
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSensitive CVG-AFS/ICP-MS label-free nucleic acid and protein assays based on a selective cation exchange reaction and simple filtration separation†
Piaopiao
Chen
a,
Ke
Huang
 b,
Rui
Dai
b,
Erica
Sawyer
a,
Ke
Sun
a,
Binwu
Ying
a,
Xiawei
Wei
a and
Jia
Geng
*a
b,
Rui
Dai
b,
Erica
Sawyer
a,
Ke
Sun
a,
Binwu
Ying
a,
Xiawei
Wei
a and
Jia
Geng
*a
aDepartment of Laboratory Medicine, State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Sichuan University and Collaborative Innovation Center for Biotherapy, Chengdu, Sichuan 610041, China. E-mail: geng.jia@scu.edu.cn
bCollege of Chemistry and Materials Science, Sichuan Normal University, Chengdu, Sichuan 610068, China
First published on 18th March 2019
Abstract
Nowadays, label-free atomic spectrometric bioassays are attracting great research interest because of their advantages of low cost, simple design and operation, etc. Herein, a novel and simple chemical vapor generation-atomic fluorescence spectrometry (CVG-AFS)/inductively coupled plasma-mass spectrometry (ICP-MS) label-free detection method is presented for highly sensitive and selective assay of DNA and proteins. This work mainly combined a phenomenon that CdTe quantum dots (QDs) can be used to selectively differentiate free Hg2+ and the T–Hg2+–T complex, with the use of simple membrane filtration separation to improve the performance of the label-free bioassay methods. Upon hybridization with the DNA/protein (carcinoembryonic antigen, CEA) target, the T–Hg2+–T hairpin structure was opened and Hg2+ was released; this initiated the cation exchange reaction between Hg2+ and CdTe QDs which released Cd2+ simultaneously. Subsequently, the free Cd2+ was separated by the filtration membrane without separating the CdTe QDs, which could then be separated from the sample matrices for the CVG-AFS/ICP-MS assay. Under the optimal conditions, this method possessed high sensitivity for DNA and CEA determination with limits of detection (LODs) of 0.2 nM and 0.2 ng mL−1, and linear dynamic ranges of 1–160 nM and 0.5–20 ng mL−1, respectively, and exhibited excellent DNA sequence specificity and protein selectivity. This method preserves the advantages of the label-free atomic spectrometric bioassay, and combined with the selective cation exchange reaction and simple filtration separation to improve the performance.
1. Introduction
Nowadays, label-free bioassays are attracting great research interest because of their advantages of simple design and operation, low cost, etc. Recently, DNA aptamers have become attractive alternative recognition elements towards target molecules in biosensor applications owing to their simple synthesis, ease of chemical modification, structural stability and wide applicability.1,2 More importantly, label-free biosensors can be successfully realized by exploiting the unique properties of DNA and thus greatly simplifying the sensing process. Owing to the distinct advantages of DNA aptamers, label-free biosensors have been able to detect a variety of biomolecules. The key principle of this methodology focuses on the interaction between biomolecules (target or auxiliary molecules) and signal molecules.3,4 For example, the colorimetric assay of DNA and proteins is dependent on the different binding properties of single-stranded DNA and double-stranded DNA toward gold nanoparticles (Au NPs).5 Furthermore, Au NP-catalyzed chemiluminescence (CL) has been used for the construction of label-free sensors.6 Biomolecules are also used as ligands or templates to regulate the generation of luminescent nanomaterials, e.g. metal nanoclusters7–9 and luminescent quantum dots (QDs).10 In view of their predominant optical properties, these nanoparticles have potential applications in numerous fields, such as sensing in the label-free mode.11–14 However, most of these reported detection schemes for label-free sensors were primarily based on molecular spectroscopy.Generating unnatural base pairing in DNA duplexes using metal ions has also attracted many research efforts.15 Thymine (T)–thymine (T) is able to selectively bind to Hg2+ and form the T–Hg2+–T base pair in DNA duplexes. The stability of metal-mediated base pairing is comparable to that of natural base pairing (e.g. A–T) in DNA duplexes.16 More interestingly, the T–T base was found to have a high specificity to Hg2+, which is the only metal ion capable of stabilizing the T–Hg2+–T mismatch.17,18
Atomic spectrometry (AS)/atomic mass spectrometry (ICP-MS) is among the most commonly used and established elemental analysis methods in analytical chemistry. As an indispensable means of elemental and speciation analysis, it has the advantages of high sensitivity and selectivity, simultaneous determination of multiple elements, etc.19,20 With the continuous development of proteomics and genomics, the advantages of atomic spectrometry have gradually been recognized by researchers and applied to bioassays.21,22 Existing methods of atomic spectrometric bioassay are mainly conducted by labeling the signal molecules (metal elements or nanoparticles, etc.) on recognition ligands (antibodies, peptides, or aptamers). After the biorecognition reaction, the detection of the labeled signal molecules provides a way to achieve the indirect detection of the targets.23–25 However, the aforementioned methods have the following shortcomings: labelling signal molecules on a recognition ligand may affect its original immunogenicity; the processes for synthesis, labelling and separation of nanomaterials are complicated, thus increasing the detection time and causing interferences. In addition, although some researchers have developed some label-free atomic spectrometric DNA detection methods, these still require complex separation steps or even an acidic solution.26,27
Herein, a novel and simple CVG-AFS/ICP-MS label-free sensing assay is presented for the detection of nucleic acids and proteins. The capture probe (T-rich DNA) can coordinate with Hg2+ to form a T–Hg2+–T hairpin structure. More interestingly, we discovered that CdTe QDs can be used as a signal molecule to selectively differentiate the Hg2+ and T–Hg2+–T structure; Hg2+ leads to the initiation of the selective cation exchange reaction and release of the free Cd2+ simultaneously (Scheme 1A).28,29 Subsequently, the free Cd2+ was separated by the filtration membrane without separating the CdTe QDs.30 Thus, the DNA and protein quantitative assay can be achieved via monitoring the CVG-AFS/ICP-MS signal of Cd2+ before and after the selective recognition reaction (as shown in Scheme 1B and C).
