
Unified enantioselective total syntheses of (−)-scholarisine G, (+)-melodinine E, (−)-leuconoxine and (−)-mersicarpine†
Yao
Liu
and
Honggen
Wang
 *
*
School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510006, China. E-mail: wanghg3@mail.sysu.edu.cn
First published on 26th February 2019
Abstract
A unified strategy enabled the enantioselective syntheses of (−)-scholarisine G, (+)-melodinine E, (−)-leuconoxine and (−)-mersicarpine from a common 2-alkylated indole intermediate bearing an all-carbon quaternary stereogenic center. The Smith-modified Madelung indole synthesis was used to couple simple o-toluidine with chiral lactone (+)-8, incorporating the key elements for further cyclizations. Lactone (+)-8 was prepared via a palladium-catalyzed intermolecular asymmetric allylic alkylation. The unified and protecting-group-free reaction sequences allowed the synthesis of these alkaloids in a maximum of 10 steps and with high efficiency.
The leuconolam–leuconoxine–mersicarpine triads are structurally complex and biologically interesting Aspido-sperma-derived monoterpene indole alkaloids (Fig. 1).1 Biosynthetically, these natural products share the same biogenetic origin from vincadifformine,2 but feature intriguingly different ring connectivities. (−)-Scholarisine G (1),3a,e (+)-melodinine E (2)3b and (−)-leuconoxine (3)3c–e are pentacyclic alkaloids comprising an interesting [5.5.6.6]diazafenestrane core4 with two or three contiguous quaternary stereogenic centers. (−)-Mersicarpine (4),3f however, has a fused tetracyclic 6/5/6/7 ring system characterized by an unusual tetrahydro-2H-azepine ring and a hemiaminal motif. The structural complexity, along with the intriguing bioactivities has rendered these alkaloids popular targets in total synthesis.5–8 Specifically, the biosynthetic interrelationship of these compounds has inspired several unified synthetic strategies towards their synthesis.6j,7f,8 Nevertheless, only a handful of enantioselective total syntheses have been reported.7,8
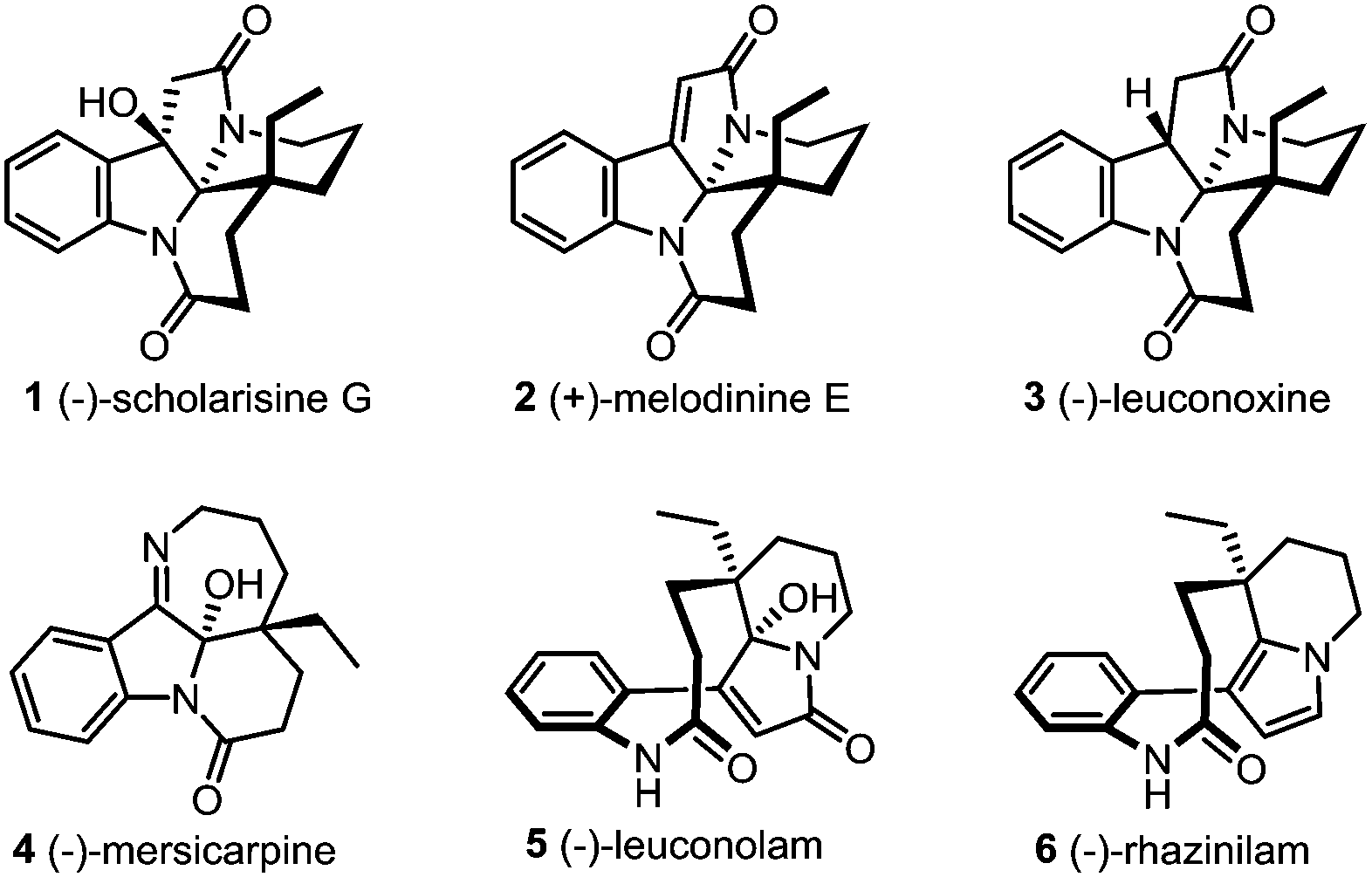 | ||
| Fig. 1 The leuconolam–leuconoxine–mersicarpine group of alkaloids. | ||
The intrinsic challenge to fulfil an enantioselective total synthesis lies in the construction of the all-carbon quaternary stereogenic carbon center.6,9 In 2010, Fukuyama and co-workers reported the first total synthesis of (−)-mersicarpine (4) (Scheme 1).7a The key chiral intermediate ketoester (B) was prepared via asymmetric Michael addition. Upon 7-step synthetic manipulations including Eschenmoser–Tanabe fragmentation, Sonogashira cross-coupling reaction and gold-catalyzed cyclization, a 2-substituted indole (C) with a chiral quaternary carbon center was assembled, which was further elaborated to the final product. Intriguingly, in an effort to synthesize (−)-rhazinal, Luo observed an unexpected aziridination/rearrangement/oxidation tandem reaction leading to the total synthesis of (−)-mersicarpine (4) based on a similar alkenylated indole intermediate (D).7d Starting from the same chiral intermediate (B), Tokuyama and co-workers accomplished a concise total synthesis of (−)-mersicarpine via the key Fischer indole synthesis and DIBAL-H-mediated reductive ring-expansion reaction.7b,c In 2013, Zhu and co-workers disclosed an enantioselective total synthesis of leuconolam–leuconoxine–mersicarpine group monoterpene indole alkaloids8 based on an elegantly integrated oxidation/reduction/cyclization (iORC) process.10 The palladium-catalyzed enantioselective decarboxylative allylation was utilized to construct the chiral center. The same strategy was utilized by Liang and Stoltz by employing an optically active allylated lactone (8),7f prepared from intramolecular palladium-catalyzed asymmetric decarboxylative allylic alkylation of N-benzyloxy cyclic imide (K),11 as a key intermediate.
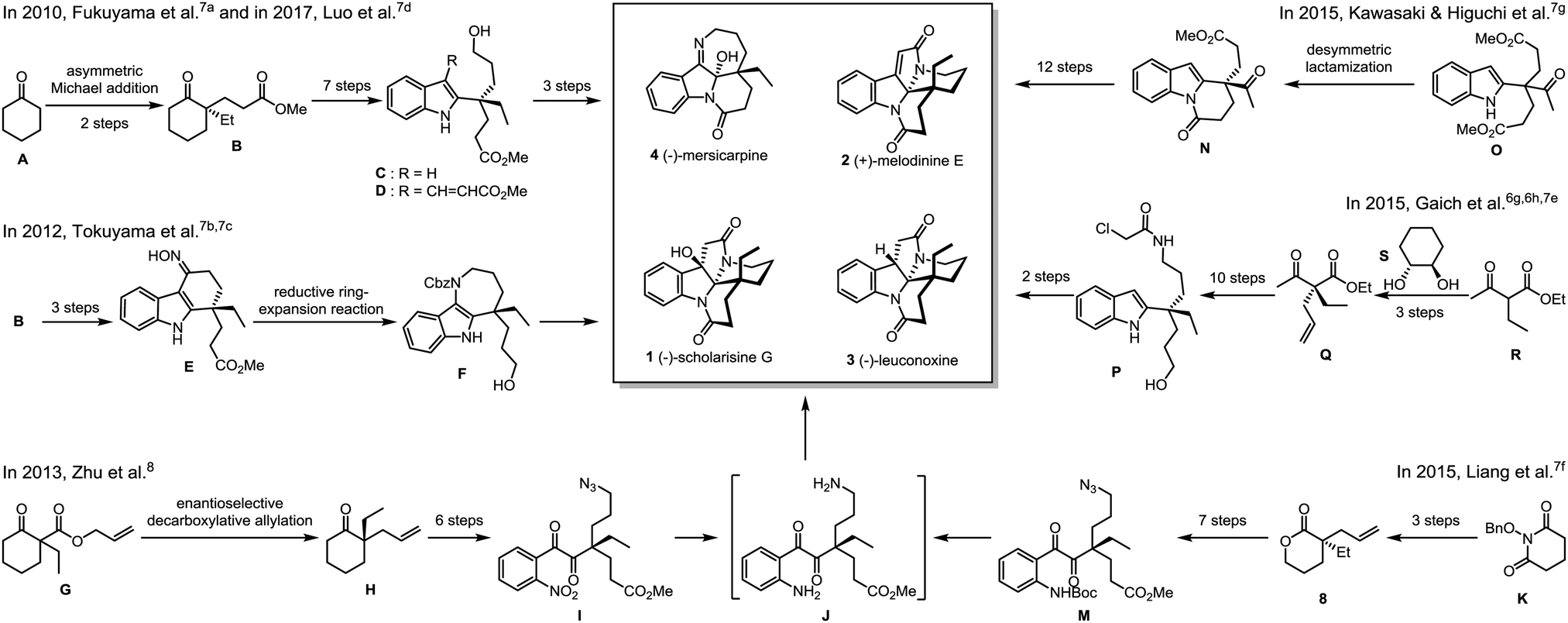 | ||
| Scheme 1 Reported enantioselective total syntheses of the leuconolam–leuconoxine–mersicarpine group alkaloids. | ||
