, S. Dara, V. Saikam, M. Kumar, S. K. Aithagani, S. Paul, R. A. Vishwakarma and P. P. Singh, Eur. J. Org. Chem., 2015, 6526–6533 CrossRef CAS; (f) D. Zhao, C. Nimphius, M. Lindale and F. Glorius, Org. Lett., 2013, 15, 4504–4507 CrossRef CAS PubMed.
- (a) N. Sauermann, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Catal., 2018, 8, 7086–7103 CrossRef CAS; (b) M. D. Kärkäs, Chem. Soc. Rev., 2018, 47, 5786–5865 RSC; (c) Y. Zhao and W. Xia, Chem. Soc. Rev., 2018, 47, 2591–2608 RSC; (d) C. Ma, P. Fang and T.-S. Mei, ACS Catal., 2018, 8, 7179–7189 CrossRef CAS; (e) S. Tang, Y. Liu and A. Lei, Chem, 2018, 4, 27–45 CrossRef CAS; (f) A. Jutand, Chem. Rev., 2008, 108, 2300–2347 CrossRef CAS PubMed.
- (a) P. Qian, Z. Yan, Z. Zhou, K. Hu, J. Wang, Z. Li, Z. Zha and Z. Wang, Org. Lett., 2018, 20, 6359–6363 CrossRef PubMed; (b) Y. Qiu, J. Struwe, T. H. Meyer, J. C. A. Oliveira and L. Ackermann, Chem. – Eur. J., 2018, 24, 12784–12789 CrossRef CAS PubMed; (c) X. Gao, P. Wang, L. Zeng, S. Tang and A. Lei, J. Am. Chem. Soc., 2018, 140, 4195–4199 CrossRef CAS PubMed; (d) X. Hu, G. Zhang, F. Bu, L. Nie and A. Lei, ACS Catal., 2018, 8, 9370–9375 CrossRef CAS; (e) S. Tang, S. Wang, Y. Liu, H. Cong and A. Lei, Angew. Chem., Int. Ed., 2018, 57, 4737–4741 CrossRef CAS PubMed; (f) Q.-L. Yang, X.-Y. Wang, J.-Y. Lu, L.-P. Zhang, P. Fang and T.-S. Mei, J. Am. Chem. Soc., 2018, 140, 11487–11494 CrossRef CAS PubMed; (g) N. Sauermann, R. Mei and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 5090–5094 CrossRef CAS PubMed; (h) M.-Y. Lin, K. Xu, Y.-Y. Jiang, Y.-G. Liu, B.-G. Sun and C.-C. Zeng, Adv. Synth. Catal., 2018, 360, 1665–1672 CrossRef CAS; (i) J. Wu, Y. Zhou, Y. Zhou, C.-W. Chiang and A. Lei, ACS Catal., 2017, 7, 8320–8323 CrossRef CAS; (j) W.-J. Gao, W.-C. Li, C.-C. Zeng, H.-Y. Tian, L.-M. Hu and R. D. Little, J. Org. Chem., 2014, 79, 9613–9618 CrossRef CAS PubMed; (k) T. Morofuji, A. Shimizu and J.-I. Yoshida, J. Am. Chem. Soc., 2014, 136, 4496–4499 CrossRef CAS PubMed; (l) S.-K. Zhang, R. C. Samanta, N. Sauermann and L. Ackermann, Chem. – Eur. J., 2018, 24, 19166–19170 CrossRef CAS PubMed; (m) Y. Yu, Y. Yuan, H. Liu, M. He, M. Yang, P. Liu, B. Yu, X. Dong and A. Lei, Chem. Commun., 2019, 55, 1809–1812 RSC.
- (a) A. Shao, N. Li, Y. Gao, J. Zhan, C.-W. Chiang and A. Lei, Chin. J. Chem., 2018, 36, 619–624 CrossRef CAS; (b) X.-Z. Tao, J.-J. Dai, J. Zhou, J. Xu and H.-J. Xu, Chem. – Eur. J., 2018, 24, 6932–6935 CrossRef CAS PubMed; (c) S. Zhang, L. Li, H. Wang, Q. Li, W. Liu, K. Xu and C. Zeng, Org. Lett., 2018, 20, 252–255 CrossRef CAS PubMed; (d) L. Li, Q. Yang, Z. Jia and S. Luo, Synthesis, 2018, 2924–2929 CAS; (e) F. Wang, M. Rafiee and S. S. Stahl, Angew. Chem., Int. Ed., 2018, 57, 6686–6690 CrossRef CAS PubMed; (f) Q.-L. Yang, Y.-Q. Li, C. Ma, P. Fang, X.-J. Zhang and T.-S. Mei, J. Am. Chem. Soc., 2017, 139, 3293–3298 CrossRef CAS PubMed; (g) F. Xu, X.-Y. Qian, Y.-J. Li and H.-C. Xu, Org. Lett., 2017, 19, 6332–6335 CrossRef CAS PubMed; (h) Y.-Q. Li, Q.-L. Yang, P. Fang, T.-S. Mei and D. Zhang, Org. Lett., 2017, 19, 2905–2908 CrossRef CAS PubMed; (i) N. Sauermann, T. H. Meyer, C. Tian and L. Ackermann, J. Am. Chem. Soc., 2017, 139, 18452–18455 CrossRef CAS PubMed; (j) L. Meng, J. Su, Z. Zha, L. Zhang, Z. Zhang and Z. Wang, Chem. – Eur. J., 2013, 19, 5542–5545 CrossRef CAS PubMed.