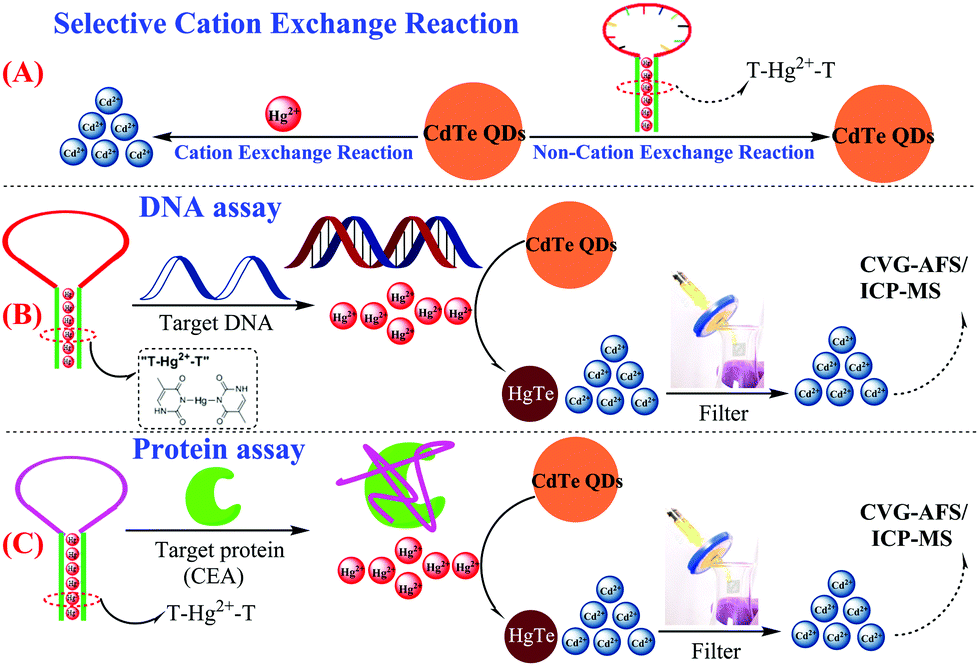 | ||
| Scheme 1 Schematic illustration of the sensitive CVG-AFS/ICP-MS label-free assay of DNA and proteins based on the selective cation exchange reaction and simple filter separation. | ||
2. Experimental
2.1 Materials and instruments
Pure water (18.2 MΩ cm) was obtained from a Milli-Q water system (Chengdu Ultrapure Technology Co., Ltd, Chengdu, China). All oligonucleotides were obtained from Shanghai Sangon Biotechnology Co., Ltd (Table 1, Shanghai, China). The high purity chemicals CdCl2, KBH4, Na3C6H5O7·2H2O, HCl, and NaOH were obtained from Kelong Reagent Factory (Chengdu, China). Na2TeO3 and 3-mercaptopropionic acid (MPA) were obtained from Aladdin Reagent Co. (Shanghai, China). Horseradish peroxidase (HRP), glucose oxidase (GOx), pectinase, trypsin, pepsin, cellulase, and carcinoembryonic antigen (CEA) were obtained from Sigma-Aldrich (St Louis, MO, USA). Human serum samples were provided by the Department of Laboratory Medicine, West China Hospital, Sichuan University (Chengdu, China). All working solutions were prepared with PBS buffer (10 mM, 12 mM/10 mM MgCl2, pH 7.4). A commercial CEA kit was obtained from Roche Pharmaceuticals (Germany).| Name | Sequence (5′–3′) |
|---|---|
| Probe 1-DNA | GTTTTTTGTCCTCGTACGTATCTATCTCCTTTTTTC |
| Target DNA | AAGGAGATAGATACGTACGAGGACAA |
| Single base mismatch | AAGGAGATAGATAC![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) TACGAGGACAA TACGAGGACAA |
| Double base mismatch | AAGGAGAAGATACGTA![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) GAGGACAA GAGGACAA |
| Probe 2-CEA | CTTTTTTATACCAGCTTATTCAATTTTTTTTG |
A CVG-AFS system (AFS-2200, Beijing Ruili Instrument Co., Beijing, China) was used for Cd2+ determination (see Table S1† for the operational parameters of the instrument). An Agilent 8900 ICP-MS system was used to detect Cd111 (Agilent Technologies, Tokyo, Japan) (see Table S2† for the operational parameters of the instrument). The fluorescence spectra were recorded on an F-7100 spectrometer (Hitachi, Japan). The UV-visible absorption spectra were recorded on a Hitachi U-1750 UV-vis spectrophotometer (Shimadzu, Kyoto, Japan). A MIX-25P shaker (Miou, Hangzhou, China) was used to mix the solution and a centrifuge (80-2B, Hunan Xinhai Instrument Co., Hunan, China) was used to centrifuge DNA and human serum. Commercially available pre-packed sterile syringe filters (0.22 μm) were purchased from Tianjin Experiment Equipment Co. Ltd (Tianjin, China). High-resolution transmission electron microscopy (HR-TEM) measurements of CdTe QDs were carried out by using a Tecnai G2F20 STWIN TEM at an accelerating voltage of 200 kV (FEI Co., USA). Energy dispersive spectroscopy (EDS) measurements of CdTe QDs + Hg2+ were carried out with a field emission scanning electron microscope (SEM, JSM-7800F, JEOL, Japan). The survey scan and Hg images of CdTe QDs + Hg2+ were recorded on a K-Alpha 1063 X-ray photoelectron spectrophotometer (XPS, Thermo Fisher Scientific, England).
