
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fullerenemalonates inhibit amyloid beta aggregation, in vitro and in silico evaluation
Martínez-Herrera Melchorab,
Figueroa-Gerstenmaier Susanacd,
García-Sierra Franciscoe,
Beltrán Hiram I. b,
Rivera-Fernández Normaf,
Lerma-Romero Jorge A.g,
López-Camacho Perla Y.b and
Basurto-Islas Gustavo*c
b,
Rivera-Fernández Normaf,
Lerma-Romero Jorge A.g,
López-Camacho Perla Y.b and
Basurto-Islas Gustavo*c
aCONACYT, Metropolitan Autonomous University, Cuajimalpa, Mexico City, 05300, Mexico
bDepartment of Natural Sciences, Metropolitan Autonomous University, Cuajimalpa, Mexico City, 05300, Mexico
cDepartment of Chemical, Electronic & Biomedical Engineering, Division of Sciences and Engineering, University of Guanajuato, Loma del Bosque No.103, Lomas del Campestre, León, 37150, Guanajuato, Mexico. E-mail: gustavo.basurto@ugto.mx
dEduard-Zintl-Institut für Anorganische und Physikalische Chemie, Technische Universität Darmstadt, D-64287 Darmstadt, Germany
eDepartment of Cell Biology, Center of Research and Advanced Studies of the National Polytechnic Institute (CINVESTAV), Mexico City, 07360, Mexico
fDepartment of Microbiology and Parasitology, School of Medicine, National Autonomous University of Mexico, Ciudad de México, 04510, Mexico
gNational Polytechnic Institute, Mexico City, 07738, Mexico
First published on 27th November 2018
Abstract
The onset of Alzheimer's disease (AD) is associated with the presence of neurofibrillary pathology such as amyloid β (Aβ) plaques. Different therapeutic strategies have focused on the inhibition of Aβ aggregate formation; these pathological structures lead to neuronal disorder and cognitive impairment. Fullerene C60 has demonstrated the ability to interact and prevent Aβ fibril development; however, its low solubility and toxicity to cells remain significant problems. In this study, we synthesized, characterized and compared diethyl fullerenemalonates and the corresponding sodium salts, adducts of C60 bearing 1 to 3 diethyl malonyl and disodium malonyl substituents to evaluate the potential inhibitory effect on the aggregation of Aβ42 and their biocompatibility. The dose-dependent inhibitory effect of fullerenes on Aβ42 aggregation was studied using a thioflavin T fluorescent assay, and the IC50 value demonstrated a low range of fullerene concentration for inhibition, as confirmed by electron microscopy. The exposure of neuroblastoma to fullerenemalonates showed low toxicity, primarily in the presence of the sodium salt-adducts. An isomeric mixture of bisadducts, trisadducts and a C3-symetrical trisadduct demonstrated the highest efficacy among the tests. In silico calculations were performed to complement the experimental data, obtaining a deeper understanding of the Aβ inhibitory mechanism; indicating that C3-symetrical trisadduct interacts mainly with 1D to 16K residues of Aβ42 peptide. These data suggest that fullerenemalonates require specific substituents designed as sodium salt molecules to inhibit Aβ fibrillization and perform with low toxicity. These are promising molecules for developing future therapies involving Aβ aggregates in diseases such as AD and other types of dementia.
Introduction
Amyloid plaques are the primary hallmark of Alzheimer's disease (AD), the most prevalent type of dementia worldwide. The aggregation of the amyloid beta (Aβ) peptide leads to the onset of extracellular plaque throughout the cortical mantle. The β- and γ-secretases sequentially proteolyze the amyloid precursor protein (APP), releasing a 40 or 42 amino acid peptide. In AD, Aβ aggregates extracellularly form soluble oligomers, insoluble β-sheet protofibrils, fibrils and plaques. Aβ plays an important role in the onset of AD, described in the amyloid cascade hypothesis, resulting from a chronic imbalance between Aβ production and Aβ clearance1 that turns into: neuronal loss, neurofibrillary tangle formation, vascular damage, and dementia that correlates directly to Aβ deposition. Despite Aβ plaques showing a low correlation with dementia, Aβ oligomers display high toxicity to neurons2,3 suggesting that Aβ fibrillogenesis plays an important role in AD-induced toxicity. The Aβ aggregation involves the C-terminus of the peptide that determines the rate of fibril formation while the N-terminus promotes Aβ–Aβ interaction for polymerization, leading to a random coil or α-helix to β-sheet transition via a nucleation mediated process.4 The extended β-sheets promote homophilic interactions and eventually lead to Aβ oligomer formation. Kinetic studies demonstrated the monomeric Aβ requirement for oligomer formation as seeds/nuclei, rich in β-sheets, for accelerated fibril growth.5Several strategies for targeting Aβ production and clearance have failed, since Aβ immunotherapies induced encephalomyelitis and possible microhemorrhages,6,7 and the inhibition of secretases disrupts important metabolic processes.8 Therefore, another strategy is based on inhibition of the Aβ peptide self-assembly. Dyes and small molecules,9–11 peptides12–14 and nanoparticles15–18 were identified as effective inhibitors of Aβ aggregation, ameliorating cell survival and cognitive deficit.
Fullerene C60, a carbon nanomaterial with a symmetric nanostructure, has been extensively used in areas of science, particularly in biomedics. Even though it has great potential in biological applications, the solubility of fullerenes has shown low compatibility in biological systems because of its hydrophobicity; so different strategies have been developed to achieve soluble fullerenes in aqueous dispersions for biological applications, including as an inhibitor of human immunodeficiency virus-1 (HIV-1) protease,19 an antioxidant,20 and a cancer therapeutic.21 C60 functions as both a reactive oxygen species (ROS) producer under UV or visible light, and a ROS scavenger in the dark. This dual property of fullerenes to either quench or generate cell-damaging ROS has been applied as a cytoprotective or cytotoxic anticancer/antimicrobial/anti-Aβ agent.22–26 Previous reports indicated that fullerenes, and certain fullerene derivatives, inhibit Aβ aggregation much more efficiently under photo-irradiation with visible light.24,25 The dual property of C60 to either scavenge or produce ROS has been used for a synergistic therapy for Alzheimer's disease (AD).26
Fullerenes have two important advantages in AD research: their structure allows them to cross the blood–brain barrier27 and they show a high capacity to inhibit Aβ fibril formation.16,17,24,25,28–30 However, there is controversy regarding the biocompatibility of fullerenes: some groups report nontoxic effects in different tested models, such as the LLC-PK1 proximal tubule cell model31 or L929 mouse subcutaneous connective cells;32 however, a high concentration of fullerenes induces toxicity in the same studies. Therefore, the correlation between the amount of fullerene necessary to perform its biological activity and biocompatibility remains unclear. Recently, impaired spatial memory with a significant decrease in BDNF protein levels and gene expression has been demonstrated in rats injected with C60, but in contrast, it showed high antioxidant capacity.33 Fullerene might be an important molecule for the treatment of neurodegenerative disorders, but the molecular design requires further research. An interesting approach to improve fullerenes for AD treatment is based on functionalization with different substituents that promote its stability, biocompatibility, capacity to cross the blood–brain barrier and ability to inhibit Aβ fibril formation. One successful method to obtain more polar fullerenes introducing functional groups consists in exohedral functionalization by the Bingel reaction.34,35 In particular, functionalization with a malonate group led to water-soluble fullerenes and in addition serves as a precursor for further functionalization, so it can be used to link a variety of functional groups to obtain different derivatives.
In this study we synthesized diethyl fullerenemalonates and the corresponding sodium salts, through the Bingel reaction to obtain adducts of C60 holding 1 to 3 diethyl malonyl and disodium malonyl substituents (C60+n(COOR)2n, where n = 1–3 and R = –CH2CH3, –Na) (Fig. 1). This is the first report to demonstrate the effect of the type and number of organic addends of fullerenemalonates on anti-Aβ activity and their biocompatibility with cells. The potential inhibitory effect of the fullerenes on Aβ42 fibril formation was shown by thioflavin fluorescence assay and electron microscopy. Low cytotoxicity was shown in neuroblastoma SH-SY5Y cells exposed to the fullerenemalonates during 24 h, and the cytotoxic effect decreased even more in the presence of the corresponding sodium salt molecules. The in silico data obtained by atomistic molecular dynamics showed that the purified C3 trisadduct binds to the Aβ42 monomer, mostly to 1D, 5R, 16K residues by means of hydrogen bonds and to 2A, 4F, 6H, 8S, 12V, 15Q residues.