In another vein from Kawasaki and Higuchi, the phosphoric acid-catalyzed desymmetric lactamization of a prochiral indole-substituted diester (O) provided the key enantiomerically enriched Kerr's intermediate with moderate ee of 74%.7g In 2015, Gaich realized enantioselective total synthesis of (−)-leuconoxine (3) by employing photoinduced domino macrocyclization/transannular cyclization involving Witkop cyclization.6g,h,7e The optically active precursor (P) was obtained via the diastereoselective alkylation of ethyl 2-ethylacetoacetate (R) using chiral 1,2-diol (S) as an acetal chiral auxiliary.
The notable feature of Smith-modified Madelung indole synthesis12 in the construction of 2-quaternary carbon substituted indole inspired us to explore novel enantioselective synthesis of leuconolam–leuconoxine–mersicarpine alkaloids starting from simple o-toluidine (9) and chiral lactone (8) (Scheme 2). The latter is commercially available and could be prepared via palladium-catalyzed intermolecular asymmetric allylation developed by Hou.13 The Smith-modified Madelung indole synthesis would provide a pivotal indole derivative with the chiral center being installed. The hydroxyl and vinyl functionality in 10 serve as valuable handles for further transformation. Therefore, upon proper functional group manipulation, lactam (13) is expected to be obtained. This species could be further elaborated to Zhu8 and Dai's6j intermediates via oxidation of the indole motif,6a,14 paving the way for (−)-scholarisine G (1) and (−)-mersicarpine (4) synthesis, respectively.
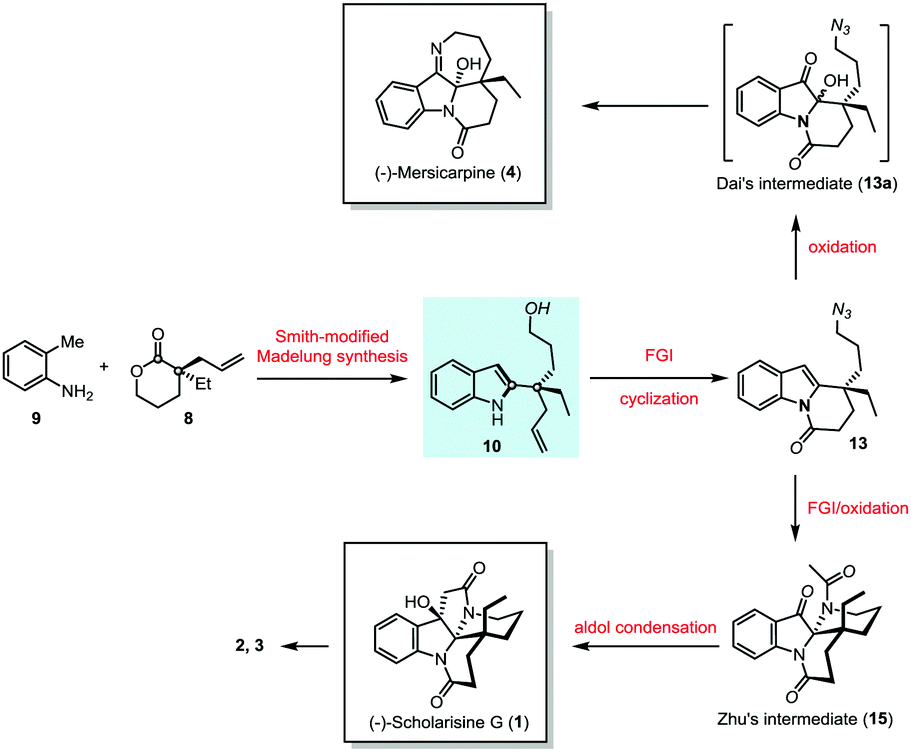 | ||
| Scheme 2 Proposed synthetic strategy. | ||
Our synthesis commenced with the preparation of the allylated lactone (+)-8 starting from 3-ethyltetrahydro-2H-pyran-2-one and allyl methyl carbonate (7). In the presence of the palladium catalyst and (R)-DM-BINAP ligand as developed by Hou,13 (+)-(8) was obtained in 72% yield and 89% ee (eqn (1)).
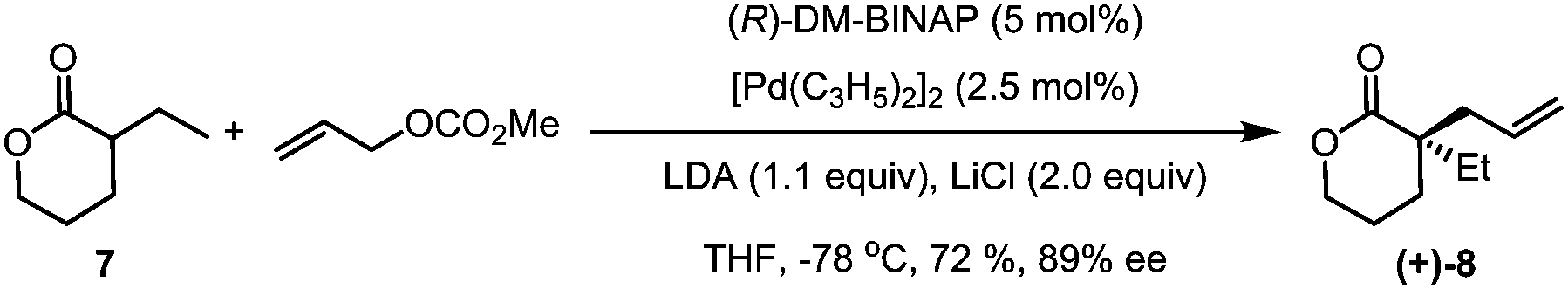 | (1) |
The key Smith-modified Madelung indole synthesis was started with the preparation of N-silylated o-toluidine (9a) via the reaction of o-toluidine (9) with a stoichiometric amount of n-butyllithium and followed by quenching with chlorotrimethylsilane (Scheme 3). Without isolation, this intermediate was exposed to 2.2 equivalents of sec-butyllithium solution at low temperature to form a reactive lithium dianion (9b). Upon slow addition of lactone (+)-8, cascade acylation/heteroatom Peterson olefination/isomerization proceeded smoothly to produce 2-quaternary carbon substituted indole (−)-10 in an overall 85% yield.
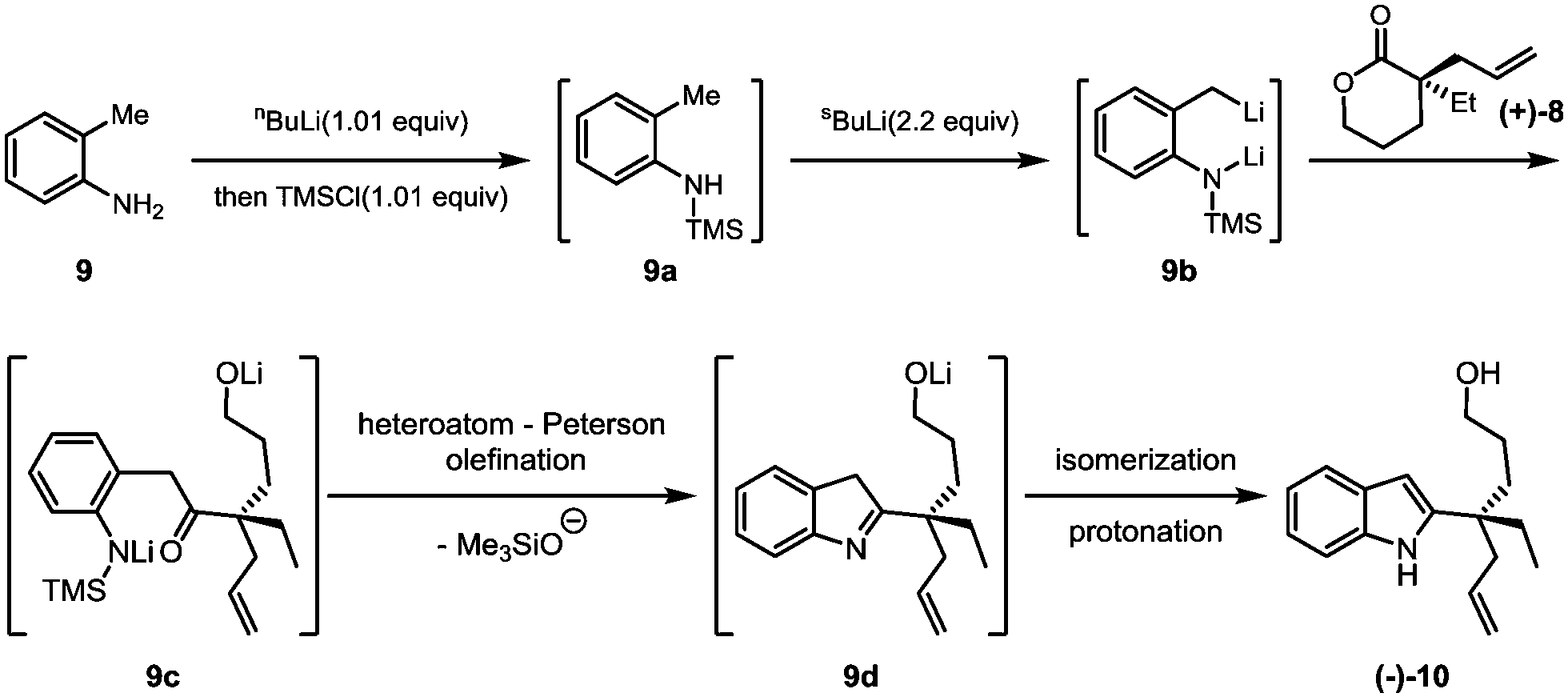 | ||
| Scheme 3 Smith-modified Madelung synthesis. | ||
The hydroxyl group in indole (−)-10 was then replaced by azido in a good yield via a Mitsunobu reaction in the presence of diisopropyl azodiformate (DIAD), triphenylphosphine and diphenylphosphonic azide (DPPA) (Scheme 4). The maintenance of low temperature (0 °C) is crucial for this step as a higher temperature (room temperature) led to a significant amount of the intramolecular nitrogen alkylation product. Following hydroboration/oxidation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, azidoindole (+)-11 was converted to (−)-12 in good efficiency (a 71% yield). Exposure of (−)-12 to Ley oxidation15 (TPAP and NMO, at rt) resulted in an intramolecular N-acylation reaction to afford N-acyl indole (+)-13 in 68% yield. With (+)-13 in hand, we next explored the synthesis of (−)-mersicarpine (4). Previous studies indicated that 2-substituted indole could easily be oxidized with various oxidants to form a keto hemiaminal structure.14 Indeed, subjection of (+)-13 to Kerr's conditions6a (oxone, acetone) afforded the desired keto hemiaminal (13a). Upon in situ treatment with PPh3, 13a underwent Staudinger-aza-Wittig cyclization to give (−)-mersicarpine (4) in 64% yield over two steps. It should be noted that the same intermediate 13a has been obtained in Dai's (±)-mersicarpine synthesis via a Witkop–Winterfeldt oxidative cleavage of an advanced indole structure.
C bond, azidoindole (+)-11 was converted to (−)-12 in good efficiency (a 71% yield). Exposure of (−)-12 to Ley oxidation15 (TPAP and NMO, at rt) resulted in an intramolecular N-acylation reaction to afford N-acyl indole (+)-13 in 68% yield. With (+)-13 in hand, we next explored the synthesis of (−)-mersicarpine (4). Previous studies indicated that 2-substituted indole could easily be oxidized with various oxidants to form a keto hemiaminal structure.14 Indeed, subjection of (+)-13 to Kerr's conditions6a (oxone, acetone) afforded the desired keto hemiaminal (13a). Upon in situ treatment with PPh3, 13a underwent Staudinger-aza-Wittig cyclization to give (−)-mersicarpine (4) in 64% yield over two steps. It should be noted that the same intermediate 13a has been obtained in Dai's (±)-mersicarpine synthesis via a Witkop–Winterfeldt oxidative cleavage of an advanced indole structure.
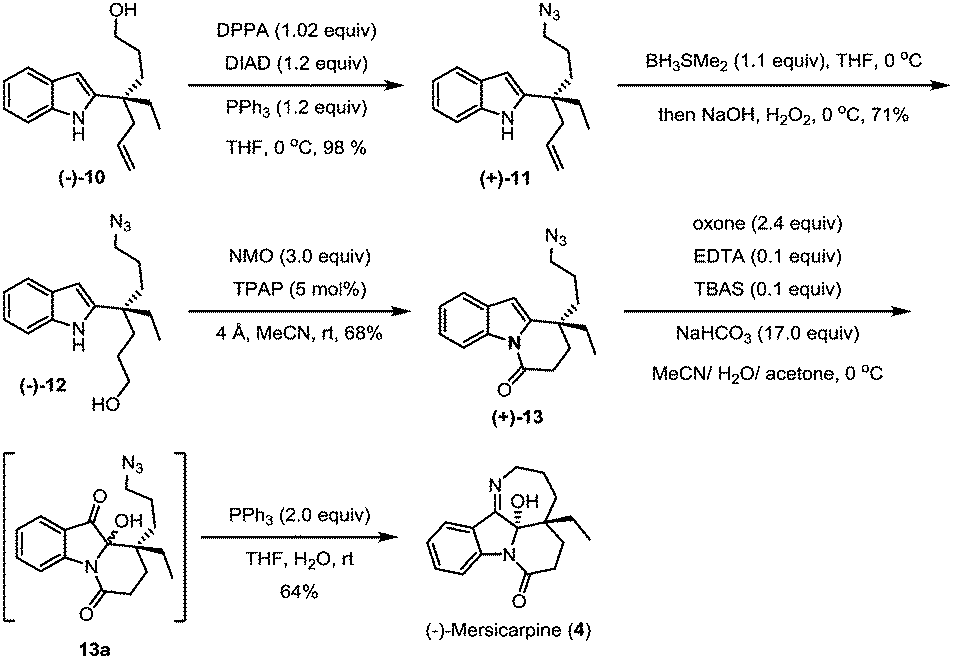 | ||
| Scheme 4 Syntheses of (−)-mersicarpine. | ||
The azide intermediate (+)-13 could also be converted to leuconoxine family alkaloids (Scheme 5). Thus, (+)-13 was first reduced using triphenylphosphine and then acetylated by a follow-up treatment with acetic anhydride to give acetamide (+)-14. Under similar indole oxidation conditions with oxone as described above, keto hemiaminal 14a was produced. Without isolation, 14a was converted under acidic conditions to Zhu's intermediate (+)-15 for their leuconolam–leuconoxine indole alkaloid syntheses in 65% yields over two steps. LDA-promoted intramolecular aldol cyclization provided leuconoxine in a good yield of 77%. Previously, mesylation of the tertiary hydroxyl group in (−)-scholarisine G (1) followed by base-promoted elimination was used to prepare (+)-melodinine E (2). We found that higher efficiency could be obtained when treating (−)-scholarisine G (1) with a Burgess reagent (2.5 equiv.) in acetonitrile at 70 °C. Finally, hydrogenation of (+)-melodinine E (2) delivered another member (−)-leuconoxine (3) in 94% yield. The spectroscopic data of (+)-melodinine E (2) and (−)-leuconoxine (3) (1H and 13C NMR) matched well with those reported in the literature. Interestingly, the NMR spectra of our synthetic (−)-scholarisine G (1) match with that of Zhu,8a but show discrepancies with the isolated samples3a,e and some other synthetic samples.6e,j,7f We assume that the differences are a result of different quality, and therefore different acidity, of CDCl3 used for the NMR studies.16
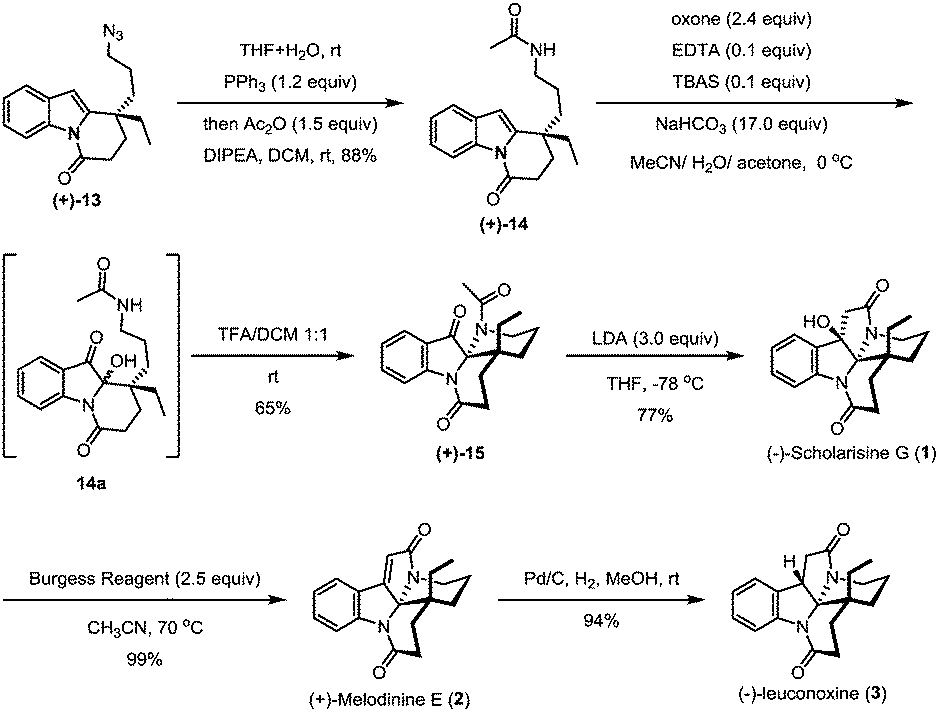 | ||
| Scheme 5 Synthesis of (−)-scholarisine G, (+)-melodinine E and (−)-leuconoxine. | ||
In conclusion, we have accomplished divergent enantioselective syntheses of four monoterpene indole alkaloids: (−)-scholarisine G (1), (+)-melodinine E (2), (−)-leuconoxine (3) and (−)-mersicarpine (4). The syntheses feature a palladium-catalyzed intermolecular asymmetric allylation to construct an optically active lactone, Smith-modified Madelung indole synthesis to quickly forge a quaternary carbon-substituted indole, and an oxone-mediated indole oxidation to form Dai’ and Zhu's intermediates, respectively. Efforts were also attempted to improve the synthetic efficiency of transforming Zhu's intermediates (15) to the leuconoxine group alkaloid. No protecting group is needed for the whole process, allowing concise syntheses of the title natural products in a maximum of 10 steps with high efficiency.
Generous financial support from the Key Project of Chinese National Programs for Fundamental Research and Development (2016YFA0602900), the National Natural Science Foundation of China (21472250), and the “1000-Youth Talents Plan” is gratefully acknowledged.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) M. Ishikura, T. Abe, T. Choshi and S. Hibino, Nat. Prod. Rep., 2013, 30, 694 RSC; (b) Y.-Y. Low, F.-J. Hong, K.-H. Lim, N. F. Thomas and T.-S. J. Kam, Nat. Prod., 2014, 77, 327 CrossRef CAS PubMed; (c) F. Abe and T. Yamauchi, Phytochemistry, 1993, 35, 169 CrossRef; (d) J. Hájíček, Collect. Czech. Chem. Commun., 2011, 76, 2023–2083 CrossRef.
- (a) G. Hugel, J. Lévy and J. L. Men, Tetrahedron Lett., 1974, 15, 3109 CrossRef; (b) G. Croquelois, N. Kunesch and J. Poisson, Tetrahedron Lett., 1974, 15, 4427 CrossRef; (c) S. H. Goh and A. R. M. Ali, Tetrahedron Lett., 1986, 27, 2501 CrossRef CAS; (d) S. H. Goh, A. R. M. Ali and W. H. Wong, Tetrahedron, 1989, 45, 7899 CrossRef CAS.
- (a) T. Feng, X.-H. Cai, P.-J. Zhao, Z.-Z. Du, W.-Q. Li and X.-D. Luo, Planta Med., 2009, 75, 1537 CrossRef CAS PubMed; (b) T. Feng, X.-H. Cai, Y.-P. Liu, Y. Li, Y.-Y. Wang and X.-D. Luo, J. Nat. Prod., 2010, 73, 22 CrossRef CAS PubMed; (c) S.-H. Lim, K.-M. Sim, Z. Abdulla, O. Hiraku, H. Masahiko, K. Komiyama and T.-S. J. Kam, Nat. Prod., 2007, 70, 1380 CrossRef CAS PubMed; (d) K.-H. Lim and T.-S. Kam, Helv. Chim. Acta, 2007, 90, 31 CrossRef CAS; (e) C.-Y. Gan, Y.-Y. Low, N. F. Thomas and T.-S. Kam, J. Nat. Prod., 2013, 76, 957 CrossRef CAS PubMed; (f) T.-S. Kam, G. Subra-maniam, K.-H. Lim and Y.-M. Choo, Tetrahedron Lett., 2004, 45, 5995 CrossRef CAS.
- For reviews, see: (a) R. Keese, Chem. Rev., 2006, 106, 4787 CrossRef CAS PubMed; (b) A. Boudhar, M. Charpenay, G. Blond and J. Suffert, Angew. Chem., Int. Ed., 2013, 52, 12786 CrossRef CAS PubMed; (c) B. Bredenkötter, B. Neumann and H.-G. Stammler, Eur. J. Org. Chem., 2014, 53 CrossRef.
- For reviews, see: (a) H. J. Tokuyama, J. Synth. Org. Chem., Jpn., 2015, 73, 1120 CrossRef CAS; (b) M. Pfaffenbach and T. Gaich, Chem. – Eur. J., 2016, 22, 3600 CrossRef CAS PubMed; (c) Q. Geng, Z. Li, Z. Lv and G. Liang, Chin. J. Org. Chem., 2016, 36, 1447 CrossRef CAS.
- (a) J. Magolan, C. A. Carson and M. A. Kerr, Org. Lett., 2008, 10, 1437 CrossRef CAS PubMed; (b) A. Biechy and S. Z. Zard, Org. Lett., 2009, 11, 2800 CrossRef CAS PubMed; (c) X. Zhong, Y. Li and F.-S. Han, Chem. – Eur. J., 2012, 18, 9784 CrossRef CAS PubMed; (d) Z. Li and G. Liang, Tetrahedron Lett., 2013, 54, 242 CrossRef CAS; (e) Z. Lv, Z. Li and G. Liang, Org. Lett., 2014, 16, 1653 CrossRef PubMed; (f) A. Umehara, H. Ueda and H. Tokuyama, Org. Lett., 2014, 16, 2526 CrossRef CAS PubMed; (g) X. Zhong, S. Qi, Y. Li, J. Zhang and F.-S. Han, Tetrahedron, 2015, 71, 3734 CrossRef CAS; (h) M. Pfaffenbach and T. Gaich, Eur. J. Org. Chem., 2015, 3427 CrossRef CAS; (i) M. Pfaffenbach, A. Roller and T. Gaich, Chem. – Eur. J., 2016, 22, 8444 CrossRef CAS PubMed; (j) Y. Yang, Y. Bai, S. Sun and M. Dai, Org. Lett., 2014, 16, 6216 CrossRef CAS PubMed.
- (a) R. Nakajima, T. Ogino, S. Yokoshima and T. Fukuyama, J. Am. Chem. Soc., 2010, 132, 1236 CrossRef CAS PubMed; (b) Y. Iwama, K. Okano, K. Sugimoto and H. Tokuyama, Org. Lett., 2012, 14, 2320 CrossRef CAS PubMed; (c) Y. Iwama, K. Okano, K. Sugimoto and H. Tokuyama, Chem. – Eur. J., 2013, 19, 9325 CrossRef CAS PubMed; (d) Y. Zhang, Y. Xue and T. Luo, Tetrahedron, 2017, 73, 4201 CrossRef CAS; (e) M. Pfaffenbach and T. Gaich, Chem. – Eur. J., 2015, 21, 6355 CrossRef CAS; (f) Z. Li, Q. Geng, Z. Lv, B. P. Pritchett, K. Baba, Y. Numajiri, B. M. Stoltz and G. Liang, Org. Chem. Front., 2015, 2, 236 RSC; (g) K. Higuchi, S. Suzuki, R. Ueda, N. Oshima, E. Kobayashi, M. Tayu and T. Kawasaki, Org. Lett., 2015, 17, 154 CrossRef CAS PubMed.
- (a) Z. Xu, Q. Wang and J. Zhu, J. Am. Chem. Soc., 2013, 135, 19127 CrossRef CAS PubMed; (b) Z. Xu, Q. Wang and J. Zhu, J. Am. Chem. Soc., 2015, 137, 6712 CrossRef CAS PubMed.
- (a) J. Magolan and M. A. Kerr, Org. Lett., 2006, 8, 4561 CrossRef CAS PubMed; (b) X. Zhong, Y. Li, J. Zhang and F.-S. Han, Org. Lett., 2015, 17, 720 CrossRef CAS PubMed; (c) H. Li, S. A. Bonderoff, B. Cheng and A. Padwa, J. Org. Chem., 2014, 79, 392 CrossRef CAS PubMed; (d) H. Li, B. Cheng, N. Boonnak and A. Padwa, Tetrahedron, 2011, 67, 9829 CrossRef CAS; (e) V. J. Colandrea, S. Rajaraman and L. S. Jimenez, Org. Lett., 2003, 5, 785 CrossRef CAS PubMed.
- For iORC examples, see: (a) Z. Xu, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2013, 52, 3272 CrossRef CAS PubMed; (b) O. Wagnieres, Z. Xu, Q. Wang and J. Zhu, J. Am. Chem. Soc., 2014, 136, 15102 CrossRef CAS PubMed; (c) C. Piemontesi, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2016, 55, 6556 CrossRef CAS PubMed and references cited therein.
- N. B. Bennett, D. C. Duquette, J. Kim, W.-B. Liu, D. C. Be-henna, S. C. Virgil and B. M. Stoltz, Chem. – Eur. J., 2013, 19, 4414 CrossRef CAS PubMed.
- (a) A. B. Smith and M. Visnick, Tetrahedron Lett., 1985, 26, 3757 CrossRef CAS; (b) A. B. Smith, M. Visnick, J. N. Haseltine and P. A. Sprengeler, Tetrahedron, 1986, 42, 2957 CrossRef CAS; (c) A. B. Smith, E. G. Nolen, R. Shi-rai, F. R. Blase, M. Ohta, N. Chida, R. A. Hartz, D. M. Fitch, W. M. Clark and P. A. Sprengeler, J. Org. Chem., 1995, 60, 7837 CrossRef CAS; (d) A. B. Smith, N. Kanoh, H. Ishiyama and R. A. Hartz, J. Am. Chem. Soc., 2000, 122, 11254 CrossRef CAS; (e) A. B. Smith, N. Kanoh, H. Ishiyama, N. Minakawa, J. D. Rainier, R. A. Hartz, Y. S. Cho, H. Cui and W. H. Moser, J. Am. Chem. Soc., 2003, 125, 8228 CrossRef CAS PubMed; (f) A. B. Smith, A. H. Davulcu and L. Kürti, Org. Lett., 2006, 8, 1665 CrossRef CAS PubMed; (g) A. B. Smith, A. H. Davulcu, Y. S. Cho, K. Ohmoto, L. Kürti and H. Ishiyama, J. Org. Chem., 2007, 72, 4596 CrossRef CAS PubMed.
- X.-H. Li, S.-L. Wan, D. Chen, R. Q. Liu, C.-H. Ding, P. Fang and X.-L. Hou, Synthesis, 2016, 1568 CAS.
- (a) L. Zhao, J. P. May, J. Huang and D. M. Perrin, Org. Lett., 2012, 14, 90 CrossRef CAS; (b) V. J. Colandrea, S. Rajaraman and L. Jimenez, Org. Lett., 2003, 5, 785 CrossRef CAS PubMed; (c) K. Higuchi, Y. Sato, M. Tsuchimochi, K. Sugiura, M. Hatori and T. Kawasaki, Org. Lett., 2009, 11, 197 CrossRef CAS PubMed; (d) A. Karadeolian and M. A. Kerr, J. Org. Chem., 2010, 75, 6830 CrossRef CAS PubMed; (e) C. I. A. Kiraz, T. J. Emge and L. S. Jimenez, J. Org. Chem., 2004, 69, 2200 CrossRef PubMed; (f) C. Zhu, Z. Liu, G. Chen, K. Zhang and H. Ding, Angew. Chem., Int. Ed., 2015, 54, 879 CrossRef CAS PubMed.
- B. E. Maki and K. A. Scheidt, Org. Lett., 2009, 11, 1651 CrossRef CAS PubMed.
- For detailed discussion, see ESI†.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc09949a |
| This journal is © The Royal Society of Chemistry 2019 |