2.2 Synthesis of CdTe QDs
The CdTe QDs were synthesised by referencing the previously reported methods.31,32 Firstly, a 50 mL solution containing CdCl2 (0.5 mmol) and trisodium citrate (0.2 g) was prepared. Then, MPA (52 μL) was instantly added into the above solution, and the pH of the solution was adjusted to 10.5 using NaOH. Later, Na2TeO3 (0.1 mmol) and KBH4 (50 mg) were added into the above solution, and refluxed for 1 h to obtain the CdTe QDs. Subsequently, high purity CdTe QDs were prepared via precipitation with n-propanol and centrifugation (11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm). The purified red CdTe QDs were redispersed in high purity water before use.33 The UV-vis absorption and fluorescence spectra are shown in Fig. S1.†
000 rpm). The purified red CdTe QDs were redispersed in high purity water before use.33 The UV-vis absorption and fluorescence spectra are shown in Fig. S1.†
2.3 Analysis procedure
For DNA and CEA detection, 40 μL of 1 μM probe1-DNA (or probe 2-DNA) and 48 μL of 5 μM Hg2+ were added to 200 μL of PBS buffer (10 mM, 12 mM/10 mM MgCl2, pH 7.4) and the T–Hg2+–T hairpin structure was formed at room temperature for 60 min. Then, target DNA or CEA (50 μL) were added into the aforesaid solutions to perform the competitive reaction, and the reaction took place over 1 h at room temperature. Then, 48 μL of CdTe QDs solution (a 15-fold diluted original CdTe QDs solution) was added into the aforementioned solution which further reacted for 75 min at room temperature to perform the cation exchange reaction. Later, the obtained solution was filtered and the filtrate solution was diluted to 5 mL with deionized water. The solution was then acidified with 3% (v/v) HCl and mixed with 2% (m/v) KBH4 to generate volatile species of Cd2+. The solution was detected by the double-channel CVG-AFS system for quantitative analysis.2.4 Real sample preparation
1.0 mL of each serum sample was placed in a 3 mL 100 kDa ultrafiltration centrifuge tube and the mixture was centrifuged at 8000 rpm for 10 min; this was repeated three times. Then, the ultrafiltrate was transferred in known aliquots to a precleaned test tube before being finally diluted with pure water.3. Results and discussion
3.1 Feasibility of using CdTe QDs to differentiate between the free Hg2+ and T–Hg2+–T complex
