
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn electrochemical MIP sensor for selective detection of salbutamol based on a graphene/PEDOT:PSS modified screen printed carbon electrode†
Decha
Dechtrirat
 *a,
Bunyarithi
Sookcharoenpinyo
b,
Pongthep
Prajongtat
a,
Chakrit
Sriprachuabwong
ac,
Arsooth
Sanguankiat
d,
Adisorn
Tuantranont
c and
Supa
Hannongbua
b
*a,
Bunyarithi
Sookcharoenpinyo
b,
Pongthep
Prajongtat
a,
Chakrit
Sriprachuabwong
ac,
Arsooth
Sanguankiat
d,
Adisorn
Tuantranont
c and
Supa
Hannongbua
b
aDepartment of Materials Science, Faculty of Science, Kasetsart University, Bangkok, Thailand. E-mail: fscidcd@ku.ac.th
bDepartment of Chemistry, Faculty of Science, Kasetsart University, Bangkok, Thailand
cNational Electronics and Computer Technology Center (NECTEC), National Science and Technology Development Agency (NSTDA), Pathumthani, Thailand
dDepartment of Veterinary Public Health, Faculty of Veterinary Medicine, Kasetsart University, Kamphaeng Saen Campus, Nakhon Pathom, Thailand
First published on 2nd January 2018
Abstract
A sensitive electrochemical MIP sensor for salbutamol detection based on a graphene nanocomposite modified screen-printed carbon electrode was successfully demonstrated. Insertion of the graphene/PEDOT:PSS layer prior to the MIP was aimed at enhancing the sensitivity of the sensor. Selection of a functional monomer was carried out using 1H-NMR titration and a computational calculation was used to further investigate the template-monomer interactions. The MIP layer was constructed on top of the highly conductive nanocomposite by co-electropolymerization of 3-aminophenylboronic acid and o-phenylenediamine in the presence of salbutamol. Using differential pulse voltammetry under optimal conditions, a linear response in the range of 1 nM to 1.2 μM, with an exceptional detection limit of 100 pM (S/N = 3) was obtained. The established sensor exhibited good reproducibility, reusability and stability. Additionally, good selectivity in discriminating salbutamol from other structurally related compounds could also be realized. Determination of salbutamol in real swine meat and feed samples was also successfully carried out with good recovery results.
Introduction
Salbutamol is a selective β2 adrenergic receptor agonist, commonly used to treat airway obstruction in patients with bronchial asthma, emphysema, chronic obstructive pulmonary and other lung diseases.1 Besides its application in humans, salbutamol is also often misused in animals as a growth promoting agent because it can induce muscle protein accretion and reduce fat deposition, thereby improving the lean meat yield.2 However, this compound can accumulate in the animal tissues and persist in the meat products. Ingestion of those contaminated foods could then possibly cause acute toxic responses in humans, such as cardiac palpitation, low blood potassium and elevated blood sugar levels.3–5 This would pose severe risks to consumers, especially to patients with heart disease, renal failure or diabetes. Due to the concern of those possible adverse effects to human health, salbutamol has been forbidden from use as a feed additive in food-producing animals by the European Union, China and many other countries. However, the illegal use of this substance is still being found in many areas across the world. Therefore, it is of particular significance to establish rapid and accurate methods to monitor the illicit use of salbutamol in animal husbandry.Several analytical techniques have been demonstrated for the determination of salbutamol, such as electrophoresis,6 colorimetry,7 fluorescence,8 electrochemiluminescence,9 spectrophotometry10 and chromatography.11,12 Some did not show either sufficient limit of detection or satisfactory selectivity. The chromatographic means including LC/MS and LC/MS/MS are, on the other hand, able to determine salbutamol quantitatively at the trace level with high precision of mass identification. However, these techniques are rather time-consuming and require expensive instruments of considerably bulky size that are not practical for on-site applications.
Electrochemical sensors, in contrast, provide much lower cost and more field portability. Additionally, they also offer fast response, high sensitivity and straightforward analysis. Among various kinds of existing electrode materials, screen-printed carbon electrodes (SPCEs) have gained increasing interest because they offer several practical advantages, such as high potential for mass production with extremely low cost, exceptional reproducibility and a fully portable platform for on-site screening with low sample/reagent consumption.13 In recent years, several nanoscale materials, such as metal nanoparticles, graphene and carbon nanotubes have been extensively applied for electrode modification to further enhance the sensitivity of the SPCE-based sensors.14,15
Graphene is a 2D carbon material with single or several atomic layers of sp2 bonded carbon atoms in a honeycomb structure. It is one of the most widely reported materials used in electrochemical sensors owing to its high surface-to-volume ratio and its excellent electrical conductivity.16 Generally, graphene is likely to form agglomerates, thereby reducing its electron-transfer efficiency. To prevent such aggregation, exfoliation of graphite rod in conductive polymers (i.e. PANI, PEDOT:PSS) has been described.17,18 The graphene/polymer nanocomposites not only showed high dispersion stability, they also exhibited extraordinary electrochemical performances. Modification of screen-printed electrodes can normally be achieved by a simple drop casting technique. However, this conventional means often results in poor reproducibility and is not realistic for large scale production. Inkjet printing, on the other hand, has undergone widespread growth and become a promising technology for biosensor fabrication due to its simple and fast manufacturing process which will enable high volume production of low cost sensors with high precision and reproducibility.19
Molecularly imprinted polymers (MIPs) have received intense interest and been widely applied as a promising alternatives to bio-receptors in electrochemical sensors.20 MIPs could be simply prepared by a strategy called molecular imprinting process, which is carried out by polymerization of functional monomers in the presence of a target molecule as a templating agent. Once the polymeric matrix has formed, the template molecule is removed, leaving behind the imprinted cavities. The binding pockets created are complementary to the target in size, molecular shape and orientation of functionalities, and are responsible for specific recognition of the resulting synthetic MIPs. To obtain a reproducible and fast sensor response, a homogeneous and thin MIP film is preferentially required. Electropolymerization, which offers the ability to grow a homogeneous film directly on the electrode surface and to control precisely the thickness of the film, is therefore considered as a method of choice for preparing electrochemical MIP sensors.21,22
Within this work, a MIP based screen-printed electrochemical sensor for selective determination of salbutamol was demonstrated. As illustrated in Scheme 1, prior to the preparation of the MIP film, a highly conductive graphene/PEDOT:PSS nanocomposite was inkjet printed onto the working area of the SPCE to further enhance the sensitivity of the established sensor. Afterwards, the imprinted film was grown on top by co-electropolymerization of 3-aminophenylboronic acid and o-phenylenediamine in the presence of salbutamol. The MIP based sensor was thus attained after template removal. Since the imprinted layer could selectively capture the target analyte from complex matrixes, no additional sample pretreatment steps were needed. This would significantly simplify the laborious process, lower the screening cost and shorten the analysis time. Although the inkjet-printed graphene/PEDOT:PSS modified SPCE has already been reported for direct electrochemical detection of salbutamol,23 this is the first time we have combined further this flexible and sensitive nanocomposite based printed electrode with a salbutamol-selective MIP layer. Since salbutamol itself exhibits only a fairly poor electrochemical activity, attaining an ultratrace level analysis of this compound is especially difficult. To overcome such a limitation, an indirect approach, with the use of a highly electroactive species as a signal probe and a non-conductive MIP film as a controlled gate, was proposed. Upon rebinding of salbutamol to the imprinted cavities/gates, a transfer of the signal probe to the electrode surface is partly blocked, thereby reducing the current responses (Scheme S1†).
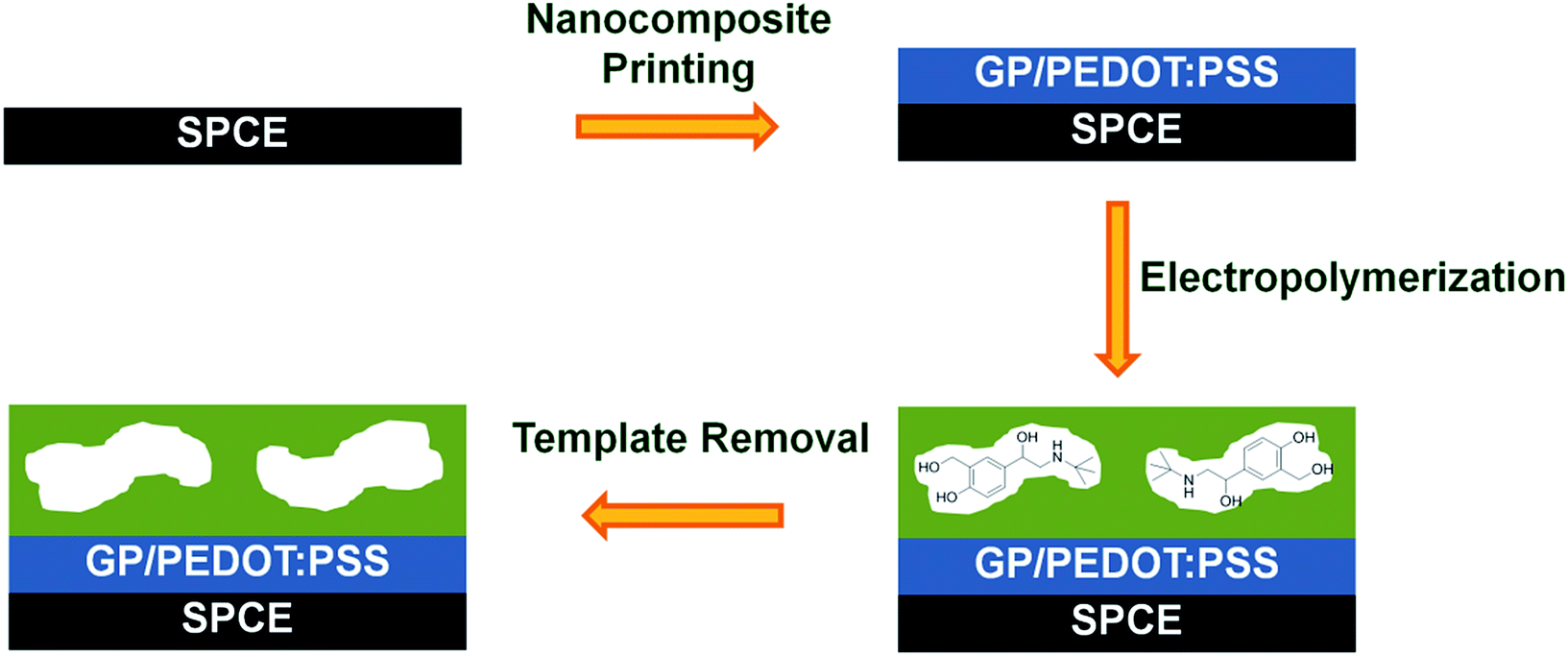 | ||
| Scheme 1 Schematic representation for the preparation process of electrosynthesized MIP modified nanocomposite based screen-printed carbon electrode (SCPE). | ||
Experimental
Materials
Clenbuterol hydrochloride and salbutamol hemisulfate were obtained from TCI chemicals, and o-phenylenediamine (o-PD), resorcinol and sibutramine hydrochloride monohydrate were from Acros. Ractopamine hydrochloride, terbutaline hemisulfate, dobutamine hydrochloride, 3-aminophenylboronic acid monohydrate, potassium hexacyanoferrate(III) (K3Fe(CN)6), potassium hexacyanoferrate(II) trihydrate (K4Fe(CN)6·3H2O), poly(3,4-ethylenedioxythiophene) polystyrene sulfonate (PEDOT:PSS) solution and sulfuric acid were purchased from Sigma-Aldrich. All buffer salts and organic solvents were purchased from Merck. All chemicals were of analytical grade or higher and used without further purification.Apparatus
The nanocomposite ink was printed onto SPCEs by a material inkjet printer from FUJIFILM (Dimatix Materials Printer, DMP-2800). The morphology of exfoliated graphene dispersed in the solution was examined by a transmission electron microscope (TEM, JEOL JEM-2010). The surface morphology of the printed composite film was characterized by a scanning electron microscope (SEM, Hitachi S-3400N). NMR measurements were carried out on a Bruker NMR spectrometer (Avance 500 MHz). All electrochemical measurements were performed using a potentiostat from PalmSens.NMR titration
Interactions between the template molecule and four different types of monomer were studied prior to polymer synthesis by means of 1H-NMR titration in DMSO-d6. To a solution of salbutamol (2 mM), increasing amounts of the guest monomer were added. The complexation-induced shifts (Δδ) were recorded and the titration curve was plotted of Δδ against the number of equivalents of guest added. The raw data was fitted to the isotherm and the binding constant was determined by non-linear regression of the isotherm.24Computational calculation
Geometry optimization of molecules was carried out using the M062X/6-31G (d,p) method,25 which is implemented in the Gaussian 09 software package. The interaction energy (EINT) between the template and the monomer was calculated by the following equation:| EINT = Ecomplex − Etemplate − ∑Emonomer | (1) |
Preparation of inkjet-printed GP/PEDOT:PSS nanocomposite electrode
A graphene/PEDOT:PSS aqueous dispersion was prepared by electrical exfoliation of graphite rod in PEDOT:PSS solution at a constant potential of 8 V for 5 h. The as-obtained graphene/PEDOT:PSS dispersed product was centrifuged to separate large aggregates. The graphene/PEDOT:PSS aqueous dispersion was then printed onto the working area of the SPCE and dried at 60 °C for 15 min.Preparation of imprinted and non-imprinted electrodes
A MIP layer was prepared on top of the nanocomposite modified SPCE by cyclic voltammetry sweeping between 0 and 1.2 V at a scan rate of 50 mV s−1 for 20 cycles in a mixture solution containing 5 mM aminophenylboronic acid, 5 mM o-PD and 2.5 mM salbutamol (100 mM acetate buffer, pH 5.6). The electrode was then immersed in 0.3 M H2SO4 at room temperature for 15 min to remove the template. A non-imprinted layer was prepared in an identical manner, but in the absence of salbutamol.Electrochemical detection of salbutamol
All standard salbutamol solutions were freshly prepared in acetate buffer (100 mM, pH 5.6) and used immediately. After the imprinted electrodes were incubated in solutions containing different concentrations of salbutamol for 10 min, the electrodes were rinsed and the electrochemical measurements were carried out in 5 mM K3[Fe(CN)6]/K4[Fe(CN)6] solution containing 100 mM KCl. Cyclic voltammetry (CV) measurements were carried out from −0.3 V to 0.5 V with a scan rate of 50 mV s−1. Differential pulse voltammetry (DPV) experiments were performed by scanning the potential from −0.3 to 0.6 V with a scan rate of 10 mV s−1.Determination of salbutamol in real samples
Swine meat and feed samples were purchased from retail markets in Thailand. These samples were finely ground. Two grams of the homogenous samples were placed into 50 mL polypropylene centrifuge tubes and spiked with sufficient volumes of salbutamol stock solution, to which 15 mL of methanol and 5 mL of distilled water were added. The mixtures were sonicated for 30 min and centrifuged for 10 min at 3000 rpm. The supernatants were filtered and the filtrates were diluted with 100 mM acetate buffer (pH 5.6) to obtain the samples for detection.Results and discussion
Selection of the functional monomer
The interactions between the template molecule and the functional monomers are the key towards the successful imprinting process. Arrays of non-covalent interactions acting in concert are known to lead to selective recognition in MIPs.26 On the other hand, they also play a crucial role in enhancing binding affinity. A suitable functional monomer was therefore selected from those electropolymerizable ones that can form multiple non-covalent bonds with the template molecule, including 3-aminophenylboronic acid, resorcinol, o-phenylenediamine and scopoletin. Within this study, a rational selection of the functional monomer was carried out by means of 1H-NMR titration. Upon addition of aminophenylboronic acid to a solution of salbutamol, downfield shifts of the –NH and –OH protons of the template were observed. The deshielding effect found in the –NH and –OH signals reveal that these binding sites are H-bond acceptors in the complex. The change in chemical shift of the –NH signal as the most-influenced binding site (Fig. S1†) gave a convincincing association constant (Ka) of 291 ± 4% M−1. The binding constants of the template with three other monomers were calculated, and are summarized in Table S1.† In comparison to aminophenylboronic acid, scopoletin, resorcinol and o-phenylenediamine showed much lower association constants. Therefore, aminophenylboronic acid was selected as the functional monomer to prepare further the MIP based receptor. Since aminophenylboronic acid has strong interactions with salbutamol, this might cause high non-specific binding when it alone is used to prepare the MIP. Therefore, o-phenylenediamine, which showed particularly low affinity towards the template molecule, was selected here as a co-monomer to suppress those undesirable interactions.A computational simulation was used to further investigate a possible conformation of the complex formed between the template and the surrounding functional monomers. The calculation revealed that the template molecule could form a stable 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 complex with two nearby aminophenylboronic acid molecules, as shown in Fig. S2,† with the lowest interaction energy of −172.17883747 kJ mol−1. Additionally, the calculation also revealed that H-bonding is a predominant interaction within the ternary complex, and thus plays a major role in creating the microenvironment for molecular recognition of the target molecule.
:2 complex with two nearby aminophenylboronic acid molecules, as shown in Fig. S2,† with the lowest interaction energy of −172.17883747 kJ mol−1. Additionally, the calculation also revealed that H-bonding is a predominant interaction within the ternary complex, and thus plays a major role in creating the microenvironment for molecular recognition of the target molecule.
Characterization of inkjet-printed GP/PEDOT:PSS electrode
The presence of exfoliated graphene in PEDOT:PSS solution was visualized using TEM. As depicted in Fig. S3,† a typical sheet-like structure with an irregular shape of graphene was observed. The as-obtained aqueous dispersion of graphene/PEDOT:PSS was further inkjet printed on top of the SPCE and the morphology of the modified electrode was characterized by SEM. The SEM images of the bare SPCE and the nanocomposite modified electrode are displayed in Fig. S4.† It can clearly be seen that the modified surface is rather smooth in contrast to that observed in the image of the bare electrode, suggesting a successful deposition of the graphene/polymer composite film.The electrochemical properties of the modified and unmodified electrodes were characterized by CV using ferri/ferrocyanide as the redox probe. As displayed in Fig. S5,† typical voltammograms of the redox couple were obtained. It was clearly shown that the redox peak currents of the nanocomposite modified electrode were enhanced. The modified electrode also showed a smaller peak potential difference (ΔEp) of 213 mV in comparison to the bare electrode. This would suggest an improvement in electron-transfer kinetics and the reversibility of the redox couple by the highly conductive graphene/PEDOT:PSS nanocomposite.
Preparation and characterization of MIP and NIP electrodes
The salbutamol-imprinted film was prepared by co-electrodeposition of 3-aminophenylboronic acid and o-phenylenediamine in the presence of the template molecule on top of the graphene/PEDOT:PSS modified SPCE. As depicted in Fig. S6,† the current intensity of the oxidation peak decreased significantly under successive scans, suggesting that a non-conductive film was gradually formed and blocked the electrode surface. This would lead to suppression of the voltammetric response.The electrochemical properties of the electrode at each modification step were characterized by CV using ferri/ferrocyanide as the redox probe. As shown in Fig. 1, the response of the electrode obtained after electropolymerization was almost suppressed. This could indicate that the polymeric film was successfully deposited and compactly covered the electrode surface. Therefore, the permeability of the redox probe to the underlying electroactive surface was hindered. After the template removal, the imprinted cavities were generated, making the redox signal reappear. Upon the rebinding of salbutamol to the imprinted sites, the number of cavities available for the redox probe to access the electrode surface decreased, which would again cause a decrease in the redox signal (Scheme S1†).
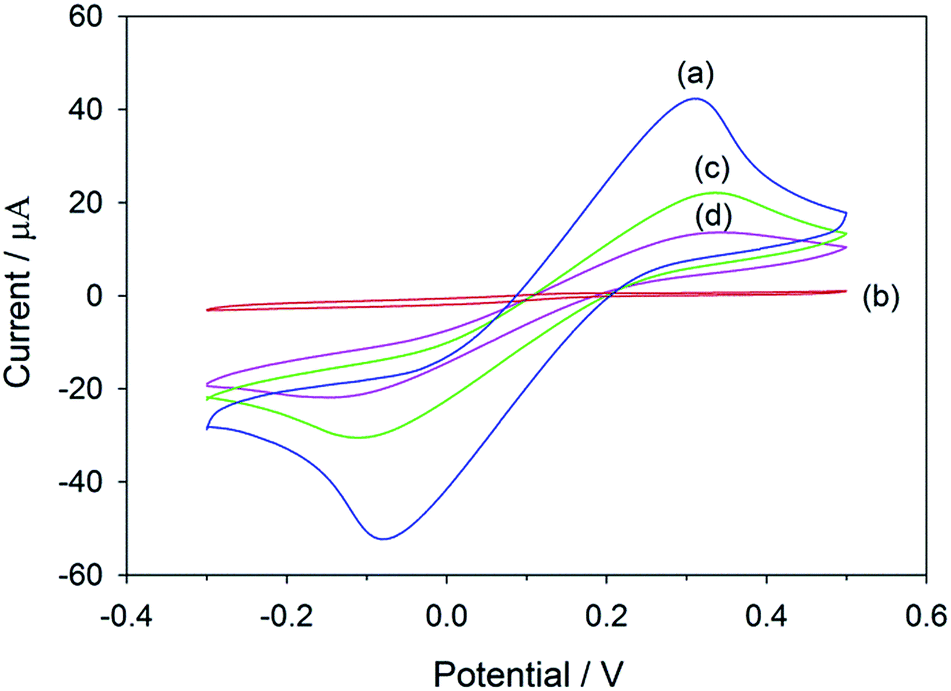 | ||
| Fig. 1 Cyclic voltammograms of 5 mM K3[Fe(CN)6]/K4[Fe(CN)6] in 100 mM KCl at (a) graphene/PEDOT:PSS/SPCE, (b) MIP electrode before template removal, (c) MIP electrode after template removal and (d) MIP electrode after target rebinding. | ||
SEM images showing surface morphologies of the MIP and NIP films are displayed in Fig. 2. In comparison to a rather compact NIP film, the imprinted layer exhibited a relatively rougher surface, which could be attributed to the successful removal of the template molecule from the MIP film.
 | ||
| Fig. 2 SEM images of (a) MIP and (b) NIP modified graphene/PEDOT:PSS/SPCEs. | ||
Linear range and detection limit
As the concentration of salbutamol increases, the current signal drops off (Fig. 3a), owing to the increasing number of cavities occupied by the target molecule. Instead of using the absolute current values, which might vary with the surface areas as well as the electrode configurations from electrode to electrode, a calibration curve was plotted between the relative current change (ΔI/I0) and salbutamol concentration.27,28 Here, ΔI = I0 − IC, where I0 and IC are the current values when the concentration of salbutamol is 0 and C nM, respectively. As shown in Fig. 3b, the relative current change increases with the increasing salbutamol concentrations. A linear relationship was observed over the range of 1 nM to 1.2 μM with a correlation coefficient (R2) of 0.992. The limit of detection of the MIP electrode was found to be 0.1 nM based on a signal-to-noise ratio (S/N = 3). To evaluate the enhancement effect of the GP-PEDOT:PSS nanocomposite on the MIP sensor performance, a control sensor (MIP/SPCE) was fabricated by direct deposition of a salbutamol imprinted layer on top of the bare screen printed carbon electrode. The same detection procedure was used to study the resulting control MIP/SPCE for comparison. As the concentration of salbutamol increases, the current response decreases (Fig. S7a†). A linear relationship between the relative current change and the concentration was observed over the range of 100 nM to 600 nM (Fig. S7b†), which was narrower than that obtained from the MIP/GP-PEDOT:PSS/SPCE. The detection limit of the MIP/SPCE was found to be 50 nM, which was two orders of magnitude higher than that from the MIP/GP-PEDOT:PSS/SPCE. The better detection limit of the MIP/GP-PEDOT:PSS/SPCE would be attributed to the modification of the electrode with the GP-PEDOT:PSS nanocomposite, which can efficiently enhance the electron-transfer rate, and eventually amplify the sensor signal.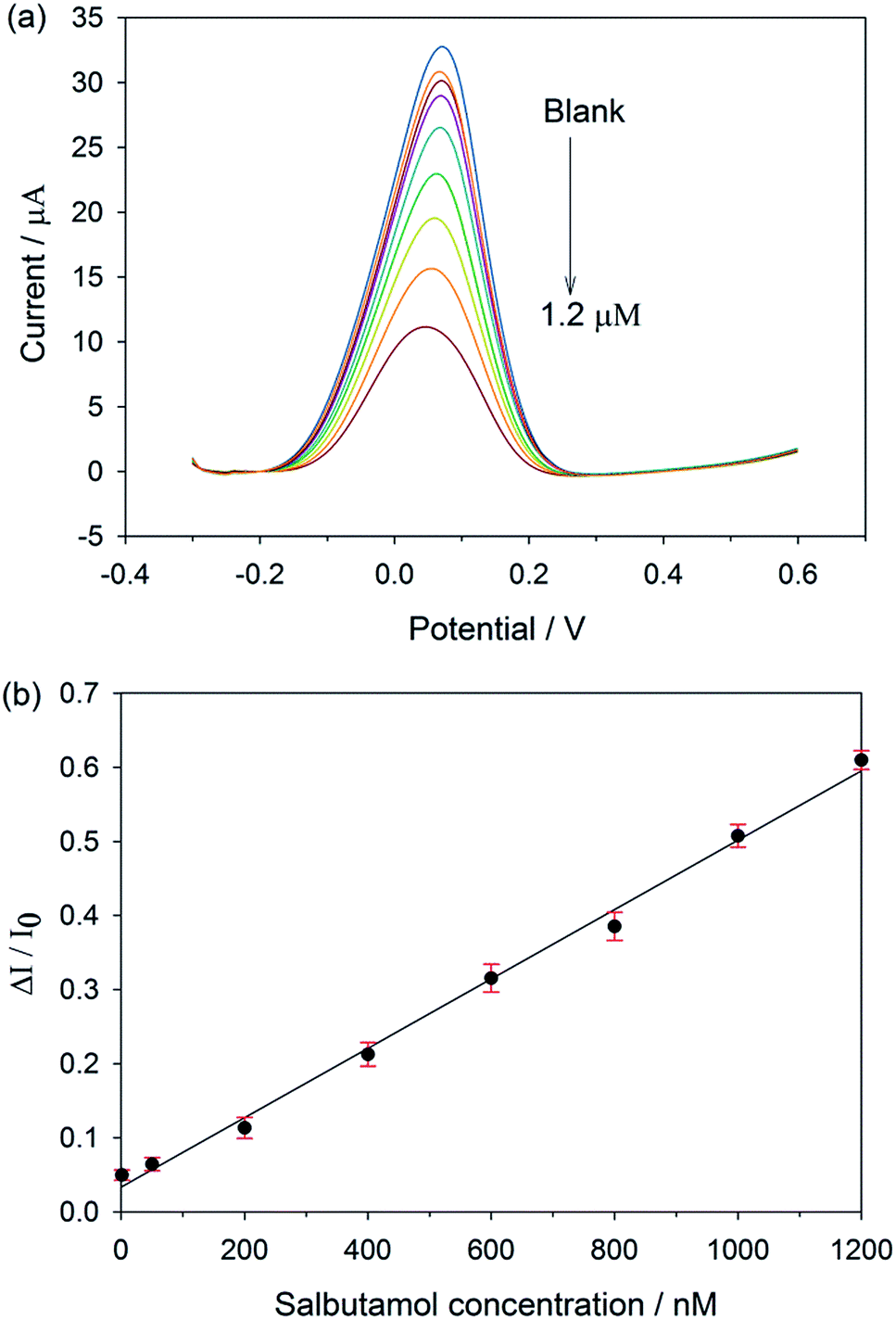 | ||
| Fig. 3 (a) Differential pulse voltammograms of MIP/GP-PEDOT:PSS/SPCE recorded in 5 mM K3[Fe(CN)6]/K4[Fe(CN)6] after incubation in different concentrations of salbutamol, (b) calibration curve of the MIP/GP-PEDOT:PSS/SPCE. The error bars represent the standard deviation obtained from 6 independent measurements. | ||
The sensor performances in terms of limit of detection and linear range of calibration were compared with other previously reported electrochemical sensors for salbutamol, and summarized in Table S2.† In comparison to most of the MIP- and nanocomposite based electrodes, our established sensor showed a better limit of detection and a wider linear range. Even though the detection limit of our sensor is not as low as those reported by Wang et al.29 and Chen et al.,30 our sensor offers a greatly broader linear response and requires no use of biologically derived materials (i.e., antibody and nucleic acid).
Selectivity
To investigate the selectivity of the prepared sensor, five compounds structurally related to salbutamol including clenbuterol, terbutaline, ractopamine, sibutramine and dobutamine (Fig. 4) were employed. The current shift (ΔI) between the peak currents of the redox probe measured after incubating the electrode in the background solution and in the analyte containing solution was used to identify the binding capacity of the analyte to the electrode.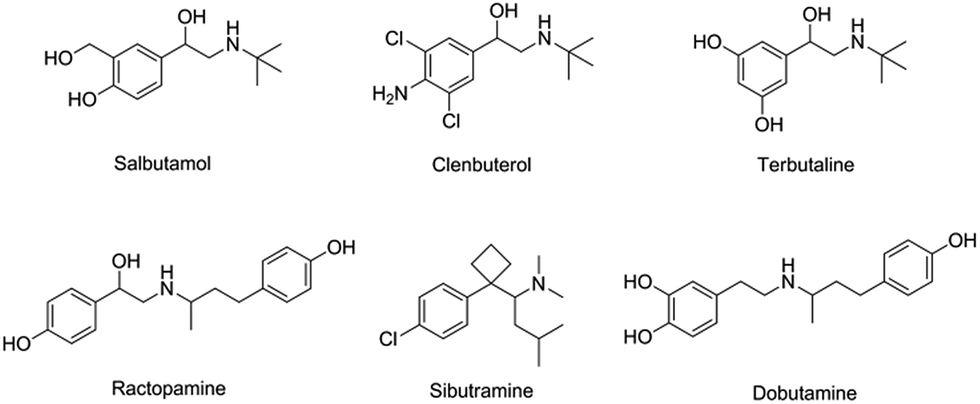 | ||
| Fig. 4 Chemical structures of salbutamol and its structurally related compounds. | ||
The change in the current response (ΔI) of the electrodes pre-incubated in 1.2 μM of each analyte was measured and is shown in Fig. 5. The current change (ΔI) of the non-imprinted sensor toward all the analytes was relatively low. This observed change would rather be due to the non-specific adsorption of the compounds to the polymer. Clearly, the current response of the MIP electrode toward salbutamol was significantly higher than those of the other five compounds. This high selectivity could be attributed to the complementary imprinted cavities within the MIP which match well with the unique structure of salbutamol in size, shape and orientation of functionalities. Ractopamine and dobutamine in this case are considerably too large in comparison to the imprinted sites. Sibutramine, on the other hand, contains a cyclobutane ring and a butyl group within its structure, making it too bulky for the binding pocket. Although salbutamol, clenbuterol and terbutaline hold an identical hydroxy–amine side chain motif, their substituents on the aromatic moiety are completely different. As a result, clenbuterol and terbutaline could not establish a fully matched hydrogen-bond network with the conserved functionalities within the imprinted sites. In the case of salbutamol, the current change observed on the imprinted electrode is significantly higher than that of the non-imprinted control, clearly because of the specific binding through the imprinting effect. This is in contrast to the other control molecules, where a relatively higher binding capacity of the MIP would rather stem from a rougher surface area of the MIP in comparison to the NIP film.
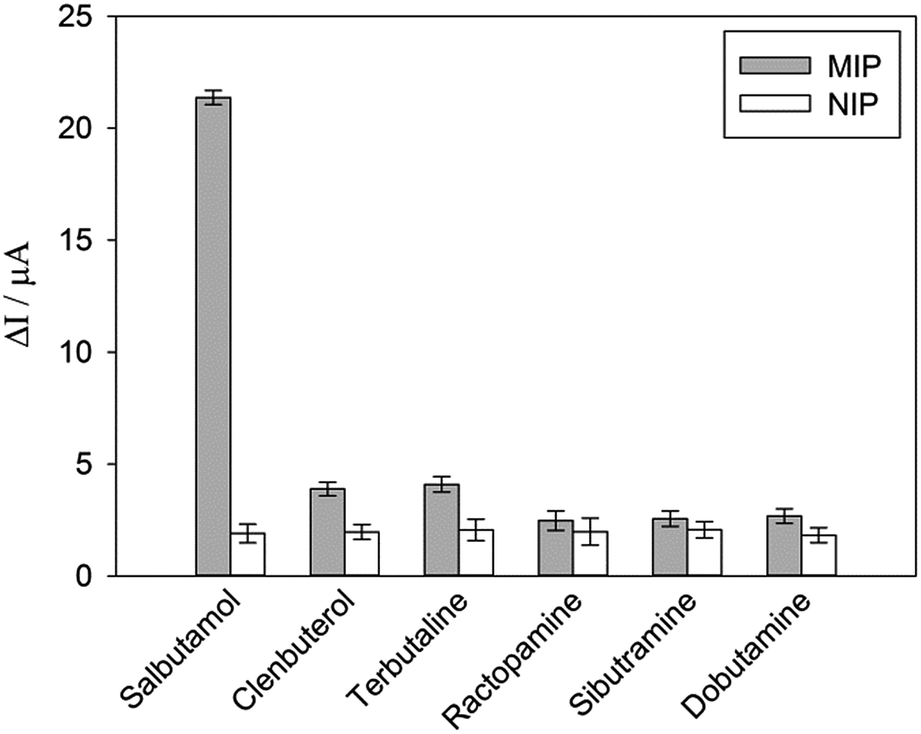 | ||
| Fig. 5 DPV responses of MIP and NIP toward 1.2 μM of salbutamol and other structurally related compounds. | ||
Reproducibility and stability
The sensor-to-sensor reproducibility was investigated using MIP electrodes prepared independently by the same procedure. The reproducibility was reported in terms of relative standard deviations (RSDs) of the current responses obtained after incubating the sensors in various concentrations of salbutamol. The RSDs of each concentration level were found to be less than 3.53%, reflecting a good sensor-to-sensor reproducibility. For assessment of reusability, the MIP electrode was regenerated using diluted sulfuric acid solution and reused for ten repeated measurements to determine 1.2 μM salbutamol. The RSD of the current response was found to be 2.73%, indicating a good reusability of the prepared sensor. The stability of the sensor was evaluated by storing the MIP electrode at 4 °C under N2 for 2 months and the sensor was then used to determine salbutamol at the concentration of 1.2 μM. It was found that the sensor lost around 6% of its initial response value, suggesting good stability of the prepared sensor.Determination of salbutamol in real samples
The capability of the sensor in real sample analysis was examined in the feed and swine meat samples. The spiked sample solutions were prepared and analyzed, and the results are shown in Table 1. The recoveries at three different concentration levels of each sample were in the range of 99.0–105.2% with RSDs of less than 3.16%. These results indicate that matrixes of real samples have no significant interference effect on the analysis. To further validate the reliability and accuracy of the developed sensor, HPLC was employed as a reference method. As shown in Table S3,† there is a good agreement between the values obtained by the established MIP sensor and HPLC, suggesting that the prepared sensor has a potential to be used as an analytical tool to detect salbutamol in complex real systems.| Sample | Spiked (nM) | Found (nM) | Recovery (%) | RSD (%) |
|---|---|---|---|---|
| Feed sample | 10 | 10.52 | 105.2 | 3.04 |
| 100 | 104.9 | 104.9 | 3.06 | |
| 1000 | 990 | 99.0 | 2.05 | |
| Swine meat | 10 | 10.18 | 101.8 | 3.16 |
| 100 | 102.0 | 102.0 | 2.95 | |
| 1000 | 1027 | 102.7 | 2.05 |
Conclusions
In this work, a MIP based electrochemical sensor for salbutamol was prepared by co-electropolymerization of aminophenylboronic acid and o-phenylenediamine in the presence of the template molecule. This MIP layer was constructed on top of the graphene/PEDOT:PSS modified SPCE. The graphene nanocomposite was used here in an attempt to enhance the current response, so that the sensitivity of the sensor could be improved. Under optimized conditions, the established sensor showed an exceptional detection limit of 100 pM and a wide dynamic response over the range of 1 nM to 1.2 μM. The sensor also exhibited high selectivity, excellent reproducibility and good stability. Reusability could be attained for at least 10 repeated measurements without any significant loss in electrochemical response. For real sample analysis, the proposed sensor showed good recovery results without the need for tedious and time-consuming pretreatment steps. This in turn indicates that our established electrochemical MIP sensor could be a potential candidate for food safety control at reduced cost.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support of the Kasetsart University Research and Development Institute (KURDI) and the Faculty of Science, Kasetsart University.Notes and references
- C. Tantucci, A. Duguet, T. Similowski, M. Zelter, J. P. Derenne and J. Milic-Emili, Eur. Respir. J., 1998, 12, 799–804 CrossRef CAS PubMed.
- P. D. Warriss, S. C. Kestin, T. P. Rolph and S. N. Brown, J. Anim. Sci., 1990, 68, 128–136 CrossRef CAS.
- G. R. Dickens, R. A. McCoy, R. West, J. S. Stapczynski and G. D. Clifton, Pharmacotherapy, 1994, 14, 729–733 CAS.
- R. Huupponen and K. Pihlajamäki, Int. J. Clin. Pharmacol., Ther. Toxicol., 1986, 24, 374–376 CAS.
- J. A. Bennett, E. T. Smyth, I. D. Pavord, P. J. Wilding and A. E. Tattersfield, Thorax, 1994, 49, 771–774 CrossRef CAS PubMed.
- W. Wang, Y. Zhang, J. Wang, X. Shi and J. Ye, Meat Sci., 2010, 85, 302–305 CrossRef CAS PubMed.
- Y. Zhou, P. Wang, X. Su, H. Zhao and Y. He, Talanta, 2013, 112, 20–25 CrossRef CAS PubMed.
- L. Oliva Mde, R. A. Olsina and A. N. Masi, Analyst, 2005, 130, 1312–1317 RSC.
- F. Cai, N. Wang, T. Dong, A. Deng and J. Li, Analyst, 2015, 140, 5885–5890 RSC.
- K. Basavaiah and H. C. Prameela, Anal. Bioanal. Chem., 2003, 376, 879–883 CrossRef CAS.
- L. Y. Zhang, B. Y. Chang, T. Dong, P. L. He, W. J. Yang and Z. Y. Wang, J. Chromatogr. Sci., 2009, 47, 324–328 CAS.
- C. Guo, F. Shi, L. Gong, H. Tan, D. Hu and J. Zhang, J. Pharm. Biomed. Anal., 2015, 25, 526–534 CrossRef.
- Z. Taleat, A. Khoshroo and M. Mazloum-Ardakani, Microchim. Acta, 2014, 181, 865–891 CrossRef CAS.
- J. N. Tiwari, V. Vij, K. C. Kemp and K. S. Kim, ACS Nano, 2016, 10, 46–80 CrossRef CAS.
- C. Zhu, G. Yang, H. Li, D. Du and Y. Lin, Anal. Chem., 2015, 87, 230–249 CrossRef CAS.
- P. Bollella, G. Fusco, C. Tortolini, G. Sanzò, G. Favero, L. Gorton and R. Antiochia, Biosens. Bioelectron., 2017, 89, 152–166 CrossRef CAS PubMed.
- H. Choi, K. J. Ahn, Y. Lee, S. Noh and H. Yoon, Adv. Mater. Interfaces, 2015, 2, 1500117 CrossRef.
- C. Sriprachuabwong, C. Karuwan, A. Wisitsorrat, D. Phokharatkul, T. Lomas, P. Sritongkham and A. Tuantranont, J. Mater. Chem., 2012, 22, 5478–5485 RSC.
- J. Li, F. Rossignol and J. Macdonald, Lab Chip, 2015, 15, 2538–2558 RSC.
- M. F. Frasco, L. A. A. N. A. Truta, M. G. F. Sales and F. T. C. Moreira, Sensors, 2017, 17, 523 CrossRef.
- C. Malitesta, E. Mazzotta, R. A. Picca, A. Poma, I. Chianella and S. A. Piletsky, Anal. Bioanal. Chem., 2012, 402, 1827–1846 CrossRef CAS.
- P. S. Sharma, A. Pietrzyk-Le, F. D'Souza and W. Kutner, Anal. Bioanal. Chem., 2012, 402, 3177–3204 CrossRef CAS.
- C. Karuwan, C. Sriprachuabwong, A. Wisitsoraat, D. Phokharatkul, P. Sritongkham and A. Tuantranont, Sens. Actuators, B, 2012, 161, 549–555 CrossRef CAS.
- A. J. Hall, F. Lanza-Sellergren, P. Manesiotis and B. Sellergren, Anal. Chim. Acta, 2005, 538, 9–14 CrossRef CAS.
- D. Liang, Y. Wang, S. Li, Y. Li, M. Zhang, Y. Li, W. Tian, J. Liu, S. Tang, B. Li and R. Jin, Int. J. Mol. Sci., 2016, 17, 1750 CrossRef.
- B. C. Iacoba, E. Bodoki, C. Farcau, L. Barbu-Tudoran and R. Oprean, Electrochim. Acta, 2016, 217, 195–202 CrossRef.
- Y. Liu, Q. J. Song and L. Wang, Microchem. J., 2009, 91, 222–226 CrossRef CAS.
- K. Kor and K. Zarei, Talanta, 2016, 146, 181–187 CrossRef CAS.
- H. Wang, Y. Zhang, H. Li, B. Du, H. Ma, D. Wu and Q. Wei, Biosens. Bioelectron., 2013, 49, 14–19 CrossRef CAS.
- D. Chen, M. Yang, N. Zheng, N. Xie, D. Liu, C. Xie and D. Yao, Biosens. Bioelectron., 2016, 80, 525–531 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra09601a |
| This journal is © The Royal Society of Chemistry 2018 |