
Selenium dioxide-promoted selective synthesis of mono- and bis-sulfenylindoles†
Samuel
Thurow
 a,
Filipe
Penteado
a,
Gelson
Perin
a,
Diego
Alves
a,
Claudio
Santi
b,
Bonifacio
Monti
b,
Carl H.
Schiesser
*c,
Thiago
Barcellos
d and
Eder J.
Lenardão
*a
a,
Filipe
Penteado
a,
Gelson
Perin
a,
Diego
Alves
a,
Claudio
Santi
b,
Bonifacio
Monti
b,
Carl H.
Schiesser
*c,
Thiago
Barcellos
d and
Eder J.
Lenardão
*a
aCentro de Ciências Químicas, Farmacêuticas e de Alimentos Universidade Federal de Pelotas – UFPel, P.O. box 354, CEP: 96010-900, Pelotas, RS, Brazil. E-mail: lenardao@ufpel.edu.br
bDepartment of Pharmaceutical Sciences, University of Perugia, Via del Liceo, 1, Perugia, PG, Italy
cSeleno Therapeutics Pty. Ltd, Brighton East, Victoria 3187, Australia. E-mail: carl@selenotherapeutics.com
dLaboratory of Biotechnology of Natural and Synthetic Products, Universidade de Caxias do Sul – UCS, Caxias do Sul, RS, Brazil
First published on 7th May 2018
Abstract
A selective method for the preparation of mono- and bis-sulfenylindoles was developed using an oxidizing agent and the I2/SeO2 system as an efficient catalyst. The selectivity was controlled by varying the amount of SeO2. This represents the first example of the application of SeO2 in this type of selective chemistry.
Introduction
Over one century after its first isolation in 1869,1 indole still is one of the most explored building blocks in organic chemistry. Because of its extensive applicability to pharmaceutical, agrochemical, and material sciences, indole has been called “the lord of the rings” by Bandini.2Regarding its biological importance, 3-sulfenylindole I and its derivative II have been studied as potent anti-cancer agents that inhibit tubulin polymerization in a more effective way than combretastatin-A-4.3 The fluorinated 3-sulfenylindoles III and IV showed selective COX-2-enzyme inhibition similar to etoricoxib and analogues4 (Fig. 1A). In addition, anti-HIV,5a anti-allergenic,5b and antiasthmatic5c properties have also been attributed to sulfur-containing indole compounds.
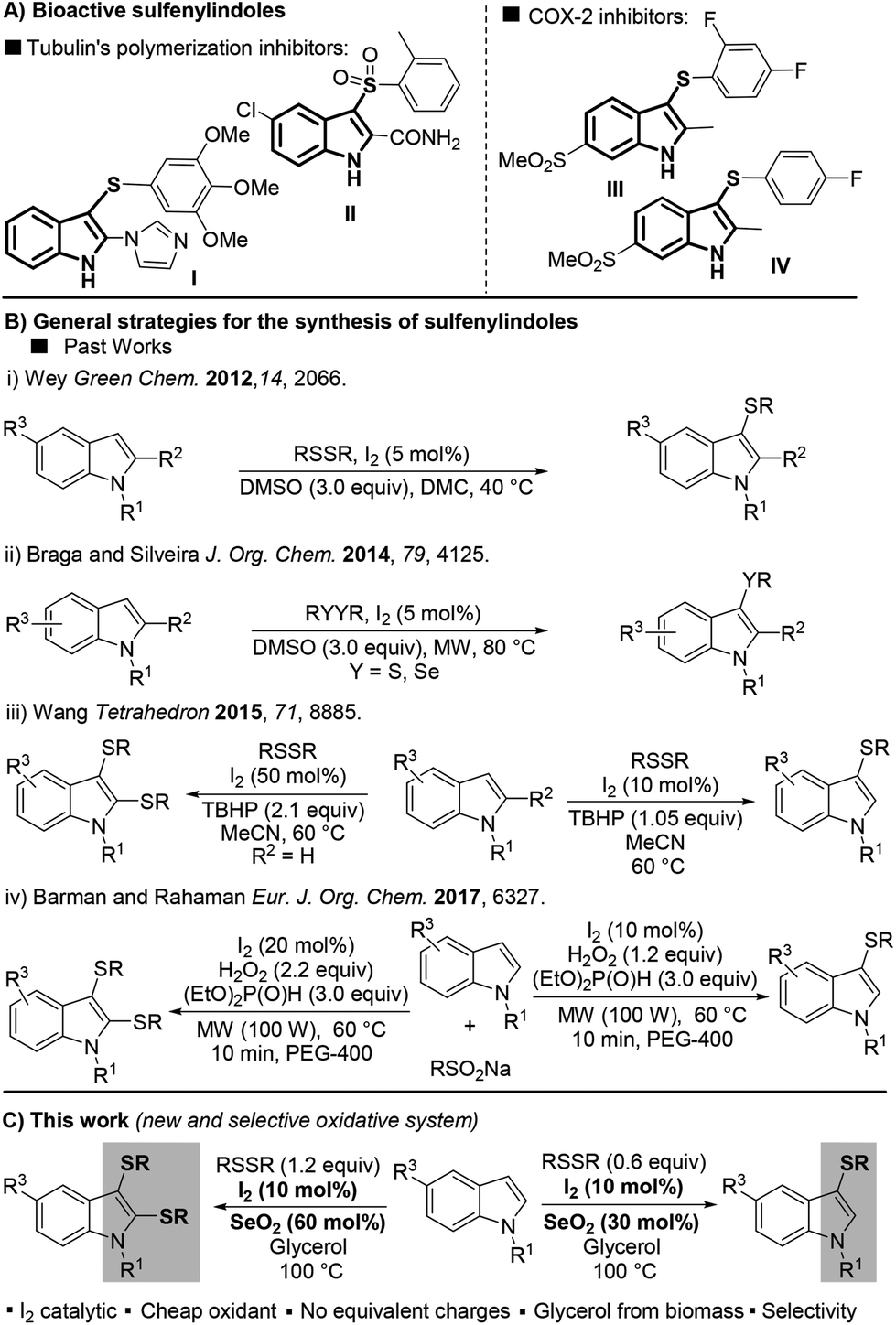 | ||
| Fig. 1 A: 3-Sulfenylindole derivatives with analogous activities to combretastatin A-4 (I and II) and etoricoxib (III and IV). B: Previous strategies for the preparation of sulfenylindoles. C: Present work. | ||
Due to the importance of sulfur-containing scaffolds in drug development, especially sulfenylindoles, the development of methods that selectively access this class of compound has increased in the past few years.6,7 Most of the current methodologies are limited to 3-sulfenylindoles and involve (A) the electrophilic cyclization of 2-alkynylanilines,7a–c or (B) the direct C–H sulfenylation of the indole ring by electrophilic sulfur species, normally generated in situ.7d–f Because it involves the use of readily available indole as the starting material, approach B is the most attractive, and efficient protocols using this strategy have been described in the past few years.
In 2012, Wey proposed a metal-free protocol for the preparation of a range of 3-sulfenylindoles using dimethyl carbonate (DMC) as a green solvent. Products were obtained in yields of up to 96% with 5 mol% I2 as the catalyst and 3 equiv. of DMSO as the oxidant (Fig. 1B-i).8
Subsequently, many attempts aimed at improving this methodology have been described;9–12 for instance through the use of microwaves (MWs) as an alternative energy source to accelerate the reaction (Fig. 1B-ii).9 The selective preparation of mono- and bis-sulfenylated derivatives is an important sulfenylindole-synthesis endeavor. In 2015, Wang and co-workers described the first protocol for the selective formation of mono and bis-arylsulfenylindoles directly from indoles and diaryl disulfides.10 Despite the selectivities, the need to use 50 mol% of I2 and over-stoichiometric amounts of TBHP (2.1 equiv.) contributed to the low atom-efficiency of the reaction (Fig. 1B-iii). Very recently, Barman and Rahaman described a MW-promoted synthesis of mono- and bis-sulfenylindoles using the I2/H2O2 system (Fig. 1B-iv).11 The diorganyl disulfide was generated in situ from the respective sodium sulfinate (RSO2Na) in the presence of I2 (10–20 mol%), H2O2 (2.2 equiv.), and an excess of diethyl phosphite (3 equiv.). Despite these recent advances, the use of sub-equivalent amounts of oxidant during the direct, selective mono- and bis-sulfenylation of indoles remains an unsolved problem, which provides challenges for any new reaction system.