As discussed above and shown in Scheme 1, the key of this CVG-AFS/ICP-MS-based bioassay was the feasibility of using CdTe QDs to differentiate between the free Hg2+ and T–Hg2+–T complex. We verified the cation exchange reaction between Hg2+ and CdTe QDs first. As shown in Fig. 1A and B, the TEM images of the CdTe QDs and the products after the cation exchange reaction indicate that the spherical CdTe QDs (with an average diameter of ∼4 nm) changed to the aggregate state of HgTe. The inset of Fig. 1A and B is the corresponding physical (a) and fluorescence emission images with UV-light excitation (b), respectively, which further confirms that the cation exchange reaction took place. Subsequently, we verified that Hg2+ exchanged with Cd2+ to incorporate into CdTe QDs in many ways. Firstly, when Hg2+ was added to CdTe QDs, the AFS signal of Hg2+ was significantly reduced (Fig. 1C); then, Hg can be found in the energy dispersive spectroscopy (EDS) pattern of the CdTe QDs after the cation exchange reaction (Fig. 1D); finally, the binding energy of Hg 4f was found to be 101.3 eV in the XPS spectra of CdTe QDs (original CdTe QDs solution) + Hg2+ (1000 mg L−1) (Fig. 1E and F); all the above results indicated that Hg2+ exchanged with Cd2+ and generated HgTe.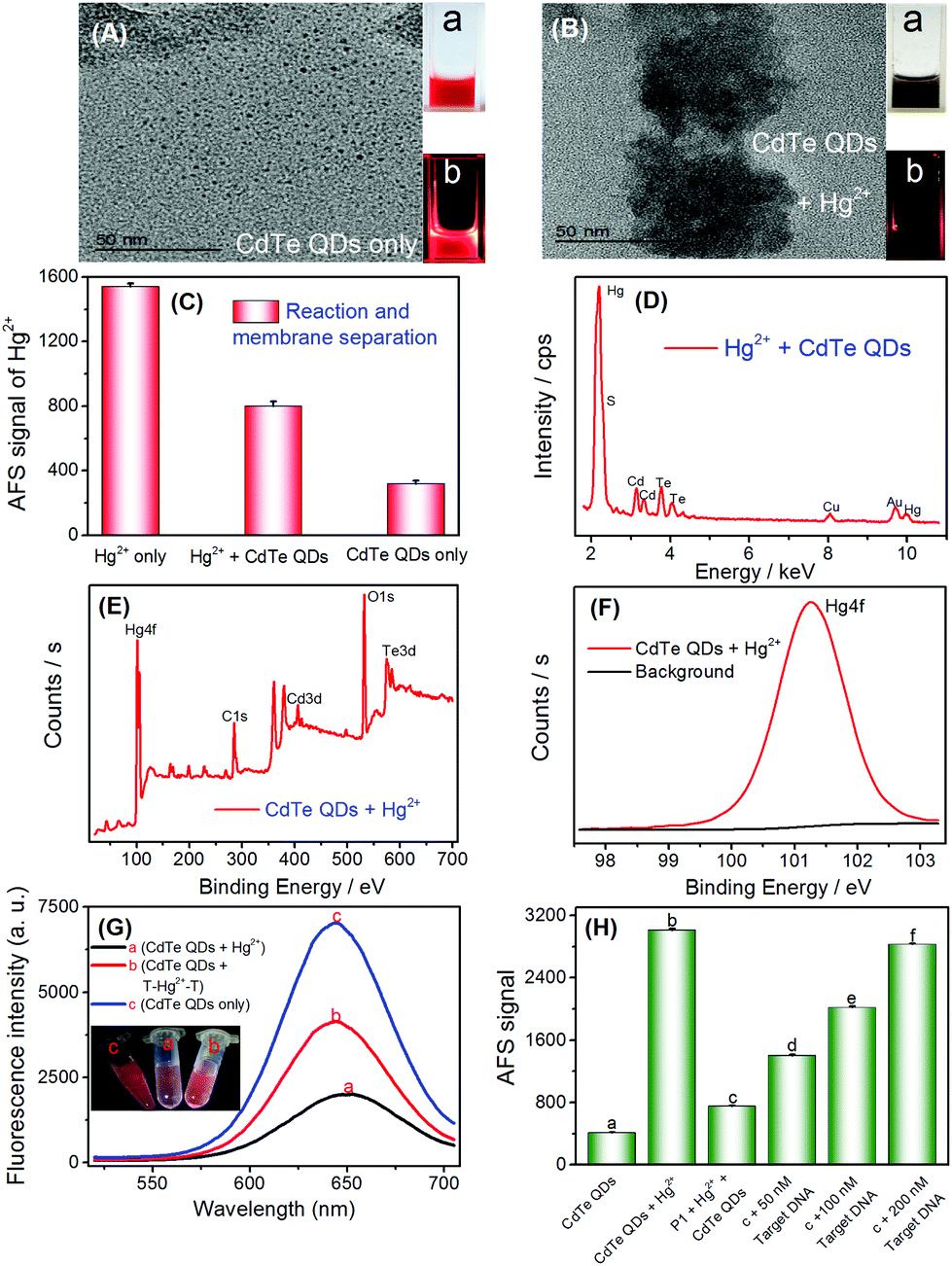 | ||
| Fig. 1 (A) TEM image of CdTe QDs. (B) TEM image of CdTe QDs + Hg2+. The inset is the corresponding physical (a) and fluorescence emission image with UV-light excitation (b). (C) Comparison of AFS signals of Hg2+ of different solutions. (D) The EDS image of CdTe QDs after the cation exchange reaction. (E) Survey scan and (F) Hg XPS spectra of CdTe QDs + Hg2+. (G) Fluorescence spectra under different conditions. The inset is the corresponding fluorescence emission image with UV-light excitation. (H) Experimental feasibility verification under different conditions. | ||
To further verify the feasibility of this method to selectively differentiate Hg2+ from the T–Hg2+–T complex using CdTe QDs, Hg2+ and the T–Hg2+–T complex were added into CdTe QDs solutions, and their fluorescence signals were detected, respectively. As shown in Fig. 1G, the fluorescence signal decreased significantly upon the addition of Hg2+ (Fig. 1G-a vs. Fig. 1G-c). However, when the T–Hg2+–T complex was added to the CdTe QDs, the fluorescence signal of the solution was significantly higher than that of the one with Hg2+ (Fig. 1G-b vs. Fig. 1G-a). When T–Hg2+–T and Hg2+ were simultaneously added and compared to the CdTe QDs solution (as shown in Fig. 1G: inset photos), highly luminescent products were obtained for the sample containing the T–Hg2+–T complex in the CdTe QDs solution (corresponding to curve b: shown in Fig. 1G, inset photo b). This solution showed intense red fluorescence under a 365 nm UV lamp. In contrast, weakly luminescent products were generated for the sample containing both Hg2+ and CdTe QDs (corresponding to curve a: shown in Fig. 1G, inset photo a). Therefore, CdTe QDs have demonstrated considerable feasibility for the selective differentiation between free Hg2+ and the T–Hg2+–T complex.
