
Optically active N-alkyl aziridines via stereospecific reductive cyclization of α-mesylated acetamides†
Duanshuai
Tian
,
Henian
Peng
,
Ziyue
Liu
and
Wenjun
Tang
 *
*
State Key Laboratory of Bio-Organic and Natural Products Chemistry, Center for Excellence in Molecular Synthesis, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, 345 Ling Ling Rd, Shanghai 200032, China. E-mail: tangwenjun@sioc.ac.cn
First published on 16th August 2018
Abstract
An efficient method for the synthesis of optically active N-alkyl aziridines has been realized for the first time by stereospecific reductive cyclization of optically active α-mesylated acetamides. A series of optically active N-alkyl aziridines are prepared in moderate to good yields and excellent ees. The employment of (9-BBN)2 as the reagent is crucial for the success of the transformation.
Optically active aziridine moieties exist in a number of biologically important alkaloids1 and therapeutic agents such as mitomycin A,2a,b madurastatin A1,2e ficellomycin,2f and azicemicin B2c,d (Fig. 1). They also belong to key structural motifs in agrochemicals and dyes.2 Structurally, the ring strain possessed by aziridine makes them highly reactive species. For example, they can undergo ring-opening reactions in the presence of nucleophiles,3 and they can also be employed for cycloaddition reactions.4 Optically active aziridines have become highly valuable building blocks for the synthesis of nitrogen-containing compounds.
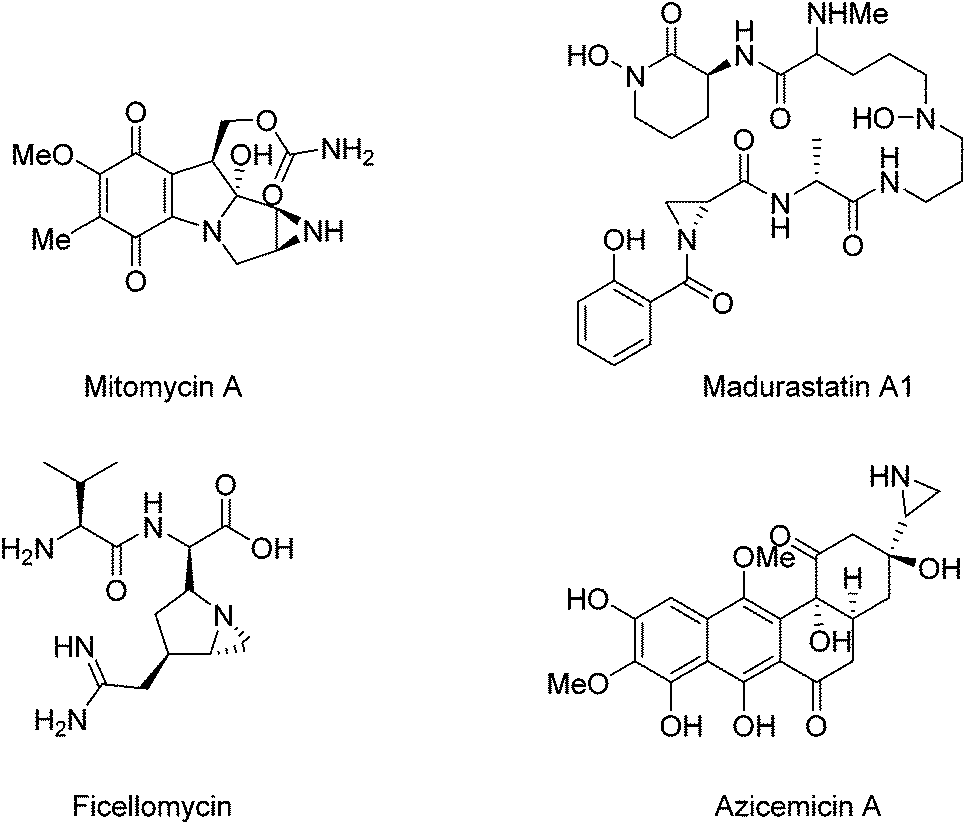 | ||
| Fig. 1 Therapeutic agents containing optically active aziridine moieties. | ||
The synthesis of optically active aziridines has thus gained significant interest. Common methods for the stereoselective synthesis of aziridines include Wenker cyclization or Mitsunobu reaction of optically active amino alcohols,5 reaction of imines with carbene compounds (aza-Darzens reaction)6 or sulfur ylides (Corey–Chaykovsky aziridination),7 or reaction of alkenes with nitrenes in situ generated from various nitrene precursors in the presence of a catalyst.8 However, most of these methods applied well to the synthesis of aziridines with N-acyl or -sulphonyl functionalities in order to achieve a good yield. So far, only a few methods are available for the synthesis of optically active N-alkyl aziridines. Lindsley and coworkers9 described a three-step protocol for the synthesis of terminal N-alkyl aziridines (Fig. 2a). An enantioselective aziridination was reported by Buchwald and coworkers from allylic hydroxylamine ester through a CuH-catalyzed regioselective intramolecular hydroamination10 (Fig. 2b). Nevertheless, a facile and straightforward synthetic methods for optically active N-alkyl aziridines from readily accessible starting material remains highly desirable. We have recently reported a stereospecific nucleophilic substitution of optically active α-aryl-α-mesylated acetamides with arylboronic acids as nuclephiles.11 The readily availability of optically active α-aryl-α-mesylated acetamides from mandelic acid derivatives or by asymmetric hydrogenation12 makes them attractive feedstocks for preparing other valuable optically active intermediates. Herein we report a facile preparation of optically active N-alkyl aziridines by stereospecific reductive cyclization of α-mesylated acetamides using (9-BBN)2 as a reducing reagent (Fig. 2c).
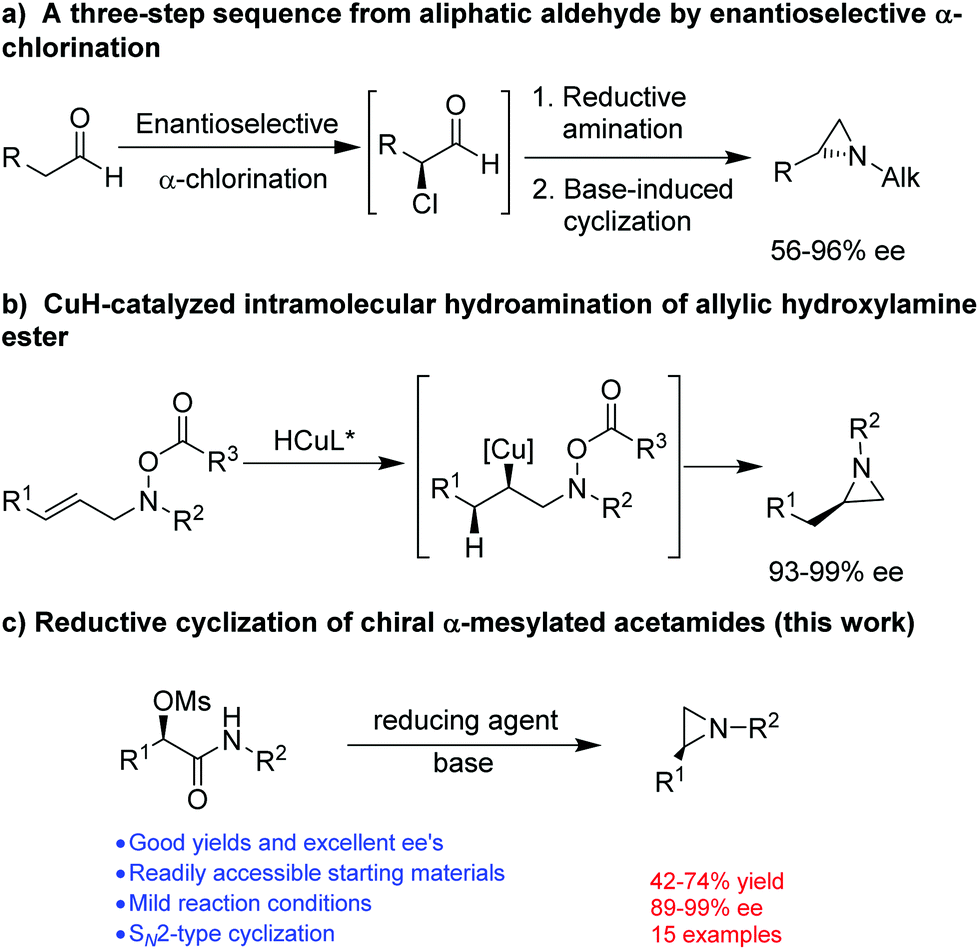 | ||
| Fig. 2 The construction of aziridines. | ||
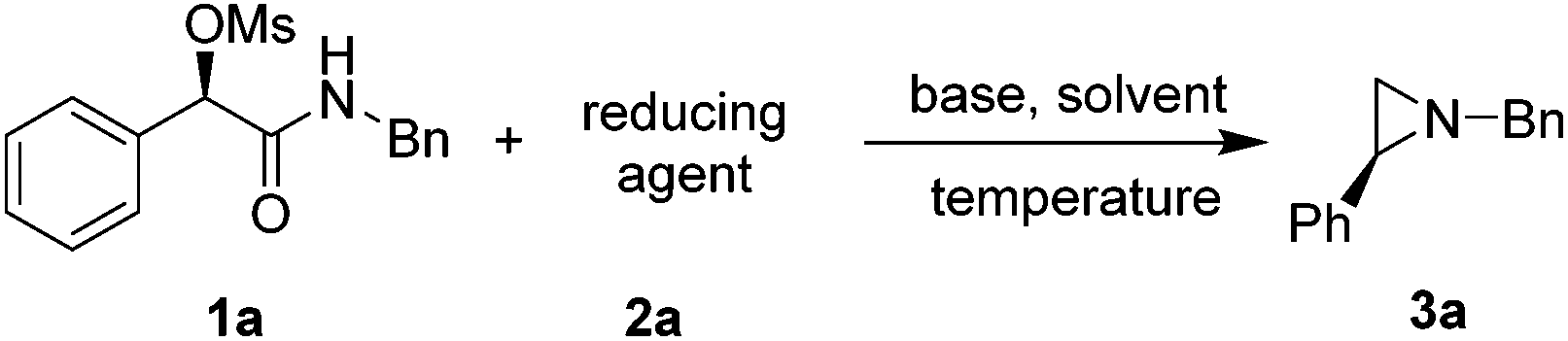
Optically active (R)-2-(benzylamino)-2-oxo-1-phenylethyl methanesulfonate (1a) was chosen as the model substrate for study of reductive cyclization (Table 1). The reaction was initially carried out in toluene with (9-BBN)2 as the reducing reagent and K2CO3 as the base. A decent yield (25%) of 3a with an excellent ee (97%) was achieved at 60 °C for 8 h (entry 1). However, no 3a was formed with other reducing reagents including BH3, LiAlH4, and DIBAL-H (entries 2–4). We thus employed (9-BBN)2 as the reducing reagent for the rest of study. When K3PO4 was used as the base, the yield of 3a increased to 62% without loss of ee (97%, entry 5). Further screening of the base including inorganic and organic bases did not further improve the yield (entries 6–11). It is noteworthy that no product of 3a was achieved in the absence of base (entry 12). We further investigated the effect of the solvents. Polar solvents such as dioxane, THF, DMF or CH3CN did not yield any product (entries 14–16 and 18). Nonpolar or less polar solvent such as DCE, cyclohexane, or mesitylene provided the desired product, albeit with lower yields (entries 13, 17 and 19). The absolute configuration of 3a was determined as S by comparing the optical rotation with reported data,13 indicating that 1a was cyclized reductively to form 3a through an SN2-type process.
|
|
|||||
|---|---|---|---|---|---|
| Entriesa | Base | Solvent | Reducing agent | Yieldb (%) | eec (%) |
| a Unless otherwise specified, all reactions were performed at 60 °C for 8 h in the selected solvent (5 mL) with 1a (0.5 mmol), 2a (1 mmol), base (2 equiv.). b Isolated yields. c Determined by chiral HPLC on a chiralcel column. | |||||
| 1 | K2CO3 | Toluene | (9-BBN)2 | 25 | 97 |
| 2 | K2CO3 | Toluene | BH3·THF | 0 | ND |
| 3 | K2CO3 | Toluene | LiAlH4 | 0 | ND |
| 4 | K2CO3 | Toluene | DABAL-H | Trace | ND |
| 5 | K3PO4 | Toluene | (9-BBN)2 | 62 | 97 |
| 6 | TEA | Toluene | (9-BBN)2 | 14 | 97 |
| 7 | KOtBu | Toluene | (9-BBN)2 | 0 | ND |
| 8 | DABCO | Toluene | (9-BBN)2 | 0 | ND |
| 9 | CsF | Toluene | (9-BBN)2 | 7 | 98 |
| 10 | KOH | Toluene | (9-BBN)2 | 0 | ND |
| 11 | Cs2CO3 | Toluene | (9-BBN)2 | 40 | 93 |
| 12 | None | Toluene | (9-BBN)2 | 0 | ND |
| 13 | K3PO4 | DCE | (9-BBN)2 | 27 | 91 |
| 14 | K3PO4 | Dioxane | (9-BBN)2 | 0 | ND |
| 15 | K3PO4 | THF | (9-BBN)2 | 0 | ND |
| 16 | K3PO4 | DMF | (9-BBN)2 | 0 | ND |
| 17 | K3PO4 | Cyclohexane | (9-BBN)2 | 22 | 94 |
| 18 | K3PO4 | CH3CN | (9-BBN)2 | 0 | ND |
| 19 | K3PO4 | Mesitylene | (9-BBN)2 | 20 | 97 |
We then looked into the substrate scope of this reaction. As depicted in Table 2, a series of optically active aziridines were formed in excellent enantioselectivities and moderate to good yields from optically active α-substituted-phenyl-α-mesylated acetamides with (9-BBN)2 as the reducing reagent with a minimum loss of enantiomeric purity. Various optically active N-alkyl aziridines including N-cyclic alkyl (3c–e), N-noncyclic primary alkyl (3f–h) and N-noncyclic secondary aziridines (1i–g) were prepared in moderate to good yields and excellent ee's. Surprisingly, no desired aziridine product was formed when an N-aryl substrate was employed. Halogen-substituted benzylic mesylates (1k–o) at various positions were all applicable. However, mesylates with electron-donating substituents at aryl groups were generally too unstable to prepare. It should be noted that ortho-halo substituted benzylic mesylates such as 1k, 1o were also suitable for the transformation, providing the optically active aziridines 3k and 3o in excellent enantioselectivities and moderate yields.
| a Unless otherwise specified, the reactions were carried in toluene under nitrogen at 60 °C for 8 h in the presence of (9-BBN)2 (2 equiv.) and K3PO4 (2 equiv.), isolated yields, ee value of the substrate in parenthesis. |
|---|
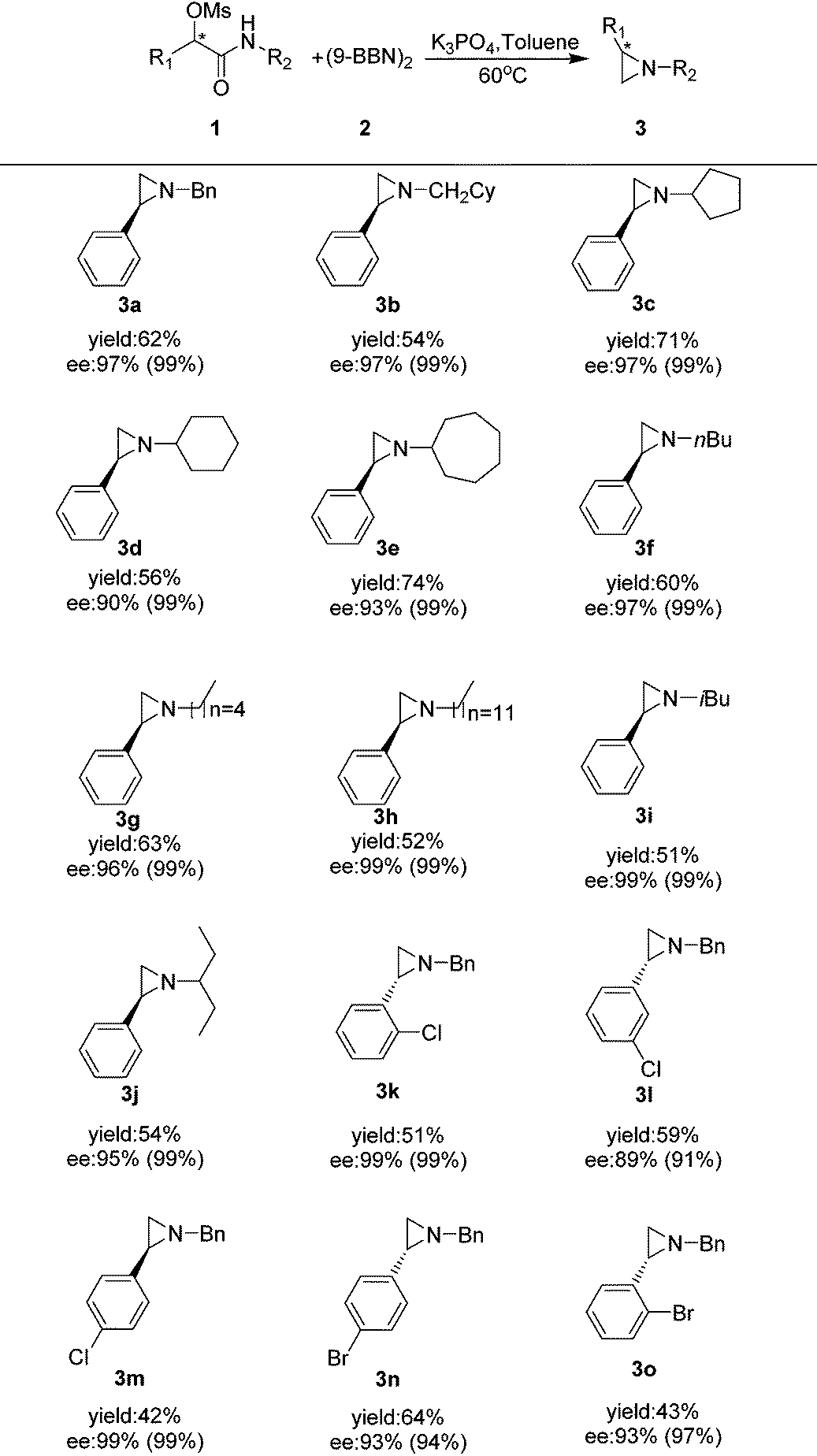
|
To shed light on the mechanism of this transformation, the following experiments were implemented (Scheme 1). Firstly, a deuterium-labeling experiment with 1c′ as the substrate proved no deprotonation occurring at the benzylic position (eqn (1)). Secondary, no deuterium was included in 3c with C6D6 as solvent, indicating that the hydrogen in the product is derived from (9-BBN)2 rather than solvent (eqn (2)). Thirdly, only a trace amount of the target product 3c was isolated when the (9-BBN)2 was reduced to 0.5 equiv., indicating that multiple equivalents of 9-BBN were needed for a complete transformation of 1c to the target product 3c (eqn (3)). Fourthly, no target product 3a was formed without base in the reaction, and only a large amount of 1a was recovered (eqn (4)), demonstrating that the base had played a crucial role in the reduction of amide. We believe that the addition of base not only enhanced the reducing ability of 9-BBN, but also promoted the formation of the aziridine ring. To understand why no aziridine was formed when DIBAL-H was used as the reducing reagent, we isolated a secondary amine as a major product from the reaction (eqn (5)), indicating that DIBAL-H was too active as the reducing agent for the transformation. When an N-phenyl substrate was employed, a demesylate product was isolated in 46% yield (eqn (6)), indicating a facile demesylation process with an N-aryl substrate. This result explained the limitation of the reductive cyclization in the preparation of N-aryl aziridines.
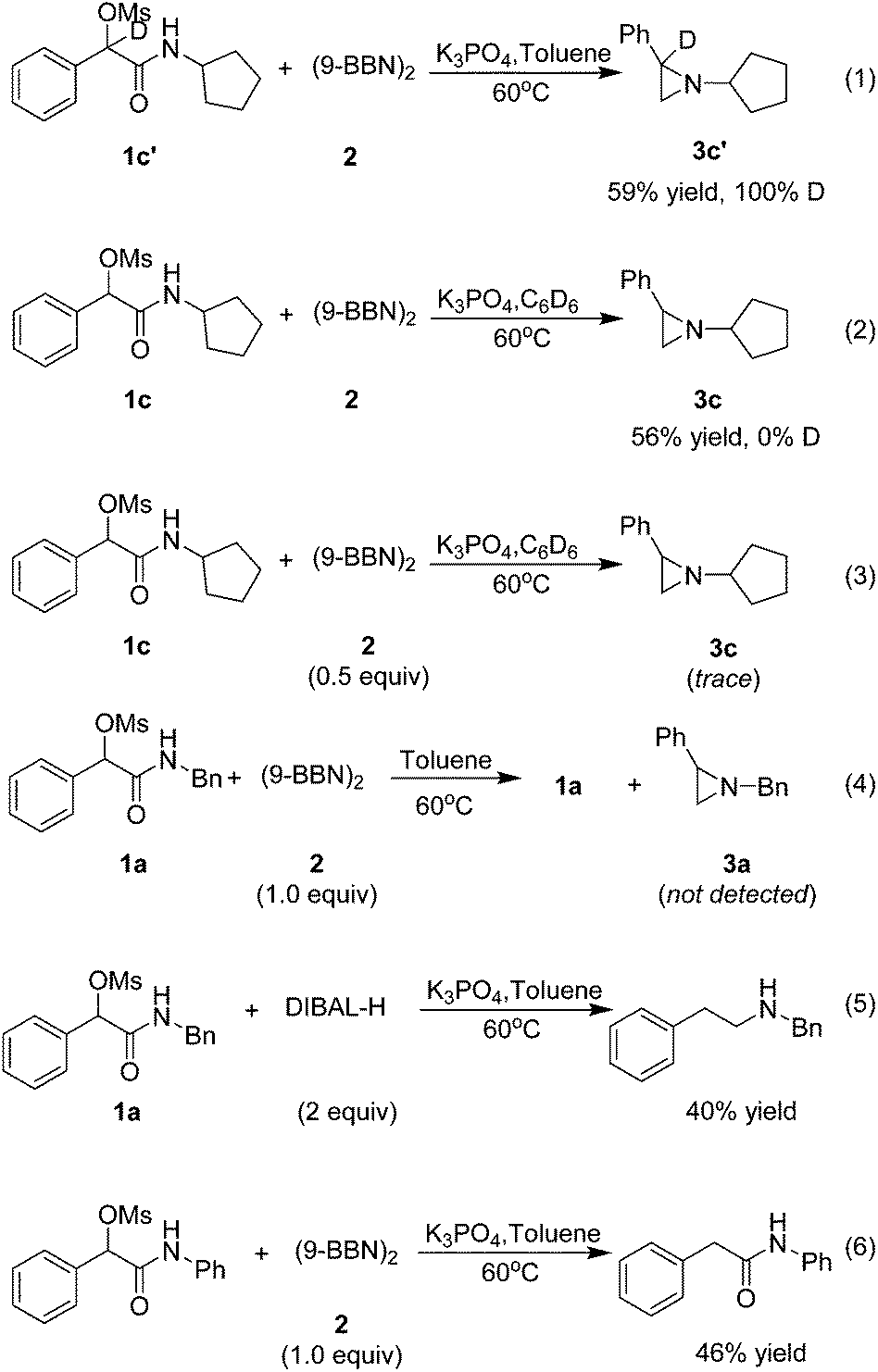 | ||
| Scheme 1 Mechanistic investigation. | ||
On the basis of the above observation, we proposed two reaction pathways for the reductive cyclization (Scheme 2). In pathway I, 9-BBN reacts with substrate 1a to generate imidate I, which further react with a 2nd equiv. of 9-BBN to form II. In the presence of a base, intramolecular cyclization at a SN2-type fashion occurs to form cyclic product III, which can break down to a cyclic iminium IV. Further reduction with a 3rd equiv. of 9-BBN to form product 3a. Alternatively in pathway II, intermediate II can give rise to the formation of imine V under basic conditions, which can further reduced by 9-BBN to form VI. In the presence of a base, cyclization of VI through a SN2-type process takes place to form product 3a.
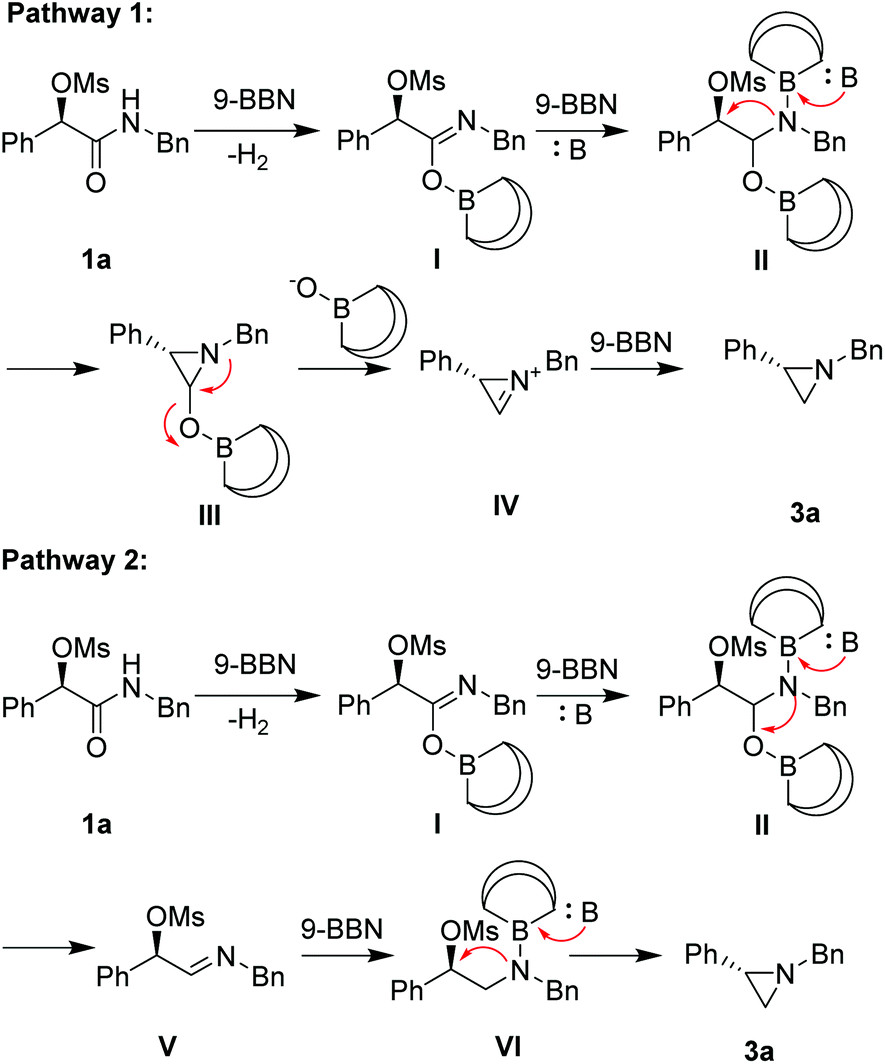 | ||
| Scheme 2 Proposed mechanism. | ||
In summary, an efficient method for the synthesis of optically active N-alkyl aziridines has been realized for the first time by stereospecific reductive cyclization of optically active α-mesylated acetamides. With this method, a series of optically active N-alkyl aziridines have been prepared in moderate to good yields with excellent ees. Mechanistic investigation has indicated an intramolecular SN2-type process under reductive conditions and the employment of (9-BBN)2 has been proven to be crucial for the success of reaction. Because of the readily availability of optically active α-hydroxy acetamides, this method offers a facile and expedite preparation of optically active aziridines complementary to other existing methods. The methodology should find a number of applications in drug discovery and process chemistry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Strategic Priority Research Program of the Chinese Academy of Sciences XDB20000000, CAS (QYZDY-SSW-SLH029), NSFC (21725205, 21432007, 21572246), STCSM-18520712200, and K.C. Wong Education Foundation.Notes and references
- For selected reviews, see: (a) J. B. Sweeney, Chem. Soc. Rev., 2002, 31, 247 RSC; (b) I. D. G. Watson, L. Yu and A. K. Yudin, Acc. Chem. Res., 2006, 39, 194 CrossRef PubMed; (c) G. S. Singh, M. DQhooge and N. De Kimpe, Chem. Rev., 2007, 107, 2080 CrossRef PubMed; (d) S. Stanković, M. DQhooge, S. Catak, H. Eum, M. Waroquier, V. Van Speybroeck, N. De Kimpe and H.-J. Ha, Chem. Soc. Rev., 2012, 41, 643 RSC; (e) G. Callebaut, T. Meiresonne, N. De Kimpe and S. Mangelinckx, Chem. Rev., 2014, 114, 7954 CrossRef PubMed; (f) B. H. Rotstein, S. Zaretsky, V. Rai and A. K. Yudin, Chem. Rev., 2014, 114, 8323 CrossRef PubMed.
- (a) T. Hata, T. Hoshi, K. Kanamori, A. Matsumae, Y. Sano, T. Shima and R. Sugawara, J. Antibiot., 1956, 9, 141 Search PubMed; (b) T. Hata and R. Sugawara, J. Antibiot., 1956, 9, 147 Search PubMed; (c) T. Tsuchida, H. Iinuma, N. Kinoshita, T. Ikeda, T. Sawa, M. Hamada and T. Takeuchi, J. Antibiot., 1995, 48, 217 CrossRef PubMed; (d) T. Tsuchida, R. Sawa, Y. Takahashi, H. Iinuma, T. Sawa, H. Naganawa and T. Takeuchi, J. Antibiot., 1995, 48, 1148 CrossRef PubMed; (e) K. I. Harada, K. Tomita, K. Fujii, K. Masuda, Y. Mikami, K. Yazawa and H. Komaki, J. Antibiot., 2004, 57, 125 CrossRef PubMed; (f) A. D. Argoudelis, F. Reusser, H. A. Whaley, L. Baczynskyj, S. A. Mizsak and R. L. Wnuk, J. Antibiot., 1976, 29, 1001 CrossRef PubMed.
- (a) X. E. Hu, Nucleophilic ring opening of aziridines, Tetrahedron, 2004, 60, 2701–2743 CrossRef; (b) T. N. Wade, J. Org. Chem., 1980, 45, 5328–5333 CrossRef; (c) J. Wu, X.-L. Hou and L.-X. Dai, J. Chem. Soc., Perkin Trans. 1, 2001, 1314–1317 RSC; (d) L. Ma and J. Xu, Prog. Chem., 2004, 16(2), 220–235 Search PubMed.
- (a) M. R. Kuszpit, W. D. Wulff and J. J. Tepe, J. Org. Chem., 2011, 76, 2913–2919 CrossRef PubMed; (b) R. A. Craig II, N. R. O'Connor, A. F. G. Goldberg and B. M. Stoltz, Chem. – Eur. J., 2014, 20, 4806–4813 CrossRef PubMed; (c) M. Sengoden and T. Punniyamurthy, Angew. Chem., Int. Ed., 2013, 52, 572–575 CrossRef PubMed; (d) Y.-M. Shen, W.-L. Duan and M. Shi, Eur. J. Org. Chem., 2004, 3080–3089 CrossRef; (e) Y. Du, Y. Wu, A.-H. Liu and L.-N. He, J. Org. Chem., 2008, 73, 4709–4712 CrossRef PubMed.
- (a) X. Y. Li, N. Chen and J. X. Xu, Synthesis, 2010, 3423–3428 Search PubMed; (b) B. R. Buckley, A. P. Patel and K. G. U. Wijayantha, J. Org. Chem., 2013, 78, 1289–1292 CrossRef PubMed.
- (a) S. E. Larson, G. L. Li, G. B. Rowland, D. Junge, R. C. Huang, H. L. Woodcock and J. C. Antilla, Org. Lett., 2011, 13, 2188–2191 CrossRef PubMed; (b) Q. Peng, D. Guo, J. Bie and J. Wang, Angew. Chem., Int. Ed., 2018, 57, 3767 CrossRef PubMed; (c) T. Akiyama, T. Suzuki and K. Mori, Org. Lett., 2009, 11, 2445 CrossRef PubMed; (d) C. De Fusco, T. Fuoco, G. Croce and A. Lattanzi, Org. Lett., 2012, 14, 4078 CrossRef PubMed; (e) B. M. Trost, T. Saget and C.-I. Hung, Angew. Chem., Int. Ed., 2017, 56, 2440 CrossRef PubMed.
- (a) J. J. Li, Name Reactions in Heterocyclic Chemistry, Wiley, Hoboken, 2005, pp. 2–14 Search PubMed; (b) A. Padwa and S. S. Murphree, ARKIVOC, 2006, 6–33 Search PubMed; (c) Aziridines and Epoxides in Organic Synthesis, ed. A. Yudin, Wiley-VCH, Weinheim, 2006 CrossRef.
- (a) M. M. Abu-Omar, Dalton Trans., 2011, 40, 3435–3444 RSC; (b) J. W. W. Chang, T. M. U. Ton and P. W. H. Chan, Chem. Rec., 2011, 11, 331–357 CrossRef PubMed; (c) D. Mansuy, J. P. Mahy, A. Dureault, G. Bedi and P. Battioni, J. Chem. Soc., Chem. Commun., 1984, 1161–1163 RSC; (d) D. A. Evans, M. M. Faul and M. T. Bilodeau, J. Org. Chem., 1991, 56, 6744–6746 CrossRef; (e) P. Müller, C. Baud and Y. Jacquier, Tetrahedron, 1996, 52, 1543–1548 CrossRef; (f) N. Jung and S. Brase, Angew. Chem., Int. Ed., 2012, 51, 5538 Search PubMed; (g) D. A. Evans, M. M. Faul, M. T. Bilodeau, B. A. Anderson and D. M. Barnes, J. Am. Chem. Soc., 1993, 115, 5328 CrossRef; (h) Z. Li, R. W. Quan and E. N. Jacobsen, J. Am. Chem. Soc., 1995, 117, 5889 CrossRef; (i) X. Wang and K. Ding, Chem. – Eur. J., 2006, 12, 4568 CrossRef PubMed; (j) J. Tao, L.-M. Jin and X. P. Zhang, Beilstein J. Org. Chem., 2014, 10, 1282 CrossRef PubMed.
- (a) O. O. Fadeyi, M. L. Schulte and C. W. Lindsley, Org. Lett., 2010, 12, 3276 CrossRef PubMed; (b) T. J. Senter, M. C. O'Reilly, K. M. Chong, G. A. Sulikowski and C. W. Lindsley, Tetrahedron Lett., 2015, 56, 1276 CrossRef PubMed.
- H. Wang, J. C. Yang and S. L. Buchwald, J. Am. Chem. Soc., 2017, 139, 8428–8431 CrossRef PubMed.
- D. Tian, C. Li, G. Gu, H. Peng, X. Zhang and W. Tang, Angew. Chem., Int. Ed., 2018, 57, 7176 CrossRef PubMed.
- G. Gu, T. Yang, O. Yu, H. Qian, J. Wang, J. Wen, L. Dang and X. Zhang, Org. Lett., 2017, 19, 5920 CrossRef PubMed.
- A. Kawamoto and M. Wills, Tetrahedron: Asymmetry, 2000, 11, 3257–3261 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qo00774h |
| This journal is © the Partner Organisations 2018 |