
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ionic-to-electronic coupling efficiency in PEDOT:PSS films operated in aqueous electrolytes†
Achilleas
Savva
 ,
Shofarul
Wustoni
and
Sahika
Inal
*
,
Shofarul
Wustoni
and
Sahika
Inal
*
Biological and Environmental Science and Engineering Division, King Abdullah University of Science and Technology, Saudi Arabia. E-mail: sahika.inal@kaust.edu.sa
First published on 11th July 2018
Abstract
Poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate), PEDOT:PSS, is a polymeric mixed conductor used in the vast majority of devices in bioelectronics, electrochromics, energy storage/generation, neuromorphic computing, and thermoelectrics. These devices operate at the interface with electrolytes and rely on the uptake of mobile ions by the film, making the coupling between electronic and ionic charges crucial. For efficient transduction of ionic charges into electronic ones, all the ions injected into the film should lead to a change in conductivity. Although extensively studied, fundamental knowledge regarding the losses during this process is lacking. In this study, we quantify the efficiency of ion-to-electron coupling in PEDOT:PSS films by measuring the number of cations taken up by the film as well as the in situ current generated as a result of their interactions with the electrically active sites. We find that not all the injected cations are used for reducing PEDOT oligomers in thick films and some of these ions remain in the film upon de-doping. The efficiency of ion-to-electron transduction thus varies with thickness, a parameter which critically affects the distribution of PEDOT and PSS in the bulk as revealed by Raman spectroscopy, X-ray photoelectron spectroscopy and scanning transmission electron microscopy studies. Tracking the traces of ions, we provide guidelines on how to maximize the coupling between ionic and electronic charges for high performance transducers. Our approach is thus fundamental to future development and optimization of mixed conductors applied at the interface with electrolytes.
Introduction
Organic mixed conductors are often soft, conjugated polymers that have garnered attention due to their ability to conduct ionic and electronic current simultaneously.1–3 When interfacing with an electrolyte, the electrical and optical properties of a conjugated polymer film can be modulated dramatically if the ions penetrate into the bulk of the film.4,5 These ions compensate for electronic charges injected from/extracted to an electrode, i.e., electrochemical doping or de-doping.6 A variety of devices ranging from electrochromic displays to capacitors, batteries and ionic-organic ratchets rely on this interplay between the ionic charges of the electrolyte and the electronic ones of the film.7 The electrochemical phenomena at the organic mixed conductor/electrolyte interface are thus of interest to multiple disciplines and have also led to state-of-the-art bioelectronic devices which yield advances in communicating with living systems.8 One such device is the organic electrochemical transistor (OECT) where the coupling between ionic and electronic charges occurs within the entire volume of the polymeric film in the channel, resulting in high performance devices.3,9 Due to transconductance values that exceed those of all other electrolyte-gated transistors, OECTs could detect low-amplitude biological signals with record-high sensitivities.10–12A typical material for OECTs is poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS), a mixed conductor utilized at the channel and sometimes also as gate electrode. Due to charge compensation by the immobile PSS chains, PEDOT:PSS films conduct holes with electronic conductivity values reported as high as 4000 S cm−1.13 When hydrated, the negatively charged macromolecular dopant enables the transport of cations through the bulk. During OECT operation, with a positive bias applied at the gate electrode, cations of the electrolyte enter the PEDOT:PSS film in the channel and compensate for the PSS sites that neutralize the holes of PEDOT. This results in a decrease in the output (channel) current. Thus, the transduction of ionic signals into electrical current relies heavily on the ability of electrolyte ions to modulate PEDOT:PSS conductivity. In other words, to ensure high performance OECTs, each cation that is injected from the electrolyte should first fully penetrate into the channel, then access the site where it replaces (electrostatically) one hole.6,14,15 The volumetric interactions of cations and holes lead to thickness dependent device performance, i.e., increasing the thickness of the channel (proportional to its capacitance) enhances the transconductance.16 This is the underlying operation principle of most electrochemical devices based on mixed conductors. It is, therefore, crucial to monitor the ion-to-electron conversion process and identify any loss mechanisms that involve ions not taking part in charge generation/depletion. However, monitoring the interplay between ionic and electronic charges in these systems is rather challenging due to the simultaneous presence of electronic and ionic species, as well as charge transport in three-dimensions and at multiple length scales.17,18
While a number of techniques has been established to build our current understanding of mixed conduction, significant gaps remain. Electrochromism, i.e. the change in optical absorption of a conjugated polymer film as a result of a change in its redox or doping state, is a typical method to indirectly investigate the interactions between ionic and electronic carriers. As ions from an electrolyte are injected into the film, the alterations in the UV-vis-NIR spectrum provide insights into the evolution of polaron/bipolaron species indicative of the delocalized electrons that generate current.19–21 Similarly, electrochromism can be used to quantify the one-dimensional transport of cations along PEDOT:PSS films by performing electrochromic moving front measurements.22,23 Using a scanning-probe imaging technique, Giridharagopal et al. identified the regions of an organic semiconductor film which take up water/anions and change their softness due to electrochemical doping.2 These techniques clearly aid in understanding ion transport and the subsequent changes in film morphology and electronic properties. Nevertheless, a method to quantify ion-to-electron coupling efficiency will help identify mechanisms that impede ion injection/charge transport at the solid–liquid interface. This can not only provide better understanding of fundamental interfacial phenomena but also guide future materials design for the development of high performance transducers.