3.2 The feasibility of the target DNA assay
To test the feasibility of the sensor for DNA determination (Scheme 1B), a thymine (T)-rich ssDNA (probe 1-DNA, Table 1) was employed as the probe, and Hg2+ as the competitor was designed. The experimental results are shown in Fig. 1H; addition of Hg2+ into the CdTe QDs solution initiated the cation exchange reaction and led to the release of free Cd2+. After the membrane separation, the AFS signal of Cd2+ increased significantly in comparison with that of CdTe QDs only (Fig. 1H-a vs. Fig. 1H-b). The results indicate that the cation exchange reaction between Hg2+ and the CdTe QDs was initiated, and Cd2+ was successfully separated rather than CdTe QDs by the filter. In the absence of the target DNA, the AFS signal of Cd2+ was very low (Fig. 1H-c vs.Fig. 1H-b), indicating that the T–Hg2+–T structure was successfully formed and the cation exchange reaction was inhibited to a certain extent. Furthermore, after incubating increasing amounts of the target DNA with the T–Hg2+–T structure, the AFS signal of Cd2+ also increased correspondingly (Fig. 1H-c, d, e and f). In addition, we conducted a stability test experiment (Fig. S2†). There are no significant changes in the atomic fluorescence signals of CdTe QDs, T–Hg2+–T + CdTe QDs, and target DNA + T–Hg2+–T + CdTe QDs within 1–6 h, respectively. These experimental results confirmed that free Hg2+ and the T–Hg2+–T complex could be differentiated by CdTe QDs; i.e., the proposed selective cation exchange reaction-based protocol for target DNA detection is feasible.3.3 Detection of target DNA
Under the optimal experimental conditions (Fig. S3–S5†), the analytical performance of this approach for DNA assays was investigated by analyzing the AFS signals of Cd2+ towards different concentrations of target DNA. The AFS signal of Cd2+ was sensitive to the DNA concentration and gradually increased with increasing concentrations of DNA (Fig. 2A). Target DNA concentrations ranging from 0 to 250 nM could be directly measured by this sensing strategy. Furthermore, the inset of Fig. 2A shows a good linear relationship between the AFS signal of Cd2+ and the DNA concentrations ranging from 1–160 nM. The correlation equation was determined to be Y = 11.3 CDNA + 981.3 (R2 = 0.996) (Y is the AFS signal intensity of Cd2+, CDNA is the DNA concentration, and R is the correlation coefficient). The LOD towards the target DNA was estimated to be 0.2 nM, which was comparable to other reported assays without signal amplification, and the comparison of the biosensor with that of several other analytical methods is summarized in Table S3.† The reproducibility of this method, expressed as the relative standard deviation (RSD) of 15 nM target DNA, was 2.8%. In order to verify the accuracy and sensitivity of this sensor, the above standard solutions were also analyzed by ICP-MS and good analytical performance results were obtained (Fig. S6†), which were comparable to or better than using the CVG-AFS as a detector.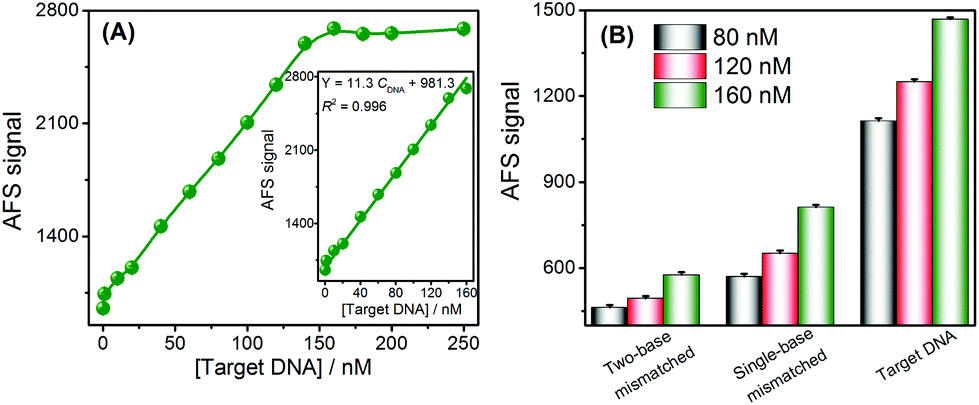 | ||
| Fig. 2 Analytical performance of the proposed method: (A) the relationship between the concentrations of the target DNA and the AFS signal of Cd2+; the inset reveals a linear relationship between the AFS signal and the concentration of the target DNA ranging from 0 to 160 nM. (B) sequence specificity investigation using different DNA targets at the same concentrations of 80, 120, and 160 nM. | ||
The specificity of the DNA sensing strategy was investigated via using different DNA sequences, including complementary, single-base mismatched and two-base mismatched, at the same concentrations of 80 nM, 120 nM and 160 nM, respectively. As shown in Fig. 2B, the AFS signal induced by single-base mismatched and two-base mismatched sequences was significantly lower than that of the complementary DNA at the same concentration. Thus, this sensor exhibited a high selectivity to discriminate the target DNA from DNA with a single-base mismatch, and showed promising potential for single nucleotide polymorphism (SNP) analysis.