Selenium dioxide (SeO2) was firstly introduced as an oxidizing agent in 1932 by Riley, Morlex, and Friend,12 and to this day remains one of the most convenient reagents for allylic oxidation13 and oxidative-elimination reactions.14 In the past few years, only a few new protocols using SeO2 have been reported, including the dihydroxylation and hydroxymethoxylation of alkenes,15 the syntheses of glyoxal,16 triarylethanones,17 azoxyarenes,18 and in oxidative couplings.19
With this previous work in mind, herein we describe the unprecedent use of the I2/SeO2 system for the preparation of mono- and bis-sulfenylindoles through the C–H sulfenylation of indoles with diaryl disulfides. The selectivity of the reaction is modulated by the amount of SeO2 (Fig. 1C).
Results and discussion
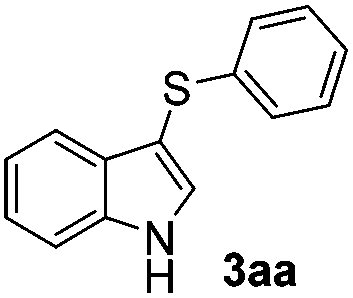
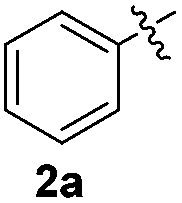
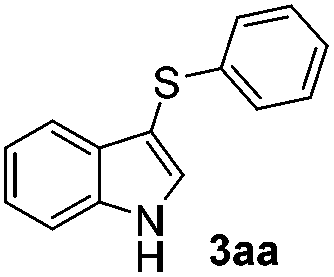
1H-Indole (1a) (0.5 mmol) and diphenyl disulfide 2a (0.3 mmol) were chosen as model substrates during the optimization of the reaction conditions and the preparation 3-(phenylsulfenyl)indole 3aa (Table 1).|
|
||||
|---|---|---|---|---|
| Entry | 2a (mmol) | SeO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (mol%) b (mol%) |
3aa (% conv.) | 4aa (% conv.) |
| a Reaction conditions: 1a (0.5 mmol), glycerol (1.0 mL), and 10 mol% of I2 with respect to disulfide 2a. The progress of the reaction was followed by TLC. b Relative to indole 1a. c Conversion values obtained by GC/MS. | ||||
| 1 | 0.3 | 20 | 84 | — |
| 2 | 0.3 | 30 | 100 | — |
| 3 | 0.3 | 40 | 55 | 45 |
| 4 | 0.3 | 50 | 24 | 76 |
| 5 | 0.3 | 60 | 61 | 39 |
| 6 | 0.3 | 70 | 45 | 55 |
| 7 | 0.3 | 80 | 40 | 60 |
| 8 | 0.3 | 90 | 53 | 47 |
| 9 | 0.3 | 100 | 52 | 48 |
| 10 | 0.6 | 100 | — | 100 |
| 11 | 0.6 | 90 | — | 100 |
| 12 | 0.6 | 80 | — | 100 |
| 13 | 0.6 | 70 | — | 100 |
| 14 | 0.6 | 60 | — | 100 |
| 15 | 0.6 | 50 | 17 | 83 |
| 16 | 0.6 | 40 | 34 | 66 |
| 17 | 0.6 | 30 | 93 | 7 |
In order to evaluate and optimize the amounts of catalyst and oxidant, we conducted an initial experiment using 10 mol% of I2 and 20 mol% of SeO2 in glycerol as the solvent. The expected product 3aa was obtained in very good conversion (84%) after heating at 100 °C for 1 h (Table 1, entry 1). Based on this result, we concentrated our efforts toward improving the yield of 3aa by gradually increasing the amount of SeO2 to 100 mol% (Table 1, entries 2–9).
Indole (1a) was quantitatively converted into the mono-sulfenylindole 3aa using 30 mol% of SeO2 (Table 1, entry 2). On the other hand, mixtures of the mono- and bis-sulfenylated indoles 3aa and 4aa, were detected in different ratios in the presence of larger amounts of SeO2 (40–100 mol%) (Table 1, entries 3–9).
Based on these results, we conclude that the optimal conditions for the preparation selective of mono-sulfenylindole 3aa involves stirring a mixture of 1a, 2a (1.2 equiv.), 10 mol% of I2, and 30 mol% of SeO2 in glycerol for 1 h at 100 °C (Table 1, entry 2).
With these interesting results in hand, our next objective involved determining the optimal amounts of SeO2 and diphenyl disulfide 2a that lead to the selective preparation of bis-sulfenylindole 4aa (Table 1, entries 10–17).
In preliminary studies, we showed that double the amount of diphenyl disulfide 2a (0.6 mmol, instead 0.3 mmol) and 100 mol% of SeO2 (0.5 mmol) led to the exclusive formation of 4aa (Table 1, entry 10; or Table S1,† entry 7). We performed a set of experiments in which the amount of SeO2 varied between 100 and 30 mol%, during which we discovered that 60 mol% was optimal for the complete conversion of indole (1a) to the bis-sulfenylindole 4aa (Table 1, entries 10–17). The use of less SeO2 resulted in an increase in the formation of 3aa, to 17, 34, and 93% with 50, 40 and 30 mol% SeO2, respectively. These data reveal that the oxidizing agent plays a fundamental role in the selectivity of this transformation (Table 1, entries 15–17).
We subsequently studied the reaction-time-dependent effect of the amount of catalyst on conversion, as monitored by GC/MS (Fig. 2 and 3). The reaction that produced bis-(phenylsulfenyl)indole 4aa was completed in 16 min at 100 °C, while at 75 °C 50 min was required. Fig. 2 reveals that indole (1a) was entirely converted into 3aa after 4 min at 100 °C, at which point 3aa begins to be converted into 4aa.