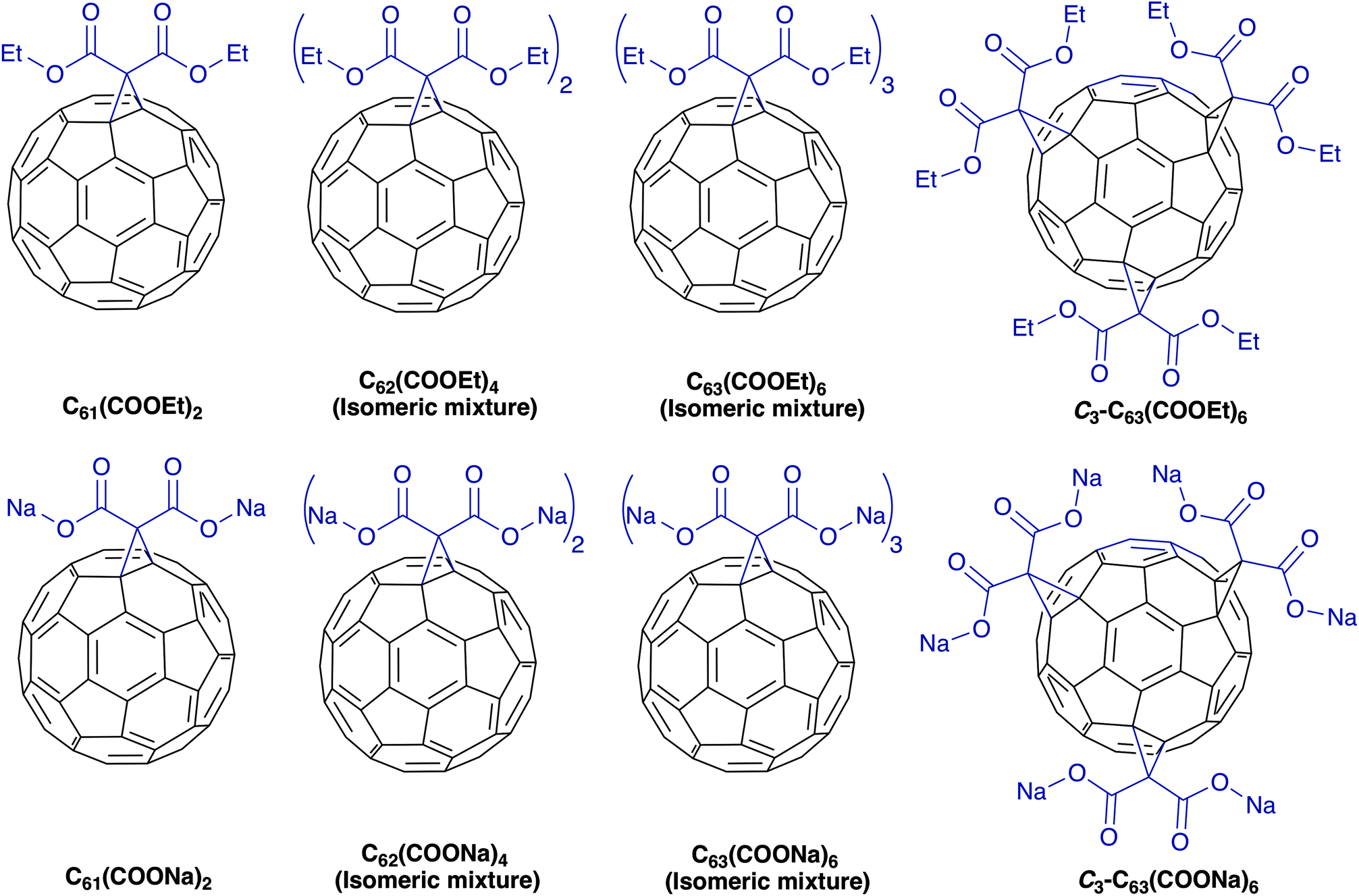 | ||
| Fig. 1 Molecular structure of diethyl fullerenemalonates and the corresponding sodium salts, synthesized and evaluated in this study. | ||
Materials and methods
Chemicals and reagents
Fullerene C60 98%, diethyl malonate 99%, carbon tetrabromide 99%, 1,8-diazabicyclo[5.4.0]-undec-7-ene (DBU), 98%, thioflavin T (ThT), and all other chemicals and solvents were purchased from Sigma-Aldrich and were used as supplied without further purification. Beta-amyloid (1–42) Human was purchase from (ANASPEC-1 mg).Fullerene synthesis
The Bingel-type adducts were synthesized by cyclopropanation of C60 with different equivalents of diethyl malonate, CBr4 and 1,8-diazabicyclo-[5,4,0]undec-7-ene (DBU) as an auxiliary base, following the procedure published by X. Camps et al. and E. Straface et al.36,37 Diethylmalonate mono- and bisadducts were isolated and purified by a chromatography column on silica gel. Chromatographic separation of the reaction mixture with n-hexane/toluene (65![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :35) generated a first fraction consisting of a residual amount of C60. Subsequently, pure toluene was used to yield a second fraction that consisted of the monoadduct contaminated with traces of C60, and finally there was a third fraction containing an isomeric mixture of the corresponding bisadducts. Seven regioisomers of the bisadducts (of the eight possible) have been isolated and characterized by Hirsch et al.38 The second and third fractions were also purified by chromatography, using n-hexane/toluene (65:35) and pure toluene to remove the traces of C60 or monoadduct to obtain pure monoadduct, C61(COOEt)2, and an isomeric mixture of bisadducts, C62(COOEt)4. The trisadducts were purified using elution of the reaction mixture with n-hexane/toluene (65:35) to pure toluene, allowing the separation of three fractions containing enriched samples of monoadduct, bisadducts and trisadducts as the major components, respectively. The third fraction containing semipure trisadducts was rechromatographed on silica gel using toluene as the mobile phase to obtain the purified isomeric mixture, C63(COOEt)6. Seven regioisomers of the trisadducts (of the 10 possible considering the restriction that only e or trans additions to bisadducts having e- and trans-positional relationships are considered) have been isolated and characterized by Djojo et al.39 A purified sample of the C3 trisadduct (C3-C63(COOEt)6) was obtained by elution of the fraction containing the isomeric mixture of trisadducts with toluene/acetonitrile (99.5:0.5), producing a last fraction (red band) enriched in the regioisomeric trisadduct with C3 symmetry. The fraction containing semipure isomer C3 was further rechromatographed twice using toluene/acetonitrile (99.5:0.5) thus obtaining a pure C3 adduct (C3-C63(COOEt)6). The identity of all compounds was confirmed by electrospray ionization time-of-flight mass spectrometry (ESI-TOF-MS), giving molecular ions identical to those calculated. The obtained data was as follow: C61(COOEt)2 (m/z) 878.059 (M−), calculated for C67H10O4 878.058; C62(COOEt)4 (m/z) 1036.117 (M−), calculated for C74H20O8 1036.116; C63(COOEt)6 (m/z) 1195.181 (M+ + 1), calculated for C81H31O12 1195.181; C3-C63(COOEt)6 (m/z) 1195.179 (M+ + 1), calculated for C81H31O12 1195.181. The samples were further characterized by 1H nuclear magnetic resonance (NMR), except for C62(COOEt)4 and C63(COOEt)6, ultraviolet visible (UV-vis) and infrared (IR) spectroscopies.
:35) generated a first fraction consisting of a residual amount of C60. Subsequently, pure toluene was used to yield a second fraction that consisted of the monoadduct contaminated with traces of C60, and finally there was a third fraction containing an isomeric mixture of the corresponding bisadducts. Seven regioisomers of the bisadducts (of the eight possible) have been isolated and characterized by Hirsch et al.38 The second and third fractions were also purified by chromatography, using n-hexane/toluene (65:35) and pure toluene to remove the traces of C60 or monoadduct to obtain pure monoadduct, C61(COOEt)2, and an isomeric mixture of bisadducts, C62(COOEt)4. The trisadducts were purified using elution of the reaction mixture with n-hexane/toluene (65:35) to pure toluene, allowing the separation of three fractions containing enriched samples of monoadduct, bisadducts and trisadducts as the major components, respectively. The third fraction containing semipure trisadducts was rechromatographed on silica gel using toluene as the mobile phase to obtain the purified isomeric mixture, C63(COOEt)6. Seven regioisomers of the trisadducts (of the 10 possible considering the restriction that only e or trans additions to bisadducts having e- and trans-positional relationships are considered) have been isolated and characterized by Djojo et al.39 A purified sample of the C3 trisadduct (C3-C63(COOEt)6) was obtained by elution of the fraction containing the isomeric mixture of trisadducts with toluene/acetonitrile (99.5:0.5), producing a last fraction (red band) enriched in the regioisomeric trisadduct with C3 symmetry. The fraction containing semipure isomer C3 was further rechromatographed twice using toluene/acetonitrile (99.5:0.5) thus obtaining a pure C3 adduct (C3-C63(COOEt)6). The identity of all compounds was confirmed by electrospray ionization time-of-flight mass spectrometry (ESI-TOF-MS), giving molecular ions identical to those calculated. The obtained data was as follow: C61(COOEt)2 (m/z) 878.059 (M−), calculated for C67H10O4 878.058; C62(COOEt)4 (m/z) 1036.117 (M−), calculated for C74H20O8 1036.116; C63(COOEt)6 (m/z) 1195.181 (M+ + 1), calculated for C81H31O12 1195.181; C3-C63(COOEt)6 (m/z) 1195.179 (M+ + 1), calculated for C81H31O12 1195.181. The samples were further characterized by 1H nuclear magnetic resonance (NMR), except for C62(COOEt)4 and C63(COOEt)6, ultraviolet visible (UV-vis) and infrared (IR) spectroscopies.
Diethyl fullerenemalonates
C61(COOEt)2: 1H NMR (400 MHz, CS2-CDCl3), δ 4.54 (q, J = 7 Hz, 4H), 1.52 (t, J = 7 Hz, 6H); UV-vis (THF) λmax/nm 327, 427, 477; IR (KBr)/cm−1 2963, 2924, 2853, 1743, 1633, 1538, 1461, 1427, 1385, 1364, 1292, 1262, 1233, 1206, 1179, 1095, 1059, 1019, 860, 804, 733, 705, 670, 579, 540, 525. C62(COOEt)4: UV-vis (THF) λmax/nm 303, 417, 428, 474; IR (KBr)/cm−1 2962, 2926, 2854, 1743, 1634, 1538, 1459, 1440, 1386, 1366, 1294, 1232, 1180, 1098, 1059, 1018, 857, 806, 733, 704, 668, 623, 574, 524. C63(COOEt)6: UV-vis (THF) λmax/nm 303, 402 (sh), 412 (sh), 462 (w); IR (KBr)/cm−1 2953, 2929, 2900, 2860, 1744, 1632, 1461, 1443, 1388, 1367, 1296, 1232, 1180, 1101, 1063, 1019, 859, 808, 755, 734, 705, 668, 573, 547, 525. C3-C63(COOEt)6: UV-vis (THF) λmax/nm 303, 378 (sh), 390 (sh), 474 (w), 562 (sh); IR (KBr)/cm−1 2962, 2923, 2854, 1743, 1653, 1634, 1462, 1446, 1387, 1369, 1281, 1241, 1214, 1174, 1099, 1064, 1022, 860, 807, 738, 707, 667, 626, 563, 525. All the samples of C60 adducts showed identical spectroscopic data to those already reported.34,40–42The disodium fullerenemalonates were synthesized by hydrolysis of the respective diethyl fullerenemalonates, with a 1.5-fold (relative to ester groups) molar amount of NaOH (1 M) in tetrahydrofuran:methanol:water for the case of monoadduct C61(COONa)2 and bisadducts C62(COONa)4 and toluene:methanol:water in the case of the trisadducts C63(COONa)6 and C3-C63(COONa)6, according to a method described already.43 The reaction was stopped after all of the starting diethyl malonate was consumed, as monitored by thin-layer chromatography. The sodium salts were triturated from toluene and water or methanol and isolated by centrifugation. Finally, the product was evaporated, and dried under vacuum. The yields were nearly quantitative. The samples were characterized by UV-vis and IR spectroscopies. Disodium fullerenemalonates were identified by the shift in the characteristic and strong 〉C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration in the IR spectrum (KBr pellet) of the ester from v = 1743 cm−1 to v = 1638 cm−1, 1621 cm−1, 1626 cm−1 and 1638 cm−1 for the monoadduct, isomeric mixture of bisadducts and trisadducts, and C3-trisadduct, respectively; due to the mesomeric weakening of the 〉CO double bond in the dicarboxylate.