3.4 Protein detection
Inspired by the results of the target DNA assay, this method was further extended to the detection of proteins, by using carcinoembryonic antigen (CEA) as a model and its aptamer as the recognition ligand. The detection principle of the CEA assay method is displayed in Scheme 1C; this principle is similar to that of DNA detection, except that the stem loop of the hairpin structure is replaced with the aptamer of CEA.First, the feasibility of the CEA assay was verified. The addition of Hg2+ into the CdTe QDs initiated the cation exchange reaction and the AFS signal of Cd2+ increased significantly (Fig. 3A-a vs. Fig. 3A-b). Furthermore, when probe 2 DNA (Table 1) reacted with Hg2+ and formed the T–Hg2+–T hairpin structure, the cation exchange reaction was inhibited, leading to a lower AFS signal intensity (Fig. 3A-c vs. Fig. 3A-b). The AFS intensity of Cd2+ also increased significantly with the increasing concentration of CEA (5, 10, and 20 ng mL−1) for incubation (shown in Fig. 3A-d, e, f). Therefore, the above experimental results indicated that the proposed protocol for CEA detection is feasible.
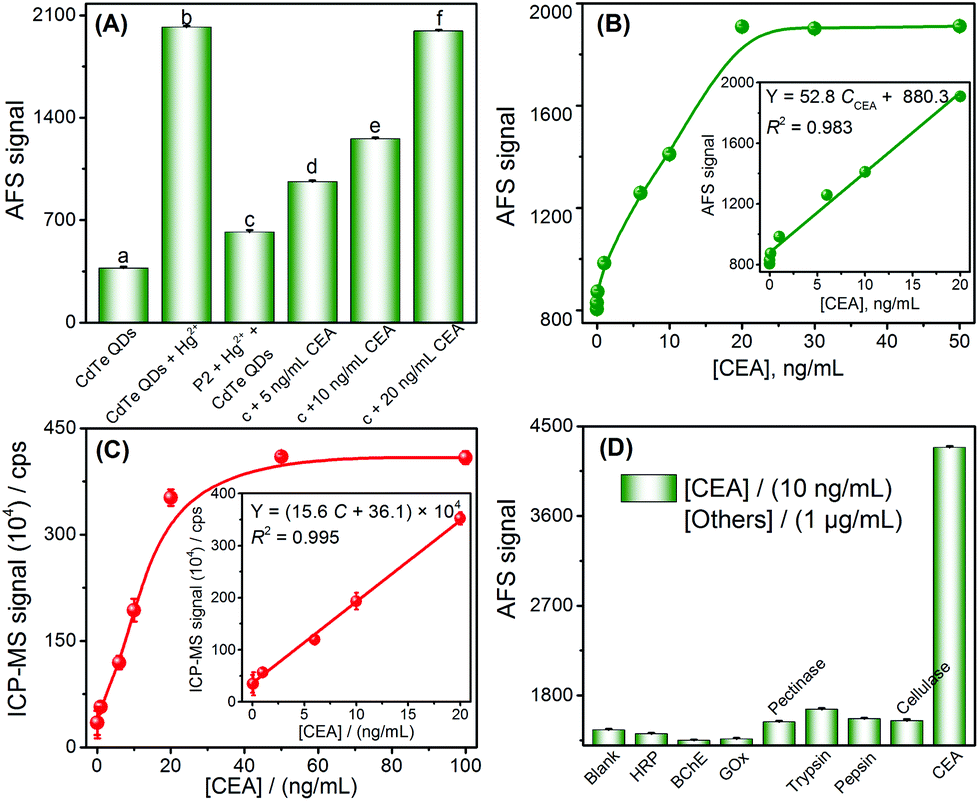 | ||
| Fig. 3 (A) Feasibility of the proposed method for CEA detection. (B) Relationship between the AFS signal and the concentration of CEA. (C) Relationship between the ICP-MS signal and the concentration of CEA. (D) The selectivity of the proposed method towards CEA detection. | ||
To test whether the proposed method could be used for the determination of CEA, the AFS signals for detecting CEA at different concentrations were first measured to evaluate the sensitivity of the sensor under optimal conditions (Fig. S7 and S8†). The AFS signals of the biosensor increased with increasing concentrations of CEA ranging from 0.5 to 20 ng mL−1. The correlation equation was Y = 52.8 CCEA + 880.3. The LOD towards the CEA was estimated to be 0.2 ng mL−1 (3σ, n = 11), and the performance of this method for the CEA assay was comparable to that of many other strategies (Table S4†). A series of eleven repetitive measurements of 2.5 ng mL−1 CEA were used for estimating the precision and the value of the RSD was 2.7%. In order to verify the accuracy and sensitivity of the sensor, the above standard solutions were analyzed by ICP-MS and good analytical performance was obtained (Fig. 3C), which was comparable or even better than using the CVG-AFS as a detector.