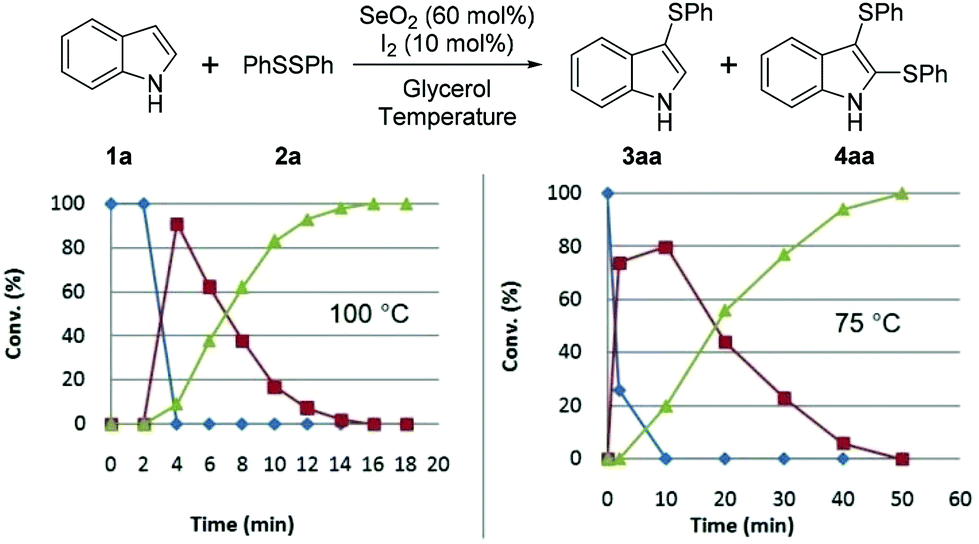 | ||
| Fig. 2 The effect of temperature on reaction. Blue line (◆): 1a, red line (■): 3aa, green line (▲): 4aa [1a (0.5 mmol), 2a (0.6 mmol), SeO2 (60 mol%, 0.033 g), I2 (10 mol%, 0.015 g) and glycerol (1.0 mL)]. | ||
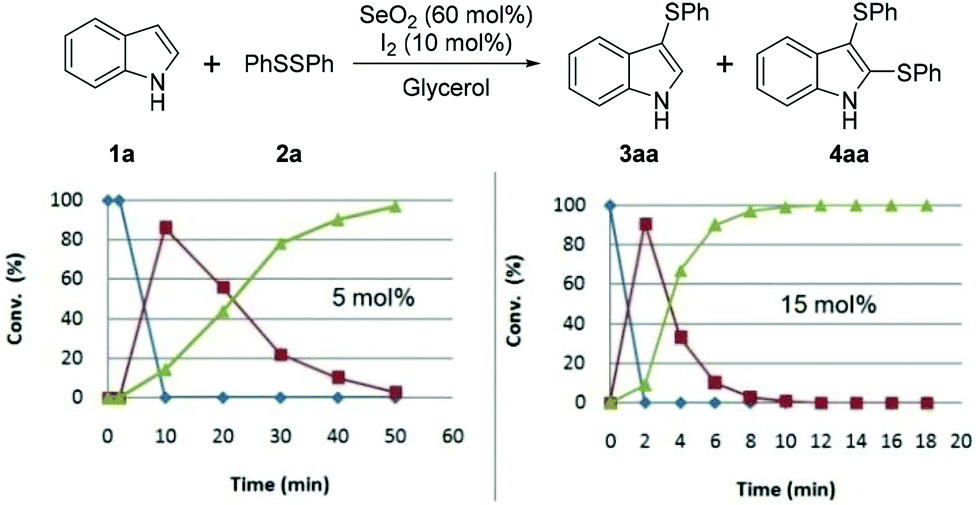 | ||
| Fig. 3 Effect of the amount of I2 (5 mol% = 0.0075 g; 15 mol% = 0.0225 g). Blue line (◆): 1a, red line (■): 3aa, green line (▲): 4aa [1a (0.5 mmol), 2a (0.6 mmol), SeO2 (60 mol%, 0.033 g) and glycerol (1.0 mL)]. | ||
Regarding the catalyst, we observed that 50 min was required for the complete consumption of 1a when 5 mol% of I2 was used, which is four times longer than that required when 10 mol% was used. When 15 mol% of I2 was used, the time required for the complete conversion to 4aa was 12 min, which does not represent a significant reduction in time compared to that required with 10 mol% of I2 (Fig. 3).
With the best reaction conditions in hands, we explored the efficiency and generality of our methodology with other diorganyl disulfides 2 and indoles 1. The optimal conditions for the synthesis of mono-substituted sulfenylindoles 3 worked well for a range of diaryl disulfides containing electron-donating (EDG) and electron-withdrawing groups (EWG). It was not possible, however, to establish a relationship between the performance of the reaction and electronic nature of the aromatic ring in 2 (Table 2).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Indole 1 | Disulfide 2 (R2) | Time (min) | Product 3 | Yieldb (%) |
|
a Reaction conditions: Indole 1 (0.5 mmol), disulfide 2 (0.3 mmol), SeO2 (30 mol%), and I2 (10 mol%) in glycerol (1.0 mL). The reaction progress was followed by TLC.
b Yields following purification by column chromatography.
c A mixture of 3ac and 4ac (89:11) was obtained.
d 20 mol% of SeO2 was used.
e The conditions described in Table 1, entry 14 were used.
|
|||||
| 1 |
|
|
4 |
|
70 |
| 2 | 1a |
|
6 |
|
76 |
| 3 | 1a |
|
4 |
|
84c |
| 4 | 1a |
|
8 |
|
71 |
| 5 | 1a |
|
10 |
|
67 |
| 6 | 1a |
|
18 |
|
23 |
| 7 | 1a |
|
30 |
|
66 |
| 8 | 1a |
|
18 |
|
80d |
| 9 | 1a |
|
20 |
|
22 (84)e |
| 10 | 1a |
|
7 |
|
31 (69)e |
| 11 |
|
2a | 7 |
|
64 |
| 12 |
|
2a | 18 |
|
52 |
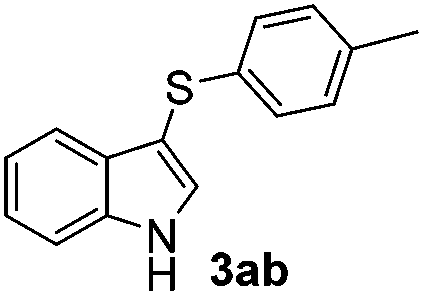
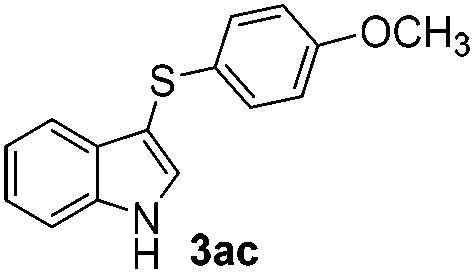
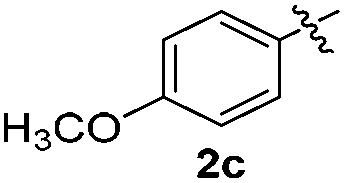
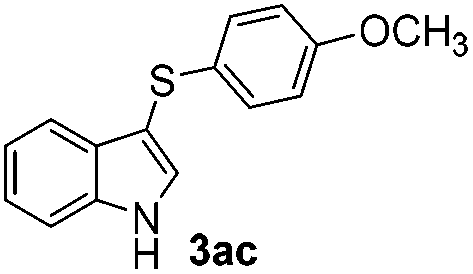
Products bearing groups such as methyl (3ab) and methoxy (3ac) were obtained in better yields than that of the neutral analogue 3aa (Table 2, entries 1–3).