O vibration in the IR spectrum (KBr pellet) of the ester from v = 1743 cm−1 to v = 1638 cm−1, 1621 cm−1, 1626 cm−1 and 1638 cm−1 for the monoadduct, isomeric mixture of bisadducts and trisadducts, and C3-trisadduct, respectively; due to the mesomeric weakening of the 〉CO double bond in the dicarboxylate.
Disodium fullerenemalonates
C61(COONa)2: UV-vis (THF) λmax/nm 318; IR (KBr)/cm−1 2955, 2926, 2852, 1726, 1638, 1560, 1457, 1407, 1383, 1097, 1066, 1027, 841, 704, 674, 615, 529. C62(COONa)4: UV-vis (CH3OH) λmax/nm 349; IR (KBr)/cm−1 2960, 2926, 2855, 1728, 1621, 1408, 1376, 1330, 1245, 1204, 1166, 1101, 1066, 841, 704, 649, 613, 526. C63(COONa)6: UV-vis (CH3OH) λmax/nm 414; IR (KBr)/cm−1 2961, 2928, 2855, 1728, 1626, 1458, 1379, 1331, 1105, 1058, 840, 703, 671, 623, 522. C3-C63(COOEt)6: UV-vis (CH3OH) λmax/nm 414; IR (KBr)/cm−1 2959, 2928, 2858, 1728, 1638, 1600, 1580, 1562, 1461, 1410, 1382, 1289, 1272, 1122, 1071, 1039, 961, 848, 796, 742, 702, 651, 602.Preparation and fibrillization of Aβ42 peptide
The Aβ42 monomer (1 mg) was resuspended in 1 ml of cold 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP), and it was kept in the dark for 30 min at 4 °C to solubilize the peptide in the monomeric stage.44,45 The solution was aliquoted (25 μl) and dried under vacuum in a rotary evaporator for 1 h at room temperature; the resultant transparent film was stored at −80 °C for further experiments. The amyloid fibrils were formed as previously described;46 briefly, the solubilized Aβ42 was dissolved in polymerization buffer containing PBS 1× (final concentration of 10 mM PO43−, 137 mM NaCl, and 2.7 mM KCl) and 1% DMSO and incubated at 37 °C at different time points. The required volume of polymerization buffer was adjusted according to each experiment.Fibril formation analysis by western blot
Western blots were performed using 16% SDS-PAGE, loading 1.5 μg of non-boiled Aβ42 fibril samples per line, followed by transfer to a nitrocellulose membrane and blocking with 5% skim milk. The blot was performed with a mouse monoclonal anti-β-amyloid antibody (Sigma-Aldrich) at 1:3000 dilution, incubated overnight and probed with goat anti-mouse Ig-G (Millipore) at 1:10000, and, finally, detected using enhanced chemiluminescence reagents (Thermo Scientific).
Fluorescence analysis
Fibrillization of Aβ42 peptide was detected by ThT fluorescence intensity that correlates with the number of fibrils formed.47 ThT was added to the Aβ42 samples to final concentrations of 10 and 20 μM, respectively, in a total volume of 200 μl of polymerization buffer. The fluorescence was measured in a 96-well plate during 24 h at 37 °C using a TECAN Infinite (M1000PRO) spectrofluorometer at 440 nm excitation and 490 nm emission. The fluorescence intensity was the normalized value of Aβ42 aggregates at 24 h (positive control). Likewise, to evaluate the inhibitory effect of the fullerenes, 13 μM (final concentration in the solution) of the respective fullerene was added to the same reaction. The relative fluorescence of fullerenemalonates without Aβ42 was measured and subtracted from the respective assay with the peptide. The IC50 for Aβ42 amyloid fibrillization inhibition were determined from the curves obtained by fitting the average fluorescence values in three independent experiments at the following fullerene concentrations: 2.5, 5, 7.5 and 10 μM. The experiments were done in triplicate.Electron microscopy
The polymerization assay solution samples and that with disodium fullerenemalonates C3-C63(COONa)6 and an isomeric mixture of C62(COONa)4 were sedimented and placed side by side onto Formvar-coated copper grids for 1 min, followed by incubation in 50 mM ammonium bicarbonate for carbon coating of the sample for 3 min and then negatively stained with 2% uranyl acetate for 1 min. This procedure was repeated twice for 2 min and 1 min, respectively. After drying, the samples were imaged with a JEOL 1400 EX transmission electron microscope (TEM). The experiment was done in duplicate.Cell viability assay
The neuroblastoma cell lines SH-SY5Y (ATCC) were grown in 25 cm2 flasks at 37 °C with 5% CO2 in advanced DMEM/F-12 medium (Sigma-Aldrich), supplemented with 10% fetal bovine serum (Invitrogen); for cytotoxicity assay, cells were seeded at a density of 1 × 104 in triplicate per sample in a 96 well microplate 24 h before the treatment. The fullerenemalonates were incubated for 24 h at the corresponding concentration for their ability to inhibit Aβ42 aggregation, 13 μM. Likewise, the vehicles of each fullerenemalonate other than water (acetonitrile and ethanol) were assessed to validate its toxicity. The cell viability assay was evaluated using Thiazolyl Blue Tetrazolium Bromide (MTT, Sigma-Aldrich); based on the conversion of MTT to water-insoluble MTT-formazan of dark blue color by the mitochondrial dehydrogenases of living cells. The absorbance of formazan was measured at a wavelength of 570 nm in a plate reader, iMark Biorad. For a cytotoxic concentration (CC50) the fullerenemalonates were incubated at different concentrations followed by a cell viability assay. The CC50 was determined from the curves obtained by fitting the average absorbance values in three independent experiments in the 10–80 μM fullerenemalonate concentration range.In silico experiments
The topology, pdb file, for the Aβ42 molecule was taken from Crescenzi O. et al.;48 the derivative fullerene was obtained by editing a pdb topology file of trisadduct with a diethyl malonyl substituents,49 and further optimized using the Automated Topology Builder.50 The atomistic force field used was CHARMM3651 together with the TIP3P model for water52 modified53 for use with this specific force field. Four systems were considered: one simulation of Aβ42 chains in vacuo, two of Aβ42 in water at two different temperatures and the last one was composed of Aβ42 chains with trisadducts of fullerene, in water. The amount of water added was enough to mimic the experimental value of the water density under thermodynamic conditions of pressure and temperature. In the case of the ternary system, the concentration of Aβ42 and fullerene was kept low, close to the experimental values, preventing an excess of water molecules. The details of the simulation boxes and thermodynamic conditions are shown in Table 1. The pure system and the binary systems were used to set the stability of the peptide structure when we use the force field CHARMM36. The initial boxes were prepared in an ensemble with a constant number of particles, constant pressure and constant temperature (NPT), using a Berendsen thermostat and barostat; the conditions were then changed to use an NVT ensemble, where V, the volume of the simulation box, is kept fixed, using a Nose–Hoover thermostat and turning off the barostat, setting the volume of the box to the average value obtained in the equilibrated NPT simulation. After equilibration was reached (around 100 ns), the simulations were sampled for 10 ns. The Verlet algorithm was used to integrate the movement equations with a time step of 0.001 ps. Long-range electrostatic interactions were calculated using the Particle Mesh Ewald method with 1.2 nm as the cut-off. The van der Waals interactions were calculated using a cut-off equal to 1.2 nm. The coupling times of temperature and pressure were fixed to 2.0 ps.