A quartz crystal microbalance with dissipation monitoring (QCM-D), which was demonstrated for the first time in the 1990s,24,25 utilizes the piezoelectric properties of a quartz crystal to measure the mass change (down to nanogram levels) on its surface.26 In these measurements, a decrease in the oscillation frequency (f) indicates an increase in the mass of the film coated on the quartz crystal due to, for instance, adsorption of species on the film surface or swelling of the film in a solvent.27 A QCM-D can, therefore, be considered as a molecular balance. When the decrease in f is accompanied by an increase in dissipation of energy (D) and spreading of its overtones, this is a strong indication of conformational changes associated with the soft nature of the film.28 These signals can be treated either with the Sauerbrey model which directly correlates the change in f to the coupled mass (applied for dry/rigid thin films) or with the Kelvin–Voigt viscoelastic (VSE) model considered typically for soft materials such as conjugated polymers.29 An electrochemical QCM-D (EQCM-D), on the other hand, allows for monitoring mass exchange between an electrically active film and an electrolyte as the film undergoes electrochemical doping/de-doping. EQCM-D applications included monitoring the evolution of the viscoelastic properties of a conjugated polymer film during its electrochemical cycling,30 electropolymerization of conducting polymers31,32 and ion-exchange in conjugated polyelectrolytes, as well as deformations of carbon electrodes under different charging conditions33,34 and charge/discharge processes in Li+ ion battery electrodes.35,36
In this study, we investigate the efficiency of ion-to-electron coupling for PEDOT:PSS films of different thicknesses. Using an EQCM-D, we quantify the number of cations that are injected into the films from an aqueous electrolyte solution under electrochemical biasing while simultaneously recording the in situ current associated with this process. Based on the number of cations injected at the maximum de-doping bias, we calculate the electronic charge carrier density of these films. We find that not all the injected cations are used for reducing PEDOT oligomers and that the efficiency of ion-to-electron coupling depends on the film thickness. For thick films, a smaller fraction of injected cations is used to compensate for PSS chains involved in the doping of PEDOT and the uptake of these ions involves more water molecules. The process is also less reversible: the film does not return to its original mass upon removal of the de-doping bias. For the thin film, on the other hand, the cations are predominantly utilized for compensating holes and leave the film when the bias polarity is reversed. X-ray photoelectron spectroscopy and scanning transmission electron microscopy studies evidence that the bulk composition of PEDOT:PSS films varies with film thickness. Inside thick films, we find a loosely interconnected network of PSS chains, an ideal environment to store excess de-dopant cations and water. This methodology, here shown for the standard material of bioelectronics, PEDOT:PSS, will be valuable to understand electrochemical phenomena at solid–liquid interfaces of other mixed conductor/electrolyte systems based on their chemical composition and microstructure.
Experimental
Film preparation
PEDOT:PSS (PH1000) was purchased from Clevios GmbH, while all the other chemicals were purchased from Sigma Aldrich and used as received. The conducting polymer solution was prepared by adding 5 vol% ethylene glycol (EG), 1 wt% 3-glycidoxypropyltrimethoxysilane (GOPS) and 0.002 vol% dodecyl benzene sulfonic acid (DBSA) to the PEDOT:PSS solution. EG, GOPS and DBSA were included in the mixture as a conductivity enhancer, crosslinker and wetting agent, respectively.37 The films were cast on Au coated QCM-D sensors (QX-338 Ti–Au QCM). The thin films were cast by spin coating the mixture at 3000 rpm for 30 s, while for the thick films a two-step coating process at 3000 rpm and 700 rpm was performed including a soft annealing break (at 100 °C for 1 minute) following the first step. Both of the films were annealed in ambient air at 120 °C for 45 minutes. The electrochemical area of the sensors/films was 0.7855 cm2 and the wet thicknesses were calculated to be 280 nm and 570 nm, for the thin and thick PEDOT:PSS (see swelling analysis below).Electrochemical quartz crystal microbalance with dissipation monitoring (EQCM-D)
We performed EQCM-D measurements using a Q-sense analyzer (QE401, Biolin Scientific AB, Sweden). Cyclic voltammetry measurements were performed using an Autolab PGstat128N potentiostat coupled with the Q-sense electrochemistry module. The three-electrode setup comprised an Ag/AgCl reference electrode, a Pt counter electrode and a polymer coated EQCM-D sensor used as the working electrode. The electrolyte solution (0.1 M, NaCl, aqueous) was filtered through a glass fiber filter with a 1.2 μm porous diameter to exclude any contribution of impurities. The electrolyte solution was pumped continuously into the chamber with a flow rate of 100 μL min−1 controlled by a peristaltic pump over the course of the measurements. EQCM-D measurements were also performed in a N2 saturated environment.Swelling analysis
After recording the QCM-D response of the bare Au sensors in air, 0.1 M aqueous NaCl solution was injected into the chamber. This resulted in large shifts in f and D, due to the density differences between the two media, which must be excluded for accurate calculations of swelling percentage. The measurements were then stopped, the sensors were removed and PEDOT:PSS layers were spun cast directly on the same sensor as explained above. We then compared the absolute difference in f for multiple overtones between the bare sensor and the Au/PEDOT:PSS coated sensor, both in air and in NaCl solution, by using the function “stitched data” of the Q-soft software. This function compares the selected datasets based on the raw frequencies measured and excludes the effect of the different densities between the two media (Fig. S1, ESI†). Thus, the difference in the f values of the stitched data is directly analogous to the thickness of the polymer in both media, which is calculated using the Sauerbrey equation (eqn (1), below). The dry film thicknesses were verified using a Dektak mechanical profilometer.EQCM-D analysis
The physical modelling of the two measured parameters, f and D, is related to the viscoelastic properties of the film. A rigid film shows zero change in D as there are theoretically no energy losses (no viscoelasticity). In this case, the Sauerbrey equation can be used to quantify the change in mass (Δm) based on the change in f (Δf) as described in eqn (1):| Δm = −17.7/nΔfn | (1) |