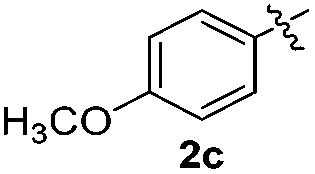
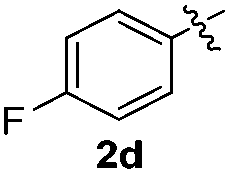
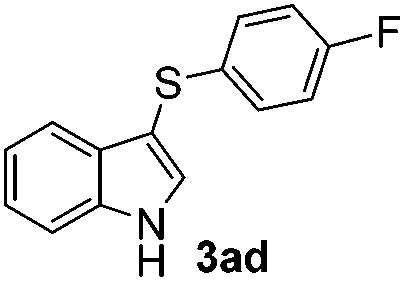
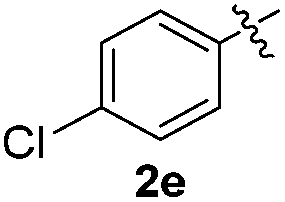
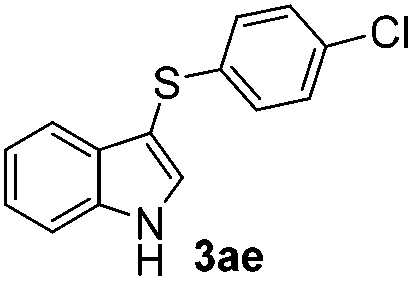
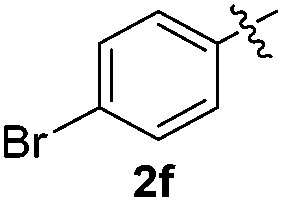
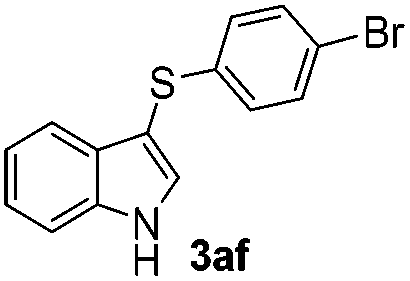
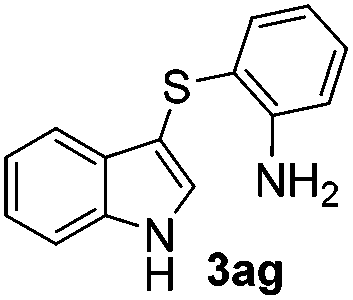
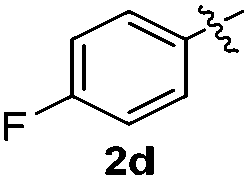
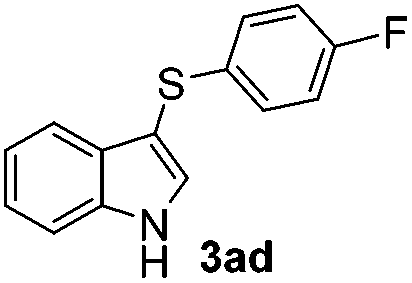
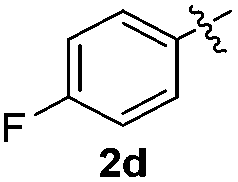
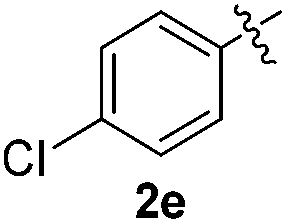
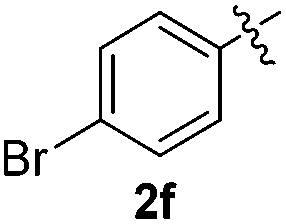
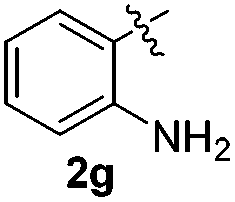
The disulfide 2c (R2 = 4-CH3OC6H4) produced a 89:11 mixture of 3ac and 4ac, while halogen-substituted diaryl disulfides, as 4-fluoro- (2d), 4-chloro- (2e), and 4-bromophenyl (2f) afforded their respective products, 3ad, 3ae, and 3af, in 71, 67 and 23% yields after 8, 10 and 18 min of reaction time, respectively (Table 2, entries 4–6). Additional time (30 min) was required for the preparation of the 2-amino derivative 3ag (Table 2, entry 7), probably due to an electron-donating effect of the amino group positioned ortho to the disulfide in 2g that reduces the electrophilicity of the sulfur (see Scheme 2 for a plausible mechanism).
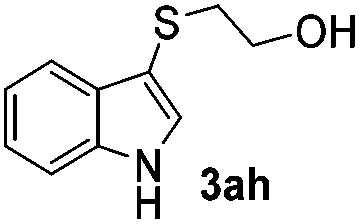
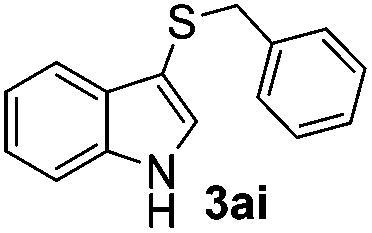
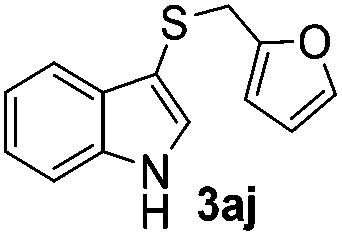
Dibenzyl disulfide (2i) and difurfuryl disulfide (2j), in which the sulfur atoms are attached to sp3-hybridized carbons, afforded lower yields of the respective products 3ai and 3aj (22 and 31%) under the conditions optimized for the formation of mono-sulfenylindoles.
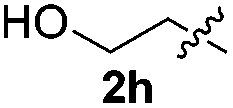
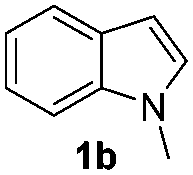
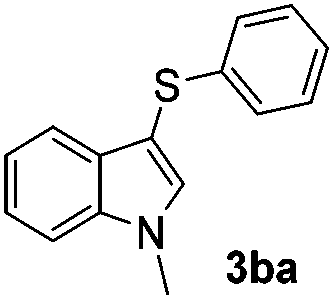
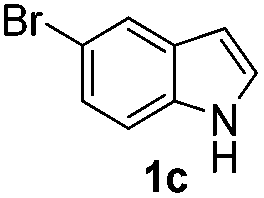
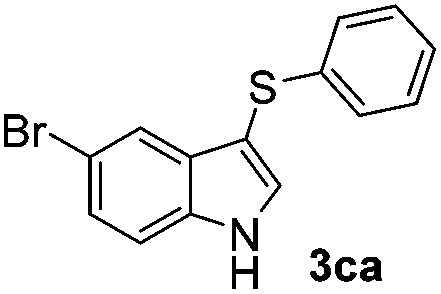
Fortunately, the yields of 3ai and 3aj improved to 84 and 69% when the optimized conditions for the preparation of the bis-sulfenylated indoles 4 (i.e., 60 mol% of SeO2 and 2.4 equiv. of the disulfide 2i or 2j) were used (Table 2, entries 9 and 10). On the other hand, the same effect was not observed for di(2-hydroxyethyl) disulfide 2h; the corresponding mono-sulfenylindole 3ah was obtained in 80% yield after 18 min with 20 mol% SeO2 (Table 2, entry 8). The protocol was successfully extended to other indoles, such as 1-methyl-1H-indole (2b), which reacted with diphenyl disulfide (2a) to give the 3-phenylsulfenylindole 3ba in 64% yield after 7 min (Table 2, entry 11). 5-Bromo-1H-indole (1c) reacted with diphenyl disulfide 2a to give the corresponding 3-sulfenylindole 3ca in 52% yield after 18 min (Table 2, entry 12).
We also observed that glycerol can be replaced by H2O without compromising the reaction yields and selectivities (Table 3). Mono-sulfenylated products 3aa–ae were obtained in almost identical yields and in similar reaction times (Table 3, entries 1–5). A better outcome was observed for the bromo-substituted sulfenylindole 3af, which was obtained in better yield in water (49%) (Table 2, entry 6 vs.Table 3, entry 5).
|
|
||||
|---|---|---|---|---|
| Entry | Disulfide 2 (R2) | Time (min) | Product 3 | Yieldb (%) |
| a Reaction conditions: Indole 1a (0.5 mmol), disulfide 2 (0.3 mmol), SeO2 (30 mol%) and I2 (10 mol%) in water (1.0 mL). The reaction progress was followed by TLC. b Yields following purification by column chromatography. | ||||
| 1 |
|
4 |
|
70 |
| 2 |
|
2 |
|
77 |
| 3 |
|
10 |
|
63 |
| 4 |
|
12 |
|
72 |
| 5 |
|
16 |
|
49 |
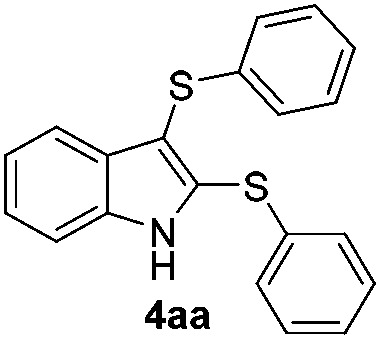
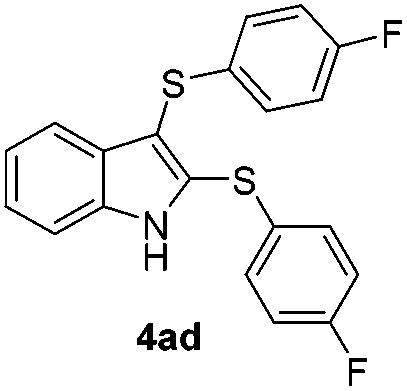
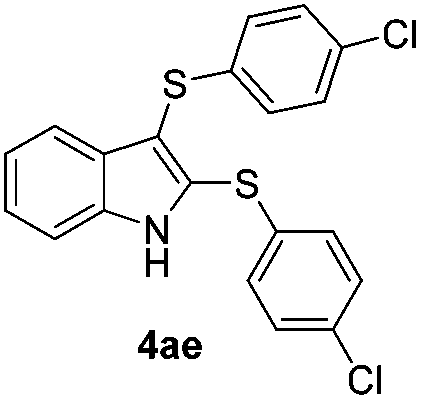
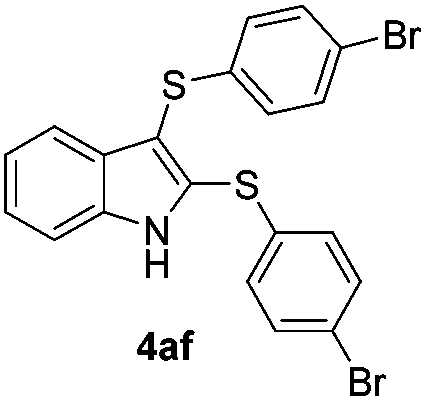
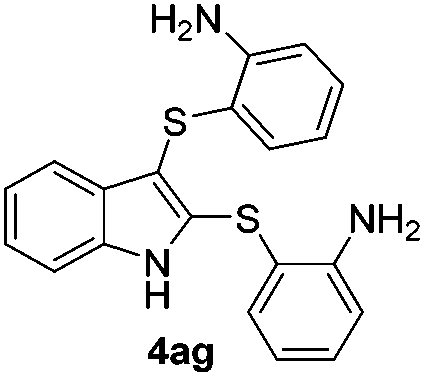
We next used the optimized conditions described in Table 1, entry 14 to prepare the 2,3-bis-sulfenylindoles 4, by reacting 1H-indole (1a) with several diaryl disulfides 2a–g (Table 4); consequently, 2,3-bis(phenylsulfenyl)indole 4aa was isolated in 85% yield after 16 min following the reaction of 1a with diphenyl disulfide 2a (Table 4, entry 1). Diaryl disulfides 2b and 2c, bearing EDGs (R2 = 4-CH3 and R2 = 4-CH3O), reacted with 1a to form 4ab and 4ac in 75 and 71% yields respectively in no more than 20 min (Table 4, entries 2 and 3). In the case of 4ac, the formation of the mono-substituted 3-sulfenylindole 3ac was also observed (4ac:3ac ratio of 76:24). The 4-fluoro derivative 4ad was isolated in 62% yield after 19 min, while moderate yields of the chloro- and bromo-analogues 4ae (39%) and 4af (53%) were obtained (Table 4, entries 4–6). As was observed for the mono-sulfenylindole 3ag, the ortho-amino derivative 4ag was obtained in low yield (33%), even increasing the amount of SeO2 to 80 mol% (Table 4, entry 7). An excellent yield (97%) of 4ba was obtained from the reaction of 1-methyl-1H-indole (2b) with 2a after 30 min (entry 8), while 5-bromo-1H-indole (2c) was converted to the respective 2,3-bis(phenylsulfenyl)indole 4ca in 77% yield after 18 min (Table 4, entry 9).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Indole 1 | Disulfide 2 (R2) | Time (min) | Product 4 | Yieldb (%) |
|
a Reaction conditions: 1 (0.5 mmol), disulfide 2 (0.6 mmol), SeO2 (60 mol%), and I2 (10 mol%) in glycerol (1.0 mL). The reaction progress was followed by TLC.
b Yields following purification by column chromatography.
c A mixture of 4ac and 3ac (76:24) was obtained.
d 80 mol% of SeO2 was used.
|
|||||
| 1 |
|
|
16 |
|
85 |
| 2 | 1a |
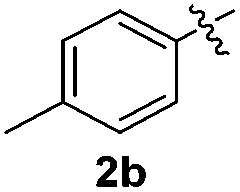
|
20 |
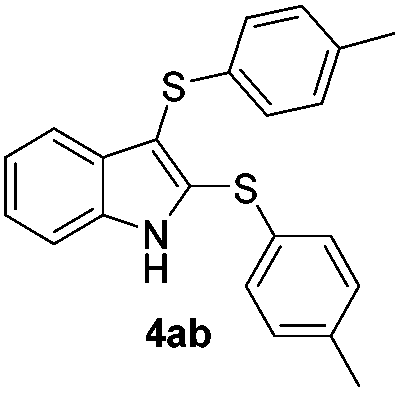
|
75 |
| 3 | 1a |
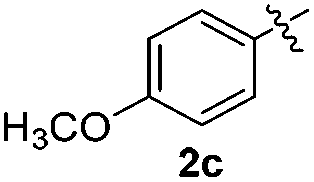
|
10 |
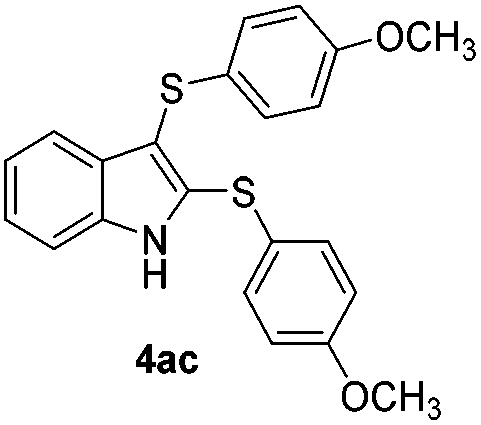
|
71c |
| 4 | 1a |
|
19 |
|
62 |
| 5 | 1a |
|
18 |
|
39 |
| 6 | 1a |
|
23 |
|
53 |
| 7 | 1a |
|
23 |
|
33d |
| 8 |
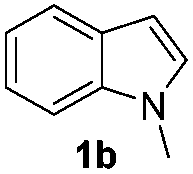
|
2a | 30 |
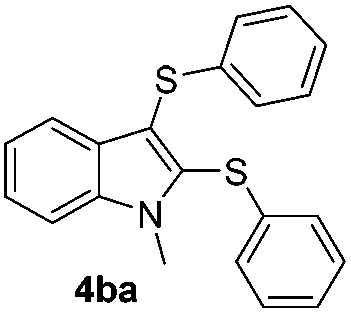
|
97 |
| 9 |
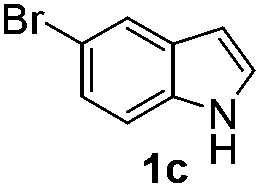
|
2a | 18 |
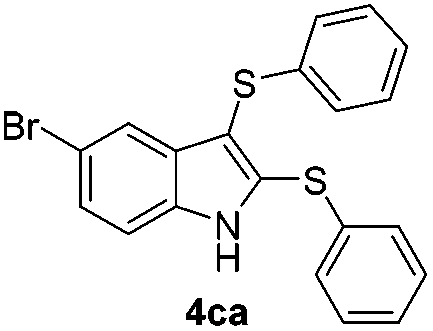
|
77 |
Interestingly, dibenzyl disulfide (2i) and difurfuryl disulfide (2j) afforded the mono-substituted products 3ai and 3aj exclusively under these disulfenylation conditions, as previously discussed (Table 2, entries 9 and 10). Increasing the amount of SeO2 to 100 mol% was ineffective at producing isolable amounts of the bis-sulfenyl partners, while di(2-hydroxyethyl) disulfide 2h afforded a complex mixture of products.
Mechanisms for the formation of mono- and bis-sulfenylindoles are already described in the literature.20 Even the reaction between indole (1a) and SeO2 has been previously described by Witkop, in 1947:21 this reaction gives 3,3′-bis-indolyl selenide 5, whose structure was elucidated by Wilshire in 1967 (Scheme 1, #1).22
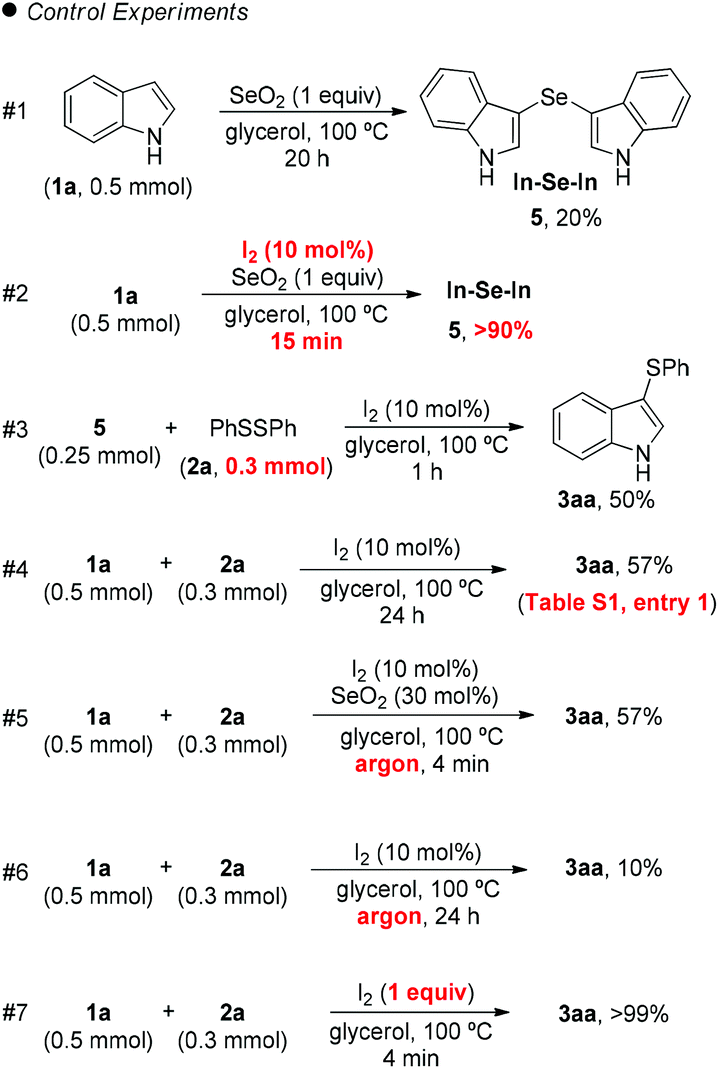 | ||
| Scheme 1 Control experiments in the synthesis of 3aa. | ||
However, SeO2 has never been explored as an oxidizing agent in this specific transformation. In order to clarify the role of the I2/SeO2 system in the promotion and selectivity of this reaction, several control experiments were performed (Scheme 1).
Initially, we hypothesized that the 3,3′-diindolyl selenide 5 could be the key intermediate in the reaction mechanism. When we reacted indole 1a with SeO2 in glycerol at 100 °C, 20% of 5 was obtained after 20 h (Scheme 1, control exp. #1). On the other hand, the addition of a catalytic amount of I2 (10 mol%) to the reaction, resulted in the formation of 5 in 90% after 15 min (Scheme 1, #2).
Considering that 3,3′-diindolyl selenide 5 may be a key intermediate in the overall transformation, we reacted 5 with diphenyl disulfide (2a) (1.2 equiv.) and I2 (30 mol%), in the absence of SeO2, which led to the formation of 3aa in 50% yield after 1 h (Scheme 1, #3). However, 24 h was necessary in order to access 3aa in 57% yield starting from indole 1a, instead intermediate 5, (Scheme 1, #4), which demonstrates that the reactivity of intermediate 5 is significant higher than that of indole 1a.
When the standard reaction conditions used to prepare 3aa were performed under argon, a decreasing in the yield, from 70% (Table 2, entry 1) to 57% was observed, after 4 min of reaction (Scheme 1, #5). The yield of 3aa was even smaller, (10% after 24 h) in the absence of SeO2 or air (Scheme 1, #6). Taken together, experiments #5 and #6 show that SeO2 plays a crucial role that guarantees the efficiency of the reaction, and that atmospheric oxygen plays a role in which it acts as a co-oxidant that improves the reaction efficiency.
Finally, the reaction was performed in the absence of SeO2 but with 1 equiv. of molecular iodine, which delivered 3aa in 99% yield after 4 min (Scheme 1, #7), which shows that the formation of an electrophilic sulfur species in the reaction medium affords an ideal reaction outcome.
Based on these experiments, we propose a mechanism for formation 3aa, which contains three catalytic cycles (Scheme 2). In Cycle A, the first step involves the reaction of 2a with I2 to form PhSI, which then reacts with indole to form 3aa and HI; this cycle is completed by the reaction of HI with SeO2 to form I2. The in situ-formed selenium by-product (perhaps elemental Se reduced by HI) facilitates Cycle B by reacting with indole (1a) to form intermediate 5.23 Intermediate I is formed after reaction with PhSI, which is generated in situ from PhSSPh (2a) and I2.24 Intermediate I then suffers disproportionation to deliver the expected product 3aa and selenone II. Nucleophilic attack of II by 1a forms more 5 and HI, which is oxidized back to I2 by SeO2, giving more Se (Cycle B). The formation of the bis-sulfenylated indole 4aa could follow a similar pathway to that shown in Cycle A, with the exception that the monosulfenylated indole 3aa is the starting material instead the parent 1a, as previously described (Cycle C).20 This mechanism accounts for the sub-stoichiometric amounts of SeO2 required in these reactions because it is involved in the formation of 3aa (or 4aa) in Cycle A (or Cycle C), and indirectly involved in the formation of additional 3aa through Cycle B.
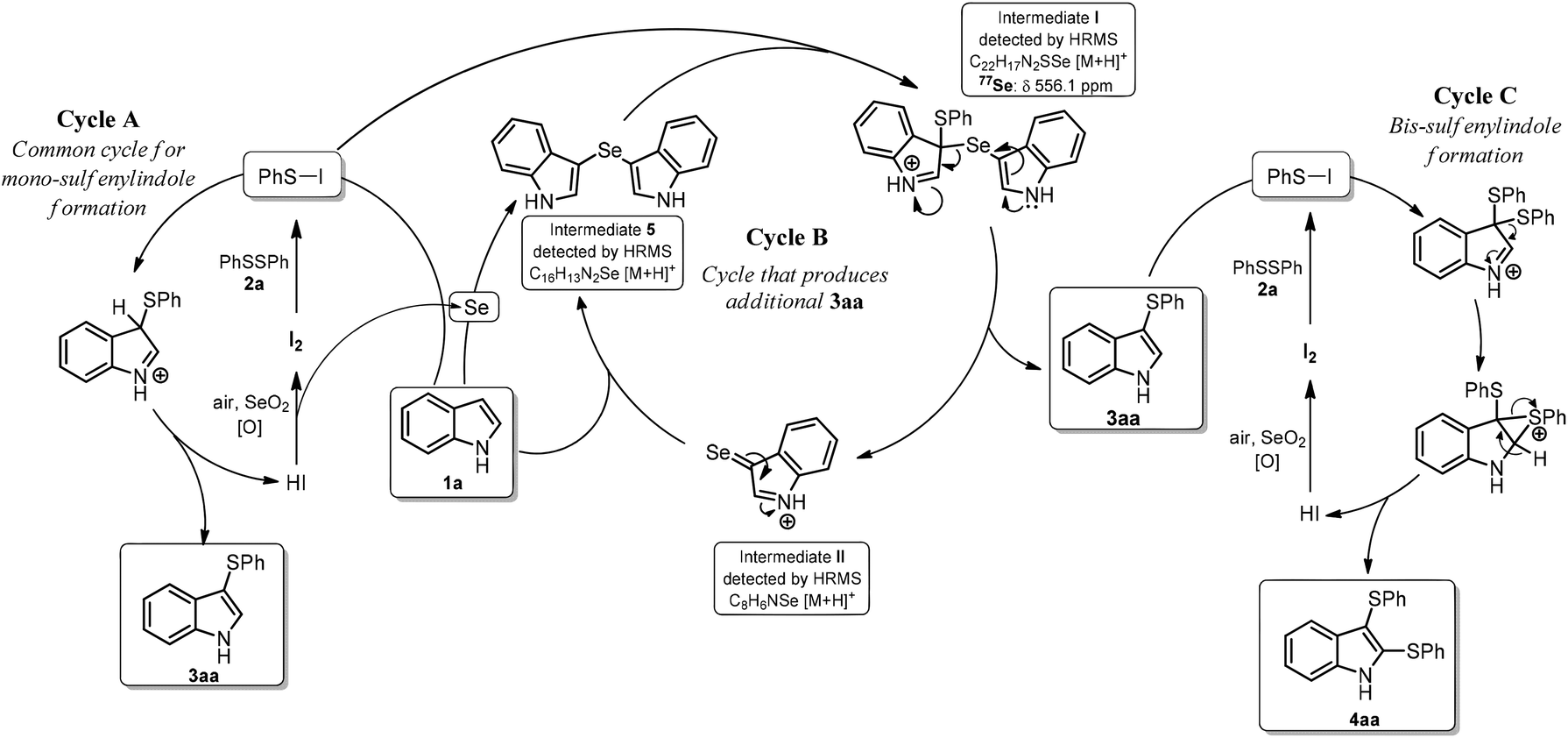 | ||
| Scheme 2 Proposed mechanism for the formation of 3aa and 4aa. | ||
To provide support for the proposed mechanism, aliquots from a 3aa-forming reaction were directly injected in a high-resolution mass spectrometer equipped with an APCI source (see ESI† for experimental details). Both key intermediates 5 and I were detected as relatively intense [M + H]+ ions with m/z values of 313.0213 and 195.9647, respectively, along with an ion at m/z 226.0671, which corresponds to product 3aa ([M + H]+) (Fig. S2–S6†). A very-low intensity signal for the proposed key intermediate II was detected at m/z 421.0256; importantly the isotopic distribution is in agreement with the simulated one (Fig. S3†).
Experimental
General information
The reactions were monitored by TLC on Merk silica gel (60 F254) visualized with UV light or with 5% of vanillin in 10% H2SO4 with heating. Merck silica gel (particle size 0.040–0.063 mm) was used for flash chromatography. 1H NMR spectra were obtained at 400 MHz on a Bruker DPX 400 spectrometer. The spectra were recorded in CDCl3. Chemical shifts are reported in ppm, referenced against tetramethylsilane (TMS) as internal standard. 1H coupling patterns are described as singlet (s), doublet (d), triplet (t) and multiplet (m). Coupling constants (J) are reported in Hz. 13C NMR spectra were obtained at 100 MHz on the above-mentioned instrument. The chemical shifts are reported in ppm, referenced to the solvent peak (CDCl3). Low-resolution mass spectra were obtained on a Shimadzu GC-MS-QP2010 mass spectrometer. GC analyses were performed using a RESTEC RTX-5MS capillary column (30 m, 0.25 mm id, 0.25 μm film thickness) using products dissolved in ethyl acetate under the following conditions: Injected sample volume, 1.0 μL; flow rate, 54.1 mL min−1; initial inlet temperature, 40 °C ramped to 72 °C at 10 °C min−1 followed by a 5 °C min−1 ramp to 100 °C (held for 10 min) and 10 °C min−1 to 280 °C and held for 20 min (total run time: 56.8 min). High-resolution mass spectra were obtained on a Bruker Daltonics micrOTOF-Q II instrument equipped with an APCI source operating in positive mode. The samples were dissolved in HPLC-grade acetonitrile and injected into the APCI source by means of a syringe pump at a flow rate of 5.0 μL min−1. The Compass 1.3 for micrOTOF-Q II software (Bruker daltonics, USA) was used for data acquisition, processing, and isotopic distribution simulations.General procedure for the synthesis of the 3-(sulfenyl)-1H-indoles 3
The diorganyl disulfide 2a–j (0.3 mmol), indole 1a–c (0.5 mmol), SeO2 (30 mol%, 0.016 g), I2 (10 mol%, 0.0076 g) and glycerol (1.0 mL) were added to a round-bottomed flask. The resultant solution was stirred at 100 °C (oil bath) for the time indicated in Table 2, after which the reaction mixture was poured into water (20 mL), extracted with ethyl acetate (3 × 10 mL), dried over MgSO4, filtered, and concentrated under vacuum. The residue was then purified by silica-gel chromatography with hexane/ethyl acetate as the eluent.General procedure for the synthesis of the 2,3-bis(sulfenyl)-1H-indoles 4
The diorganyl disulfide 2a–g (0.6 mmol), indole 1a–c (0.5 mmol), SeO2 (60 mol%, 0.033 g), I2 (10 mol%, 0.015 g) and glycerol (1.0 mL) were added to a round-bottomed flask. The resulting mixture was stirred at 100 °C (oil bath) for the time indicated in Table 3, after which the reaction mixture was poured into water (20 mL), extracted with ethyl acetate (3 × 10 mL), dried over MgSO4, filtered, and concentrated under vacuum. The residue was purified by silica-gel chromatography with hexane/ethyl acetate as the eluent.![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) . 13C NMR (100 MHz, CDCl3) δ 136.6, 134.9, 133.7, 133.3, 129.8, 127.8, 127.4, 127.1, 125.3, 124.6, 123.7, 119.9, 112.5, 112.3, 106.0. MS (relative intensity, %) m/z: 413 (23), 303 (4), 255 (5), 223 (100), 207 (10), 119 (4), 77 (5). HRMS (APCI-TOF) m/z: calculated for C20H14BrNS2 [M + H]+ 411.9825, found 411.9824.
. 13C NMR (100 MHz, CDCl3) δ 136.6, 134.9, 133.7, 133.3, 129.8, 127.8, 127.4, 127.1, 125.3, 124.6, 123.7, 119.9, 112.5, 112.3, 106.0. MS (relative intensity, %) m/z: 413 (23), 303 (4), 255 (5), 223 (100), 207 (10), 119 (4), 77 (5). HRMS (APCI-TOF) m/z: calculated for C20H14BrNS2 [M + H]+ 411.9825, found 411.9824.
Conclusions
In summary, we describe herein an easy, inexpensive, rapid, and efficient method for the controlled preparation of 3-sulfenylindoles and 2,3-bis-sulfenylindoles using I2/SeO2 as catalyst/oxidizing agent and glycerol as the solvent. This new protocol uses readily available diorganyl disulfides and indoles as starting materials and involves S–S bond cleavage and the formation of new S–Csp2 (indole) bonds. The products were selectively obtained in good yields in only a few minutes in air and with good substituent tolerance on both the disulfide and the indole.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank FAPERGS, CNPq, and CAPES for financial support. CNPq is also acknowledged for Fellowships to G. P., D. A., and E. J. L. University of Perugia is acknowledged for an “accordi quadro” Fellowship to B. M. and financial support through “Ricerca di Base 2017” to C. S. The work described in this manuscript is part of the scientific activity of the international multidisciplinary “SeS Redox and Catalysis” network.Notes and references
- A. Baeyer and A. Emmerling, Ber. Dtsch. Chem. Ges., 1869, 2, 679 CrossRef.
- M. Bandini and A. Eichholzer, Angew. Chem., Int. Ed., 2009, 48, 9608 CrossRef PubMed.
- G. La Regina, R. Bai, W. M. Rensen, E. Di Cesare, A. Coluccia, F. Piscitelli, V. Famiglini, A. Reggio, M. Nalli, S. Pelliccia, E. Da Pozzo, B. Costa, I. Granata, A. Porta, B. Maresca, A. Soriani, M. L. Iannitto, A. Santoni, J. Li, M. M. Cona, F. Chen, Y. Ni, A. Brancale, G. Dondio, S. Vultaggio, M. Varasi, C. Mercurio, C. Martini, E. Hamel, P. Lavia, E. Novellino and R. Silvestri, J. Med. Chem., 2013, 56, 123 CrossRef PubMed.
- J. A. Campbell, V. Bordunov, C. A. Broka, M. F. Browner, J. M. Kress, T. Mirzadegan, C. Ramesha, B. F. Sanpablo, R. Stabler, P. Takahara, A. Villasenor, K. A. M. Walker, J.-H. Wang, M. Welch and P. Weller, Bioorg. Med. Chem. Lett., 2004, 14, 4741 Search PubMed.
- (a) R. Silvestri, G. D. Martino, G. La Regina, M. Artico, S. Massa, L. Vargiu, M. Mura, A. G. Loi, T. Marceddu and P. La Colla, J. Med. Chem., 2003, 46, 2482 CrossRef PubMed; (b) P. C. Unangst, D. T. Connor, S. R. Stabler, R. J. Weikert, M. E. Carethers, J. A. Kennedy, D. O. Thueson, J. C. Chestnut, R. L. Adolphson and M. C. Conroy, J. Med. Chem., 1989, 32, 1360 CrossRef PubMed; (c) C. D. Funk, Nat. Rev. Drug Discovery, 2005, 4, 664 CrossRef PubMed.
- For reviews, see: (a) M. Feng, B. Tang, S. H. Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200 CrossRef PubMed; (b) H. Liu and X. Jiang, Chem. – Asian J., 2013, 8, 2546 CrossRef PubMed.
- (a) H. Du, R. Tang, C. Deng, Y. Liu, J. Li and X. Zhang, Adv. Synth. Catal., 2011, 353, 2739 Search PubMed; (b) Y. Chen, C. Cho, F. Shi and R. C. Larock, J. Org. Chem., 2009, 74, 6802 CrossRef PubMed; (c) Y. Chen, C. Cho, F. Shi and R. C. Larock, Org. Lett., 2009, 11, 173 CrossRef PubMed; (d) E. G. Zimmermann, S. Thurow, C. S. Freitas, S. R. Mendes, G. Perin, D. Alves, R. G. Jacob and E. J. Lenardão, Molecules, 2013, 18, 4081 CrossRef PubMed; (e) C. C. Silveira, S. R. Mendes, L. Wolf, G. M. Martins and L. von Mühlen, Tetrahedron, 2012, 68, 10464 CrossRef; (f) X. Fang, R. Tang, P. Zhong and J. Li, Synthesis, 2009, 4183 Search PubMed.
- W. Ge and Y. Wei, Green Chem., 2012, 14, 2066 RSC.
- J. B. Azeredo, M. Godoi, G. M. Martins, C. C. Silveira and A. L. Braga, J. Org. Chem., 2014, 79, 4125 CrossRef PubMed.
- H. Zhang, X. Bao, Y. Song, J. Qu and B. Wang, Tetrahedron, 2015, 71, 8885 CrossRef.
- R. Rahaman and P. Barman, Eur. J. Org. Chem., 2017, 6327 CrossRef.
- (a) H. L. Hiley, J. F. Morley and N. A. C. Friend, J. Chem. Soc., 1932, 1875 Search PubMed; (b) H. L. Hiley and N. A. C. Friend, J. Chem. Soc., 1932, 2342 Search PubMed.
- (a) D. Chen, W.-D. Xu, H.-M. Liu, M.-M. Li, Y.-M. Yan, X.-X. Li, Y. Li, Y.-X. Cheng and H.-B. Qin, Chem. Commun., 2016, 52, 8561 RSC; (b) R. M. Patel, V. G. Puranik and N. P. Argade, Org. Biomol. Chem., 2011, 9, 6312 RSC; (c) U. Kauhl, L. Andernach, S. Weck, L. P. Sandjo, S. Jacob, E. Thines and T. Opatz, J. Org. Chem., 2016, 81, 215 CrossRef PubMed.
- (a) Y. Wang, B. Zhu, Q. Xu, Q. Zhu and L. Yu, RSC Adv., 2014, 4, 49170 RSC; (b) B. A. Sousa and A. A. dos Santos, Eur. J. Org. Chem., 2012, 3431 CrossRef.
- C. Santi, R. D. Lorenzo, C. Tidei, L. Bagnoli and T. Wirth, Tetrahedron, 2012, 68, 10530 CrossRef.
- R. M. Young and M. T. Davies-Coleman, Tetrahedron Lett., 2011, 52, 4036 CrossRef.
- B. M. Laloo, H. Mecadon, M. R. Rohman, I. Kharbangar, I. Kharkongor, M. Rajbangshi, R. Nongkhlaw and B. Myrboh, J. Org. Chem., 2012, 77, 707 CrossRef PubMed.
- M. R. Rohman, I. Kharkongor, M. Rajbangshi, H. Mecadon, B. M. Laloo, P. R. Sahu, I. Kharbangar and B. Myrboh, Eur. J. Org. Chem., 2012, 320 CrossRef.
- C. Gebhardt, B. Priewisch, I. Irran and K. Rück-Braun, Synthesis, 2008, 1889 Search PubMed.
- (a) K. J. Anzai, Heterocycl. Chem., 1979, 16, 567 CrossRef; (b) R. Plate, H. C. J. Ottenheijm and R. J. F. Nivard, J. Org. Chem., 1984, 49, 540 CrossRef; (c) R. Plate, H. C. J. Ottenheijm and R. J. F, Tetrahedron, 1986, 42, 4511 CrossRef; (d) P. Hamel and P. Préville, J. Org. Chem., 1996, 61, 1573 CrossRef PubMed; (e) P. Hamel, Tetrahedron Lett., 1997, 38, 8473 CrossRef; (f) P. Hamel, Y. Girard and J. G. Atkinson, J. Chem. Soc., Chem. Commun., 1989, 63 RSC.
- B. Witkop, Ann., 1947, 558, 106 Search PubMed.
- J. F. K. Wilshire, Aust. J. Chem., 1967, 20, 359 CrossRef.
- We speculate that SeO2 is reduced to Se by HI (SeO2 + 4HI → Se + 2I2 + 2H2O). The in situ-formed Se then acts as an electrophile for indole (1a) to form a selenide (RSe−) that may react with I2 to form RSeI, and then with another equivalent of 1a to form 5. HI has been reported to be an efficient reducing agent in organic synthesis; for reviews, see: (a) I. D. Entwistle and W. W. Wood, Hydrogenolysis of Allyl and Benzyl Halides and Related Compounds, in Comprehensive Organic Synthesis: selectivity, strategy and efficiency in modern organic chemistry, ed. B. M. Trost and I. Fleming, Pergamon Press, Oxford, 1991, vol. 8, pp. 955–982 Search PubMed; (b) G. W. Breton, P. J. Kropp and R. G. Harvey, Hydrogen Iodide, in Encyclopedia of Reagents for Organic Synthesis, ed. L. Paquette, J. Wiley & Sons, New York, 2004, DOI:10.1002/047084289X.rh039.
- When equimolar amounts of 3,3′-diindolyl selenide 5 and PhSSPh 2a were mixed in MeOD-d3, after 12 h it was observed in the 77Se-NMR the incipient formation of a product having a chemical shift of 556 ppm that can be speculated to be due to intermediate I. This is in agreement also to the data reported in literature for some selenoketals. See: H. Duddeck, P. Wagner and A. Biallab, Magn. Reson. Chem., 1991, 29, 248 CrossRef.
- C. D. Prasad, S. Kumar, M. Sattar, A. Adhikary and S. Kumar, Org. Biomol. Chem., 2013, 11, 8036 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and figures of NMR spectra. See DOI: 10.1039/c8qo00360b |
| This journal is © the Partner Organisations 2018 |