| System | Box type | Box volume (nm3) | Temperature (K) | Pressure (bar) | N water | N Aβ | N fullerene |
|---|---|---|---|---|---|---|---|
| (1) Pure in vacuo | Rhombic dodecahedron | 422.9 | 310.15 | 1 | 0 | 64 | |
| (2) In water | Rhombic dodecahedron | 11941.5 |
310.15 | 1 | 389005 |
64 | |
| (3) In water | Rhombic dodecahedron | 12214.6 |
333.15 | 1 | 389005 |
64 | |
| (4) Ternary | Cubic | 679.34 | 310.15 | 1 | 21250 |
8 | 6 |
Results
Aβ polymer formation
The study of Aβ aggregation inhibition requires an assay with Aβ monomers that interact with molecules that prevent further aggregation. To validate that Aβ monomers without pre-aggregated formation were appropriate for evaluating the adducts of C60, we used HFIP that breaks the beta sheet structures, preventing Aβ aggregation. The monomer and fibril formation were observed by western blot. The Aβ monomer without pre-aggregates is shown in Fig. 2A with a single band between 2 and 4 kDa, and aggregates between 40 and 160 kDa were formed under the same conditions except for HFIP treatment, demonstrating the requirements of the treatment. Moreover, we evaluated fibril formation during 24 h. Following 3 h of polymerization, low and high molecular weight polymers were seen as well as a reduction in the monomer at 6 h, validating the efficacy of the assay (Fig. 2B).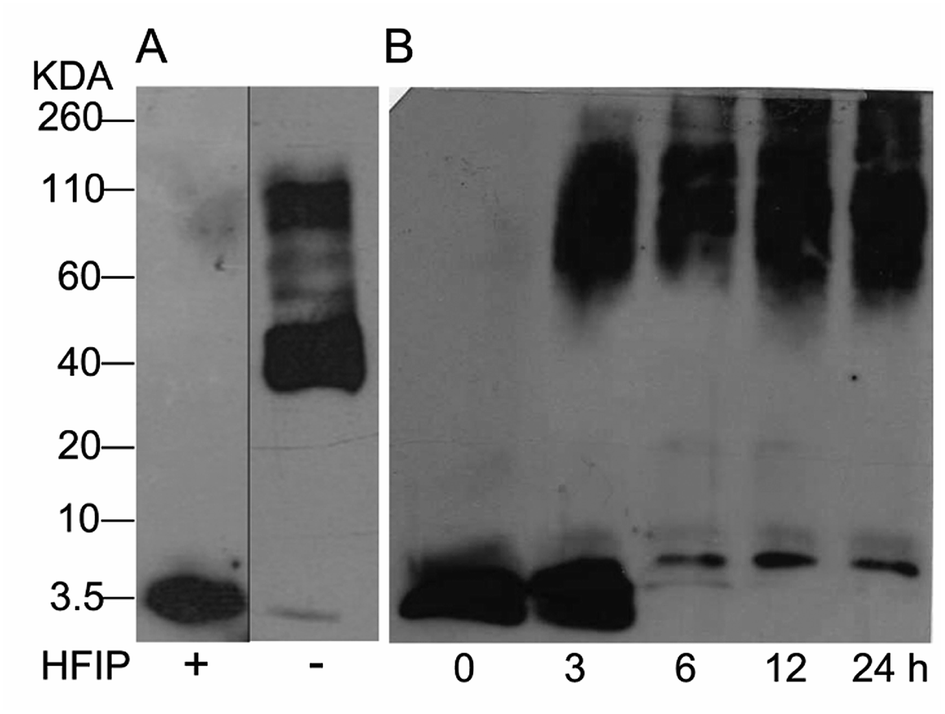 | ||
| Fig. 2 Western blot analysis from Aβ42 monomers resuspended or not with HFIP, panel (A). Aβ42 polymerization assay for 24 h at 37 °C (B). | ||
Fullerenemalonates inhibit amyloid β peptide aggregation
The inhibition of Aβ aggregation by fullerene derivatives has been previously demonstrated; however, it has shown solubility complications in water and high toxicity in cells. In this study, we synthesized adducts of C60 with one to three diethylmalonate substituents and their corresponding sodium salts to increase their biocompatibility and capacity to inhibit Aβ aggregation, evaluated by a ThT fluorescence assay. In Fig. 3A, the normalized values of the fluorescence signal of the Aβ aggregates showed significantly lower aggregation of Aβ in the presence of eight different fullerenemalonates compared to the control, in three independent experiments. The highest Aβ aggregation inhibition was shown by both isomeric mixtures of bisadducts, C62(COONa)4 and C3-symmetrical trisadduct (C3-C63(COONa)6), with 97% and with 80%, respectively (Fig. 3B). To confirm these results, the polymerization assay was analyzed by TEM, showing scattered fibrils (Fig. 4A). In the presence of either C62(COONa)4 or C3-C63(COONa)6 (Fig. 4B and C, respectively) Aβ fibrils were not found. These data indicate that fullerenemalonates inhibit and/or delay Aβ aggregation. The functionalization of C60 with two or three disodium malonate substituents increased the efficacy compared to monoaddition.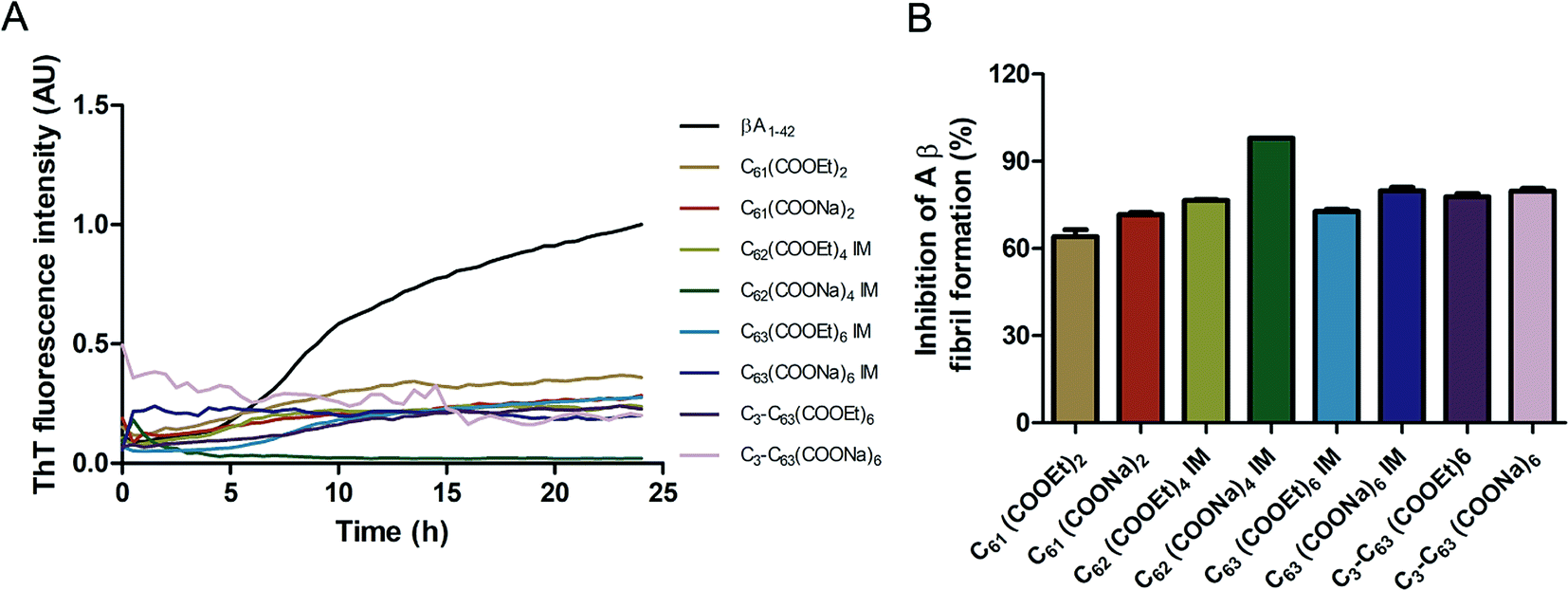 | ||
| Fig. 3 Fullerenemalonates inhibit amyloid β peptide aggregation, analysed by ThT assay during 24 h (A), comparison at 24 h as a percentage of inhibition (B). | ||
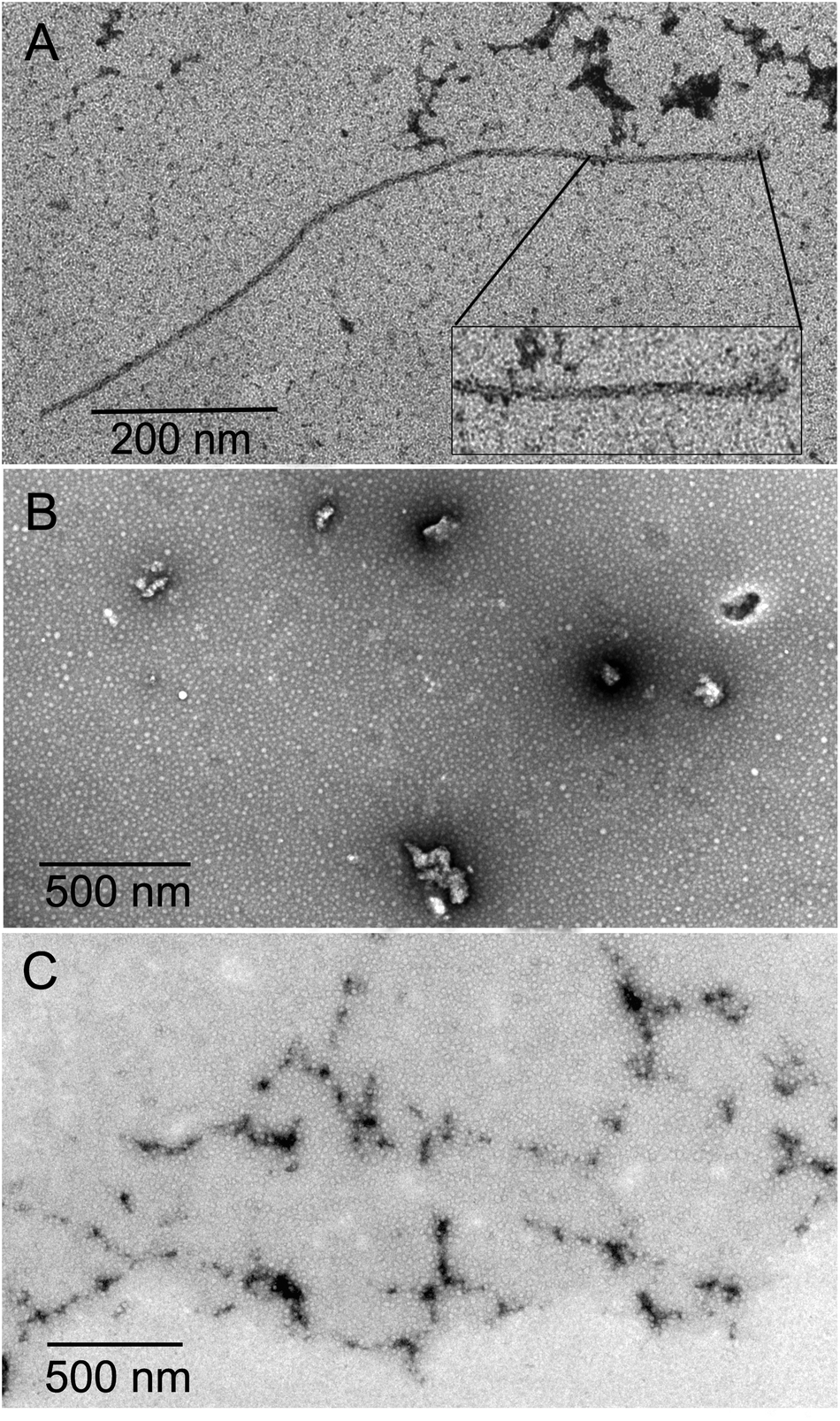 | ||
| Fig. 4 Aβ polymerization assay analysed by TEM. (A) Representative micrograph of identified Aβ polymers. Aβ polymerization assay in the presence of either C62(COONa)4 or C3-C63(COONa)6 (B and C, respectively). | ||
Inhibitory activity of the fullerenemalonates by IC50
To evaluate the Aβ anti-aggregatory capacity of C62(COONa)4, the most efficient inhibitory fullerene, we determined the IC50 value at 24 h by ThT assay, in the concentration range from 2.5 to 13 μM at a fixed peptide concentration of 20 μM. The relative fluorescence spectra showed that C62(COONa)4 (Fig. 5) inhibits the process of Aβ aggregation in a dose-dependent manner and the concentration to inhibit 50% of Aβ fibril formation determined by IC50 value is equal to 6.7 μM.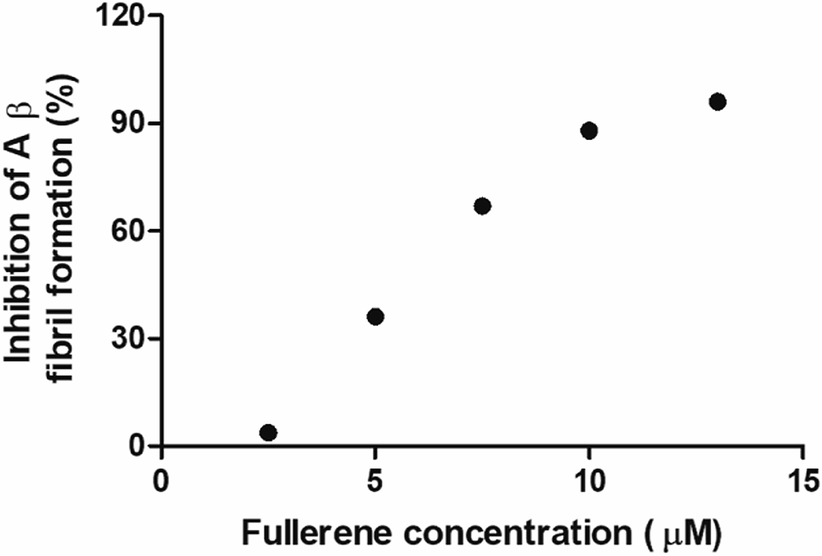 | ||
| Fig. 5 C62(COONa)4 inhibitory activity on Aβ fibril formation by IC50. | ||
Biocompatibility of fullerenemalonates
To evaluate the cytotoxic effect of the fullerenemalonates, we used the SH-SY5Y cell line that is used as a model for neurodegenerative diseases including AD;54 they can be differentiated from a dominantly cholinergic phenotype suitable for AD studies55 and they have been tested for Aβ toxicity in a large number of studies.56–58 The cells were incubated for 24 h in the presence of fullerenemalonates at 13 μM, corresponding to an Aβ fibril inhibition concentration. The viability of the cells was evaluated by MTT assay. The values normalized to control showed the highest cell viability for fullerenemalonates bearing two (C62(COONa)4) or three (C63(COONa)6 and C3-C63(COONa)6) disodium malonyl groups (Table 2). Likewise, disodium fullerenemalonates demonstrated higher solubility compared to diethyl fullerenemalonates, indicating that this molecular modification is important for biocompatibility. To determine the cytotoxic concentration of C62(COONa)4 and C3-C63(COONa)6 to reduce cell viability by 50%, CC50 was performed (Fig. 6). The dependence of cell viability on fullerene derivative concentration values: CC50: 38.8 μM and 69.6 μM for C62(COONa)4 and C3-C63(COONa)6, respectively, confirmed that the toxic concentrations for these molecules are 7 and 12 times higher than the required concentration for Aβ aggregation inhibition.| C60 fullerene adducts (13 μM) | Cell survival rate (%) |
|---|---|
| C3-C63(COONa)6 | 99.34 ± 0.22 |
| C63(COONa)6 (isomeric mixture) | 96.89 ± 0.22 |
| C62(COONa)4 (isomeric mixture) | 91.17 ± 0.44 |
| C3-C63(COOEt)6 | 78.59 ± 0.22 |
| C63(COOEt)6 (isomeric mixture) | 66.43 ± 0.22 |
| C62(COOEt)4 (isomeric mixture) | 66.07 ± 0.08 |
| C61(COOEt)2 | 65.65 ± 0.29 |
| C61(COONa)2 | 64.34 ± 0.30 |
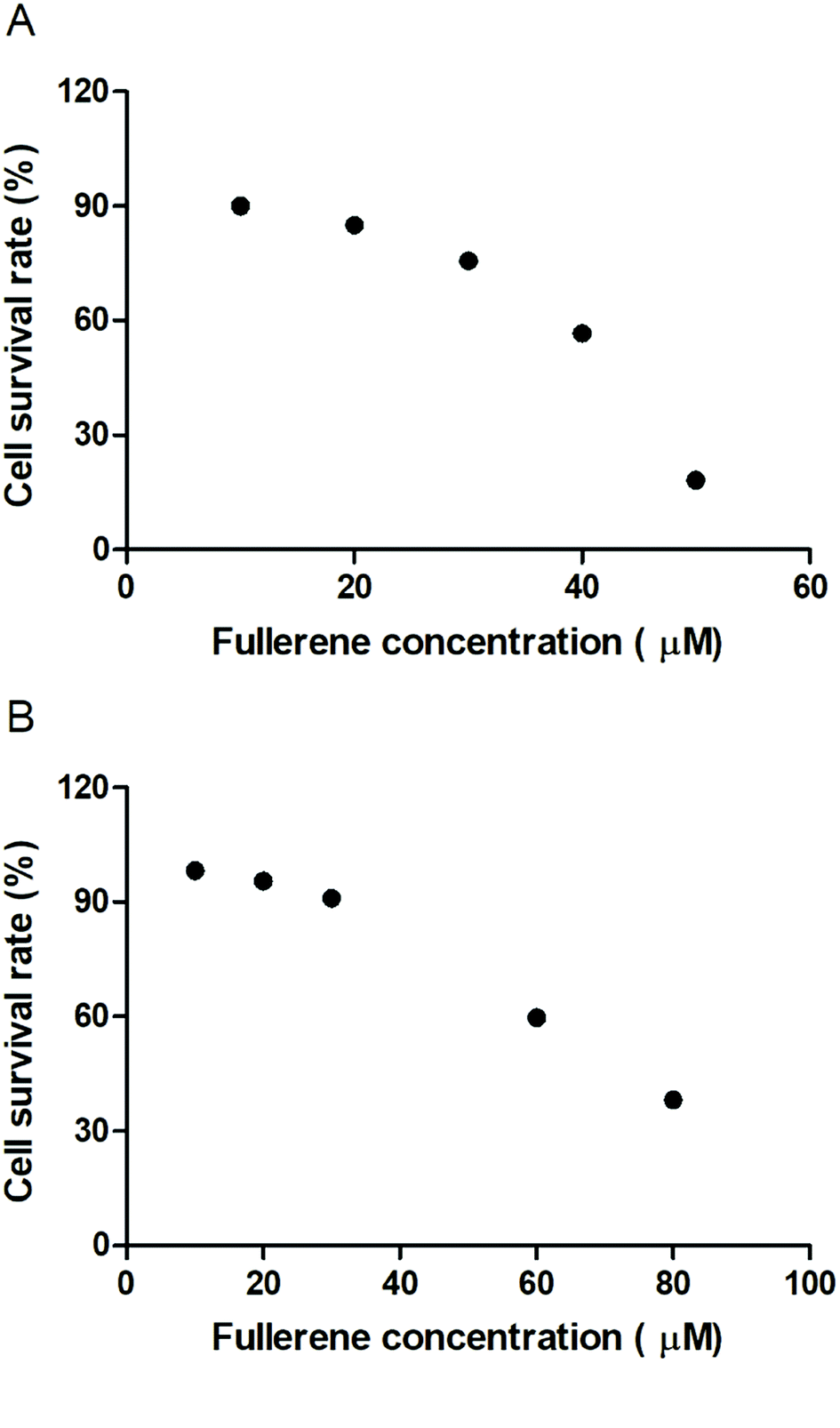 | ||
| Fig. 6 Cytotoxic concentration of C62(COONa)4 (A) and C3-C63(COONa)6 (B) by CC50. | ||
In silico interaction of trisadduct with Aβ42
The in vitro inhibition of the Aβ42 fibril formation demonstrated an interaction between the fullerene molecules and Aβ42. To propose a mechanism for this effect, the only isolated fullerene, C3-symmetrical trisadduct (C3-C63(COONa)6), was analysed in silico in the presence of Aβ42 and water to determine the possible interactions leading to inhibition of Aβ42 fibril formation. Herein atomistic molecular dynamics simulations of the ternary system were performed and the GROMACS 5.1.4 software package59 was used to generate the trajectory files. In Fig. 7 a snapshot of the ternary system can be found; this figure was prepared using the VMD software package60 using different colors for different residues; water molecules are not shown for clarity. Radial distribution functions (rdf) were calculated between every residue from Aβ42, analysing the center of the mass corresponding to each aminoacid together with that of C3-C63(COONa)6. The analysis of the data indicated that residue 1D shows a high pick at 0.83 nm; residue 2A at 0.82 nm; residue 4F has the highest pick at 0.86 nm and a second one at 1.05 nm; 5R has a pick at 0.86 nm, in the same position; residue 6H shows its highest pick at 0.84 nm and a second at 1.16 nm; 8S at 0.88 nm; 12V at 0.82 nm; and finally, residue 15Q shows its highest pick at 0.81 nm. A representative plot of the 5R residue is shown in Fig. 8A. These data suggest a remarkable structure at very short distance (less than 1 nm) between these residues and the fullerene trisadduct, probably binding to the carboxylate group. On average, the fullerene molecules kept their positions during the simulation time. For all the other residues, the picks had an rdf signal lower than 20. The interactions between the residue and the carboxylate group were assessed; the rdf between the center of mass of the two oxygen atoms in the carboxylate group and the center of mass of each residue were also calculated. The analysis of these results indicated that the residues where the rdf was higher than 15 were the 1D, 2A, 4F and 5R residues. Only the plot of the 1D residue is shown in Fig. 8B. 6H, 8S, 12V, 15Q, and 16K residues presented rdf values lower than 20, but they were still well-defined structures. The number of hydrogen bonds and the hydrogen bond distribution as a function of donor–acceptor distance between them and the residues, during the simulation time, were also calculated. The residues that showed hydrogen bonds were 1D, 5R, and 16K. In Fig. 9A and B we show results of the 5R and 16K residues, respectively. 2A and 15Q residues contain a weaker structure of hydrogen bonds, so that they are not retained during the whole simulation time. The minimal distances between each residue and the fullerenemalonate adduct were calculated using the center of mass as reference, and the systems with the shortest distances were the 1D, 5R, and 16K residues. The results for 16K are shown in Fig. 9C. The other residues that retained shorter distances lower than 0.35 nm were 2A, 3E, 4F, 6H, 8S, 11E, 12V, 14H, 15H, 32 G and 35V. These results demonstrated which residues contain strong interactions with C3-C63(COONa)6.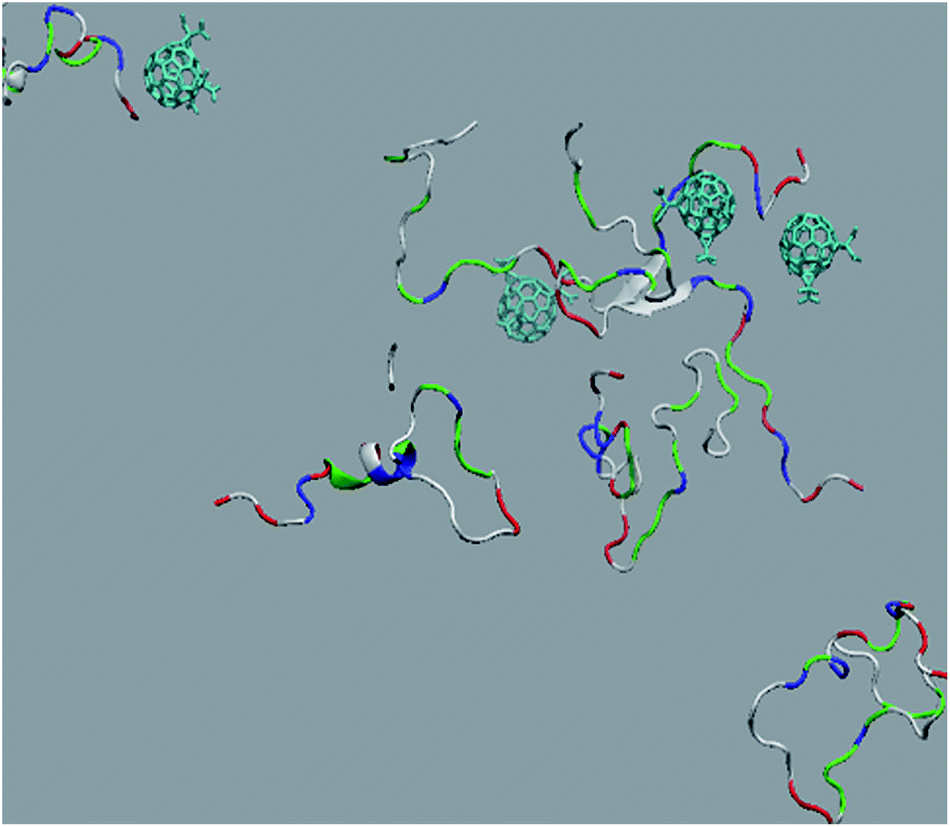 | ||
| Fig. 7 Snapshot of an equilibrated configuration of the ternary system: fullerene trisadduct in blue, Aβ42 is represented using different colours for different residues, water molecules are not shown for clarity. | ||
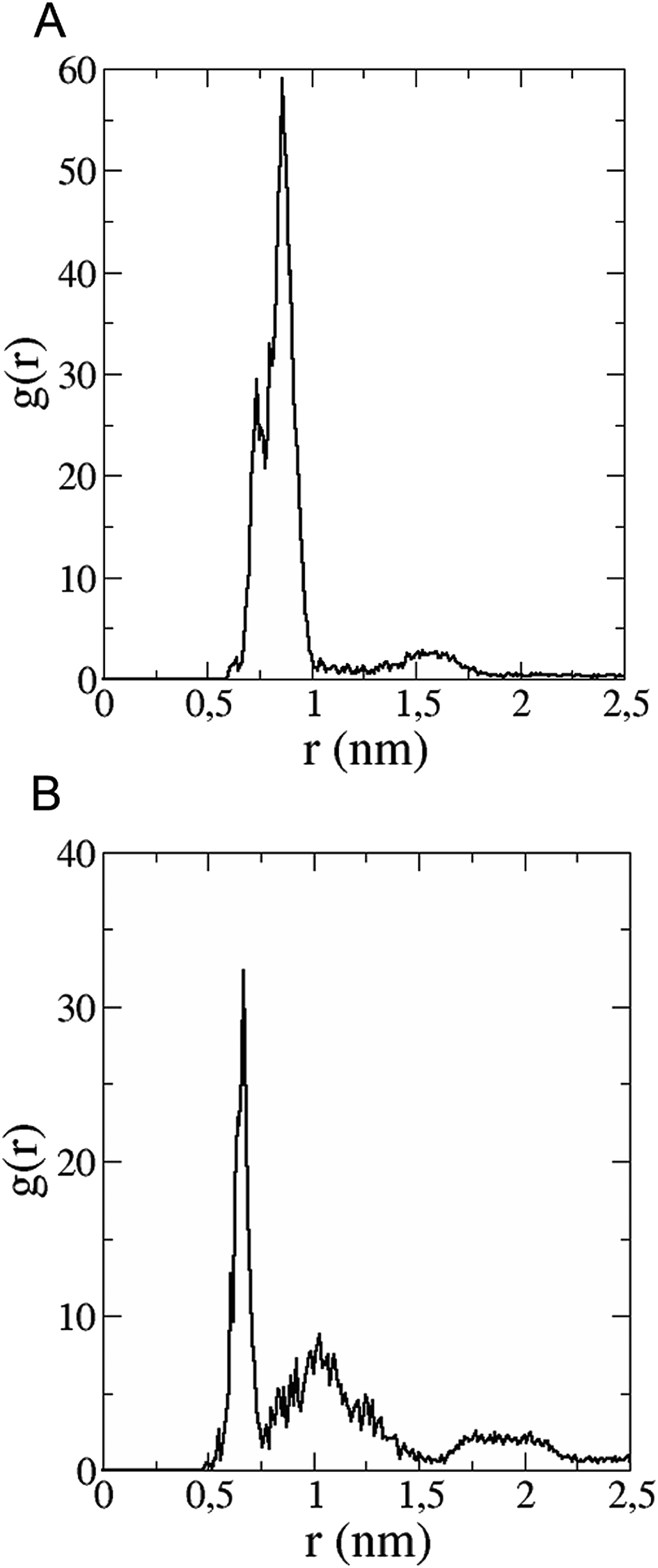 | ||
| Fig. 8 (A) Radial distribution function versus distance of 5R residue (Aβ42) between centres of mass of residue and fullerene trisadduct. (B) Radial distribution function as a function of the distance between the centre of mass of 1D residue and the two oxygen atoms in the COO-group. | ||
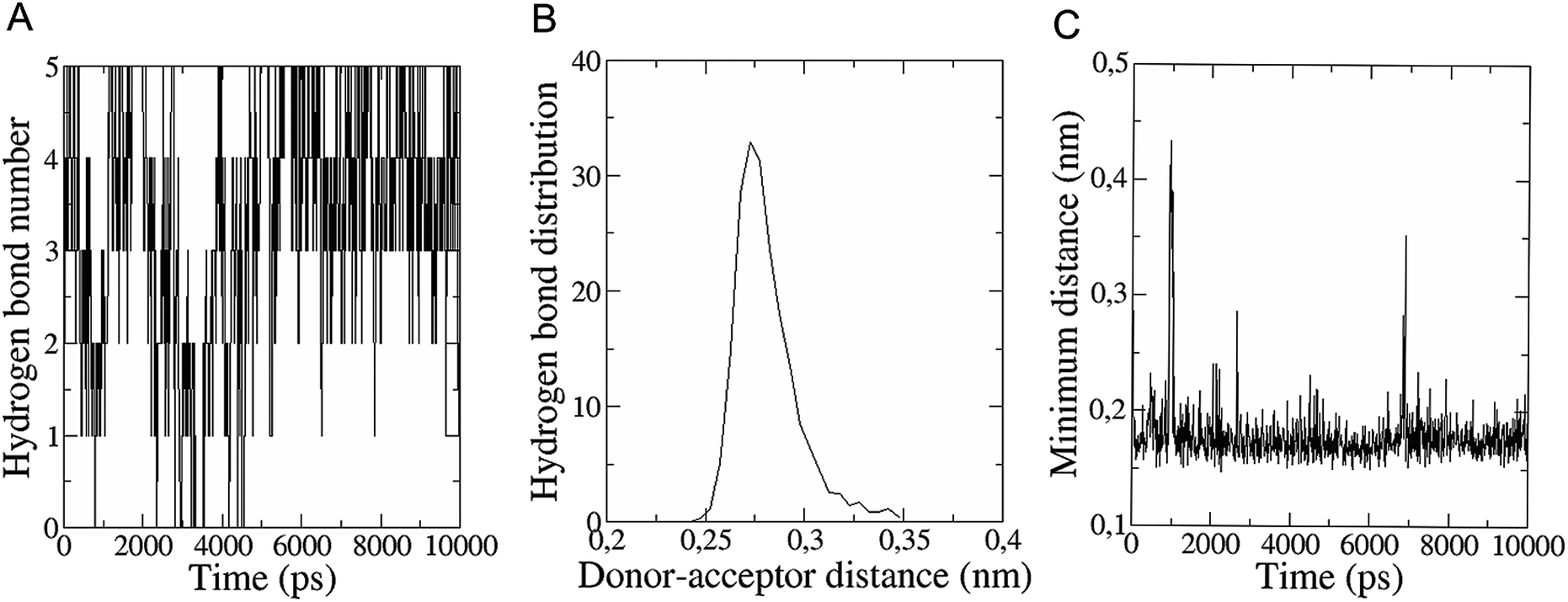 | ||
| Fig. 9 (A) Number of hydrogen bonds during the simulation time between 5R residue (Aβ42) and the fullerene trisadduct. (B) Hydrogen bond distribution as a function of time calculated for the 16K residue. (C) Minimal distance during the simulation time between the 16K residue and the fullerene trisadduct using the centres of mass as reference. | ||
Discussion
In this study, we demonstrated the capacity of fullerenemalonates to inhibit Aβ fibril formation. Our polymerization assay contains mostly monomers at the starting point, which confirms that the interaction of the fullerenes is not directly with pre-aggregates. This is important in terms of the molecular dynamics, since we evaluated the interaction of C3-C63(COONa)6 specifically with the Aβ monomer in silico. Several experimental and computational studies have demonstrated that the type of substituent inserted on the fullerene surface has a remarkable influence on their anti-amyloid activity. For example, in vitro experiments showed that 1,2-(dimethoxymethano) fullerene strongly inhibits Aβ peptide aggregation in the early stages (IC50 ∼ 9 μM, Aβ42 concentration 20 μM).16 Another group has studied the anti-amyloid activity of the sodium salt of the fullerene polycarboxylic derivative C60Cl(C6H4CH2COONa), the sodium fullerenolate and complexes of fullerene with polyvinylpyrrolidone and revealed the existence of a strong anti-amyloid activity in Aβ42, concluding that the latter two had the most effective Aβ inhibitory effect in both Aβ42 and muscle amyloid X-protein.29,61,62 Recently, Bednarikova et al. demonstrated through in vitro and in silico experiments that fullerenol, fullerene C60 modified with 16 OH groups (C60(OH)16), inhibits the fibrillization of Aβ1–40 in a dose-dependent manner (IC50 ∼ 32 μg ml−1, Aβ1–40 concentration 10 μM) and the inhibition of β-sheet formation results from the strong electrostatic interactions of the fullerenol OH groups with the polar, negatively charged amino acids.28 Zhou et al. performed multiple all-atom explicit solvent molecular dynamics simulations to study the effect of fullerene substituents and concluded that functionalization with a dimethoxymethane group on the fullerene surface retarded the rotation of the fullerene, thus enhancing the binding stability of the 1,2-(dimethoxymethano)fullerene.17 Xie et al. found by molecular dynamics simulations that the contact surface area between the fullerenes and the Aβ16–22 octamers is an important factor that affects the Aβ–fullerene interaction and a large contact surface area usually implies strong interactions.63The results obtained in this study show that all fullerenemalonates inhibit Aβ42 aggregation and their activity depends highly on the number and the nature of the substituents attached to the fullerene surface. In addition, an in silico approach demonstrated that the inhibition of Aβ42 fibrillization by C3-C63(COONa)6 results from strong electrostatic interactions and hydrogen bonding of the fullerenemalonate carboxylate groups predominantly with amino acid groups (residues 1D, 2A, 4F and 5R) and residues 1D, 5R, and 16K, respectively. The higher anti-aggregatory effect of C62(COONa)4 (98% at 24 h, IC50 6.7 μM, Aβ42 concentration 20 μM) may be explained by a balanced relationship between the number of organic addends and the contact surface area available compared to those of the monoadduct and trisadducts. Moreover, disodium fullerenemalonates exhibit higher anti-Aβ activity compared to diethyl fullerenemalonates, which suggests that the addends/surface area ratio, besides the nature of the addends, plays an important role in the anti-Aβ activity of the fullerene derivatives. This finding is consistent with previous studies, suggesting that the strong Aβ–fullerene derivative interaction, due to introduced addends and the contact area available in the fullerene, significantly weakens the Aβ–Aβ interaction and thus inhibits β-sheet formation.17,28,63
Likewise, the appended organic addends on the carbon cage in fullerenemalonates make them truly amphiphilic and induce a strong tendency to self-assemble in polar solvents to form stable solutions of nanoaggregates.25,43,64–68 Specifically, the self-assembly of sodium carboxylated fullerenes (C61(COONa)2 and C62(COONa)4) in aqueous solution produces solid spherical particles with an average hydrodynamic radius Rh ≈ 32 nm for C61(COONa)2 and hollow shells with mainly two different size scales of Rh ≈ 23 nm and Rh ≈ 104 nm for the isomeric mixture of bisadducts, C62(COONa)4.66 Therefore, it is not surprising that aqueous solutions of disodium fullerenemalonates (C61(COONa)2, C62(COONa)4 and C63(COONa)6) and other water-soluble fullerene derivatives evaluated as inhibitors of Aβ aggregation comprise nanoclusters rather than individual solvated molecules.16,24,25,29,30,61,62,69 Nevertheless, the results obtained in this study and other reports show that self-assembly in polar solvents to form stable nanoaggregates does not affect its anti-Aβ activity.
Regarding the biocompatibility of fullerenemalonates, our results showed that modification of the fullerene surface with diethyl malonyl groups causes higher cytotoxicity compared to those with disodium malonyl groups. Moreover, it was found that, excluding the monoadduct sodium salt (C61(COONa)2), disodium fullerenemalonates with two or three substituents were not toxic for the SH-SY5Y neuroblastoma cell line at the half maximal inhibitory concentration (IC50 6.7 μM). Likewise, it was found that the cytotoxicity is reduced as the number of disodium malonyl substituents attached to the fullerene surface and their attachment symmetry increase. The higher biocompatibility of C3-symmetrical triadduct (C3-C63(COONa)6, viability 99%) compared to those of monoadduct (C61(COONa)2, viability 64%) and the isomeric mixture of bisadducts (C62(COONa)4, viability 91%) and triadducts (C63(COONa)6, viability 97%) may be attributed to the combined effect of the decrease in hydrophobic surface due to hydrophilic addends attached to the fullerene core and the e,e,e-symmetrical addition pattern of C3-C63(COONa)6. This study agrees with the reported literature, which suggests that a higher abundance of hydrophilic addends on the fullerene surface and a high-symmetry addition pattern, result in a decrease in cytotoxicity.29,70,71
Conclusions
In this study we have demonstrated that fullerenemalonates bearing 1 to 3 diethyl malonyl substituents and their corresponding sodium salts interact and effectively reduce Aβ fibril formation in vitro. The bisadduct salts (C62(COONa)4) and trisadduct (C63(COONa)6) inhibit 98 and 83% of Aβ aggregation, respectively. The 6.7 μM IC50 value of a C62(COONa)4 mixture confirmed one of the highest anti-amyloid capacities that has been reported. The anti-aggregatory effect of the bisadduct salts, C62(COONa)4, is mostly attributed to the balance between the hydrophobic surface and the number of substituents bound to the fullerene, promoting stability in the interaction with the Aβ peptide. The sodium salts C62(COONa)4, C63(COONa)6 and C3-C63(COONa)6 showed low toxicity in neuroblastoma SH-SY5Y cell viability, suggesting that these molecules are highly biocompatible at concentrations which are able to effectively inhibit Aβ aggregation. The lowest toxicity presented in the trisadduct salt, C3-C63(COONa)6, is associated with the combined effect of the reduction in the fullerene hydrophobic surface through the addition of hydrophilic substituents and the fullerene symmetry. The effective anti-amyloid activity and low toxicity of the bisadduct isomeric mixture (C62(COONa)4) and trisadduct (C63(COONa)6) could be promising candidates for further animal studies, and potential therapeutic molecules for the treatment of Alzheimer's disease.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial supports provided by Mexico's National Science and Technology Board (CONACyT) for the CATEDRA CONACYT no. 953 of M.-H. M. G.-S. F. and P. Y. L.-C. were supported by CONACYT grants CB-255224 and CB-2012/182003 respectively. And project CB 2016-287067-F. The Universidad de Guanajuato, through the Convocatoria Institucional, project no. 262. S. F.-G. thanks to Professor Müller-Plathe for computational time in his laboratory during her sabbatical stay. We would like to thank Dr Aarón Rojas Aguilar (Department of Chemistry at CINVESTAV) for help with the Mass Spectrometry.References
- J. Hardy, Trends Neurosci., 1997, 20, 154–159 CrossRef CAS PubMed.
- L. Mucke, E. Masliah, G. Q. Yu, M. Mallory, E. M. Rockenstein, G. Tatsuno, K. Hu, D. Kholodenko, K. Johnson-Wood and L. McConlogue, J. Neurosci., 2000, 20, 4050–4058 CrossRef CAS PubMed.
- W. L. Klein, G. A. Krafft and C. E. Finch, Trends Neurosci., 2001, 24, 219–224 CrossRef CAS PubMed.
- J. T. Jarrett and P. T. Lansbury Jr, Cell, 1993, 73, 1055–1058 CrossRef CAS PubMed.
- S. Kumar and J. Walter, Aging, 2011, 3, 803–812 Search PubMed.
- J. M. Orgogozo, S. Gilman, J. F. Dartigues, B. Laurent, M. Puel, L. C. Kirby, P. Jouanny, B. Dubois, L. Eisner, S. Flitman, B. F. Michel, M. Boada, A. Frank and C. Hock, Neurology, 2003, 61, 46–54 CrossRef CAS PubMed.
- E. Uro-Coste, G. Russano de Paiva, C. Guilbeau-Frugier, N. Sastre, P. J. Ousset, N. A. da Silva, V. Lavialle-Guillotreau, B. Vellas and M. B. Delisle, Clin. Neuropathol., 2010, 29, 209–216 CrossRef CAS PubMed.
- S. S. Sisodia and P. H. St George-Hyslop, Nat. Rev. Neurosci., 2002, 3, 281–290 CrossRef CAS PubMed.
- S. Fuse, K. Matsumura, Y. Fujita, H. Sugimoto and T. Takahashi, Eur. J. Med. Chem., 2014, 85, 228–234 CrossRef CAS PubMed.
- Y. Nakagami, S. Nishimura, T. Murasugi, I. Kaneko, M. Meguro, S. Marumoto, H. Kogen, K. Koyama and T. Oda, Br. J. Pharmacol., 2002, 137, 676–682 CrossRef CAS PubMed.
- E. O'Hare, D. I. Scopes, J. M. Treherne, K. Norwood, D. Spanswick and E. M. Kim, Behav. Brain Res., 2010, 210, 32–37 CrossRef PubMed.
- M. H. Baig, K. Ahmad, G. Rabbani and I. Choi, Front. Aging Neurosci., 2018, 10, 21 CrossRef PubMed.
- M. H. Viet, K. Siposova, Z. Bednarikova, A. Antosova, T. T. Nguyen, Z. Gazova and M. S. Li, J. Phys. Chem. B, 2015, 119, 5145–5155 CrossRef CAS PubMed.