| G* = μ + 2πifη | (2) |
X-ray photoelectron spectroscopy (XPS)
XPS measurements were performed on identically prepared films using an AMICUS/ESCA 3400 KRATOS instrument equipped with an achromatic Al Kα X-ray source (1468.6 eV). We operated the source at a voltage of 10 kV and a current of 10 mA, corresponding to a power of 100 W. The elemental narrow scan region was acquired with a step of 0.1 eV. The pressure in the analysis chamber was on the order of 10−7 Pa during the course of measurements. The obtained spectra were calibrated to the reference C 1s at 284.8 eV. The spectra were deconvoluted using Gaussian and Lorentzian methods and background subtraction was carried out using the Tougaard method.Raman spectroscopy
Raman spectra were recorded using a LabRAM Aramin Raman spectrometer from Horiba Jobin Yvon with an Olympus 50× objective on an automated x–y translation stage. A 633 nm helium–neon laser was used as the excitation source with a grating of 1800 g mm−1. The data were collected using an exposure time of 3 s and 5 cycles at room temperature (21 °C). The Raman spectra were baseline corrected.Scanning transmission electron microscopy (STEM) measurements
The cross section of both samples was prepared with a focused ion beam (FIB). TEM lamellas were prepared with a FEI Helios NanoLab 400S FIB/SEM dual-beam system equipped with a Ga+ ion source. C/Pt layers were deposited on the surface region of interest with an electron & ion beam for sample protection. Samples were thinned down to a relative thickness of 80 nm using progressively decreasing ion beam energies in the FIB down to 5 keV. Scanning transmission electron microscopy (STEM) images were taken with a FEI Themis-Z transmission electron microscope operated at 200 kV. Energy Dispersive X-ray Spectroscopy (EDS) studies were performed using a Bruker super-X EDS detector.Results and discussion
Swelling of a mixed conductor in an aqueous solution, i.e., the ability to absorb water and expand its volume, is considered to be essential for efficient ion injection and transport.38 When exposed to aqueous NaCl solution (0.1 M), an increase in the mass of PEDOT:PSS films was measured, i.e., a thin (170 nm) and a thick one (300 nm), translated into a thickness increase of 110 nm and 270 nm, respectively (Fig. S1a, ESI†). This corresponds to 86% swelling in both cases. Regardless of its initial thickness, PEDOT:PSS swells to nearly double its size due to the diffusion of water molecules and ions into the film. Once the films are stabilized in the electrolyte (no further mass exchange), a potentiostat is coupled to the electrochemical QCM-D cell. The films are cycled first over doping potentials, i.e., the voltage is above the open circuit potential (V > Voc ∼ 0.3 V), and then through de-doping potentials (V < Voc). Fig. 1 (top row) presents the cyclic voltammograms of PEDOT:PSS films recorded in our system. During the cyclic voltammetry (CV) measurements, f and D signals change significantly (Fig. S1b, ESI†), indicating continuous mass exchange between the electrolyte and the polymer. Applying the VSE model, we calculate the mass coupled to and decoupled from the film as ions are drifting in and out, respectively. The voltammetry data (current vs. electrode potential) are collected simultaneously with the corresponding changes in mass as depicted in Fig. 1 (bottom row).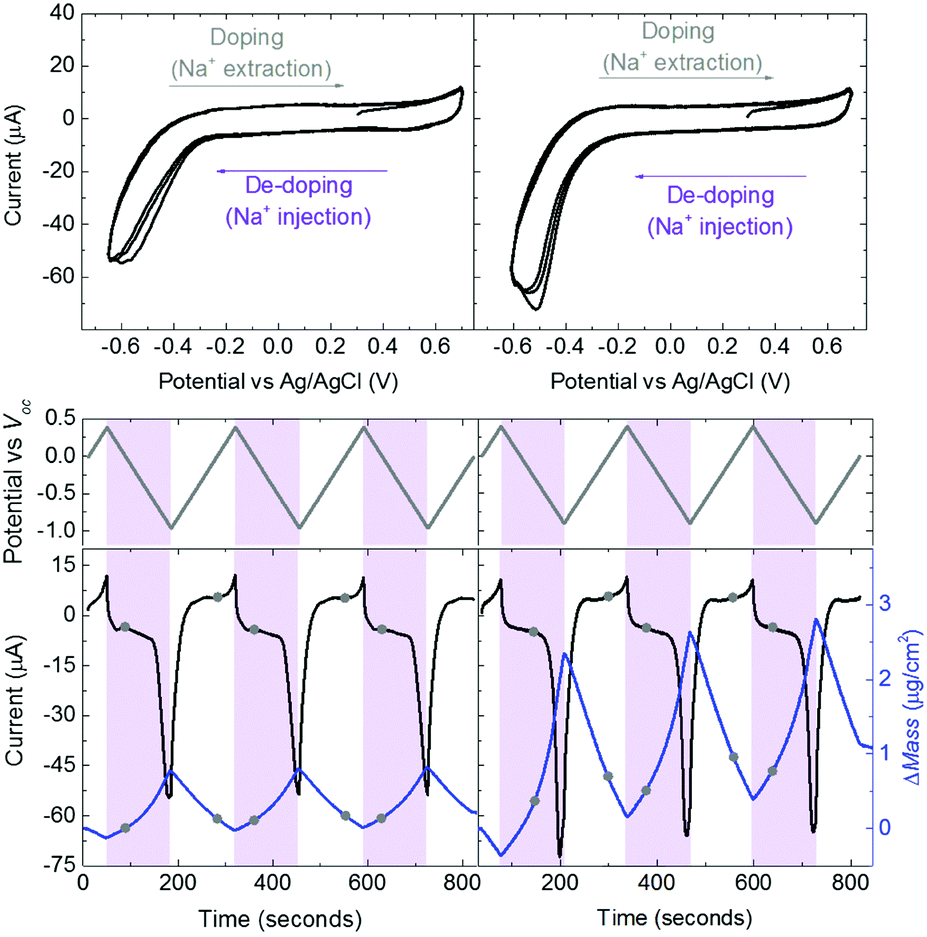 | ||
| Fig. 1 CV curves of thin (left column) and thick (right column) PEDOT:PSS films in aqueous NaCl solution (0.1 M) (top row). The potential scan starts at V = Voc = 0.29 V towards +0.4 V and then back towards −0.9 V vs. Voc with a rate of 0.01 V s−1 (middle row). Potentials above Voc oxidize the films accompanied with Na+ and H+ extraction (grey shaded areas). Potentials below Voc result in de-doping of the films by the injected Na+ ions (purple shaded areas). The mass which moves into or out of the films was calculated using the VSE model (blue lines, bottom row). The black lines represent the corresponding current recordings. The circles on mass/current–time curves mark the potential when it hits Voc. | ||
At doping potentials, we observe a slight increase in the oxidation current and an initial mass loss (grey areas, Fig. 1, bottom row). In this regime, while Cl− ions are injected into the film, Na+ ions already diffused into the film or the positively charged mobile counterions (often in the form of protons) that are inherently present alongside PSS chains are ejected. The QCM-D signal is therefore dominated by the cations leaving the film. Note that the electrolyte on top of the sensors is constantly replenished by fresh NaCl solution, therefore we ensure that any ions that are expelled from the films during the first doping cycle are washed away. Sweeping to potentials lower than Voc leads to a substantial increase in the mass (negative frequency shift) as well as in the reduction current (purple areas, Fig. 1, bottom row). These results agree well with our understanding of the de-doping process of PEDOT:PSS.14,39 At de-doping potentials, cations (predominantly Na+) are injected into the film, drifting to the sites consisting of hole/PSS pairs while holes are extracted through the metal contact (Au) at the other side of the film. The mass increase scales with the de-doping potential, reaching a maximum of 0.9 μg cm−2 for the thin and 2.6 μg cm−2 for the thick film. This corresponds to 53 μA and 64 μA reduction current, respectively. When the potential sweep is reversed, the Δf trace of the thick film does not return to its original value (Fig. S1b, ESI†), indicating that not all the cations that entered the film drift away from the electrode. In contrast, the current does not change significantly, indicating that these trapped cations are not interacting with the parts of the film responsible for current generation (e.g. PSS coupled to percolating PEDOT chains). The mass remaining on the sensors and the corresponding current at the end of each CV cycle are summarized in Table S1 (ESI†). Furthermore, for the thick film, the dissipation change (ΔD) is higher than for the thin one (ΔDthin ∼ 2 and ΔDthick ∼ 10 at V = −0.6 vs. Ag/AgCl, Fig. S1b, ESI†) accompanied with considerable spreading of its overtones. These findings suggest large changes in the viscoelastic properties of the thick film upon de-doping with cations, in analogy to artificial muscles made of similar organic mixed conductors.41–43 The evolution of the film volume during EQCM-D measurements is sketched in Fig. 2.