- (a) Y. Yuan, Y. Cao, J. Qiao, Y. Lin, X. Jiang, Y. Weng, S. Tang and A. Lei, Chin. J. Chem., 2019, 37, 49–52 CrossRef CAS; (b) Y. Yuan, Y. Yu, J. Qiao, P. Liu, B. Yu, W. Zhang, H. Liu, M. He, Z. Huang and A. Lei, Chem. Commun., 2018, 54, 11471–11474 RSC; (c) A. A. Folgueiras-Amador, X.-Y. Qian, H.-C. Xu and T. Wirth, Chem. – Eur. J., 2018, 24, 487–491 CrossRef CAS PubMed; (d) X.-Y. Qian, S.-Q. Li, J. Song and H.-C. Xu, ACS Catal., 2017, 7, 2730–2734 CrossRef CAS; (e) P. Wang, S. Tang, P. Huang and A. Lei, Angew. Chem., Int. Ed., 2017, 56, 3009–3013 CrossRef CAS PubMed; (f) P. Wang, S. Tang and A. Lei, Green Chem., 2017, 19, 2092–2095 RSC; (g) M.-L. Feng, L.-Y. Xi, S.-Y. Chen and X.-Q. Yu, Eur. J. Org. Chem., 2017, 2746–2750 CrossRef CAS; (h) S. H. Kim, K. H. Kim, J. W. Lim and J. N. Kim, Tetrahedron Lett., 2014, 55, 531–534 CrossRef CAS; (i) V. A. Kokorekin, V. L. Sigacheva and V. A. Petrosyan, Tetrahedron Lett., 2014, 55, 4306–4309 CrossRef CAS.
- (a) Z.-J. Wu, S.-R. Li, H. Long and H.-C. Xu, Chem. Commun., 2018, 54, 4601–4604 RSC; (b) Q.-L. Yang, C.-Z. Li, L.-W. Zhang, Y.-Y. Li, X. Tong, X.-Y. Wu and T.-S. Mei, Organometallics, 2019 DOI:10.1021/acs.organomet.8b00550; (c) C. Ma, C.-Q. Zhao, Y.-Q. Li, L.-P. Zhang, X.-T. Xu, K. Zhang and T.-S. Mei, Chem. Commun., 2017, 53, 12189–12192 RSC; (d) Z.-J. Wu and H.-C. Xu, Angew. Chem., Int. Ed., 2017, 56, 4734–4738 CrossRef CAS PubMed; (e) N. Fu, L. Li, Q. Yang and S. Luo, Org. Lett., 2017, 19, 2122–2125 CrossRef CAS PubMed; (f) H. Gao, Z. Zha, Z. Zhang, H. Ma and Z. Wang, Chem. Commun., 2014, 50, 5034–5036 RSC; (g) Y. B. Dudkina, D. Y. Mikhaylov, T. V. Gryaznova, O. G. Sinyashin, D. A. Vicic and Y. H. Budnikova, Eur. J. Org. Chem., 2012, 2114–2117 CrossRef CAS.
- (a) A. Wiebe, S. Lips, D. Schollmeyer, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2017, 56, 14727–14731 CrossRef CAS PubMed; (b) X. Zhang, C. Wang, H. Jiang and L. Sun, Chem. Commun., 2018, 54, 8781–8784 RSC.
- (a) F. Effenberger and H. Kottmann, Tetrahedron, 1985, 41, 4171–4182 CrossRef CAS; (b) T. V. Grayaznova, Y. B. Dudkina, D. R. Islamov, O. N. Kataeva, O. G. Sinyashin, D. A. Vicic and Y. H. Budnikova, J. Organomet. Chem., 2015, 785, 68–71 CrossRef CAS.
- (a) C. Hamdouchi, J. de Blas, M. del Prado, J. Gruber, B. A. Heinz and L. Vance, J. Med. Chem., 1999, 42, 50–59 CrossRef CAS PubMed; (b) L. Almirante, L. Polo, A. Mugnaini, E. Provinciali, P. Rugarli, A. Biancotti, A. Gamba and W. Murmann, J. Med. Chem., 1965, 8, 305–312 CrossRef CAS PubMed; (c) S. Boggs, V. I. Elitzin, K. Gudmundsson, M. T. Martin and M. J. Sharp, Org. Process Res. Dev., 2009, 13, 781–785 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc00975b |
| ‡ Yong Yuan and Jin Qiao contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |