
First total synthesis of ent-asperparaline C and assignment of the absolute configuration of asperparaline C†
Irena
Dokli
 ,
Radek
Pohl
,
Blanka
Klepetářová
and
Ullrich
Jahn
*
,
Radek
Pohl
,
Blanka
Klepetářová
and
Ullrich
Jahn
*
Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo Náměstí 2, 16610 Prague 6, Czech Republic. E-mail: ullrich.jahn@uochb.cas.cz
First published on 8th March 2019
Abstract
The first asymmetric total synthesis of a member of the asperparaline family was accomplished and the unknown absolute configuration of asperparaline C has been determined to be all-(S). The key steps of the synthesis are an oxidative radical cyclization, a selective reduction of one of the tertiary amides, a singlet oxygen Diels–Alder reaction and a reductive spirocyclization.
The asperparalines A–C and 16-oxoaspergillimide (1–4) are a small subfamily of fungal metabolites possessing a bridged diazabicyclo[2.2.2]octanone core, which are a privileged part of the large family of alkaloids with a central diketopiperazine unit (Fig. 1).1 The asperparalines are special, since they contain a unique 3-spirosuccinimide motif instead of the common indole or spirooxindole unit, which is biosynthetically presumably formed by oxidative degradation of an initially present indole ring.2 Asperparalines A–C were isolated by Hayashi and co-workers from Aspergillus japonicus JV-23.3,4 Asperparaline A (aspergillimide) and 16-oxoaspergillimide were independently isolated by Everett and co-workers from Aspergillus sp. IMI 337664.5 Asperparalines A–C have strong paralytic effects on silkworms whereas asperparaline A and 16-oxoaspergillimide show anthelmintic activity against Trichostrongylus colubriformis. A recent study to determine their mode of action revealed that asperparaline A strongly and selectively blocks insect nicotinic acetylcholine receptors.6,7
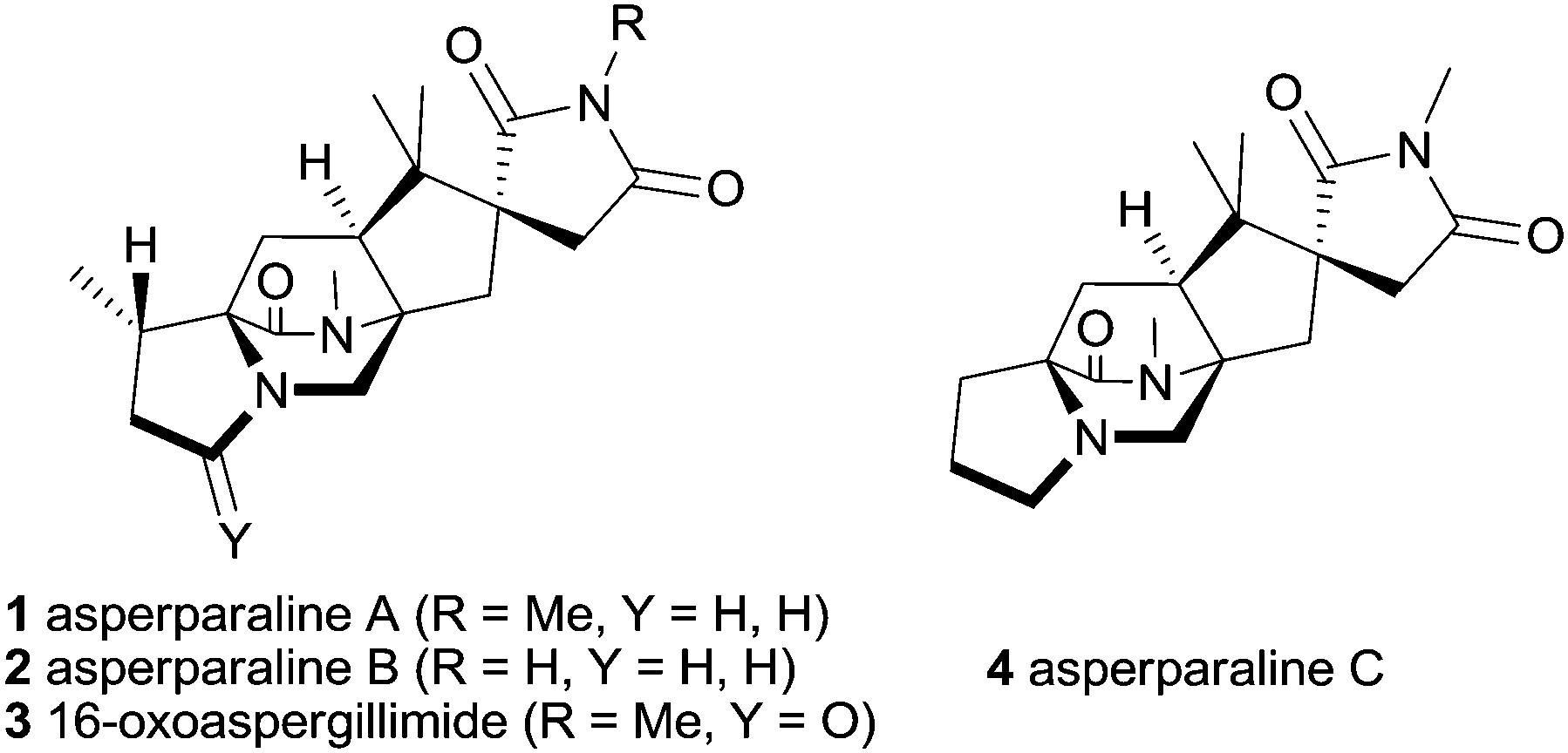 | ||
| Fig. 1 Asperparalines A–C and 16-oxoaspergillimide (1–4). | ||
The three-dimensional structure and relative configuration of asperparaline A (1) was determined by X-ray crystallography, and the structure of 2–4 was deduced on this basis.4 However, their absolute configurations are still unassigned. That relates to their proposed biosynthesis, which is related to the paraherquamides,2 in which both amino acid-derived stereogenic centers are planarized to an azadiene unit, which subsequently undergoes an intramolecular Diels–Alder reaction with a reverse prenyl group located at the indole unit of tryptophan forming the diazabicyclo[2.2.2]octane core. Therefore, total synthesis remains the only strategy to determine their absolute configuration, but so far, all members of the asperparaline family withstood multiple attempts toward their total synthesis.8–11 This can be traced to their challenging structural elements, such as the three quaternary carbon atoms at the spiro and annulated cyclopentane ring bearing three stereogenic centers, the unusual spiro-succinimide unit, the syn configuration at the bridging bicycle, and the partially reduced 2,5-diketopiperazine scaffold at the more hindered position of the tetracyclic core.
The (di)ketopiperazine motif as a privileged scaffold in biology or medicinal chemistry12 attracted our attention not only from the viewpoint of total synthesis of diketopiperazine alkaloids.13 We recently developed a unified approach to diverse DKP frameworks exploiting the persistent radical effect (PRE). This atom-economic process was shown to work efficiently for a formal total synthesis of bicyclomycin,14 several stephacidin intermediates,15 and a synthetic approach to asperparaline C, in which we failed to reduce the bridged diketopiperazine core to the crucial diazabicyclo[2.2.2]octanone unit.11 This necessitated a revision of the synthetic approach and we hypothesized that the crucial selective monoreduction at the more hindered position of the diazabicyclooctane unit had to be performed before assembly of the strongly rigidifying cyclopentane ring. Except for Tanimori's model studies,9 no approach considered the diazabicyclo[2.2.2]octanone skeleton instead of the more common DKP core, thus the different and increased reactivity of a tertiary amine function has to be taken into account, which is much more sensitive in oxidative transformations and may thwart transition metal-mediated reaction steps.
We report here an asymmetric total synthesis of ent-asperparaline C (ent-4) based on L-proline (8). The retrosynthesis is based on a late and challenging disconnection between two quaternary centers at the cyclopentane ring for the assembly of the spirosuccinimide–cyclopentane unit by an iron-mediated reductive radical cyclization (Scheme 1). This leads to a 1,6-diene embedded in the diazabicyclooctanone and butenolide units of intermediate 5. Further disconnection calls for selective monoreduction and oxidative furan modification of tricyclic derivative 6, which will be approached by using a PRE-based radical cyclization of furylmethyl diketopiperazine 7. This can be traced to natural L-proline (8) as the starting material. The PRE-based radical cyclization is distinct from Simpkins’ radical10,16 and carbocation-based17 approaches using the same disconnection since it avoids radical translocation or overall reductive conditions, which prevented elaboration of the properly substituted cyclopentane ring.10,16
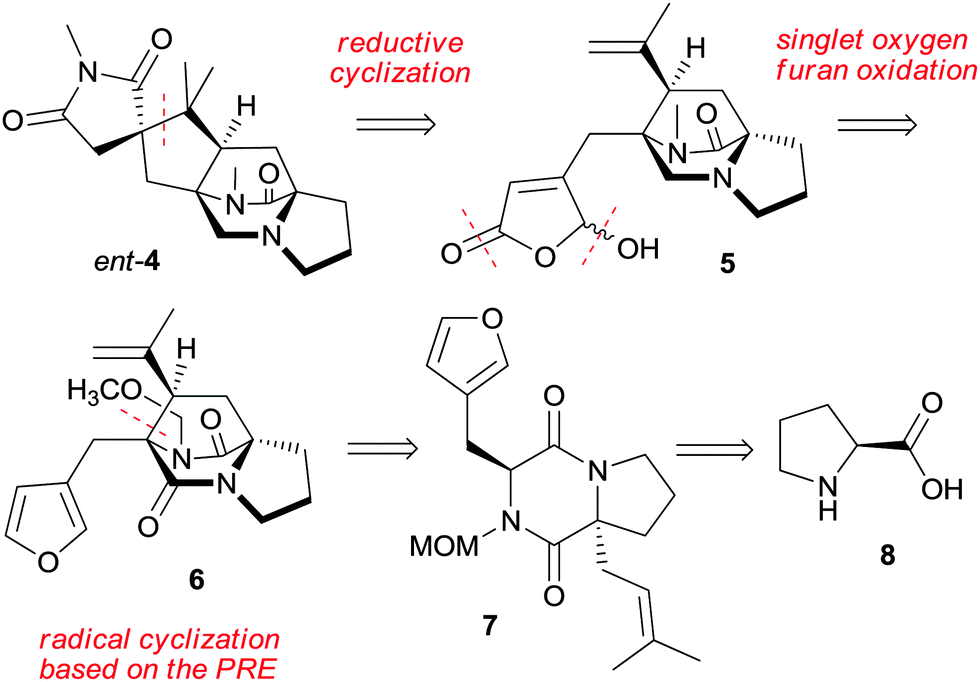 | ||
| Scheme 1 Retrosynthetic analysis of asperparaline C. | ||
L-Proline (8) was transformed via the trichloromethyl analog of Seebach's oxazolidinone 9 by deprotonation and alkylation with prenyl bromide to prenyloxazolidinone 10 (Scheme 2).18 Oxazolidinone 10 was opened with NH3 in MeOH to give a mixture of ester 11 and amide 12 in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio together with traces of formylated amide 13, which was not separated but directly transformed with bromoacetyl bromide to bromoacetamides 14 and 15. These compounds were also not isolated but converged to diketopiperazine 16 upon treatment with ammonia in methanol in 76% yield based on bicyclic oxazolidinone 10. At this point, temporary protection of the remaining free amide function was necessary to promote the subsequent alkylation efficiently. Acyl groups were not applicable because of competing Chan-type rearrangements15,19 and extensive experimentation with the PMB protecting group failed (see the ESI†), which is in line with results by Williams and Simpkins in their campaigns toward DKP alkaloids.20–22 The most suitable protecting group proved to be the MOM group;23 thus deprotonation by sodium hydride and reaction with MOMCl provided fully protected DKP derivative 17. Alkylation of 17 using nBuLi and 3-(bromomethyl)furan in THF at −78 °C proceeded smoothly furnishing furyl DKP 7 as a single trans diastereomer in 82% yield.
:1 ratio together with traces of formylated amide 13, which was not separated but directly transformed with bromoacetyl bromide to bromoacetamides 14 and 15. These compounds were also not isolated but converged to diketopiperazine 16 upon treatment with ammonia in methanol in 76% yield based on bicyclic oxazolidinone 10. At this point, temporary protection of the remaining free amide function was necessary to promote the subsequent alkylation efficiently. Acyl groups were not applicable because of competing Chan-type rearrangements15,19 and extensive experimentation with the PMB protecting group failed (see the ESI†), which is in line with results by Williams and Simpkins in their campaigns toward DKP alkaloids.20–22 The most suitable protecting group proved to be the MOM group;23 thus deprotonation by sodium hydride and reaction with MOMCl provided fully protected DKP derivative 17. Alkylation of 17 using nBuLi and 3-(bromomethyl)furan in THF at −78 °C proceeded smoothly furnishing furyl DKP 7 as a single trans diastereomer in 82% yield.
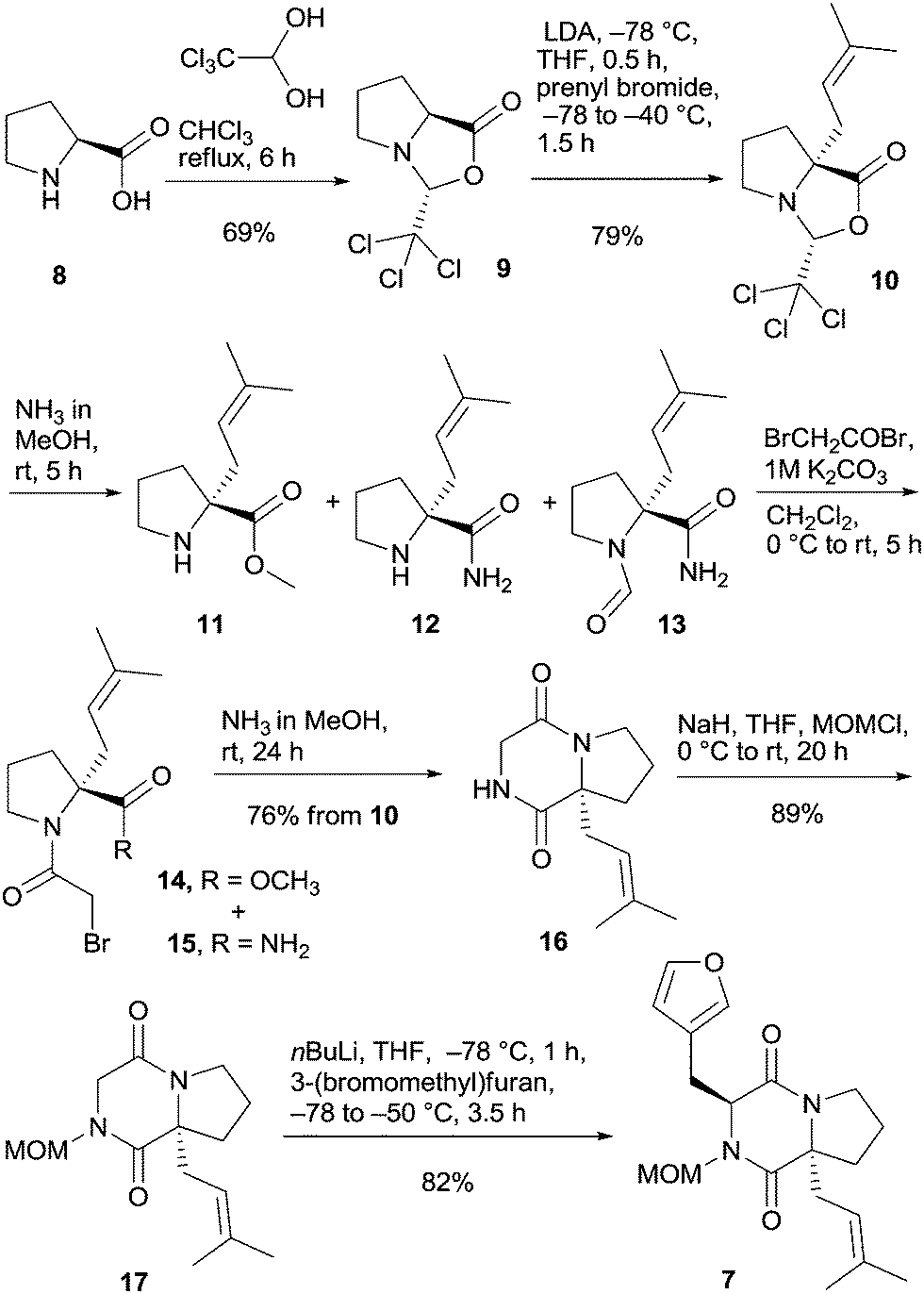 | ||
| Scheme 2 Synthesis of furylmethyl DKP 7 starting from L-proline. | ||
The efficient access to bicyclic DKP 7 set the stage for the crucial oxidative radical cyclization to approach the tricyclic diazabicyclo[2.2.2]octane skeleton (Scheme 3). Deprotonation by nBuLi at −78 °C,24 followed by oxygenation with ferrocenium hexafluorophosphate and TEMPO at −40 °C generated quaternary alkoxyamine 18in situ, which could not be isolated, but had to be directly subjected to heating at 100 °C to promote the radical cyclization. Under these conditions tricyclic isopropenyldiazabicyclo[2.2.2]octandione 6 was obtained in 87% yield as a single syn diastereoisomer at the bridge stereocenter, whose structure and absolute configuration were unambiguously confirmed to be (R) at all three stereogenic centers by X-ray analysis.25 This result is in line with very recent computational results revealing that the formation of quaternary alkoxyamines, such as 18, can be endergonic and that homolysis may occur below room temperature, however heating is mandatory to overcome the considerable cyclization barrier.13 The high diastereoselectivity of the cyclization of radical A is remarkable, given that the MOM group is sterically not very demanding, but still directs the prenyl group very efficiently to the syn orientation in the transition state of the radical 6-exo cyclization. The cyclization conditions enabled at the same time effective introduction of the alkene unit, which is formed either (a) by direct β-hydrogen abstraction of the cyclized radical B by TEMPO or (b) by coupling of the cyclized radical with TEMPO and subsequent concerted elimination from intermediate C (Scheme 3).
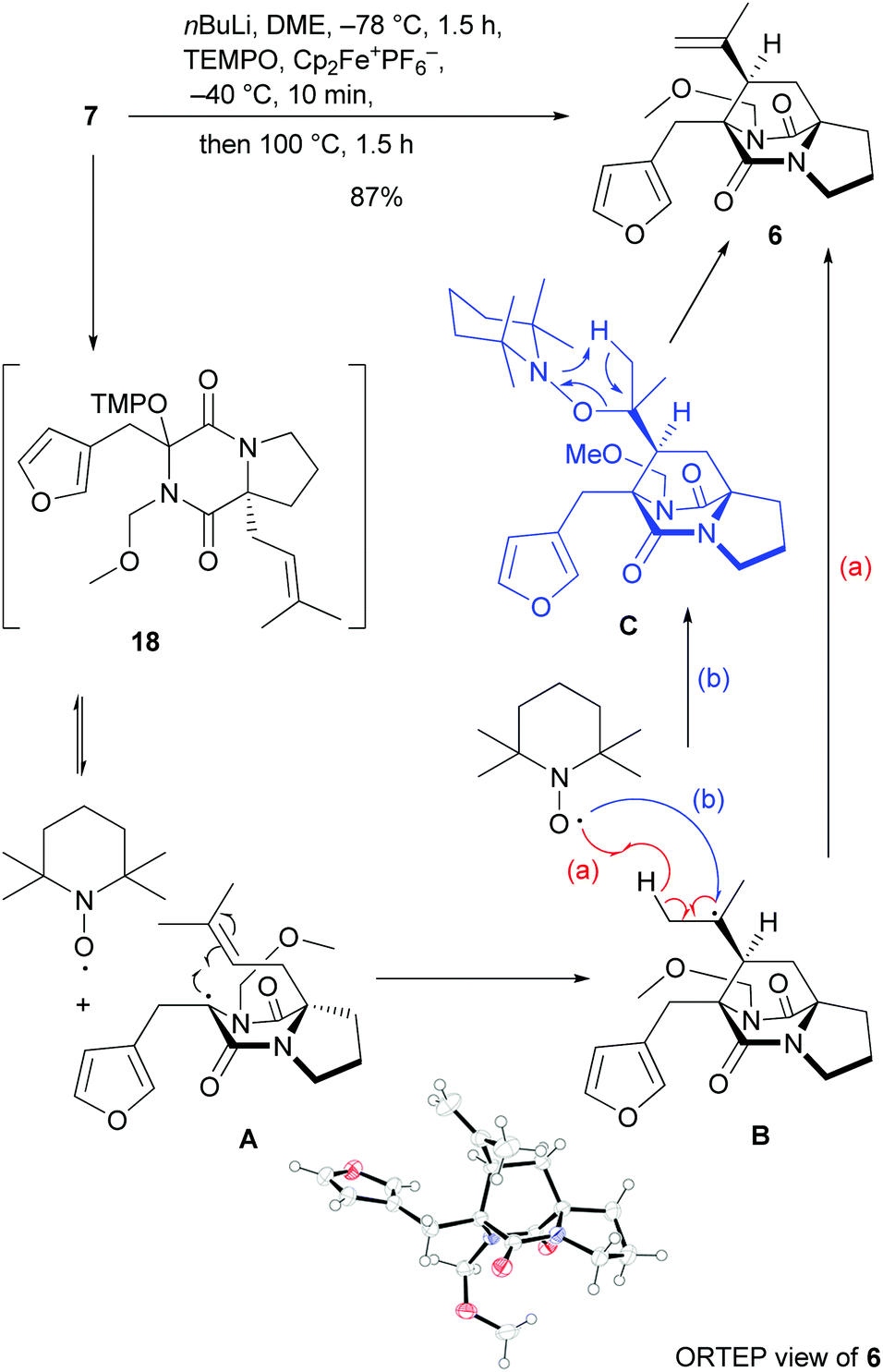 | ||
| Scheme 3 Oxidative radical cyclization with mechanistic rationalization and ORTEP view of 6 (displacement ellipsoids shown at 50% probability level). | ||
With tricyclic 6 at hand, the MOM group was removed using B-bromocatecholborane giving free amide 19 in 96% yield (Scheme 4). This enabled selective reduction of the tertiary amide group at C-8 in the presence of the secondary. Although implementation required some experimentation the reduction proceeded reproducibly with 3.6–3.8 equivalents of DIBAL-H in toluene at room temperature, furnishing bridged piperazinone 20 in 79% yield.26 With more than four equivalents of DIBAL-H, both groups were reduced, and with less than 3.6 equivalents, some 19 remained. Bridged piperazinone 20 was N-methylated and the furan unit in amide 21 was subsequently subjected to oxidative dearomatization via a hetero-Diels–Alder reaction with singlet oxygen and subsequent Kornblum–DeLaMare rearrangement27 in the presence of Hünig's base providing γ-hydroxybutenolide 5 in 85% yield with high regioselectivity as an epimeric mixture at the hemiacetal center. The tertiary amine present in the structure remained intact and no oxidation or cleavage of the core tricycle was observed. Acetalization with methanol using a small excess of (+)-CSA provided acetal 22 as a 1:1 mixture of epimers (Scheme 4). The excess was necessary because the first equivalent protonates the tertiary amine. However, the chiral acid did not induce diastereomeric preference for a particular acetal isomer. The use of a chiral alcohol to provide diastereomerically enriched acetals was not successful (not shown).
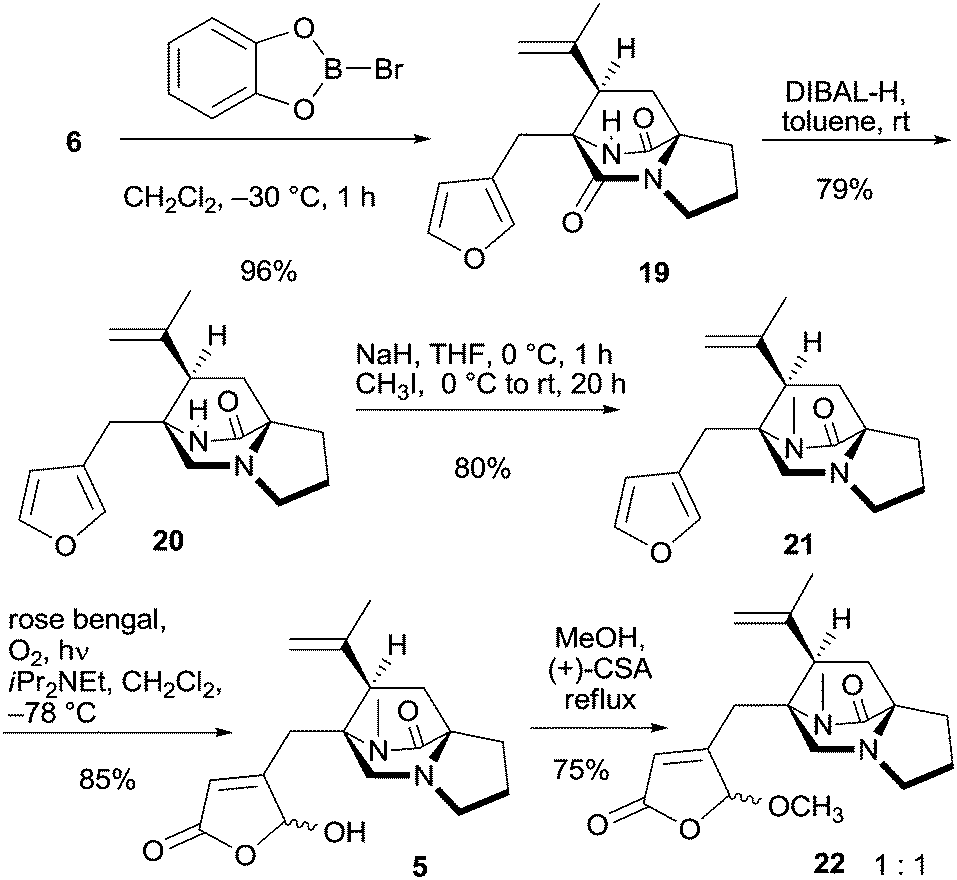 | ||
| Scheme 4 Synthesis of acetal 22 from tricycle 6. | ||
Reductive cyclization of isopropenyl butenolide 22 using Fe(acac)3 and PhSiH3 as developed by Baran and coworkers28 efficiently provided a partly separable 1:1 mixture of pentacyclic spirobutyrolactones 23a and 23b (Scheme 5). The configuration of both isomers was determined by ROESY experiments. The outcome of the cyclization can be rationalized by assuming that the tertiary alkyl radical is steered with full control to the butenolide face, which is opposite to the residing methoxy group. Spirocycle 23a was reacted with N-methylamine solution in methanol to form hemiaminal 24a, which was not isolated but was directly oxidized with PCC; acidic impurities were subsequently removed by filtration through a pad of Amberlite IRN-78, providing ent-asperparaline C (ent-4) in 66% yield from 23a (Scheme 5). Diastereomeric spiroimide 23b was similarly transformed to asperparaline C analogue 25 in 77% yield, differing in the configuration at the spiro center. Synthetic asperparaline C (4) displayed identical NMR spectra to that of isolated 4 (see the ESI† for detailed comparison).4 However, the optical rotation of synthetic 4 was with [α]20D = +26.3 (c 0.316, MeOH) opposite to that of isolated asperparaline C, for which a specific optical rotation of [α]20D = −20 (c 0.05, MeOH) was determined.4 The spectral and optical rotation data prove that natural asperparaline C has absolute (all-S) configuration whereas the synthetic material proved to have (R) configuration at all stereogenic centers and is thus the enantiomer of natural asperparaline C, ent-4.
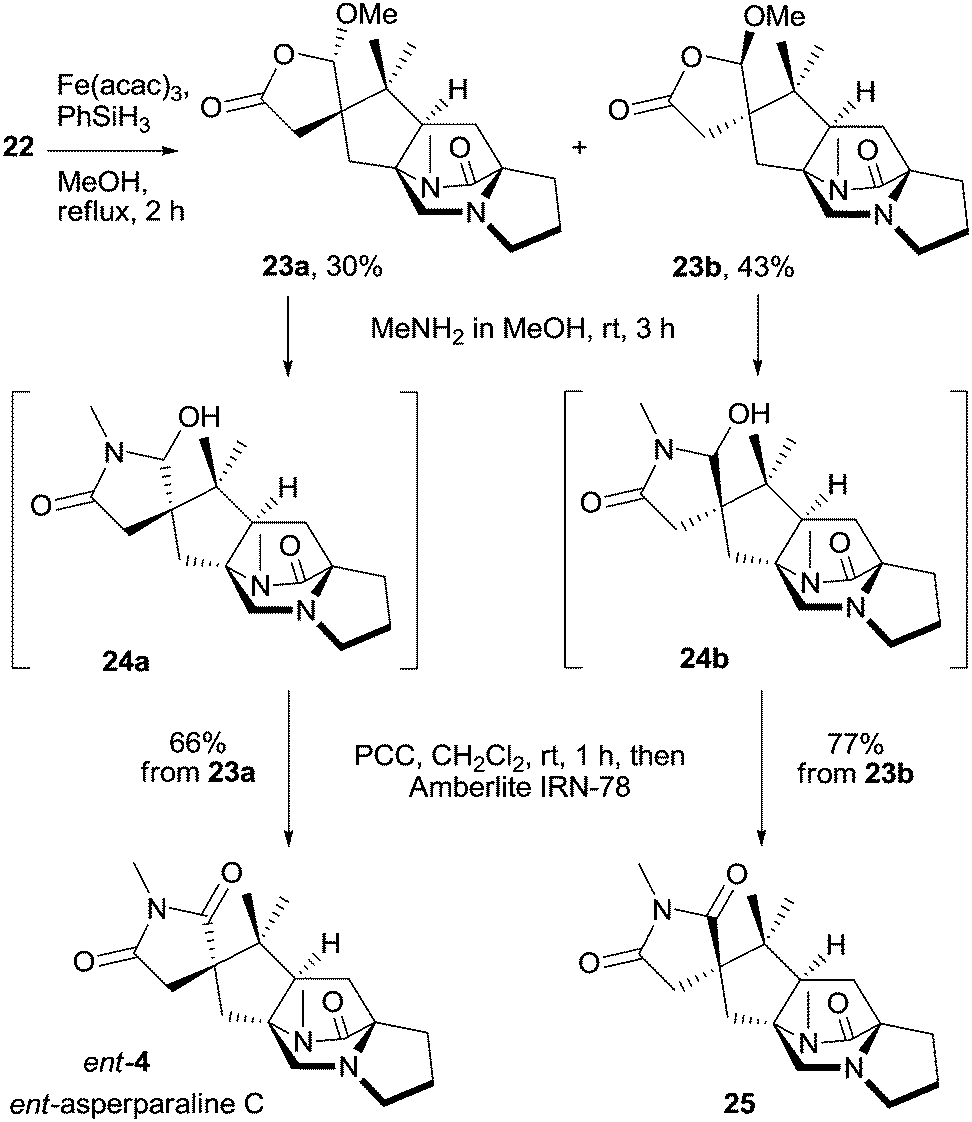 | ||
| Scheme 5 Reductive cyclization of dienes 22 to spirobutyrolactones 23a and 23b and completion of the total synthesis of ent-asperparaline C (ent-4) and its diastereoisomer 25. | ||
In summary, we present the first enantioselective total synthesis of ent-asperparaline C starting from L-proline in overall 2.0% yield over 16 linear steps. The key steps were a rare, but highly stereoselective radical cyclization of an in situ generated quaternary alkoxyamine to form the central diazabicyclo[2.2.2]octanone core with the required syn configuration at the bridge stereogenic center, selective reduction of the tertiary amide at C-8 position, singlet oxygen Diels–Alder reaction and a reductive spirocyclization. This asymmetric total synthesis enabled the assignment of the absolute configuration of asperparaline C to be (S) at all four stereogenic centers in the molecule. Based on these results it can be assumed that the absolute configuration of the other family members 1–3 is similarly (all-S) at the pentacyclic core and also (S) at the additional methyl group. The determined absolute configuration of the diazabicyclo[2.2.2]octane skeleton supports the biosynthetic relation of the asperparalines to the paraherquamides.2 The presented strategy is modular and will be applicable with appropriate modifications to total syntheses of all other members of the asperparaline family and also other alkaloids bearing the diazabicyclo[2.2.2]octane skeleton.
Generous financial support by the Institute of Organic Chemistry and Biochemistry, Czech Academy of Sciences (RVO:61388963) and the Gilead Sciences & IOCB Research Center is gratefully acknowledged. I. D. thanks the Ruđer Bošković Institute, Zagreb (Croatia) for partial financial postdoctoral support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. M. Finefield, J. C. Frisvad, D. H. Sherman and R. M. Williams, J. Nat. Prod., 2012, 75, 812–833 CrossRef CAS PubMed.
- (a) C. R. Gray, J. F. Sanz-Cervera, L. A. Silks and R. M. Williams, J. Am. Chem. Soc., 2003, 125, 14692–14693 CrossRef CAS PubMed; (b) Y. Ding, S. Gruschow, T. J. Greshock, J. Finefield, D. H. Sherman and R. M. Williams, J. Nat. Prod., 2008, 71, 1574–1578 CrossRef CAS PubMed.
- H. Hayashi, Y. Nishimoto and H. Nozaki, Tetrahedron Lett., 1997, 38, 5655–5658 CrossRef CAS.
- H. Hayashi, Y. Nishimoto, K. Akiyama and H. Nozaki, Biosci., Biotechnol., Biochem., 2000, 64, 111–115 CrossRef CAS PubMed.
- R. M. Banks, S. E. Blanchflower, J. R. Everett, B. R. Manger and C. Reading, J. Antibiot., 1997, 50, 840–846 CrossRef CAS PubMed.
- K. Hirata, S. Kataoka, S. Furutani, H. Hayashi and K. Matsuda, PLoS One, 2011, 6, e18354 CrossRef CAS PubMed.
- K. Hirata, J. Pestic. Sci., 2016, 41, 87–94 CrossRef PubMed.
- L. A. Adams, C. R. Gray and R. M. Williams, Tetrahedron Lett., 2004, 45, 4489–4493 CrossRef CAS.
- (a) S. Tanimori, K. Fukubayashi and M. Kirihata, Tetrahedron Lett., 2001, 42, 4013–4016 CrossRef CAS; (b) S. Tanimori, T. Sunami, K. Fukubayashi and M. Kirihata, Tetrahedron, 2005, 61, 2481–2492 CrossRef CAS.
- P. J. Crick, N. S. Simpkins and A. Highton, Org. Lett., 2011, 13, 6472–6475 CrossRef CAS PubMed.
- T. Amatov, R. Pohl, I. Cisařová and U. Jahn, Org. Lett., 2017, 19, 1152–1155 CrossRef CAS PubMed.
- Selected review: A. D. Borthwick, Chem. Rev., 2012, 112, 3641–3716 CrossRef CAS PubMed.
- T. Amatov, H. Jangra, R. Pohl, I. Cisařová, H. Zipse and U. Jahn, Chem. – Eur. J., 2018, 24, 15336–15345 CrossRef CAS PubMed.
- T. Amatov, R. Pohl, I. Cisařová and U. Jahn, Angew. Chem., Int. Ed., 2015, 54, 12153–12157 CrossRef CAS PubMed.
- T. Amatov, M. Gebauer, R. Pohl, I. Cisařová and U. Jahn, Free Radical Res., 2016, 50, S6–S17 CrossRef CAS PubMed.
- (a) N. S. Simpkins, I. Pavlakos, M. D. Weller and L. Male, Org. Biomol. Chem., 2013, 11, 4957–4970 RSC; (b) N. Simpkins, I. Pavlakos and L. Male, Chem. Commun., 2012, 48, 1958–1960 RSC.
- F. Frebault, N. S. Simpkins and A. Fenwick, J. Am. Chem. Soc., 2009, 131, 4214–4215 CrossRef CAS PubMed.
- G. D. Artman, R. J. Rafferty and R. M. Williams, Org. Synth., 2009, 86, 262–273 CrossRef CAS.
- T. Coursindel, A. Restouin, G. Dewynter, J. Martinez, Y. Collette and I. Parrot, Bioorg. Chem., 2010, 38, 210–217 CrossRef CAS PubMed.
- R. M. Williams, R. W. Armstrong and J.-S. Dung, J. Am. Chem. Soc., 1985, 107, 3253–3266 CrossRef CAS.
- R. M. Williams and T. Glinka, Tetrahedron Lett., 1986, 27, 3581–3584 CrossRef CAS.
- F. C. Frebault and N. S. Simpkins, Tetrahedron, 2010, 66, 6585–6596 CrossRef CAS.
- P. S. Baran, C. A. Guerrero, N. B. Ambhaikar and B. D. Hafensteiner, Angew. Chem., Int. Ed., 2005, 44, 606–609 CrossRef CAS PubMed.
- When LiHMDS or LDA (ref. 11 and 13–15) were applied as bases, starting DKP 7 was fully recovered.
- Crystallographic data of compound 6 were deposited at the Cambridge Crystallographic Data Centre under the number CCDC 1875571†.
- Earlier use: E. M. Stocking, J. F. Sanz-Cervera and R. M. Williams, J. Am. Chem. Soc., 2000, 122, 1675–1684 CrossRef CAS.
- T. Montagnon, D. Kalaitzakis, M. Triantafyllakis, M. Stratakis and G. Vassilikogiannakis, Chem. Commun., 2014, 50, 15480–15498 RSC.
- (a) J. C. Lo, Y. Yabe and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 1304–1307 CrossRef CAS PubMed; (b) J. C. Lo, J. Gui, Y. Yabe, C.-M. Pan and P. S. Baran, Nature, 2014, 516, 343–348 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical data, copies of 1H and 13C NMR spectra and crystallographic data. CCDC 1875571. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc00945k |
| This journal is © The Royal Society of Chemistry 2019 |