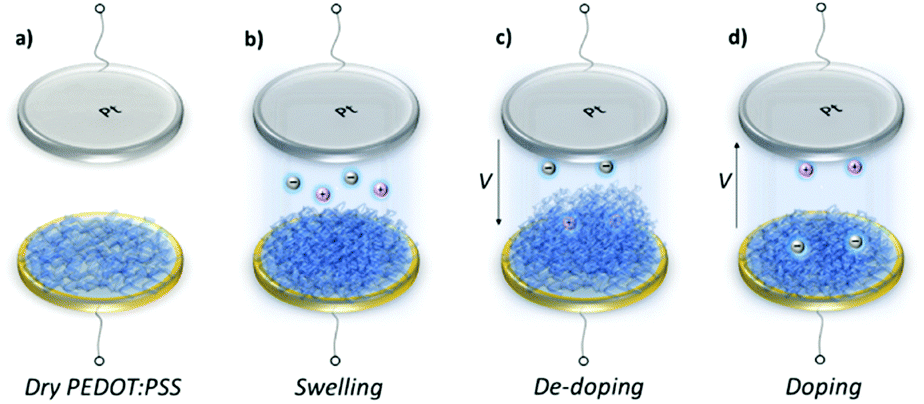 | ||
| Fig. 2 Voltage induced swelling/shrinkage of PEDOT:PSS film. (a) Dry film on a gold coated EQCM-D sensor, (b) swelling when in contact with the electrolyte, (c) additional swelling during de-doping due to up-take of Na+ ions and water, (d) shrinkage when the potential is reversed. We record significant differences in the volumetric expansion as well as the reversibility of the de-doping process depending on the film thickness. | ||
These results show that an EQCM-D can be used to directly monitor the voltage-triggered ion up-take and the corresponding current change in organic mixed conductors. To quantify the coupling of the ions-to-holes, we begin with converting the mass accumulated on the film into the number of cations (“Na+ injected”) and calculate the number of holes (“holes (h+) extracted”) from the reduction current (Fig. 3a and b). The injected cations should, however, be hydrated and the amount of water involved in the transfer of each Na+ ion is unknown.40 We, therefore, calculate the number of Na+ ions considering three different scenarios: when Na+ injection does not involve water and when each Na+ ion is coupled to 2 or 4 water molecules (namely, Na+, Na+:2H2O and Na+:4H2O, from darker to lighter blue lines in Fig. 3a and b, respectively). In order to estimate the amount of water coupled to the cations, we follow this argument: PEDOT:PSS contains a certain number of “sites” comprising h+/PSS− pairs. When a Na+ ion is injected, it is attracted to one of these sites to replace (electrostatically) one hole. At the maximum de-doping potential (−0.9 V vs. Voc), all of these sites are occupied by Na+ ions. Therefore, the number of Na+ ions injected at this voltage should correspond to the maximum number of sites that can host holes, that is the “hole density”. We thus derive the hole density using the number of injected cations. For the thin film, when we consider the transfer of 2H2O molecules per each Na+ ion, we derive a site density of 1.91 × 1020 sites cm−3, matching the theoretical value.14 For the thick film, we obtain this value only when we consider the transfer of 4H2O molecules with each cation. The hole densities calculated taking into account all hydration scenarios are summarized in Table S2 (ESI†). Overall, more water is transferred into the thick PEDOT:PSS film compared to the thin one during the de-doping process. Notably, as the voltage exceeds Voc, we observe an immediate increase in the number of cations incorporated into the film evidencing that ion injection involves no energy barriers or accumulation at the surface (Fig. 3a and b).14,39
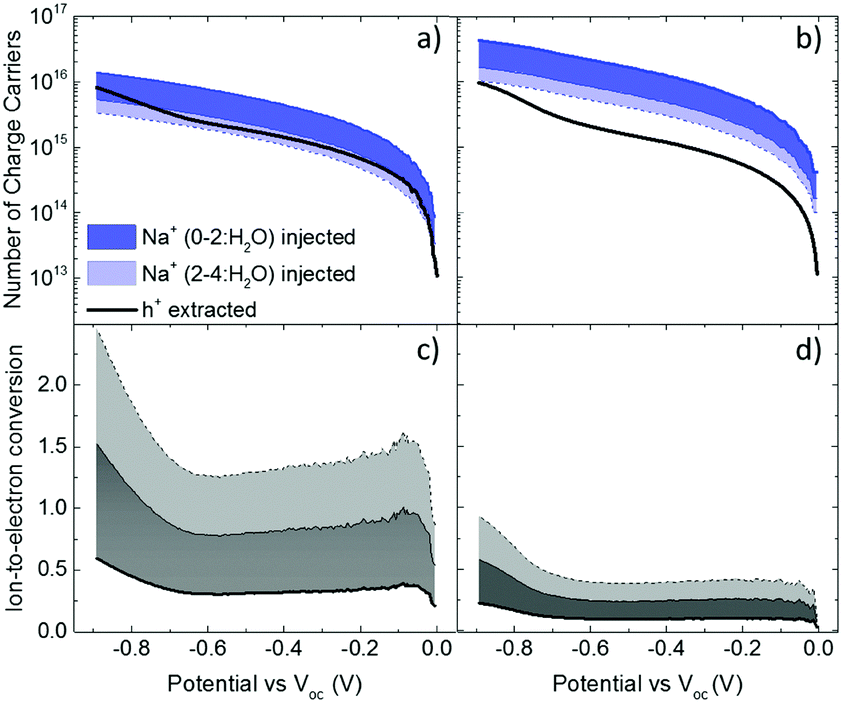 | ||
| Fig. 3 The evolution of the number of charge carriers, i.e. holes (h+, black lines) and cations (blue lines), during the backward voltage scan of the 2nd CV cycle for the (a) thin and (b) thick film. The ratio of these two parameters (h+ extracted:Na+ injected) is the efficiency of ion-to-electron conversion calculated for the (c) thin and (d) thick film. The color code represents the case when Na+ injection does not involve any water molecules (darkest color, blue or grey) and when each Na+ ion is coupled to 2 or 4 water molecules (the color of the shaded area gets lighter with more water involved, blue or grey). | ||
The next step is to quantify the efficiency of ion-to-electron conversion by correlating the number of cations incorporated into the film to the number of extracted charges. If all the injected cations are coupled to holes, then the ratio of ions to electrons should reach one (Fig. 3c and d). For instance, for the thin film, when 2H2O molecules are injected alongside each Na+ ion, the efficiency is constant for almost the whole voltage range, with an average value close to one (Fig. 3c). This means that approximately one Na+ ion and 2H2O molecules are injected for each hole that is extracted. On the contrary, for the thick film, the ratio at low de-doping voltages (−0.7 < V < Voc) is 0.2: for each h+ that is extracted, we inject about 3Na+ ions along with 4H2O molecules (Fig. 3d). In other words, for thick films, not all the injected cations are used for reducing PEDOT oligomers. Thus, although the film generates more current, the incorporated cations are not utilized as “efficiently” as in the thin film. Moreover, more water is transferred along with cations, in agreement with the higher damping of the sensor oscillations during the de-doping cycle (Fig. S1, ESI†). These results evidence thickness-dependent differences in the interaction of Na+ ions with PEDOT:PSS films. Furthermore, this trend is not limited to the current electrolyte: when the films are exposed to 0.1 M KCl solution, the discrepancy between the number of injected cations and that of extracted holes remains the same (Fig. S3, ESI†). Under the same applied voltage, the number of K+ ions taken up by the film matches the number of Na+ ions. As is the case for Na+ ions, K+ ions are utilized more efficiently in thinner PEDOT:PSS films.