To examine the selectivity of the aptamer probe for the detection of CEA, a blank solution containing the T–Hg2+–T hairpin structure was incubated with other proteins, such as horseradish peroxidase (HRP), butylacetylcholinesterase (BChE), glucose oxidase (GOx), pectinase, trypsin, pepsin, and cellulose. Only CEA (10 ng mL−1) induced a significant increase in the AFS signal; no obvious signal changes were observed in the presence of high concentrations of other species (1 μg mL−1, Fig. 3D). The excellent selectivity of the method is attributed to the high affinity between the aptamer and CEA.
To test the applicability of the sensor to clinical samples, this sensor was used in a complex sample matrix of human serum. To eliminate potential interferences, the serum samples were filtered using a centrifugal filtration device (MWCO = 100 kDa) to remove small molecules (e.g. GSH), ions (e.g. Cu2+), and other abundant proteins. The experimental results of the two methods (our method and the commercial CEA kit electrochemiluminescence method) are comparable; moreover, satisfactory recoveries were obtained (97% to 102%, Table 2). These results imply that this atomic spectrometry-based strategy has a promising future in the analysis of complex biological samples.
| Samples | CEA kit, ng mL−1 | This method, ng mL−1 | Added, ng mL−1 | Found,a ng mL−1 | Recovery,b % |
|---|---|---|---|---|---|
| a Mean and standard deviation of the results (n = 3). b Recovery (%) = (CFound/CAdded) × 100%. | |||||
| 1 | 0.64 | 0.58 | 1.00 | 1.61 ± 0.06 | 102 |
| 2 | 2.15 | 2.10 | 5.00 | 6.95 ± 0.10 | 97 |
| 3 | 2.82 | 2.58 | 8.00 | 10.70 ± 0.18 | 101 |
| 4 | 1.46 | 1.56 | 10.00 | 11.32 ± 0.18 | 98 |
4. Conclusions
In conclusion, a simple CVG-AFS/ICP-MS label-free bioassay method was presented for the highly sensitive and selective detection of DNA and CEA. This work is primarily based on the phenomenon that CdTe QDs can be used to selectively differentiate free Hg2+ and the T–Hg2+–T hairpin structure, and simple filter membrane separation. The experimental design is simple; the procedure is easy to operate and low cost. This method will not only broaden the scope of application of atomic spectrometry in label-free bioassay, but also provide insights into the revelation that the special interaction between biomolecules and metal ions (T–Hg2+–T, C–Ag+–C, nucleic acid-templated metal nanoparticles, etc.) can provide a theoretical basis for the development of various label-free atomic spectrometry bioassay methods. Considering the versatility of aptamers and peptides as recognition ligands,34,35 and various nucleic acids involved in signal amplification (strand displacement reaction, hybrid chain reaction, rolling circle amplification, etc.),36–39 the proposed CVG-AFS/ICP-MS-based bioassay platform is expected to have a broad spectrum of applications in bioanalysis (enzyme, cancer cell, etc.) with improved analytical performance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (Grant No. 2016YFA0201400 and Grant No. 2016YFC1200300) and the National Natural Science Foundation of China (No. 21605108).Notes and references
- H. M. Meng, H. Liu, H. L. Kuai, R. Z. Peng, L. T. Mo and X. B. Zhang, Chem. Soc. Rev., 2016, 45, 2583–2602 RSC.
- Y. Jiang, X. S. Pan, J. Chang, W. J. Niu, W. J. Hou, H. L. Kuai, Z. L. Zhao, J. Liu, M. Wang and W. H. Tan, J. Am. Chem. Soc., 2018, 140, 6780–6784 CrossRef CAS PubMed.
- J. H. Monserud, K. M. Macri and D. K. Schwartz, Angew. Chem., Int. Ed., 2016, 55, 13710–13713 CrossRef CAS PubMed.
- Y. P. Chen, B. F. Yin, M. L. Dong, Y. L. Xianyu and X. Y. Jiang, Anal. Chem., 2018, 90, 1234–1240 CrossRef CAS PubMed.
- X. X. Zheng, Q. Liu, C. Jing, Y. Li, D. Li, W. J. Luo, Y. Q. Wen, Y. He, Q. Huang, Y. T. Long and C. H. Fan, Angew. Chem., Int. Ed., 2011, 123, 12200–12204 CrossRef.
- Y. Y. Qi, B. X. Li and Z. J. Zhang, Biosens. Bioelectron., 2009, 24, 3581–3586 CrossRef CAS PubMed.
- N. Goswami, F. X. Lin, Y. B. Liu, D. T. Leong and J. P. Xie, Chem. Mater., 2016, 28, 4009–4016 CrossRef CAS.
- W. J. Zhou, J. B. Zhu, D. Q. Fan, Y. Teng, X. Q. Zhu and S. J. Dong, Adv. Funct. Mater., 2017, 27, 1704092 CrossRef.
- A. Rotaru, S. Dutta, E. Jentzsch, K. Gothelf and A. Mokhir, Angew. Chem., Int. Ed., 2010, 49, 5665–5667 CrossRef CAS PubMed.
- X. W. He and N. Ma, Small, 2013, 9, 2527–2531 CrossRef CAS PubMed.
- X. W. He and N. Ma, Anal. Chem., 2014, 86, 3676–3681 CrossRef CAS PubMed.
- Y. Zhang, Z. W. Chen, Y. Tao, Z. Z. Wang, J. S. Ren and X. G. Qu, Chem. Commun., 2015, 51, 11496–11499 RSC.
- L. L. Zhang, J. J. Zhao, M. Duan, H. Zhang, J. H. Jiang and R. Q. Yu, Anal. Chem., 2013, 85, 3797–3801 CrossRef CAS PubMed.
- G. L. Liu, J. J. Li, D. Q. Feng, J. J. Zhu and W. Wang, Anal. Chem., 2017, 89, 1002–1008 CrossRef CAS PubMed.
- I. Okamoto, K. Iwamoto, Y. Watanabe, Y. Miyake and A. Ono, Angew. Chem., Int. Ed., 2009, 48, 1648–1651 CrossRef CAS PubMed.
- P. P. Chen, P. Wu, J. B. Chen, P. Yang, X. F. Zhang, C. B. Zheng and X. D. Hou, Anal. Chem., 2016, 88, 2065–2071 CrossRef CAS PubMed.
- A. Porchetta, A. Vallée-Bélisle, K. W. Plaxco and F. Ricci, J. Am. Chem. Soc., 2013, 135, 13238–13241 CrossRef CAS PubMed.
- P. P. Chen, P. Yang, R. X. Zhou, X. Yang, J. B. Chen and X. D. Hou, Chem. Commun., 2018, 54, 4696–4699 RSC.
- K. Huang, K. L. Xu, W. Zhu, L. Yang, X. D. Hou and C. B. Zheng, Anal. Chem., 2016, 88, 789–795 CrossRef CAS PubMed.
- C. B. Zheng, L. G. Hu, X. D. Hou, B. He and G. B. Jiang, Anal. Chem., 2018, 90, 3683–3691 CrossRef CAS PubMed.
- R. Liu, S. X. Zhang, C. Wei, Z. Xing, S. C. Zhang and X. R. Zhang, Acc. Chem. Res., 2016, 49, 775–783 CrossRef CAS PubMed.
- P. P. Chen, P. Wu, Y. X. Zhang, J. B. Chen, X. M. Jiang, C. B. Zheng and X. D. Hou, Anal. Chem., 2016, 88, 12386–13292 CrossRef CAS PubMed.
- G. J. Han, S. C. Zhang, Z. Xing and X. R. Zhang, Angew. Chem., Int. Ed., 2013, 52, 1466–1471 CrossRef CAS PubMed.
- S. H. Hu, S. C. Zhang, Z. C. Hu, Z. Xing and X. R. Zhang, Anal. Chem., 2007, 79, 923–929 CrossRef CAS.
- X. W. Yan, L. M. Yang and Q. Q. Wang, Angew. Chem., Int. Ed., 2011, 50, 5130–5133 CrossRef CAS.
- R. Liu, C. Q. Wang, Y. M. Xu, J. Y. Hu, D. Y. Deng and Y. Lv, Anal. Chem., 2017, 89, 13269–13274 CrossRef CAS PubMed.
- Y. Li, S. K. Sun, J. L. Yang and Y. Jiang, Analyst, 2011, 136, 5038–5045 RSC.
- D. H. Son, S. M. Hughes, Y. D. Yin and A. P. Alivisatos, Science, 2004, 306, 1009–1012 CrossRef CAS PubMed.
- J. S. Li, T. R. Zhang, J. P. Ge, Y. D. Yin and W. W. Zhong, Angew. Chem., Int. Ed., 2009, 121, 1616–1619 CrossRef.
- K. Huang, W. Q. Deng, R. Dai, X. Wang, X. Xiong, Q. Q. Yuan, X. Jiang, X. Yuan and X. L. Xiong, Microchem. J., 2017, 135, 74–80 CrossRef CAS.
- Z. H. Sheng, H. Y. Han, X. F. Hu and C. Chi, Dalton Trans., 2010, 39, 7017–7020 RSC.
- K. Huang, K. L. Xu, J. Tang, L. Yang, J. R. Zhou, X. D. Hou and C. B. Zheng, Anal. Chem., 2015, 87, 6584–6591 CrossRef CAS PubMed.
- W. W. Yu, L. H. Qu, W. Z. Guo and X. G. Peng, Chem. Mater., 2003, 15, 2854–2860 CrossRef CAS.
- Y. Du and S. J. Dong, Anal. Chem., 2016, 89, 189–215 CrossRef PubMed.
- Q. Liu, H. Wang, X. H. Shi, Z. G. Wang and B. Q. Ding, ACS Nano, 2017, 11, 7251–7258 CrossRef CAS PubMed.
- S. Bi, S. Z. Yue and S. S. Zhang, Chem. Soc. Rev., 2017, 46, 4281–4297 RSC.
- Y. Jiang, B. L. Li, J. N. Milligan, S. Bhadra and A. D. Ellington, J. Am. Chem. Soc., 2013, 135, 7430–7433 CrossRef CAS PubMed.
- M. X. Li, C. H. Xu, N. Zhang, G. S. Qian, W. Zhao, J. J. Xu and H. Y. Chen, ACS Nano, 2018, 12, 3341–3350 CrossRef CAS PubMed.
- K. X. Zhang, R. J. Deng, Y. P. Sun, L. Zhang and J. H. Li, Chem. Sci., 2017, 8, 7098–7105 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8an01926f |
| This journal is © The Royal Society of Chemistry 2019 |