- K. L. Sciarretta, D. J. Gordon and S. C. Meredith, Methods Enzymol., 2006, 413, 273–312 CAS.
- A. Bellova, E. Bystrenova, M. Koneracka, P. Kopcansky, F. Valle, N. Tomasovicova, M. Timko, J. Bagelova, F. Biscarini and Z. Gazova, Nanotechnology, 2010, 21, 065103 CrossRef PubMed.
- J. E. Kim and M. Lee, Biochem. Biophys. Res. Commun., 2003, 303, 576–579 CrossRef CAS PubMed.
- X. Zhou, W. Xi, Y. Luo, S. Cao and G. Wei, J. Phys. Chem. B, 2014, 118, 6733–6741 CrossRef CAS PubMed.
- Z. Yang, C. Ge, J. Liu, Y. Chong, Z. Gu, C. A. Jimenez-Cruz, Z. Chai and R. Zhou, Nanoscale, 2015, 7, 18725–18737 RSC.
- S. H. Friedman, D. L. DeCamp, R. P. Sijbesma, G. Srdanov, F. Wudl and G. L. Kenyon, J. Am. Chem. Soc., 1993, 115, 6506–6509 CrossRef CAS.
- L. Xiao, H. Aoshima, Y. Saitoh and N. Miwa, Free Radic. Biol. Med., 2011, 51, 1376–1389 CrossRef CAS PubMed.
- J. Meng, X. Liang, X. Chen and Y. Zhao, Integr. Biol., 2013, 5, 43–47 RSC.
- Z. Markovic and V. Trajkovic, Biomaterials, 2008, 29, 3561–3573 CrossRef CAS PubMed.
- S. Tanimoto, S. Sakai, S. Matsumura, D. Takahashi and K. Toshima, Chem. Commun., 2008, 5767–5769, 10.1039/b811726h.
- Y. Ishida, T. Fujii, K. Oka, D. Takahashi and K. Toshima, Chem.–Asian J., 2011, 6, 2312–2315 CrossRef CAS PubMed.
- N. Hasunuma, M. Kawakami, H. Hiramatsu and T. Nakabayashi, RSC Adv., 2018, 8, 17847–17853 RSC.
- Z. Du, N. Gao, X. Wang, J. Ren and X. Qu, Small, 2018, 14, 1801852 CrossRef PubMed.
- M. Raoof, Y. Mackeyev, M. A. Cheney, L. J. Wilson and S. A. Curley, Biomaterials, 2012, 33, 2952–2960 CrossRef CAS PubMed.
- Z. Bednarikova, P. D. Huy, M. M. Mocanu, D. Fedunova, M. S. Li and Z. Gazova, Phys. Chem. Chem. Phys., 2016, 18, 18855–18867 RSC.
- A. G. Bobylev, A. B. Kornev, L. G. Bobyleva, M. D. Shpagina, I. S. Fadeeva, R. S. Fadeev, D. G. Deryabin, J. Balzarini, P. A. Troshin and Z. A. Podlubnaya, Org. Biomol. Chem., 2011, 9, 5714–5719 RSC.
- Y. Ishida, S. Tanimoto, D. Takahashi and K. Toshima, MedChemComm, 2010, 1, 212–215 RSC.
- D. N. Johnson-Lyles, K. Peifley, S. Lockett, B. W. Neun, M. Hansen, J. Clogston, S. T. Stern and S. E. McNeil, Toxicol. Appl. Pharmacol., 2010, 248, 249–258 CrossRef CAS PubMed.
- Y. Su, J. Y. Xu, P. Shen, J. Li, L. Wang, Q. Li, W. Li, G. T. Xu, C. Fan and Q. Huang, Toxicology, 2010, 269, 155–159 CrossRef CAS PubMed.
- A. B. Kraemer, G. M. Parfitt, D. D. S. Acosta, G. E. Bruch, M. F. Cordeiro, L. F. Marins, J. Ventura-Lima, J. M. Monserrat and D. M. Barros, Toxicol. Appl. Pharmacol., 2018, 338, 197–203 CrossRef CAS PubMed.
- C. Bingel, Chem. Ber., 1993, 126, 1957–1959 CrossRef CAS.
- W. E. Billups, J. Am. Chem. Soc., 2005, 127, 11876–11876 CrossRef CAS.
- X. Camps and A. Hirsch, J. Chem. Soc., Perkin Trans. 1, 1997, 1595–1596, 10.1039/a702055d.
- E. Straface, B. Natalini, D. Monti, C. Franceschi, G. Schettini, M. Bisaglia, C. Fumelli, C. Pincelli, R. Pellicciari and W. Malorni, FEBS Lett., 1999, 454, 335–340 CrossRef CAS PubMed.
- A. Hirsch, I. Lamparth and H. R. Karfunkel, Angew. Chem., Int. Ed. Engl., 1994, 33, 437–438 CrossRef.
- F. Djojo, A. Hirsch and S. Grimme, Eur. J. Org. Chem., 1999, 1999, 3027–3039 CrossRef.
- A. Hirsch, I. Lamparth, T. Groesser and H. R. Karfunkel, J. Am. Chem. Soc., 1994, 116, 9385–9386 CrossRef CAS.
- M. Martínez-Herrera, P. Amador and A. Rojas, J. Phys. Chem. C, 2011, 115, 20849–20855 CrossRef.
- Q. Lu, D. I. Schuster and S. R. Wilson, J. Org. Chem., 1996, 61, 4764–4768 CrossRef CAS PubMed.
- D. M. Guldi, H. Hungerbuehler, E. Janata and K. D. Asmus, J. Phys. Chem., 1993, 97, 11258–11264 CrossRef CAS.
- B. Zhang-Haagen, R. Biehl, L. Nagel-Steger, A. Radulescu, D. Richter and D. Willbold, PLoS One, 2016, 11, e0150267 CrossRef PubMed.
- W. B. Stine Jr, K. N. Dahlgren, G. A. Krafft and M. J. LaDu, J. Biol. Chem., 2003, 278, 11612–11622 CrossRef PubMed.
- W. B. Stine, L. Jungbauer, C. Yu and M. J. LaDu, Methods Mol. Biol., 2011, 670, 13–32 CrossRef CAS PubMed.
- H. Naiki, K. Higuchi, M. Hosokawa and T. Takeda, Anal. Biochem., 1989, 177, 244–249 CrossRef CAS PubMed.
- O. Crescenzi, S. Tomaselli, R. Guerrini, S. Salvadori, A. M. D'Ursi, P. A. Temussi and D. Picone, Eur. J. Biochem., 2002, 269, 5642–5648 CrossRef CAS PubMed.
- E. F. Paulus and C. Bingel, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1995, 51, 143–146 CrossRef.
- A. K. Malde, L. Zuo, M. Breeze, M. Stroet, D. Poger, P. C. Nair, C. Oostenbrink and A. E. Mark, J. Chem. Theory Comput., 2011, 7, 4026–4037 CrossRef CAS PubMed.
- J. Huang and A. D. MacKerell Jr, J. Comput. Chem., 2013, 34, 2135–2145 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar and J. D. Madura, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiórkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef CAS PubMed.
- H. Xicoy, B. Wieringa and G. J. Martens, Mol. Neurodegener., 2017, 12, 10 CrossRef PubMed.
- A. Edsjo, E. Lavenius, H. Nilsson, J. C. Hoehner, P. Simonsson, L. A. Culp, T. Martinsson, C. Larsson and S. Pahlman, Lab. Invest., 2003, 83, 813–823 CrossRef PubMed.
- Z. Datki, A. Juhasz, M. Galfi, K. Soos, R. Papp, D. Zadori and B. Penke, Brain Res. Bull., 2003, 62, 223–229 CrossRef CAS PubMed.
- N. G. Milton, A. Chilumuri, E. Rocha-Ferreira, A. N. Nercessian and M. Ashioti, ACS Chem. Neurosci., 2012, 3, 706–719 CrossRef CAS PubMed.
- J. Krishtal, O. Bragina, K. Metsla, P. Palumaa and V. Tougu, PLoS One, 2017, 12, e0186636 CrossRef PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- A. G. Bobylev, L. G. Marsagishvili and Z. A. Podlubnaia, Biofizika, 2010, 55, 780–784 CAS.
- A. G. Bobylev, M. D. Shpagina, L. G. Bobyleva, A. D. Okuneva, L. B. Piotrovskii and Z. A. Podlubnaia, Biofizika, 2012, 57, 416–421 CAS.
- L. Xie, Y. Luo, D. Lin, W. Xi, X. Yang and G. Wei, Nanoscale, 2014, 6, 9752–9762 RSC.
- G. Angelini, P. De Maria, A. Fontana, M. Pierini, M. Maggini, F. Gasparrini and G. Zappia, Langmuir, 2001, 17, 6404–6407 CrossRef CAS.
- R. Partha, M. Lackey, A. Hirsch, S. W. Casscells and J. L. Conyers, J. Nanobiotechnol., 2007, 5, 6 CrossRef PubMed.
- S. Q. Zhou, J. Y. Ouyang, P. Golas, F. Wang and Y. Pan, J. Phys. Chem. B, 2005, 109, 19741–19747 CrossRef CAS PubMed.
- V. Georgakilas, F. Pellarini, M. Prato, D. M. Guldi, M. Melle-Franco and F. Zerbetto, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5075–5080 CrossRef CAS PubMed.
- F. Y. Hsieh, A. V. Zhilenkov, Voronov II, E. A. Khakina, D. V. Mischenko, P. A. Troshin and S. H. Hsu, ACS Appl. Mater. Interfaces, 2017, 9, 11482–11492 CrossRef CAS PubMed.
- Y. Y. Zha, B. Yang, M. L. Tang, Q. C. Guo, J. T. Chen, L. P. Wen and M. Wang, Int. J. Nanomed., 2012, 7, 3099–3109 Search PubMed.
- G. Andrievsky, V. Klochkov and L. Derevyanchenko, Fullerenes, Nanotubes, Carbon Nanostruct., 2005, 13, 363–376 CrossRef CAS.
- C. M. Sayes, J. D. Fortner, W. Guo, D. Lyon, A. M. Boyd, K. D. Ausman, Y. J. Tao, B. Sitharaman, L. J. Wilson, J. B. Hughes, J. L. West and V. L. Colvin, Nano Lett., 2004, 4, 1881–1887 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |