
Iodine–DMSO-promoted divergent reactivities of arylacetylenes†
Suhail A.
Rather
ab,
Atul
Kumar
ab and
Qazi Naveed
Ahmed
 *ab
*ab
aMedicinal Chemistry Division, CSIR-Indian Institute of Integrative Medicine, Jammu 180001, India. E-mail: naqazi@iiim.ac.in
bAcademy of Scientific and Innovative Research (AcSIR), Jammu-180001, India
First published on 7th March 2019
Abstract
An unprecedented set of efficient, economical, atom-economic and exceedingly selective I2–DMSO-promoted methods is described for the generation of different structures. The reaction represents the first of its kind, involving the use of different iodine concentrations, temperatures, acids and salt to adjust the selectivity for the synthesis of different alkenes, α-functionalized ketones and α-ketomethylthioesters.
DMSO is a well-known polar aprotic organosulfur compound reported for its diverse roles as a solvent, oxidant, ligand, inhibitor of DNA primer, cryoprotectant, etc.1 Furthermore, the combination of iodine and DMSO has astoundingly altered the view of chemists in seeking prolific oxidative reactions.2 This unique combination has caught researchers’ attention owing to (a) its inexpensive protocols, (b) their stability towards air and moisture, (c) their environmentally benign nature, (d) efficient atom and step economy, (e) no need for pre-functionalization of starting materials, (f) high throughput results and (g) good functional group tolerance.3 The I2–DMSO system with acetylenes, alkenes and acetophenones has been broadly classified to act in two ways: (i) I2-catalyzed reactions in DMSO medium and (ii) I2–DMSO-mediated oxidative transformations.4 Previously, in either pathway, this system involved prior iodination of substrates, inclusive of different sp, sp2 and sp3 functionalities in the presence of molecular iodine, followed by Kornblum oxidation to 2-oxoaldehydes (OAs). Kornblum oxidation represents the conversion of primary halides into aldehydes by the action of DMSO with the simultaneous production of DMS and it is predominantly accessible at higher temperatures.5 These reactions of arylacetylenes in I2–DMSO to form aldehydes (OAs) mechanistically bear a resemblance to the reactions of alkenes and acetophenones and are well-documented.6 However, alkynes are recognized to exemplify divergent behaviours because the triple bonds can be functionalized into a myriad of constructions.7 In this regard, we present a different array of novel I2–DMSO-promoted selective reactions that are possible through the use of terminal alkynes and justify their divergent behaviour. On the one hand, at room temperature different arylacetylenes were exclusively transformed into tri-iodinated alkenes that were earlier reported to be obtained through the use of multiple expensive reagents/additives.8 Interestingly, on the other hand, the same reaction under heated conditions engendered unique – Sme-substituted regioselective di-iodinated alkenes. Such compounds are known to exhibit a wide variety of applications as key synthetic precursors and building blocks in organic synthesis.9 Further, we established direct conversion of alkynes into α-functionalized ketones such as α-mono and di-chlorinated ketones at room temperature in a regioselective manner using 2 N HCl and NaCl, respectively. Mono- and bis-α-chloro ketones are established as bis-electrophiles, which accounts for their extensive use, particularly in cyclocondensations to furnish different valuable heterocycles.10 Consequently, a number of synthetic methods for the construction of mono and bis-α-chloro ketones have been established in the past decades.11 Nevertheless, most of these procedures require dealing with toxic reagents, challenging waste streams, and corrosivity.12 Another divergence in the reactivity of arylacetylenes under heated conditions using TFA was illustrated for the generation of different novel α-ketomethylthioesters, which had not been previously reported (Scheme 1).13 Here as well, DMSO served as a cheap and efficient source of oxygen and SMe. As is evident, DMSO acted as an alternative for methanethiol gas, which is quite expensive and difficult to handle. In general, all these compounds as such are useful synthons for the generation of different complex organic architectures or can be used in situ to generate different structures.
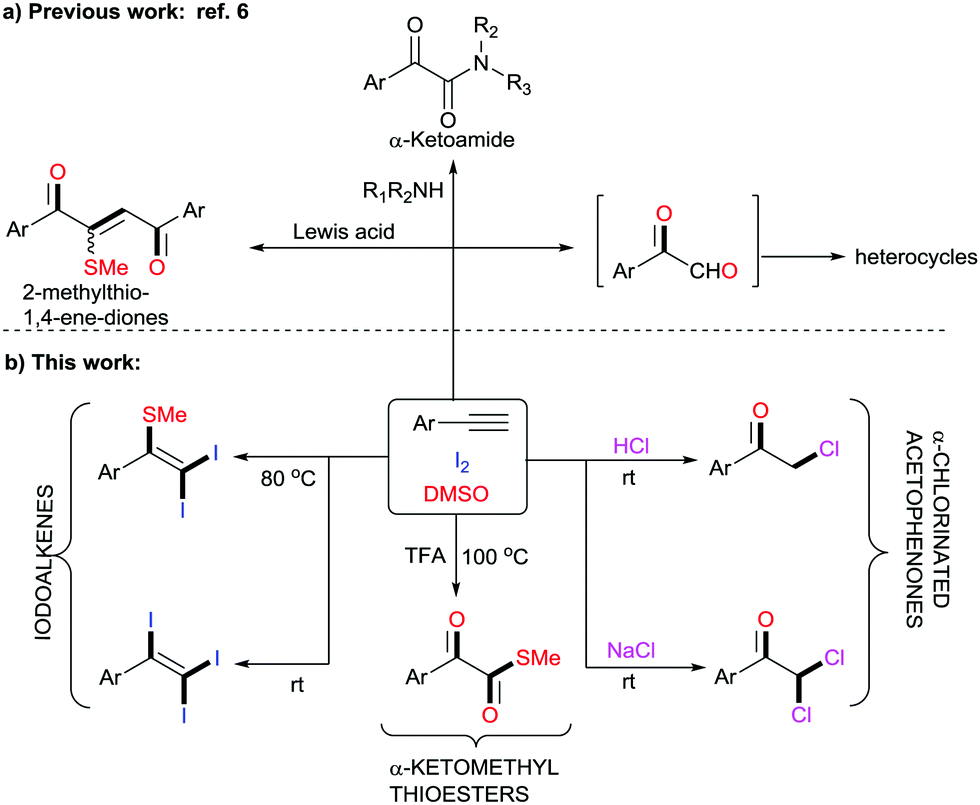 | ||
| Scheme 1 Summary of this work. | ||
In our initial investigation, we endeavoured to examine the nature of the products obtained on reacting phenylacetylene 1a and glycinemethylester hydrochloride (Table 1, entry 1). As per our previous work entitled “Divergent Reactivity of Amino Acid Alkyl Ester Hydrochlorides with 2-oxoaldehydes: Role of Selenium Dioxide To Promote Regioselective Synthesis of Imidazoles”, we expected methyl-2-(2-benzoyl-5-phenyl-1H-imidazol-1-yl)acetate as the major product.14 However, surprisingly, the reaction produced a mixture of unexpected products 2a, 3a, 4a, 5a, and 6a along with phenylglyoxal 7a. In order to attain better yields and product selectivity, a preliminary set of reactions was carried out under different conditions (Table 1, entries 2–15). Primarily, we examined the reaction of phenylacetylene 1a (1 mmol) with 1.2 mmol of I2 in DMSO at 80 °C for 2 h (entry 2). We observed that the (2,2-diiodo-1-phenylvinyl)(methyl)sulfane 2a was produced predominantly in 67% yield. Later, screening of our reaction at different concentrations of I2 was performed (entries 3–5), wherein we predominantly isolated 2a in 79% yield. Further, we observed an increase in the yield of triiodinated product 3a with a decrease in temperature (entries 6–8). However, the best yields were obtained when 1a (1 mmol) was stirred with I2 (2.2 mmol) in DMSO at r.t. for 4 h (entry 8). Further, in the reaction of phenylacetylene 1a and glycine methyl ester hydrochloride (entry 1, Table 1), we noticed α-functionalized ketones and α-ketomethylthioester as well. Thereby, we conducted different reactions in the presence of 2 N HCl (entries 9 and 10). Surprisingly, 2-chloro-1-phenylethan-1-one 4a was isolated in 72% yield when 1a (1 mmol) was stirred with I2 (2.2 mmol) and 2 N HCl in DMSO for 16 h at r.t. (entry 9). No further increase in yield was observed when the reaction was carried out with 2.5 mmol of I2 (entry 10). Later, a reaction was conducted with 1 mmol of NaCl and we noticed predominant conversion of 1a to 2,2-dichloro-1-phenylethan-1-one 5a (entry 11). In order to attain the maximum yield of 5a, reaction of 1a (1 mmol) with 2.2 mmol of I2 and 2 mmol of NaCl was conducted in DMSO at r.t. for 4 h and we successfully isolated 5a in 78% yield (entry 12). Next, three different test reactions of 1a (1 mmol) with I2 in DMSO were monitored in the presence of AcOH/TFA (1.0 mmol, entries 13–15). We succeeded in the isolation of S-methyl 2-oxo-2-phenylethanethioate 6a in 69% yield when stirred with TFA (1.0 mmol) at 100 °C (entry 14).
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | I2 (mmol) | Reagent (mmol) | Temp. (°C) | Time (h) | Yielda (%) | |||||
| 2a | 3a | 4a | 5a | 6a | 7a | |||||
| Reaction condition:a Phenylacetylene 1a (1.0 mmol) and I2 (2.2 mmol) in 3 mL of DMSO at 80 °C for 2 h.b Phenylacetylene 1a (1.0 mmol) and I2 (2.2 mmol) in 3 mL of DMSO at r.t. for 4 h.c Phenylacetylene 1a (1.0 mmol), I2 (2.2 mmol), and 2 N HCl (0.5 mL) in 3 mL of DMSO at r.t. for 16 h.d Phenylacetylene 1a (1.0 mmol), I2 (2.2 mmol), and NaCl (2.0 mmol) in 3 mL of DMSO at r.t. for 4 h.e Phenylacetylene 1a (1.0 mmol), I2 (2.2 mmol), and TFA (1.0 mmol) in 3 mL of DMSO at 100 °C for 4 h. | ||||||||||
| 1 | 1.2 | HCl·NH2CH2COOCH3 | 80 | 2 | 12 | 19 | 23 | 1 | 7 | 22 |
| 2 | 1.2 | — | 80 | 2 | 67 | 16 | — | — | — | 11 |
| 3 | 1.5 | — | 80 | 2 | 71 | 11 | — | — | — | 12 |
| 4 | 2.0 | — | 80 | 2 | 78 | 8 | — | — | — | Trace |
| 5 | 2.2 | — | 80 | 2 | 79 | 7 | — | — | — | Traces |
| 6 | 2.2 | — | 60 | 2 | 56 | 19 | — | — | — | Traces |
| 7 | 2.2 | — | 40 | 2 | 27 | 59 | — | — | — | Traces |
| 8 | 2.2 | — | r.t. | 4 | 9 | 81 | — | — | — | Trace |
| 9 | 2.2 | 2 N HCl (0.5 mL) | r.t. | 16 | — | — | 72 | 11 | — | Trace |
| 10 | 2.5 | 2 N HCl (0.5 mL) | r.t. | 16 | — | — | 73 | 19 | — | Trace |
| 11 | 2.2 | NaCl (1) | r.t. | 2 | — | — | 11 | 37 | — | 34 |
| 12 | 2.2 | NaCl (2) | r.t. | 4 | — | — | 9 | 78 | — | Trace |
| 13 | 2.2 | AcOH (1.0) | 100 | 2 | 14 | 19 | — | — | 27 | 37 |
| 14 | 2.2 | TFA (1.0) | 100 | 4 | Traces | — | — | — | 69 | 11 |
| 15 | 1.0 | TFA (1.0) | 100 | 4 | — | — | — | — | 19 | 27 |

Following these optimized procedures for the selective synthesis of each product, different sets of reactions were carried out to examine the substrate scope. In one set of experiments, different reactions were conducted between phenylacetylenes 1 with iodine in DMSO at 80 °C (Scheme 2, entries 2a–2k). It was observed that both electron-rich and electron-deficient phenylacetylenes could be smoothly transformed into the desired products 2. However, reactions with phenylacetylenes bearing electron-withdrawing groups, i.e., −F (2h), −Cl (2j), −Br (2i) and −CF3 (2k), produced slightly lower yields than those of unsubstituted 2a and those containing a donating group, 2b–2g. Another set of experiments was performed at room temperature with different acetylenes (3a–3k) as per the optimized procedure for the preparation of different tri-iodinated products 3 (Scheme 2, 3a–3k). As observed, the electronic environment of the phenyl ring in 1 affected the product yields to some extent. It was observed that the phenylacetylenes bearing electron-withdrawing groups, for example, −F (3h), −Cl (3j) and −CF3 (4m–4o), afforded slightly lower yields in comparison to unsubstituted acetylenes 3a and those containing electron-donating groups (3b–3g).
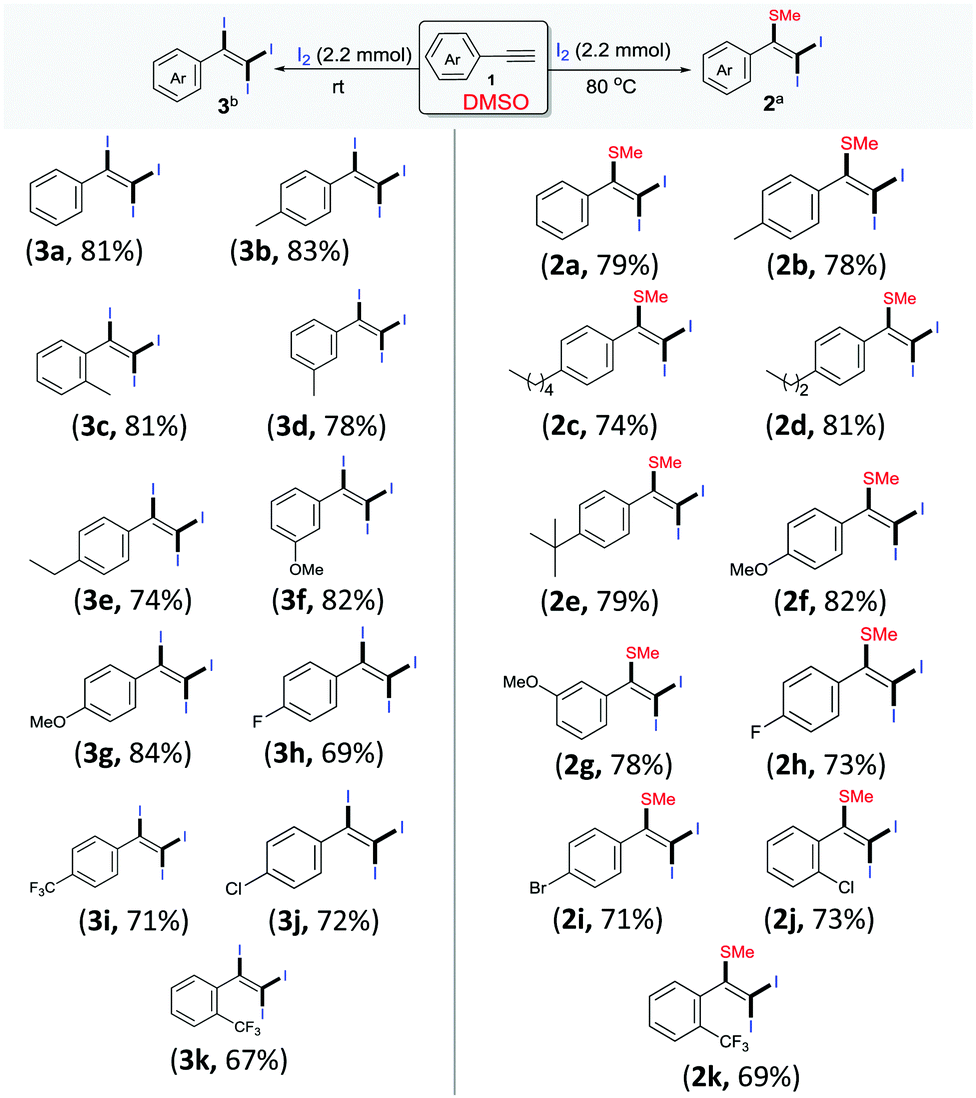 | ||
| Scheme 2 Scope of the reaction. Reaction conditions: aarylacetylene 1 (1.0 mmol) and I2 (2.2 mmol) in 3 mL of DMSO at 80 °C for 2 h. bArylacetylene 1 (1.0 mmol) and I2 (2.2 mmol) in 3 mL of DMSO at r.t. for 4 h. | ||
Having observed that the reaction of phenylacetylene 1a (1 mmol) with 2 N HCl and NaCl (2 mmol), respectively, as per the optimized procedure (Table 1, entries 9 and 12) promoted the selective synthesis of mono- and di-chlorinated acetophenone, we then decided to examine the substrate scope of each process (Scheme 3, 4a–4k and 5a–5k). As compiled, a variety of arylacetylenes 1 with diverse steric and electronic properties were tested and we were gratified to find that in all reactions tested the desired products 4 and 5 were produced predominantly in good yields (66–83%). In general, we observed that both electron-rich and electron-deficient acetylenes could be smoothly transformed into the desired product. However, in these cases the yields were also slightly better for unsubstituted and electron-donating acetylenes.
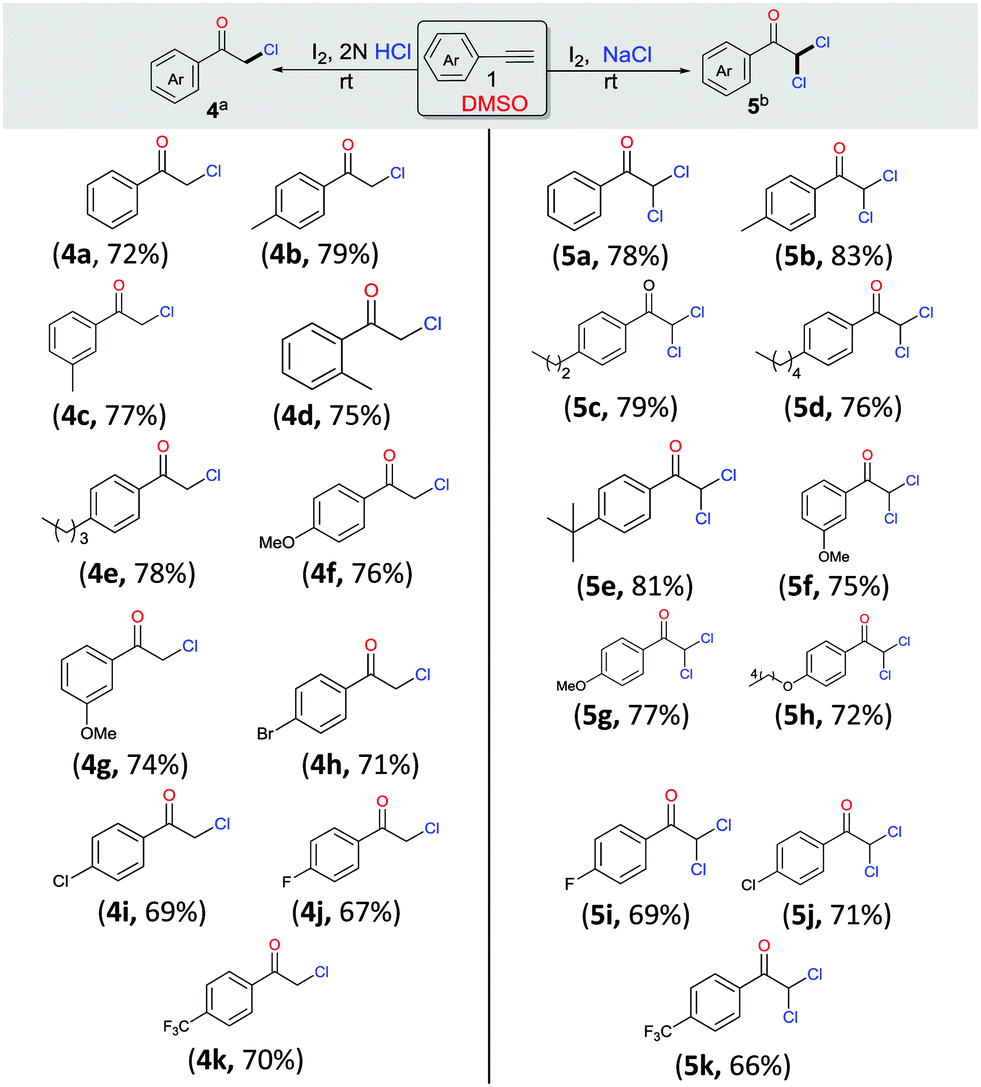 | ||
| Scheme 3 Scope of mono- and di-oxychlorination. Reaction conditions: aarylacetylene 1 (1.0 mmol), I2 (2.2 mmol), and 2 N HCl (0.5 mL) in 3 mL of DMSO at r.t. for 16 h. bArylacetylene 1 (1.0 mmol), I2 (2.2 mmol), and NaCl (2.0 mmol) in 3 mL of DMSO at r.t. for 4 h. | ||
Next, we performed different sets of experiments for the synthesis of α-ketomethylthioesters as per the optimized conditions. In all the reactions, we isolated the corresponding products 6 in good yields (64–72%) (Scheme 4a, 6a–6f). In addition, gram scale reaction with phenylacetylene was also performed that resulted in the isolation of 6a in 57% yield (Scheme 4b).
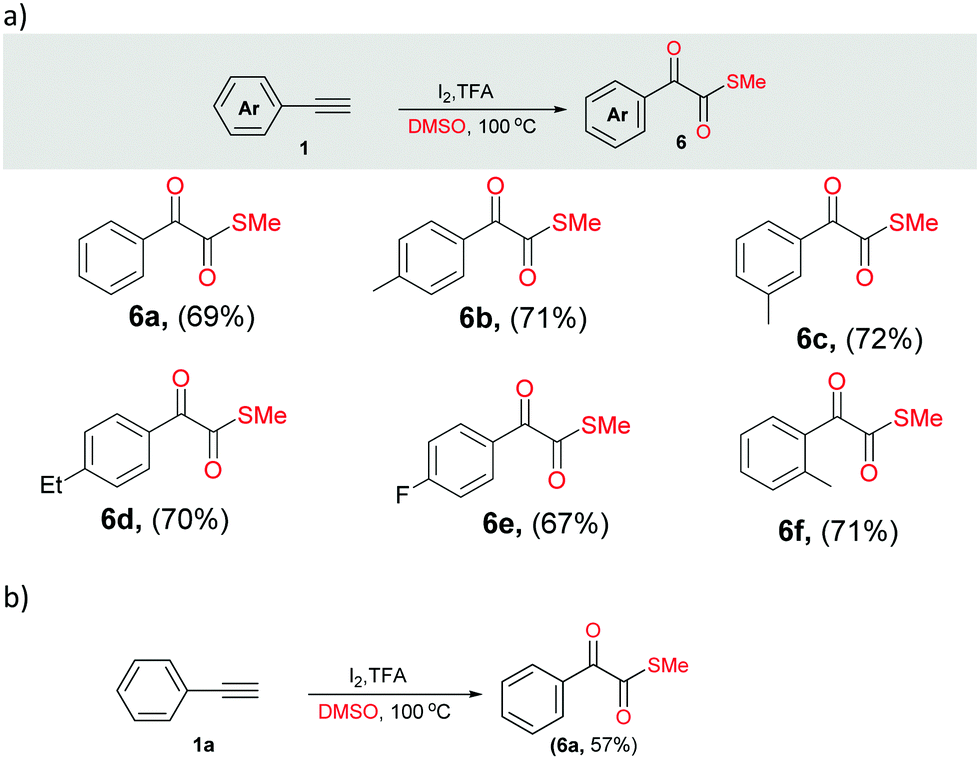 | ||
| Scheme 4 Studies related to 2-oxomethylthioesters. (a) Substrate scope. Reaction condition: arylacetylene 1 (1.0 mmol), I2 (2.2 mmol), TFA (1.0 mmol) in 3 mL of DMSO at 100 °C for 4 h. (b) Gram scale synthesis of S-methyl-2-oxo-2-phenylethanethioate. Reaction condition: arylacetylene 1 (9.8 mmol), I2 (21.6 mmol), TFA (9.8 mmol) in 30 mL of DMSO at 100 °C for 8 h. | ||
In order to probe the mechanism of these reactions, we performed a few controlled experiments using (iodoethynyl)benzene A as the starting material (Scheme 5a). In experiment 1, the reaction of A with I2 as per the optimization in entry 5, Table 1 at 80 °C generated 2a in 83% yield. Similarly, the reaction of 1a at r.t. as per entry 8, Table 1 selectively generated 3a in 86% yield. On the basis of these observations, we speculated that all the reactions are feasible through a common intermediate A. In addition, experiments 3, 4, and 5 also justified that these syntheses primarily involve iodination to A. Further, in experiment 6 reaction of phenylacetylene 1a with 1 mmol of iodine and 1 mmol of TFA at 100 °C as per Shah's procedure generated predominantly product 8.7a However, in our reactions, we used 2.2 mmol of iodine, which clearly indicates the role of excess iodine in the generation of DMS in higher concentration, which stops the phenylglyoxal pathway for generation of 8.
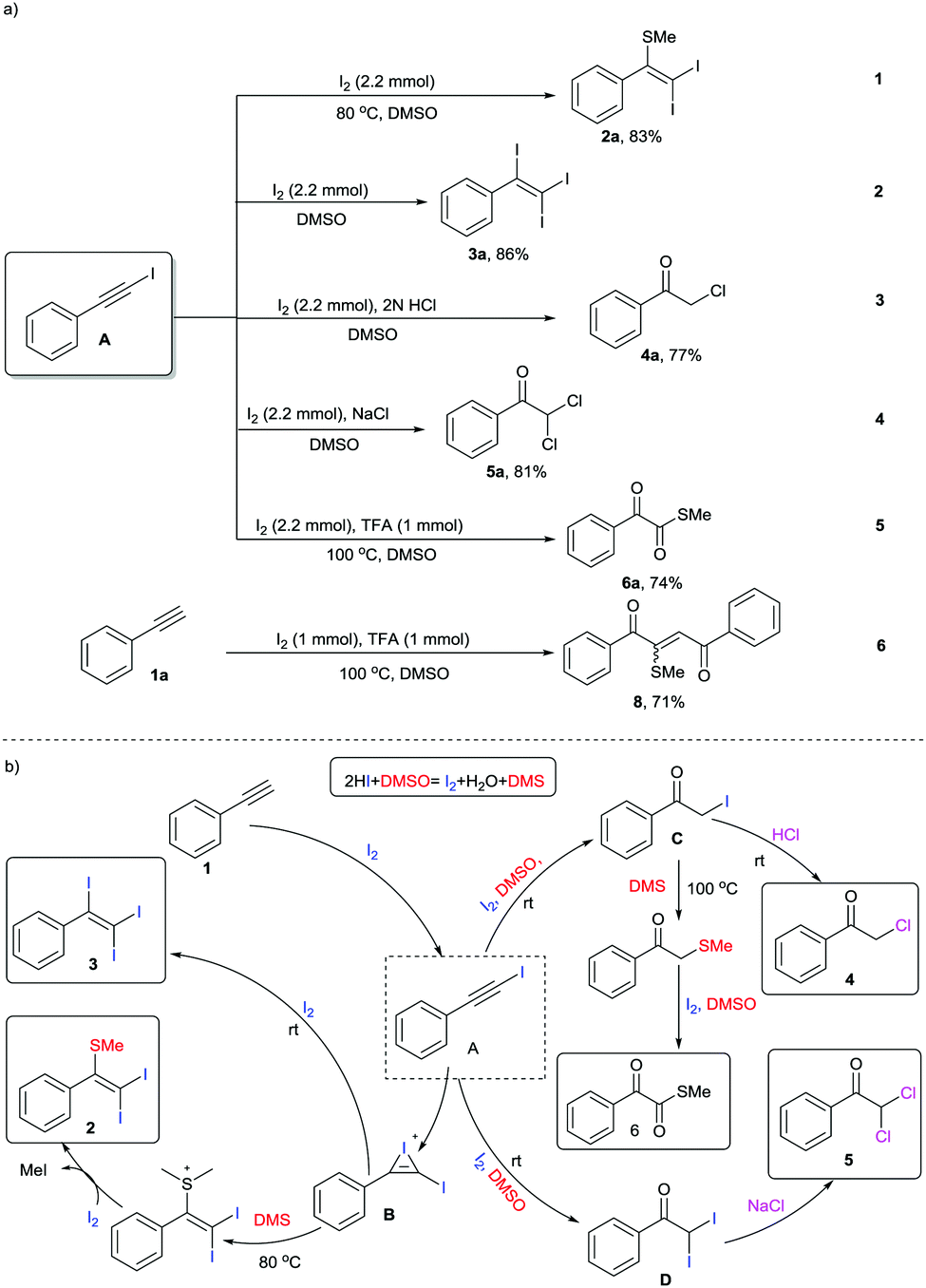 | ||
| Scheme 5 (a) Control experiments. (b) Proposed mechanism. | ||
Based on the preceding literature reports and the above-mentioned studies, we propose the mechanism of these reactions as depicted in Scheme 5b. The initial step for the synthesis of each product 2, 3, 4, 5, and 6 primarily involves the reaction of acetylene 1 and iodine to give intermediate A. This intermediate behaves differently under different conditions. In the additive-free pathway for the synthesis of alkenes 2 and 3, intermediate A undergoes iodination to common iodonium intermediate B, which either undergoes conversion to 2 at 80 °C or adds one more mole of iodine to 3 at r.t. However, in the presence of acid/NaCl, intermediate A reacts to form mono- C and di-iodinated acetophenone D, which ultimately led to the synthesis of 4 and 5, respectively. On the other hand, in the presence of TFA, intermediate A through C can undergo S-methylation followed by I2–DMSO-mediated oxidation to compound 6.
In conclusion, we have revealed a set of different, efficient, atom-economic, economical, mild and novel synthetic methods based around arylacetylenes for the generation of different isolable selective products via unconventional I2–DMSO-promoted reaction in different environments. Despite the variation in the nature of acetylenes, the reactions presented a broad substrate scope. Further study to expand the scope in one pot for generation of different coupled products is currently underway in our lab.
S. A. R and A. K. thank CSIR and University Grants Commission (UGC) for the award of a JRF and SRF. This work was generously supported by a CSIR network project through grant no. HCP-0008. IIIM communication no. IIIM.2277/2019.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) X. F. Wu and K. Natte, Adv. Synth. Catal., 2016, 358, 336–352 CrossRef CAS; (b) D. Martin, A. Weise and H. J. Niclas, Angew. Chem., 1967, 79, 318–334 CrossRef PubMed; (c) W. Zierkiewicz and T. Privalov, Organometallic, 2005, 24, 6019–6028 CrossRef CAS; (d) C. N. Yiannios and J. V. Karabinos, J. Org. Chem., 1963, 28, 3246–3248 CrossRef CAS.
- (a) Y. F. Liang, X. Li, X. Wang, M. Zou, C. Tang, Y. Liang, S. Song and N. Jiao, J. Am. Chem. Soc., 2016, 138, 12271–12272 CrossRef CAS PubMed; (b) Y. F. Liang, S. Song, L. Ai, X. Lia and N. Jiao, Green Chem., 2016, 18, 6462–6467 RSC; (c) J. B. Azeredo, M. Godoi, G. M. Martins, C. C. Silveira and A. L. Braga, J. Org. Chem., 2014, 79, 4125–4130 CrossRef CAS PubMed; (d) R. R. Addada, V. RRegalla, V. N. Vema and V. R. Anna, Tetrahedron Lett., 2016, 57, 2838–2841 CrossRef; (e) N. Mupparapu, S. Khan, S. Battula, M. Kushwaha, A. P. Gupta, Q. N. Ahmed and R. A. Vishwakarma, Org. Lett., 2014, 16, 1152–1155 CrossRef CAS PubMed.
- (a) I. Soroko, Y. Bhole and A. G. Livingston, Green Chem., 2011, 13, 162–168 RSC; (b) M. Doble and A. Kumar, Green Chemistry and Engineering, Elsevier Inc., New York, 2007, ch. 5, pp. 93–104 Search PubMed; (c) W. M. Nelson, Green Solvents for Chemistry: Perspectives and Practice in Green Chemistry, 1st edn, Oxford University Press, USA, 2003, ch. 3, pp. 60–62 and ch. 5, pp. 116–132 Search PubMed; (d) B. A. Trofimov, Sulfur Rep., 1992, 74, 207 CrossRef.
- (a) Y. Jianga and T. P. Loh, Chem. Sci., 2014, 5, 4939–4943 RSC; (b) S. Kolita, P. Borah, P. S. Naidu and P. J. Bhuyan, Tetrahedron, 2016, 72, 532–538 CrossRef CAS; (c) R. Mishra, A. Jana, A. K. Panday and L. H. Choudhury, Org. Biomol. Chem., 2018, 16, 3289–3302 RSC.
- (a) N. Kornblum, W. J. Jones and G. J. Anderson, J. Am. Chem. Soc., 1959, 81, 4113–4114 CrossRef CAS; (b) X. Wu, X. Geng, P. Zhao, Y. d. Wu and A. x. Wu, Org. Lett., 2017, 19, 4584–4587 CrossRef CAS PubMed.
- (a) B. Sis, M. Zirak and A. Akbari, Chem. Rev., 2013, 113, 2958–3043 CrossRef PubMed; (b) K. K. D. R. Viswanadham, M. P. Reddy, P. Sathyanarayana, b. Ravi, b. Kantc and S. R. Bathula, Chem. Commun., 2014, 50, 13517–13520 RSC.
- (a) S. Devari, A. Kumar, R. Deshidi and B. A. Shah, Chem. Commun., 2015, 51, 5013–5016 RSC; (b) M. Phanindrudu, D. K. Tiwari, V. K. Aravilli, K. C. Bhardwaj, G. Sabapathi, P. R. Likhar and D. K. Tiwari, Eur. J. Org. Chem., 2016, 4629–4634 CrossRef CAS.
- (a) Y. Liu, D. Huang, J. Huang and K. Maruoka, J. Org. Chem., 2017, 82, 11865–11871 CrossRef CAS PubMed; (b) V. Sriramoju, R. Jillella, S. Kurva and S. Madabhushi, Chem. Lett., 2017, 46, 560–562 CrossRef CAS.
- (a) S. Ni, W. Sha, L. Zhang, C. Xie, H. Mei, J. Han and Y. Pan, Org. Lett., 2016, 18, 712–715 CrossRef CAS PubMed; (b) J. R. Falck, P. K. Patel and A. Bandyopadhyay, J. Am. Chem. Soc., 2007, 129, 790–793 CrossRef CAS PubMed; (c) W. Shen and A. Kunzer, Org. Lett., 2002, 4, 1315–1317 CrossRef CAS PubMed; (d) J. Barluenga, M. A. Rodriguez, P. J. Campos and G. Asensio, J. Am. Chem. Soc., 1988, 110, 5567–5568 CrossRef CAS; (e) W. Shen and S. A. Thomas, Org. Lett., 2000, 2, 2857–2860 CrossRef CAS PubMed; (f) J. S. Panek and T. J. Hu, J. Org. Chem., 1997, 62, 4912–4913 CrossRef CAS; (g) M. L. N. Rao, P. Dasgupta, B. S. Ramakrishna and V. N. Murty, Tetrahedron Lett., 2014, 55, 3529–3533 CrossRef CAS.
- (a) M. A. Ganiek, M. V. Ivanova, B. Martin and P. Knochel, Angew. Chem., Int. Ed., 2018, 57, 1–6 CrossRef PubMed; (b) N. D. Kimpe and R. Verhe, in The Chemistry of α-Haloketones, α-Haloaldehydes and α-Haloimines, ed. S. Patai and Z. Rappoport, Wiley, Chichester, 1988 CrossRef; (c) Name Reactions in Heterocyclic Chemistry, ed. J. J. Li and E. J. Corey, Wiley, Hoboken, 2005 Search PubMed.
- (a) C. Valente, S. Calimsiz, K. H. Hoi, D. Mallik, M. Sayah and M. G. Organ, Angew. Chem., Int. Ed., 2012, 51, 3314–3332 CrossRef CAS PubMed; (b) M. A. Dfert, K. L. Billingsley and S. L. Buchwald, J. Am. Chem. Soc., 2013, 135, 12877–12885 CrossRef PubMed; (c) M. A. Larsen and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 4287–4299 CrossRef CAS PubMed.
- (a) S. Madabhushi, R. Jillella, K. K. R. Mallu, K. R. Godala and V. S. Vangipuram, Tetrahedron Lett., 2013, 54, 3993–3996 CrossRef CAS; (b) J. Liu, W. Li, C. Wang, Y. Li and Z. Li, Tetrahedron Lett., 2011, 52, 4320–4323 CrossRef CAS; (c) Y. Xing, M. Zhang, S. Ciccarelli, J. Lee and B. Catano, Eur. J. Org. Chem., 2017, 781–785 CrossRef CAS.
- (a) N. Mupparapu, M. Khushwaha, A. P. Gupta, P. P. Singh and Q. N. Ahmed, J. Org. Chem., 2015, 80, 11588–11592 CrossRef CAS PubMed; (b) B. Hu, P. Zhou, Q. Zhang, Y. Wang, S. Zhao, L. Lu, S. Yan and F. Yu, J. Org. Chem., 2018, 83, 14978–14986 CrossRef CAS PubMed.
- A. K. Padala, R. R. Kumar, S. Athimoolam and Q. N. Ahmed, Org. Lett., 2016, 18, 96–99 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc00346k |
| This journal is © The Royal Society of Chemistry 2019 |