Recent reports show that PEDOT based electrodes undergo the oxygen reduction reaction (ORR) in aqueous electrolytes at potentials below −1 V.44,45 Mitraka et al. demonstrated that electrochemically reduced PEDOT is more conducting at low negative applied potentials in the presence of O2 (due to the ORR) than in its absence.45 To elucidate the influence of the ORR in our measurements, we cycled the films in a deoxygenated NaCl(aq.) 0.1 M solution (Fig. S4, ESI,† top panel). Indeed, we observe lower reduction currents with an onset around −0.25 V vs. Ag/AgCl in a N2-purged environment compared with air saturated conditions, regardless of the film thickness. The difference between the two environments, that is the current contribution due to the ORR, becomes more evident above −0.4 V. While the cathodic process appears to be irreversible in air, the current profiles at the opposite polarities of the voltage scan are more comparable in a deoxygenated environment. Together, these features point to the faradaic phenomena of the ORR in air. Nevertheless, although the reduction currents are lower in N2-saturated NaCl solution, the mass that gets inside (out of) the films upon de-doping (doping) potentials is the same as that calculated from the measurements performed in air (Fig. S4, lower panel, and see Fig. S5, ESI† for raw EQCM-D data). This means that during reduction, the number of cations that are injected into the films is the same, independent of the ORR. On the other hand, due to lower reduction currents, we find a modest drop in ion-to-electron conversion efficiency when the measurements are performed in a N2 saturated environment (Fig. S6, ESI†). The thickness dependent differences in cation-film interactions are, however, consistent: the thick film takes up more water, along with cations, while a smaller fraction of these cations is used for reducing PEDOT oligomers compared with the thin film.
In order to verify the differences in the electrochemical behavior of these films, we record their Raman spectra before and after de-doping in NaCl solution. As shown in Fig. 4a, upon electrochemical reduction, the intensities of the Raman bands increase accompanied with shifts in the peak positions. Such changes are typically attributed to polymorphic alterations and the reduction of PEDOT chains.46,47 For instance, the strong band at 1425 cm−1 assigned to the ring Cα![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Cβ stretching vibrations becomes narrower and increases in intensity, indicating a longer conjugation length for PEDOT. For the thin film, the doublet peaks of asymmetrical vibrations of CαCβ at 1538 and 1536 cm−1 become a single peak at 1517 cm−1, whereas for the thick film, the peaks transform from a doublet at 1529 and 1533 cm−1 into single peak at 1515 cm−1. These shifts are ascribed to the replacement of polarons with Na+ ions.48 There is also a shift in the Cβ–Cβ peak for the thin film, from 1369 to 1367 cm−1, which is not the case for the thick film. Overall, the changes in the relative intensity, peak shapes and wavelength positions are more pronounced for the thin film (Fig. 4b), verifying that that ion-to-electron conversion is in fact more efficient. The cations injected into the thin film reach available h+/PSS− sites and compensate for holes.
Cβ stretching vibrations becomes narrower and increases in intensity, indicating a longer conjugation length for PEDOT. For the thin film, the doublet peaks of asymmetrical vibrations of CαCβ at 1538 and 1536 cm−1 become a single peak at 1517 cm−1, whereas for the thick film, the peaks transform from a doublet at 1529 and 1533 cm−1 into single peak at 1515 cm−1. These shifts are ascribed to the replacement of polarons with Na+ ions.48 There is also a shift in the Cβ–Cβ peak for the thin film, from 1369 to 1367 cm−1, which is not the case for the thick film. Overall, the changes in the relative intensity, peak shapes and wavelength positions are more pronounced for the thin film (Fig. 4b), verifying that that ion-to-electron conversion is in fact more efficient. The cations injected into the thin film reach available h+/PSS− sites and compensate for holes.
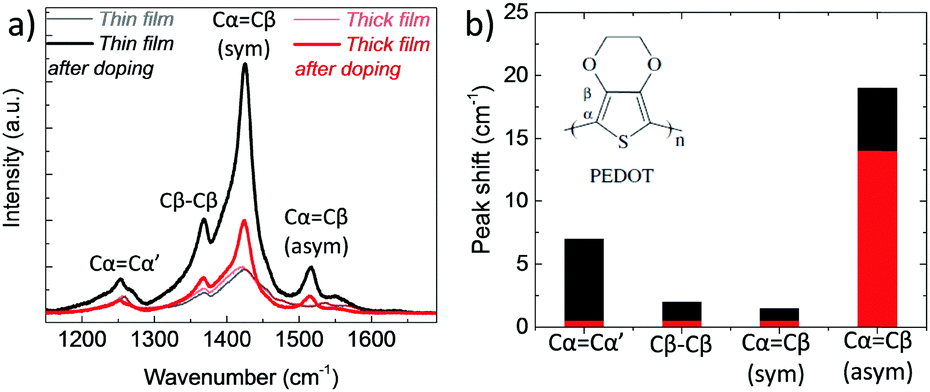 | ||
| Fig. 4 (a) Raman spectra of PEDOT:PSS films measured before and directly after electrochemical reduction (at −0.6 V vs. Ag/AgCl for 10 min in 0.1 M NaCl). The spectra were recorded at an excitation wavelength of 633 nm at room temperature. The vibration modes of PEDOT chains are located between 1200 to 1600 cm−1 and attributed to CαCβ asymmetrical stretching (1536 cm−1), CαCβ symmetrical stretching (1425 cm−1), Cβ–Cβ intra-thiophene ring (1369 cm−1) and to Cα–Cα’ inter-ring (1260 cm−1) stretching vibrations.49 (b) The absolute values of the wavelength shifts for the thin (black bars) and thick (red bars) film extracted from (a). | ||
Although we use the same dispersion and processing conditions, the two films differ in their interactions with cations. A variation in the film composition or microstructure could lead to a change in the number of injected cations, their hydration load, as well as their route inside the film. We know that, upon processing into films, PEDOT:PSS phase separates into PEDOT-rich clusters surrounded by larger PSS-rich areas.39 The film comprises PEDOT and PSS percolating networks and PSS rich regions as well as PEDOT:PSS isolated clusters that do not percolate within the conductive pathways.3 In order to explore the film composition, we first perform X-ray photoelectron spectroscopy (XPS) measurements on two identically prepared films (Fig. S7, ESI†). The relative ratio of PSS to PEDOT is calculated to be 2.74 and 2.19 for the thin and thick film, respectively, suggesting that the thick film has a higher amount of PEDOT on its surface – note that the investigated area with XPS is limited to a depth of maximum 10 nm from the surface. Since both films have the same amount of PEDOT and PSS in their composition, we postulate that the majority of PSS chains are accumulated within the bulk of the thick film. Moreover, at the surface of the thick film, we observe a smaller population of PSS chains that neutralize the doping charge compared with a thin film (Table S3, ESI†).50 These results suggest that PEDOT:PSS adopts a different composition, both at the surface and in its bulk, governed by its thickness.
In order to probe the bulk of the film, we then perform scanning transmission electron microscopy (STEM) measurements. Fig. 5a and b show the cross-sectional images of the thick and thin samples prepared for this analysis, respectively. Energy-dispersive X-ray spectroscopy (EDS) was applied on multiple spots within these films and the elemental analysis spectra are shown in Fig. S8 (ESI†). Note that the films were de-doped prior to these measurements as for the Raman spectroscopy studies above. Fig. 5c demonstrates the elemental composition of the films in terms of atomic percentages and highlights two striking differences in bulk composition governed by film thickness. First, we observe a substantial number of Na+ ions remaining inside the thick film upon de-doping compared to the thin film. This is in accordance with the finding that more mass remains in thicker films as suggested by the EQCM-D measurements (Fig. 1). Second, a higher silicon (Si) atomic percentage was found in thin films compared to thick ones. Si is a trace of GOPS, the crosslinker in the composition, which forms chemical bonds with the –SO3H groups of, mostly, excess PSS.51 In the thin film, the average atomic percentage of Si is 31.2%, while the atomic percentage of all other elements (C, O, and S, which are PEDOT and PSS traces) is 68.2%. In contrast, for the thick film, the Si atomic percentage is 18.7% and the sum of C, O, and S atomic percentages is 78%. A loosely connected PSS network, such as the one of the thick films, enables more water uptake as well as facile ion injection.22 This environment should be ideal to capture ions and facilitate their transport.
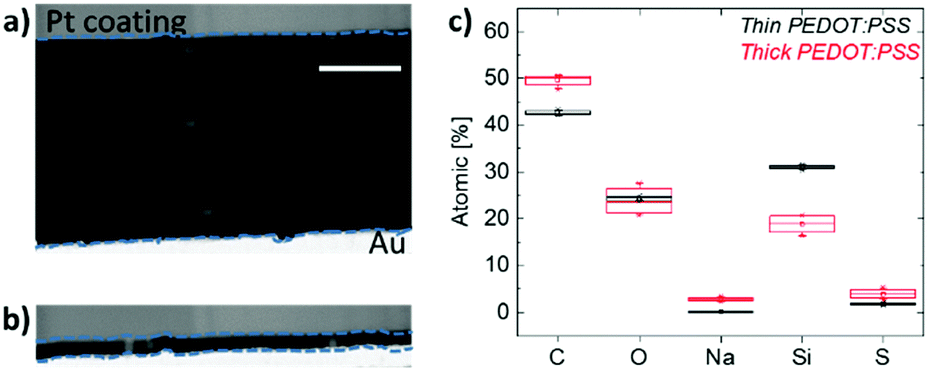 | ||
| Fig. 5 Cross section scanning transmission electron microscopy (STEM) images of (a) thick and (b) thin PEDOT:PSS films de-doped at −0.6 V vs. Ag/AgCl for 10 min in 0.1 M NaCl. The samples were cross sectioned by using a focused ion beam (FIB). The blue dashed lines serve as a guide to the eye marking the film sandwiched between the Pt coating and Au. The scale bar is 500 nm. (c) Box plot demonstration of the chemical composition of the films in terms of element atomic percentages. The compositional analysis was performed on 6 different spots within the cross section of the samples using energy-dispersive X-ray spectroscopy (EDS). The beam positions are marked by the bright areas in (a and b). | ||
Our results suggest that for electrochemical devices such as OECTs, while hydration of the film is required for ion penetration, a bulky hydrophilic phase (e.g. PSS) may act as sites for accumulation of ions that are not involved in charging/discharging the polymer. These ions contribute to the off-current (for intrinsic semiconductors) and may influence device stability and reversibility. Hütter et al. recently showed that thick PEDOT:PSS films in the OECT channel lead to less efficient use of the polymer (as well as higher charge consumption for switching the transistor off).52 They suggested the presence of PEDOT chains that do not participate in the electrochemical de-doping and that their proportion scales with the volume of the film. It was reasoned that not all the sites comprising of h+/PSS− pairs can be accessed by Na+ ions due to the partly coiled conformation of the chains. Our EQCM-D and Raman studies suggest the same thickness dependence of de-doping efficiency. We show, however, strong evidence for “un-utilized” Na+ ions, which are accumulated inside thicker films together with water. For thick films, the ions that get inside are many more than the number of accessible sites. Loosely interconnected PSS chains, present as a result of poor crosslinking with GOPS, enable greater water/cation uptake and introduce traps for cations. This hypothesis is also supported by high dissipation changes of thick PEDOT:PSS films upon de-doping. The agreement of these different techniques highlights the role of film microstructure and composition in determining the efficiency of coupling between ionic and electronic charges in PEDOT:PSS. Such aspects that have remained uncovered so far can be explored using EQCM-D.
Conclusions
Direct measurements of the mass that accumulates inside PEDOT:PSS films subject to electrochemical de-doping enable monitoring interactions of cations with the film. Correlating the number of cations to that of the extracted holes, we determine the efficiency of ion-to-electron conversion and reveal loss mechanisms for films of different thicknesses. In particular, we find that the number of cations which are not involved with charge neutralization is significantly high for a thick film. For thick films, the cations bring more water molecules along and do not fully leave the film. These results are intriguing as thickness is an easy parameter to control yet one that effects the performance of electrochemical devices. Raman studies reveal that the changes in the conjugation length upon de-doping are more pronounced in a thin film. XPS results suggest a different surface composition for the films, dictated by their thickness. STEM measurements reveal that the thickness of the film influences its bulk composition: thick films contain a lower amount of the crosslinker, i.e. a loosely connected PSS network. As a result of this particular composition, the thick films undergo a less efficient and reversible de-doping process compared to thin films. The methodology developed in this study for quantifying ion-to-electron coupling efficiency can be adapted to other mixed conductor–electrolyte systems and will therefore aid in understanding ion/polymer interactions as long as ion injection barriers are well verified.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Jokubas Surgailis for his assistance in EQCM-D analysis and Jonathan Rivnay for fruitful discussions.Notes and references
- S. Inal, G. G. Malliaras and J. Rivnay, Nat. Commun., 2017, 8, 1767 CrossRef PubMed.
- R. Giridharagopal, L. Q. Flagg, J. S. Harrison, M. E. Ziffer, J. Onorato, C. K. Luscombe and D. S. Ginger, Nat. Mater., 2017, 16, 737 CrossRef PubMed.
- J. Rivnay, S. Inal, B. A. Collins, M. Sessolo, E. Stavrinidou, X. Strakosas, C. Tassone, D. M. Delongchamp and G. G. Malliaras, Nat. Commun., 2016, 7, 11287 CrossRef PubMed.
- O. Inganas, Chem. Soc. Rev., 2010, 39, 2633–2642 RSC.
- L. Groenendaal, G. Zotti, P.-H. Aubert, S. M. Waybright and J. R. Reynolds, Adv. Mater., 2003, 15, 855–879 CrossRef.
- S. Fabiano, N. Sani, J. Kawahara, L. Kergoat, J. Nissa, I. Engquist, X. Crispin and M. Berggren, Sci. Adv., 2017, 3, e1700345 CrossRef PubMed.
- O. V. Mikhnenko, S. D. Collins and T.-Q. Nguyen, Adv. Mater., 2015, 27, 2007–2012 CrossRef PubMed.
- J. Rivnay, R. M. Owens and G. G. Malliaras, Chem. Mater., 2014, 26, 679–685 CrossRef.
- J. Rivnay, P. Leleux, M. Ferro, M. Sessolo, A. Williamson, D. A. Koutsouras, D. Khodagholy, M. Ramuz, X. Strakosas, R. M. Owens, C. Benar, J.-M. Badier, C. Bernard and G. G. Malliaras, Sci. Adv., 2015, 1, e1400251 Search PubMed.
- D. Khodagholy, T. Doublet, P. Quilichini, M. Gurfinkel, P. Leleux, A. Ghestem, E. Ismailova, T. Hervé, S. Sanaur, C. Bernard and G. G. Malliaras, Nat. Commun., 2013, 4, 1575 CrossRef PubMed.
- M. Ramuz, A. Hama, J. Rivnay, P. Leleux and R. M. Owens, J. Mater. Chem. B, 2015, 3, 5971–5977 RSC.
- P. Lin and F. Yan, Adv. Mater., 2012, 24, 34–51 CrossRef PubMed.
- H. Shi, C. Liu, Q. Jiang and J. Xu, Adv. Electron. Mater., 2015, 1, 1500017 CrossRef.
- C. M. Proctor, J. Rivnay and G. G. Malliaras, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 1433–1436 CrossRef.
- K. Tybrandt, I. V. Zozoulenko and M. Berggren, Sci. Adv., 2017, 3, eaao3659 CrossRef PubMed.
- J. Rivnay, S. Inal, A. Salleo, M. R. Owens, M. Berggren and G. G. Malliaras, Nat. Rev. Mater., 2018, 3, 17086 CrossRef.
- M. Rudolph and E. L. Ratcliff, Nat. Commun., 2017, 8, 1048 CrossRef PubMed.
- D. C. Martin and G. G. Malliaras, ChemElectroChem, 2016, 3, 686–688 CrossRef.
- C. M. Pacheco-Moreno, M. Schreck, A. D. Scaccabarozzi, P. Bourgun, G. Wantz, M. M. Stevens, O. J. Dautel and N. Stingelin, Adv. Mater., 2017, 29, 1604446 CrossRef PubMed.
- A. A. Argun, P.-H. Aubert, B. C. Thompson, I. Schwendeman, C. L. Gaupp, J. Hwang, N. J. Pinto, D. B. Tanner, A. G. MacDiarmid and J. R. Reynolds, Chem. Mater., 2004, 16, 4401–4412 CrossRef.
- M. M. Schmidt, M. ElMahmoudy, G. G. Malliaras, S. Inal and M. Thelakkat, Macromol. Chem. Phys., 2018, 219, 1700374 CrossRef.
- E. Stavrinidou, P. Leleux, H. Rajaona, D. Khodagholy, J. Rivnay, M. Lindau, S. Sanaur and G. G. Malliaras, Adv. Mater., 2013, 25, 4488–4493 CrossRef PubMed.
- S. Inal, G. G. Malliaras and J. Rivnay, J. Mater. Chem. B, 2016, 4, 3942–3947 Search PubMed.
- M. Rodahl, F. Höök, A. Krozer, P. Brzezinski and B. Kasemo, Rev. Sci. Instrum., 1995, 66, 3924–3930 CrossRef.
- M. Rodahl and B. Kasemo, Rev. Sci. Instrum., 1996, 67, 3238–3241 CrossRef.
- P. J. Molino, Z. Yue, B. Zhang, A. Tibbens, X. Liu, R. M. I. Kapsa, M. J. Higgins and G. G. Wallace, Adv. Mater. Interfaces, 2014, 1, 1300122 CrossRef.
- H.-S. Lee, M. Q. Yee, Y. Y. Eckmann, N. J. Hickok, D. M. Eckmann and R. J. Composto, J. Mater. Chem., 2012, 22, 19605–19616 RSC.
- F. Höök, M. Rodahl, P. Brzezinski and B. Kasemo, Langmuir, 1998, 14, 729–734 CrossRef.
- S. Inal, J. Rivnay, A. I. Hofmann, I. Uguz, M. Mumtaz, D. Katsigiannopoulos, C. Brochon, E. Cloutet, G. Hadziioannou and G. G. Malliaras, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 147–151 CrossRef.
- W. Gao, O. Sel and H. Perrot, Electrochim. Acta, 2017, 233, 262–273 CrossRef.
- L. Niu, C. Kvarnström and A. Ivaska, J. Electroanal. Chem., 2004, 569, 151–160 CrossRef.
- A. Baba, S. Tian, F. Stefani, C. Xia, Z. Wang, R. C. Advincula, D. Johannsmann and W. Knoll, J. Electroanal. Chem., 2004, 562, 95–103 CrossRef.
- M. D. Levi, L. Daikhin, D. Aurbach and V. Presser, Electrochem. Commun., 2016, 67, 16–21 CrossRef.
- Z. Yang, A. A. Gewirth and L. Trahey, ACS Appl. Mater. Interfaces, 2015, 7, 6557–6566 CrossRef PubMed.
- V. Dargel, N. Shpigel, S. Sigalov, P. Nayak, M. D. Levi, L. Daikhin and D. Aurbach, Nat. Commun., 2017, 8, 1389 CrossRef PubMed.
- M. D. Levi, N. Shpigel, S. Sigalov, V. Dargel, L. Daikhin and D. Aurbach, Electrochim. Acta, 2017, 232, 271–284 CrossRef.
- M. ElMahmoudy, S. Inal, A. Charrier, I. Uguz, G. G. Malliaras and S. Sanaur, Macromol. Mater. Eng., 2017, 302, 1600497 CrossRef.
- R. Merkle, P. Gutbrod, P. Reinold, M. Katzmaier, R. Tkachov, J. Maier and S. Ludwigs, Polymer, 2017, 132, 216–226 CrossRef.
- A. V. Volkov, K. Wijeratne, E. Mitraka, U. Ail, D. Zhao, K. Tybrandt, J. W. Andreasen, M. Berggren, X. Crispin and I. V. Zozoulenko, Adv. Funct. Mater., 2017, 27, 1700329 CrossRef.
- H. Ohtaki, Chem. Rev., 1993, 93, 1157–1204 CrossRef.
- T. Mirfakhrai, J. D. W. Madden and R. H. Baughman, Mater. Today, 2007, 10, 30–38 CrossRef.
- E. W. H. Jager, O. Inganäs and I. Lundström, Adv. Mater., 2001, 13, 76–79 CrossRef.
- E. Smela and N. Gadegaard, Adv. Mater., 1999, 11, 953–957 CrossRef.
- S. K. Singh, X. Crispin and I. V. Zozoulenko, J. Phys. Chem. C, 2017, 121, 12270–12277 CrossRef.
- E. Mitraka, M. J. Jafari, M. Vagin, X. Liu, M. Fahlman, T. Ederth, M. Berggren, M. P. Jonsson and X. Crispin, J. Mater. Chem. A, 2017, 5, 4404–4412 RSC.
- S. Satoshi, M. Okumura, Z. Zhao and Y. Furukawa, Chem. Phys. Lett., 2005, 412, 395–398 CrossRef.
- S. Garreau, J. L. Duvail and G. Louarn, Synth. Met., 2001, 125, 325–329 CrossRef.
- P. A. Kilmartin, K.-C. Li, G. A. Bowmaker, N. A. Vigar, R. P. Cooney and J. Travas-Sejdic, Curr. Appl. Phys., 2006, 6, 567–570 CrossRef.
- W. W. Chiu, J. Travas-Sejdic, R. P. Cooney and G. A. Bowmaker, J. Raman Spectrosc., 2006, 37, 1354–1361 CrossRef.
- X. Crispin, S. Marciniak, W. Osikowicz, G. Zotti, A. W. D. van der Gon, F. Louwet, M. Fahlman, L. Groenendaal, F. De Schryver and W. R. Salaneck, J. Polym. Sci., Part B: Polym. Phys., 2003, 41, 2561–2583 CrossRef.
- A. Håkansson, S. Han, S. Wang, J. Lu, S. Braun, M. Fahlman, M. Berggren, X. Crispin and S. Fabiano, J. Polym. Sci., Part B: Polym. Phys., 2017, 55, 814–820 CrossRef.
- P. C. Hütter, A. Fian, K. Gatterer and B. Stadlober, ACS Appl. Mater. Interfaces, 2016, 8, 14071–14076 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Swelling of thin and thick PEDOT:PSS films; changes in EQCM-D frequency and dissipation signals of PEDOT:PSS coated crystals during CV measurements; mass added on the sensors calculated via the Sauerbrey equation and viscoelastic modelling; EQCM-D measurements and analysis in KCl, and in N2 saturated NaCl; XPS measurements on the surface of thin and thick PEDOT:PSS films; and EDS elemental analysis data. See DOI: 10.1039/c8tc02195c |
| This journal is © The Royal Society of Chemistry 2018 |