
Visible light mediated, metal-free carbene transfer reactions of diazoalkanes with propargylic alcohols†
Feifei
He
and
Rene M.
Koenigs
 *
*
RWTH Aachen University, Institute of Organic Chemistry, Landoltweg, D-52074 Aachen, Germany. E-mail: rene.koenigs@rwth-aachen.de
First published on 2nd April 2019
Abstract
The development of efficient carbene transfer reactions under mild reaction conditions is very important to access small molecules for applications in organic synthesis and drug discovery. Herein, we describe the application of blue light in photochemical carbene transfer reactions of donor acceptor diazoalkanes under mild reaction conditions with propargylic alcohols, which provides – in contrast to conventional metal-catalyzed carbene transfer reactions – an access to cyclopropenes.
Carbene transfer reactions are an important fundamental transformation in organic synthesis and open up different reactivity patterns ranging from cycloadditions, insertion reactions to rearrangement reactions.1–4 Today, the majority of these carbene transfer processes are conducted in the presence of expensive precious metal1–3 catalysts and more lately catalysts based earth-abundant iron or cobalt have emerged as a sustainable alternative.4 Most of these transformations require the strict exclusion of moisture and air, which is far from being operationally simple.
The UV photolysis of diazoalkanes is a classical alternative to metal-catalyzed carbene transfer reactions, yet applications are very limited due to side reactions related to the high energy UV light.4 Although diazoalkanes are intensely colored and absorb light in the visible region, the application of low-energy visible light as a sustainable and metal-free alternative to carbene transfer reactions has received only little interest until now.5–9 Only recently, the Davies,7 He8 and our group9 reported on the use of low-energy blue light to liberate carbenes from donor–acceptor diazoalkanes for applications cycloaddition, X–H and C–H insertion, olefination and Doyle–Kirmse rearrangement reactions. Most remarkably, visible light mediated carbene transfer reactions are operationally very straightforward can be conducted using simple p.A. grade solvents in the presence of air without the need of any additional precautions.9
The carbene transfer reaction of diazoalkanes with propargylic alcohols is a valuable transformation to access a variety of different rearrangement reactions.10,11 To date, different gold(I),10a palladium(II)10b and rhodium(II)10c catalysts have been described in this transformation and provide, depending on the nature of the catalyst, an access to allenes10c or dihydrofurans.10a,b Mechanistically, these rearrangements proceed via formation of a metal carbene complex that delivers an electrophilic metal-bound carbene to the nucleophilic alcohol substrate under formation of an ylide intermediate, which undergoes either a 2,3-sigmatropic rearrangement or a migratory insertion reaction (Scheme 1a and b).
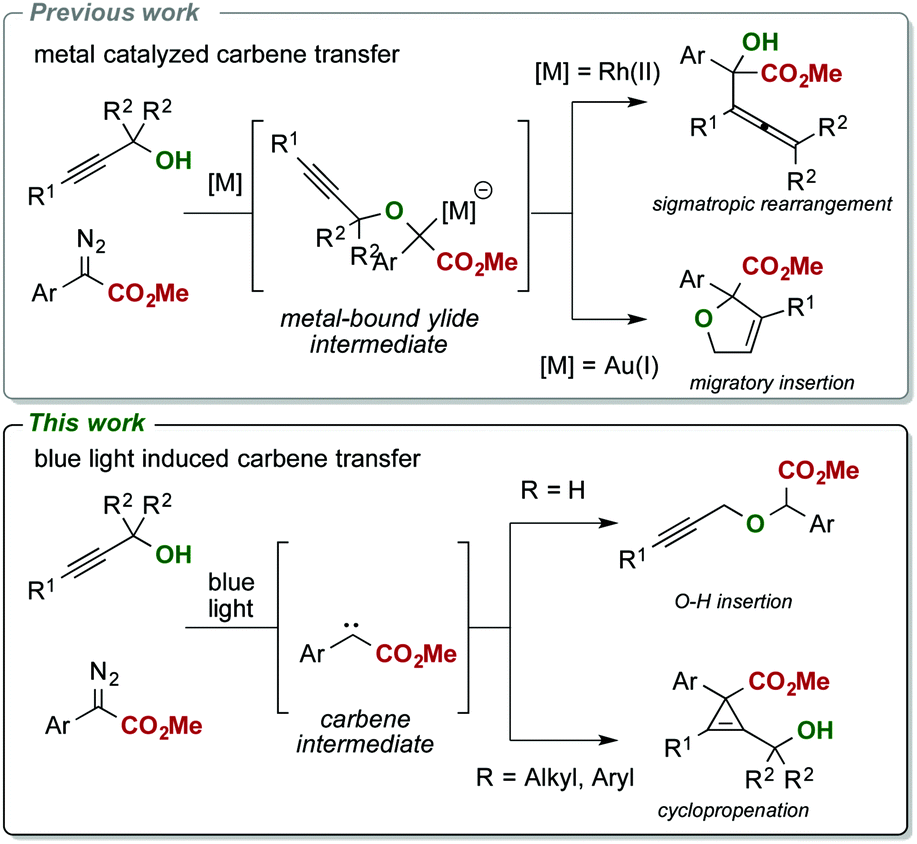 | ||
| Scheme 1 Reaction of propargylic alcohols with metal carbene complexes and free carbenes. | ||
Against this background and building on our experience in carbene transfer reactions,12 we became intrigued in studying the reaction of propargylic alcohols with diazoalkanes under irradiation with low-energy blue light and set out our studies by examining the reaction of propargylic alcohols with donor–acceptor diazoalkanes.
Surprisingly, we observed a markedly altered reactivity in this metal-free transformation and could observe a highly chemoselective cyclopropenation reaction of the tertiary alcohol 1a (Table 1, entry 1). The formation of this tetra-substituted cyclopropene is remarkable; in general, cyclopropenation reactions13–19 can be readily performed in the presence of different Rh(II),14 Au(I),15 Ir(III),16 Ag(I)17 or other precious metal based catalysts, yet applications are mostly limited towards alkynes bearing only a single sterically demanding substituent. In particular, a systematic study on the synthesis of such densely substituted cyclopropenes bearing an adjacent fully substituted carbon atom is not reported even today. Initial applications, dating back to 1960 describe limited examples of photocatalytic cyclopropenation reactions, yet limited in both substrate scope and yield.20
|
|
|||||||
|---|---|---|---|---|---|---|---|
| #a | Solvent | R1 | R2 | Substrate | Ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 :2 |
Yield (3) | Yield (4) |
| a Propargyl alcohol 1a–c (0.3 mmol) and diazoalkane 2 (2.0 eq., 0.4 mmol) were dissolved in 3 mL of solvent and stirred at room temperature for 12 hours under irradiation with blue LEDs (470 nm, 3 W). Yields were reported for isolated products. b Reaction scale: 10 mmol. c Reaction in the dark. | |||||||
| 1 | DCE | Me | Me | 1a | 1:1 |
51% | — |
| 2 | DCE | Me | Me | 1a | 1:2 |
82% | — |
| 3 | DCE | Me | Me | 1a | 1:4 |
86% | — |
| 4 | DCE | Me | Me | 1a | 4:1 |
59% | — |
| 5 | EtOAc | Me | Me | 1a | 1:4 |
71% | — |
| 6 | THF | Me | Me | 1a | 1:4 |
Traces | — |
| 7 | DCM | Me | Me | 1a | 1:4 |
86% | |
| 8 | CHCl3 | Me | Me | 1a | 1:4 |
91% | — |
| 9b | CHCl3 | Me | Me | 1a | 1:4 |
82% | |
| 10 | Hexane | Me | Me | 1a | 1:4 |
44% | — |
| 11 | Toluene | Me | Me | 1a | 1:4 |
55% | — |
| 12 | CHCl3 | H | H | 1b | 1:4 |
— | 54% |
| 13 | CHCl3 | Me | H | 1c | 1:4 |
53% | — |
| 14c | CHCl3 | Me | Me | 1a | 1:4 |
no rct. | |
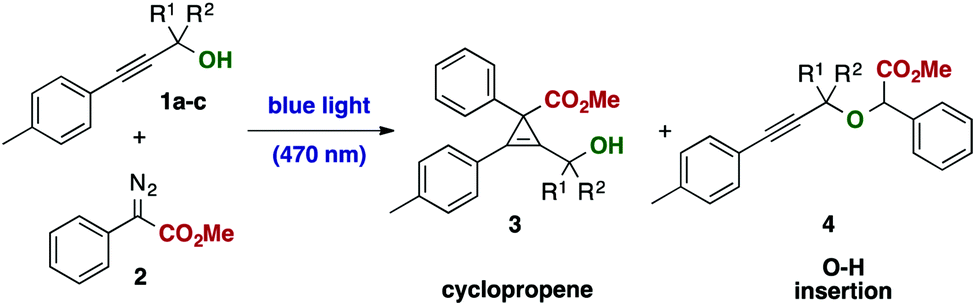
Inspired by these unexpected observations, we decided to study the influence of reaction parameters, such as stoichiometry and solvent to alter the course of the reaction. Under optimized conditions, cyclopropene 3a could be obtained using propargylic alcohol 1a in excellent yield using chloroform as solvent without the need of alcohol protection, including the gram-scale application (Table 1, entry 9). In a control experiment in the dark, no reaction was observed (Table 1, entry 14).
Notably, we were unable to observe the formation of the O–H insertion product 4 or identify any product arising from a rearrangement reaction under all reaction conditions investigated. This observation emphasizes important differences in reactivity of light-mediated and metal-catalyzed carbene transfer reactions and highlights complementary reactivities of these carbene species. Switching to the primary alcohol 1b resulted in exclusive formation of the propargylic ether 4via O–H insertion of the carbene intermediate without formation of the cyclopropene or rearrangement product (Table 1, entry 12). The secondary alcohol 1c, however, reacted under formation of the cyclopropene product and formation of both diastereoisomers in a 1:1 mixture (Table 1, entry 13). Protection of the alcohol as a methyl ether or a silylether resulted in no product formation. Cinnamyl alcohols did not undergo the respective cyclopropanation product in satisfactory selectivity and provided either no cyclopropanation or only sluggish cyclopropanation reactions.21
In subsequent investigations, we examined the applicability of different donor–acceptor diazoalkanes in this cyclopropenation reaction of unprotected propargylic alcohols. Different electron-donating substituents and halogens in para- and meta-positions of the aromatic ring were well-tolerated. In the case of an ortho substitution of the aromatic ring of the aryl diazoacetate, the yield of the corresponding cyclopropene was only moderate, which might be attributed to overcrowding due to the high steric demand of the alkyne substrate and the carbene reaction partner. Similarly, different carbocycles were tolerated or ester diazoalkanes (Scheme 2). Importantly, trifluoromethyl-substituted donor–acceptor diazoalkanes reacted sluggishly to the corresponding cyclopropene and only a mixture of compounds could be obtained.
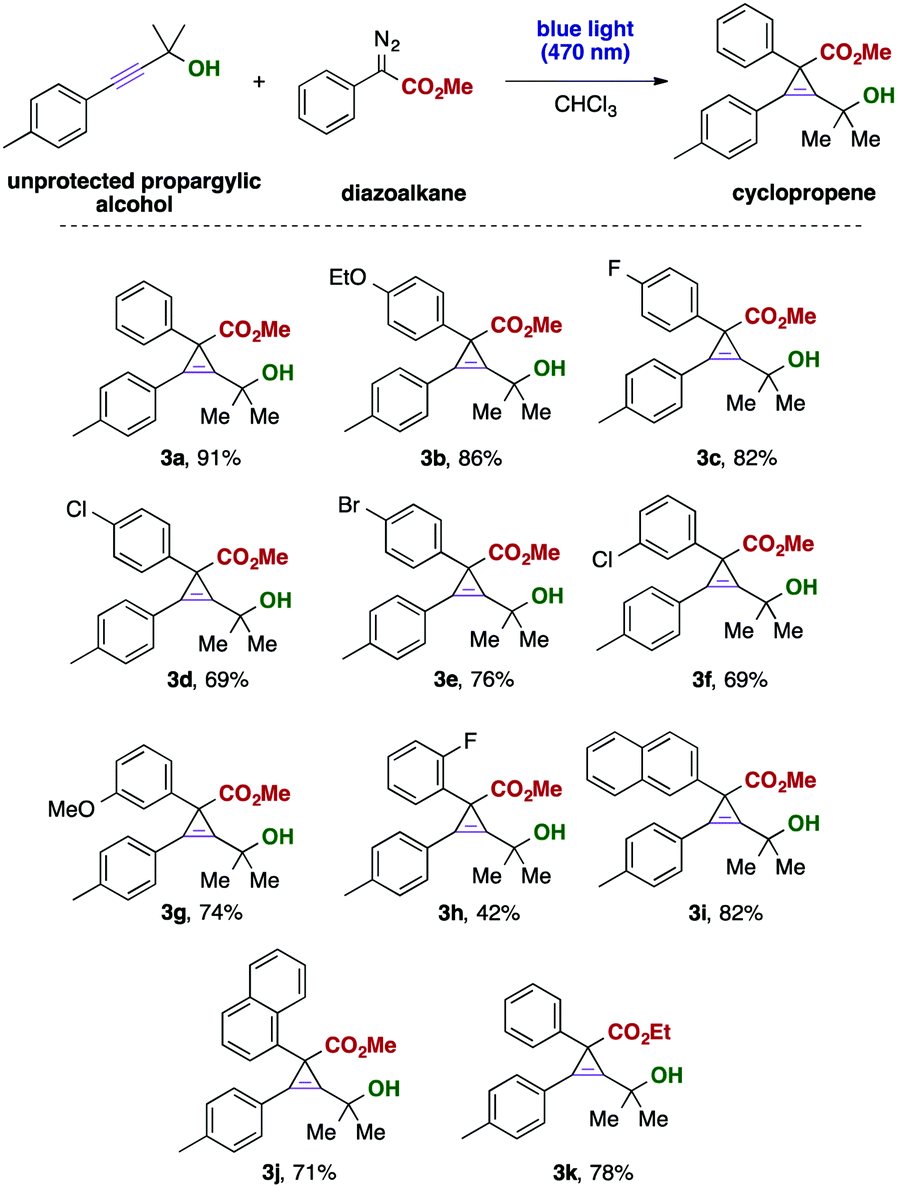 | ||
| Scheme 2 Visible light mediated carbene transfer reactions with propargylic alcohols. Propargyl alcohol 1a (0.3 mmol) and diazoalkane (4.0 eq., 1.2 mmol) were dissolved in 3 mL of CHCl3 and stirred at room temperature for 12 hours under irradiation with blue LEDs (470 nm, 3 W). Yields were reported for isolated products. | ||
Subsequently, we examined the substrate scope of unprotected propargylic alcohols. Gratifyingly, a broad range of functional groups is compatible with the present reaction conditions and the desired cyclopropenes were isolated in good yields. Even reactive and labile or strongly electron-withdrawing and thus deactivating functional groups, such as iodide (3p), nitrile (3r), trifluoromethoxy (3t) or an acetyl (3s) group, are well tolerated and the desired cyclopropenes could be obtained as the only product. The position of the substituent at the aromatic ring only had a minor effect on the reaction outcome and in all cases the desired products were obtained in good yield. Interestingly, also heterocyclic alkynes, such as the thiophene substrate (Scheme 3, 3aa) smoothly reacted to the desired cyclopropene without formation of unwanted byproducts.
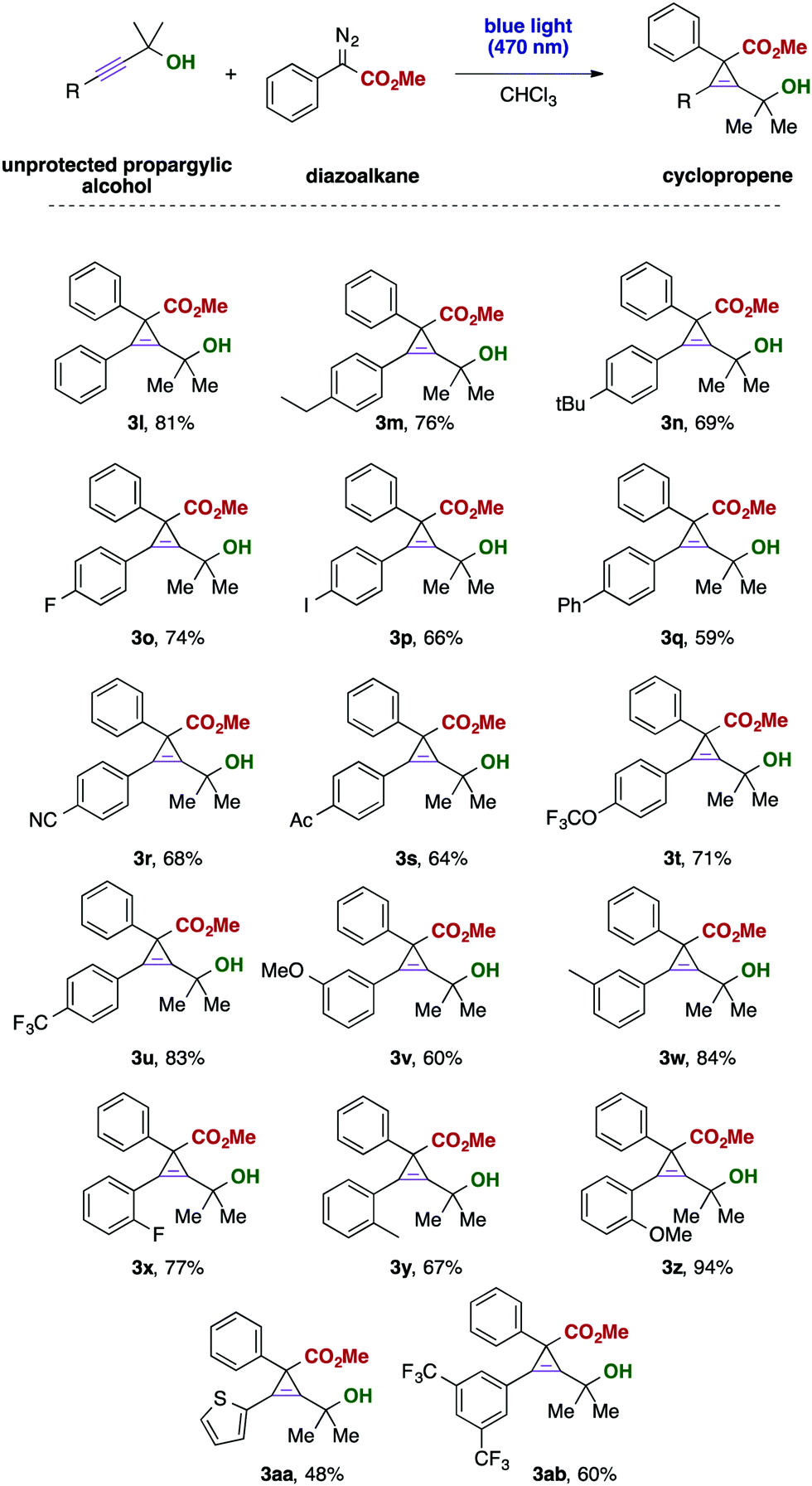 | ||
| Scheme 3 Investigations with different propargylic alcohols. Propargyl alcohol 1 (0.3 mmol) and diazoalkane 2 (4.0 eq., 1.2 mmol) were dissolved in 3 mL of CHCl3 and stirred at room temperature for 12 hours under irradiation with blue LEDs (470 nm, 3 W). Yields were reported for isolated products. | ||
Finally, we investigated different aliphatic substituents at the propargyl moiety. Different cyclic substrates reacted in this visible light mediated cyclopropenation reaction with moderate yield, giving access to interesting polycyclic building blocks containing one tetra-substituted cyclopropene ring. We furthermore examined a phenyl, methyl disubstituted alkyne, which gave the desired cyclopropene in almost quantititative yield, yet the product was obtained only in a 1:1 diastereomeric mixture (Scheme 4).
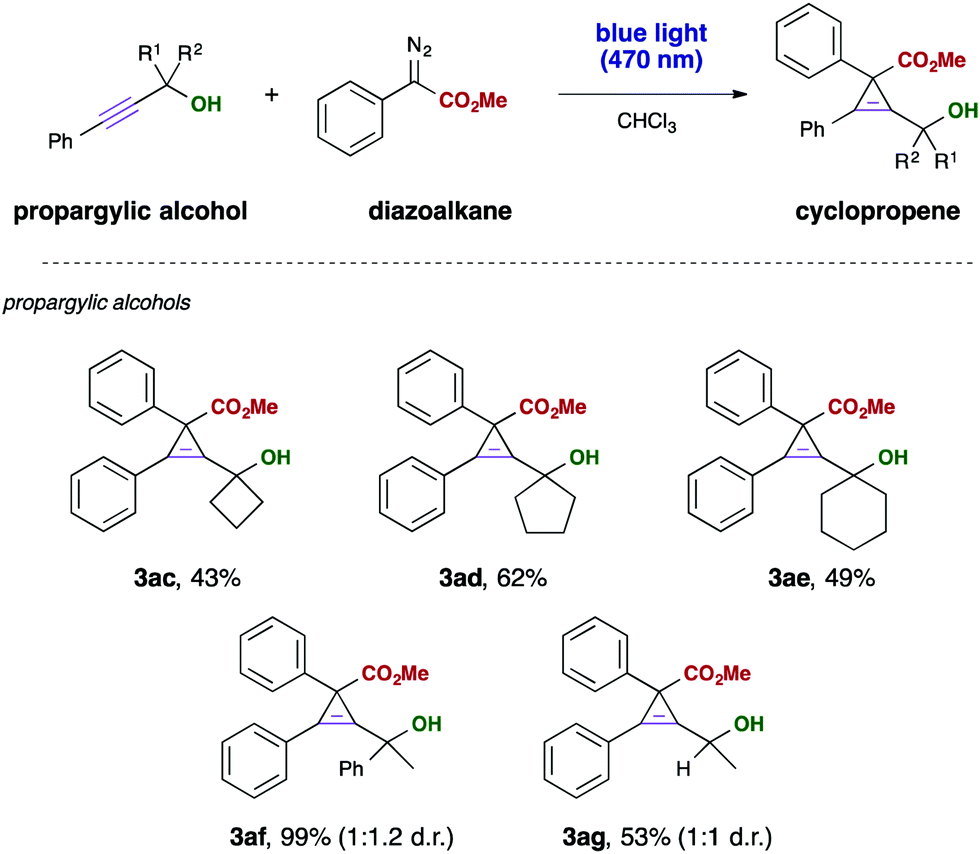 | ||
| Scheme 4 Investigations on with different substitution patterns at the alcohol position. Propargyl alcohol 1 (0.3 mmol) and diazoalkane 2 (4.0 eq., 1.2 mmol) were dissolved in 3 mL of CHCl3 and stirred at room temperature for 12 hours under irradiation with blue LEDs (470 nm, 3 W). Yields were reported for isolated products. | ||
Finally, we examined the derivatization of the cyclopropene 3a in methylation reactions. Using sodium hydride as a base, the methylation product 5 was obtained in 53% yield (Scheme 5).
 | ||
| Scheme 5 Reaction of propargylic alcohols with metal carbene complexes and free carbenes. | ||
In summary, we herein report on photochemical carbene transfer reactions that open up orthogonal reaction pathways to metal-catalyzed carbene transfer reactions. Under metal-free conditions and without the need to exclude moisture or air, diazoalkanes readily undergo a high-yielding photochemical cyclopropenation reaction with sterically demanding propargylic alcohols, which underlines the unique reactivity of carbenes generated under photochemical conditions. Under the present reaction conditions, a broad variety of functional groups are well tolerated and tetra-substituted cyclopropenes (33 examples) can be accessed in up to 99% isolated yield.
RMK gratefully acknowledges the Dean's Seed Fund of RWTH Aachen University and the Fonds der Chemischen Industrie for generous support (Sachkostenzuschuss).
Conflicts of interest
There are no conflicts to declare.Notes and references
- Selected review articles: (a) H. M. L. Davies and J. R. Manning, Nature, 2018, 451, 417 CrossRef PubMed; (b) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981 CrossRef CAS PubMed; (c) M. P. Doyle, R. Duffy, M. Ratnikov and Z. Zhou, Chem. Rev., 2010, 110, 704 CrossRef CAS PubMed; (d) L. Mertens and R. M. Koenigs, Org. Biomol. Chem., 2016, 14, 10547 RSC; (e) Y. Xia, D. Qiu and J. Wang, Chem. Rev., 2017, 117, 13810 CrossRef CAS PubMed; (f) K. J. Hock and R. M. Koenigs, Angew. Chem., Int. Ed., 2017, 56, 13566 CrossRef CAS PubMed; (g) Z. Sheng, Z. Zhang, C. Chu, Y. Zhang and J. Wang, Tetrahedron, 2017, 73, 4011 CrossRef CAS.
- Selected review articles: (a) H.-J. Knölker and I. Bauer, Chem. Rev., 2015, 115, 3170 CrossRef PubMed; (b) S.-F. Zhu and Q.-L. Zhou, Natl. Sci. Rev., 2014, 1, 580 CrossRef CAS; (c) R. Shang, L. Ilies and E. Nakamura, Chem. Rev., 2017, 117, 9086 CrossRef CAS PubMed; (d) O. F. Brandenberg, R. Fasan and F. H. Arnold, Curr. Opin. Biotechnol., 2017, 47, 102 CrossRef CAS PubMed.
- Selected references: (a) B. Morandi and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 938 CrossRef CAS PubMed; (b) M. S. Holzwarth, I. Alt and B. Plietker, Angew. Chem., Int. Ed., 2012, 51, 5351 CrossRef CAS PubMed; (c) J. Day, B. McKeever-Abbas and J. Dowden, Angew. Chem., Int. Ed., 2016, 55, 5809 CrossRef CAS PubMed; (d) J. R. Griffin, C. I. Wendell, J. A. Garwin and M. C. White, J. Am. Chem. Soc., 2017, 139, 13624 CrossRef CAS PubMed; (e) D. M. Carminati, D. Intrieri, A. Caselli, S. Le Gac, B. Boitrel, L. Toma, L. Legnani and E. Gallo, Chem. – Eur. J., 2016, 22, 13599 CrossRef CAS PubMed; (f) K. J. Hock, L. Mertens, R. Hommelsheim, R. Spitzner and R. M. Koenigs, Chem. Commun., 2017, 53, 6577 RSC; (g) C. Empel, K. J. Hock and R. M. Koenigs, Org. Biomol. Chem., 2018, 16, 7129 RSC; (h) C. Empel, K. J. Hock and R. M. Koenigs, Chem. Commun., 2019, 55, 338 RSC.
- Selected references: (a) N. R. Candeias and C. A. M. Afonso, Curr. Org. Chem., 2009, 13, 763 CrossRef CAS; (b) O. S. Galkina and L. L. Rodina, Russ. Chem. Rev., 2016, 85, 537 CrossRef CAS; (c) J. Brunner, H. Senn and F. M. Richards, J. Biol. Chem., 1980, 255, 3313 CAS; (d) Y. Liang, L. Jiao, S. Zhang and J. Xu, J. Org. Chem., 2005, 70, 334 CrossRef CAS PubMed; (e) J. Wang, G. Burdzinski, J. Kubicki and M. S. Platz, J. Am. Chem. Soc., 2008, 130, 11195 CrossRef CAS PubMed; (f) K. Nakatani, S. Maekawa, K. Tanabe and I. Saito, J. Am. Chem. Soc., 1995, 117, 10635 CrossRef CAS.
- L. W. Ciszewski, K. Rybicka-Jasińska and D. Gryko, Org. Biomol. Chem., 2019, 17, 432 RSC.
- Selected references on metal-free carbene transfer reactions: (a) Z. Wang, A. G. Herraiz, A. M. del Hoyo and M. G. Suero, Nature, 2018, 554, 86 CrossRef CAS PubMed; (b) Z. Liu, H. Tan, L. Wang, T. Fu, Y. Xia, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2015, 54, 3056 CrossRef CAS PubMed; (c) Y. Zheng, R. Bian, X. Zhang, R. Yao, L. Qiu, X. Bao and X. Xu, Eur. J. Org. Chem., 2016, 3872 CrossRef CAS; (d) C. Tortoreto, D. Rackl and H. M. L. Davies, Org. Lett., 2017, 19, 770 CrossRef CAS PubMed; (e) X. Zhang, Y. Zheng, L. Qiu and X. Xu, Org. Biomol. Chem., 2017, 16, 70 RSC; (f) Y. S. Vaske, M. E. Mahoney, J. P. Konopelski, D. L. Rogow and W. J. McDonald, J. Am. Chem. Soc., 2010, 132, 11379 CrossRef CAS PubMed.
- I. Jurberg and H. M. L. Davies, Chem. Sci., 2018, 9, 5112 RSC.
- T. Xiao, M. Mei, Y. He and L. Zhou, Chem. Commun., 2018, 54, 8865 RSC.
- (a) R. Hommelsheim, Y. Guo, Z. Yang, C. Empel and R. M. Koenigs, Angew. Chem., Int. Ed., 2019, 58, 1203 CrossRef CAS PubMed; (b) S. Jana and R. M. Koenigs, Asian J. Org. Chem. DOI:10.1002/ajoc.201900099; (c) Z. Yang, Y. Guo and R. M. Koenigs, Chem. - Eur. J. DOI:10.1002/chem.201900597.
- Selected references on rearrangement reactions: with Au catalysts: (a) J. Wang, X. Yao, T. Wang, J. Han, J. Zhang, X. Zhang, P. Wang and Z. Zhang, Org. Lett., 2015, 17, 5124 CrossRef CAS PubMed ; with Pd catalysts: ; (b) J. Wang, X. Yao, T. Wang, J. Han, J. Zhang, X. Zhang, P. Wang and Z. Zhang, Chem. Commun., 2015, 51, 15204 RSC . With Rh catalysts: ; (c) Z. Li, V. Boyarskikh, J. H. Hansen, J. Autschbach, D. G. Musaev and H. M. L. Davies, J. Am. Chem. Soc., 2012, 134, 15497 CrossRef CAS PubMed.
- Selected further references: (a) Y. Sha, S. Feng, Z. Liu, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2015, 54, 7891 CrossRef PubMed; (b) Z.-S. Chen, X.-H. Duan, P.-X. Zhou, S. Ali, J.-Y. Luo and Y.-M. Liang, Angew. Chem., Int. Ed., 2012, 51, 1370 CrossRef CAS PubMed.
- Selected references on our work: (a) K. J. Hock, A. Knorrscheidt, R. Hommelsheim, J. Ho, M. Weissenborn and R. M. Koenigs, Angew. Chem., Int. Ed., 2019, 58, 3660 CrossRef PubMed; (b) K. J. Hock, L. Mertens and R. M. Koenigs, Chem. Commun., 2016, 52, 13783 RSC.
- Selected review articles: (a) M. Rubin, M. Rubina and V. Gevorgyan, Synthesis, 2006, 1221 CAS; (b) I. Marek, S. Simaan and A. Masarwa, Angew. Chem., Int. Ed., 2007, 46, 7364 CrossRef CAS PubMed.
- References on cyclopropenation reactions of alkynes using Rh catalysts: (a) J. F. Briones and H. M. L. Davies, J. Am. Chem. Soc., 2012, 134, 11916 CrossRef CAS PubMed; (b) T. Goto, K. Takeda, N. Shimada, H. Nambu, M. Anada, M. Shiro, K. Ando and S. Hashimoto, Angew. Chem., Int. Ed., 2011, 50, 6803 CrossRef CAS PubMed; (c) Y. Lou, M. Horikawa, R. A. Kloster, N. A. Hawryluk and E. J. Corey, J. Am. Chem. Soc., 2004, 126, 8916 CrossRef CAS PubMed; (d) H. M. L. Davies and G. H. Lee, Org. Lett., 2004, 6, 1233 CrossRef CAS PubMed; (e) M. K. Palleria and J. M. Fox, Org. Lett., 2007, 9, 5625 CrossRef PubMed; (f) B. Morandi and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 4294 CrossRef CAS PubMed; (g) M. P. Doyle, M. Protopopova, P. Müller, D. Ene and E. A. Shapiro, J. Am. Chem. Soc., 1994, 116, 8492 CrossRef CAS; (h) M. P. Doyle, D. G. Ene, C. S. Peterson and V. Lynch, Angew. Chem., Int. Ed., 1999, 38, 700 CrossRef CAS; (i) S. Chuprakov and V. Gevorgyan, Org. Lett., 2007, 9, 4463 CrossRef CAS PubMed; (j) K. J. Hock, R. Spitzner and R. M. Koenigs, Green Chem., 2017, 19, 2118 RSC; (k) U. P. N. Tran, R. Hommelsheim, Z. Yang, C. Empel, K. J. Hock, T. V. Nguyen and R. M. Koenigs, ChemRxiv DOI:10.26434/chemrxiv.7545623.v1.
- Cyclopropenation reactions with Au catalysts: J. F. Briones and H. M. L. Davies, J. Am. Chem. Soc., 2012, 134, 11916 CrossRef CAS PubMed.
- Cyclopropenation reactions with Ir catalysts: M. Uehara, H. Suematsu, Y. Yasutomi and T. Katsuki, J. Am. Chem. Soc., 2011, 133, 170 CrossRef CAS PubMed.
- Cyclopropenation reactions with Ag-catalysts: (a) J. F. Briones and H. M. L. Davies, Org. Lett., 2011, 13, 3984 CrossRef CAS PubMed; (b) Z. Liu, Q. Li, P. Liao and X. Be, Chem. – Eur. J., 2017, 23, 4756 CrossRef CAS PubMed.
- Other catalytic cyclopropenation reactions: (a) L. Maestre, E. Ozkal, C. Ayats, A. Beltran, M. M. Diaz-Requejo, P. J. Perez and M. A. Pericas, Chem. Sci., 2015, 6, 1510 RSC; (b) X. Cui, X. Xu, H. Lu, S. Zhu, L. Wojtas and P. X. Zhang, J. Am. Chem. Soc., 2011, 133, 3304 CrossRef CAS PubMed.
- Metal-free thermal cyclopropenations: R. Barroso, A. Jimenez, M. C. Perez-Aguilar, M.-P. Cabal and C. Valdes, Chem. Commun., 2016, 52, 3677 RSC.
- Selected references: (a) W. E. von Doering and T. Mole, Tetrahedron, 1960, 10, 65 CrossRef; (b) W. B. DeMore, H. O. Pritchard and N. Davidson, J. Am. Chem. Soc., 1959, 81, 5874 CrossRef CAS.
- For details on other alcohols investigated, please see the ESI†.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc00927b |
| This journal is © The Royal Society of Chemistry 2019 |