
Efficient Co@CoPx core–shell nanochains catalyst for the oxygen evolution reaction†
Xiaotao
Yuan‡
a,
Zhe
Zhang‡
a,
Zichao
Liu
a,
Xin
Wang
a,
Chenlong
Dong
a,
Muhammad Sohail
Riaz
a and
Fuqiang
Huang
 *ab
*ab
aState Key Laboratory of Rare Earth Materials Chemistry and Applications, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, P.R. China
bState Key Laboratory of High Performance Ceramics and Superfine Microstructures.Shanghai Institute of Ceramics, Chinese Academy of Sciences, 1295 Dingxi Road, Shanghai 200050, P.R. China. E-mail: huangfq@pku.edu.cn
First published on 30th May 2018
Abstract
Co@CoPx core–shell nanochains were prepared via a direct-current arc-discharge method and subsequent phosphorization at 350 °C. The nanochains comprise nanospheres connected to each other with a length of several micrometers. The amount of CoPx can be easily changed by varying the phosphorization time. In the Co@CoPx core–shell structure, Co serves as a conductive channel, which improves the reaction kinetics of the oxygen evolution process on CoPx. Co metal can also inject electrons into CoPx, thus changing the work function of CoPx and improving its oxygen evolution efficiency. Based on the optimized structure, the Co@CoPx nanochains catalyst exhibits excellent oxygen evolution performance. Co@CoPx achieves an overpotential of 310 mV at a current density of 10 mA cm−2, which is lower than that of CoPx (418 mV), Co (408 mV), and RuO2 (317 mV). Furthermore, Co@CoPx exhibits good durability during the long-term electrochemical test.
Introduction
Electrochemical water splitting has drawn significant attention since it is an efficient way to obtain clean hydrogen fuel.1–3 In electrochemical processes, overpotential is required to drive the sluggish oxygen evolution reaction (OER), which leads to significant energy loss.4–7 Electrocatalysts with high activity and durability are anticipated to accelerate the oxygen evolution process. To date, RuO2 and IrO2 have been proven as the best catalysts for the OER; however, their high cost and elemental scarcity limit their use in commercial applications.8–10Over the past decade, several abundant 3d transition metal-derived materials have shown promising applications in OER.11–18 Cobalt-based materials, particularly cobalt phosphides, have been widely studied because of their low-cost and noble metal-like catalytic performance.19–23 However, cobalt phosphides alone show limited OER catalytic activity because of their relatively low specific area and charge transfer efficiency. The preparation of carbon-supported materials is a strategy generally used to improve the catalytic performance of cobalt phosphides.24–27 However, the synthesis of carbon materials is sometimes complicated. Coupling with metals can also improve the catalytic activity of the OER catalysts, as presented in our previous studies.28–31 Conductive metals can accelerate the electron exchange process between the catalyst and electrolyte, thus improving the kinetics of the reaction. Metals can also inject electrons into the catalyst, thus changing its work function and improving the efficiency of OER. Preparing nanomaterials with one-dimensional structure is also an effective way to enhance the activity of OER catalysts since one-dimensional materials possess high specific surface areas and charge transfer efficiency.32–34
Herein, one-dimensional Co@CoPx core–shell nanochains were prepared via an arc-discharge method and subsequent phosphorization. The nanochains consist of uniform nanospheres with diameter of 30–50 nm. The length of the nanochains can extend to several micrometers. The amount of CoPx in Co@CoPx can be controlled by varying the phosphorization time. The Co@CoPx nanochains catalyst exhibits excellent catalytic activity for OER. The optimal sample achieves an overpotential of 310 mV at a current density of 10 mA cm−2, which is superior to that of Co nanochains (408 mV), CoPx (418 mV), and RuO2 (317 mV). Co@CoPx also exhibits good durability during the long-term test.
Experimental
Synthesis
The chemicals used in this study were of analytical reagent grade and used without further purification.Synthesis of Co nanochains
Co nanochains were synthesized via a direct current arc-discharge method, which was applied in our previous research studies.31,32,35 The anode was a cobalt rod with diameter of 8 mm and the cathode was a pure graphite rod. The two electrodes were installed horizontally. The chamber was first evacuated to 3 Pa, and then filled with a gas mixture of He and H2 to 0.08 MPa (pressure ratio, He![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2 = 2:1). The reaction was carried out at 40 A, and the two electrodes were kept 2 mm away from each other by controlling the cathode.
:H2 = 2:1). The reaction was carried out at 40 A, and the two electrodes were kept 2 mm away from each other by controlling the cathode.
Synthesis of Co@CoPx nanochains
Initially, 8 mg Co nanochains and 28.8 mg NaH2PO2·H2O were placed separately in an evacuated quartz tube. The reaction was carried out at 350 °C for 15 min, 0.5 h, 1 h, 1.5 h and 2 h to obtain a series of Co@CoPx core–shell nanochains.Synthesis of CoPx
CoPx was synthesized the same way as Co@CoPx, except the amount of NaH2PO2·H2O was increased to 57.6 mg and the reaction time was extended to 12 h.Characterization
Transmission electron microscope images (TEM) were acquired using a JEM-2100 at an accelerating voltage of 200 kV. Scanning electron microscope (SEM) images were acquired on a Hitachi S-4800 working at 10 kV. X-ray diffraction (XRD) was carried out on a Bruker D2 diffractometer using Cu Kα radiation (λ = 1.5418 Å). X-ray photoelectron spectroscopy (XPS) was carried out on an Axis Ultra Photoelectron Spectrometer (Kratos Analytical Ltd) using a monochromatized Al Kα anode (225 W, 15 mA, and 15 kV). The C 1s peak with binding energy of 284.8 eV was used for calibration.Electrochemical measurement
The catalyst ink was prepared by dispersing 5 mg sample in 1 mL 0.5% Nafion solution under ultrasonic treatment. Then, 10 μL catalyst ink was casted onto a glassy carbon electrode with diameter of 5 mm The tests were performed on a CHI660E electrochemical workstation using a three-electrode system in 0.1 M KOH. The catalyst-modified electrode was used as the working electrode and Pt wire and Hg/HgO (1 M NaOH) were used as the counter and reference electrodes, respectively.Cyclic voltammetry (CV) was carried out at a scan rate of 50 mV s−1. Linear sweep voltammetry (LSV) data was collected at a scan rate of 5 mV s−1. Both CV and LSV were performed in the range of 0 V to 1.0 V. Electrochemical impedance spectroscopy (EIS) was carried in the frequency ranging from 0.01 Hz to 100 kHz. Chronopotentiometry was conducted to evaluate the stability of the catalysts, and the current density was fixed at 10 mA cm−2.
The electrochemical active surface area (ECSA) of the catalysts was acquired from a CV test in the region where no redox reaction occurs (0.05–0.1 V vs. Hg/HgO).36,37 The ECSA is linearly proportional to the double-layer capacitance (Cdl), which can be calculated from the CV data.
Since Hg/HgO was used as the reference electrode, the data was calibrated to the RHE referring to the literature.38 CV was performed in hydrogen saturated 0.1 M KOH at a scan rate of 1 mV s−1. Platinum wire was used as the working and counter electrodes and Hg/HgO was used as the reference electrode. The average of the two potentials at zero current, which was determined to be 0.866 V, was used to calibrate the data:
| E (vs. RHE) = E (vs. Hg/HgO) + 0.866 |
Results and discussion
The Co@CoPx core–shell nanochains were prepared via the phosphorization of arc-discharge derived Co nanochains (Fig. 1a). The Co nanochains and NaH2PO2·H2O were placed separately in a quartz tube and the experiment was carried out at 350 °C. Fig. 1b shows the XRD patterns of Co@CoPx and Co nanochains (prepared under 1 h). The XRD pattern of the Co nanochains show diffraction peaks at 44.2°, 51.5° and 75.8°, which are indexed to the (111), (200) and (220) planes of Co with a face-centered cubic structure, respectively (JCPDS Card no. 15-0806). After phosphorization, new diffraction peaks appeared at 40.7°, 43.3° and 52.0°, which are assigned to the (121), (211) and (002) planes of Co2P, respectively (JCPDS Card no. 32-0306). The small peaks at 31.6° and 48.1° can be assigned to CoP (JCPDS Card no. 29-0497).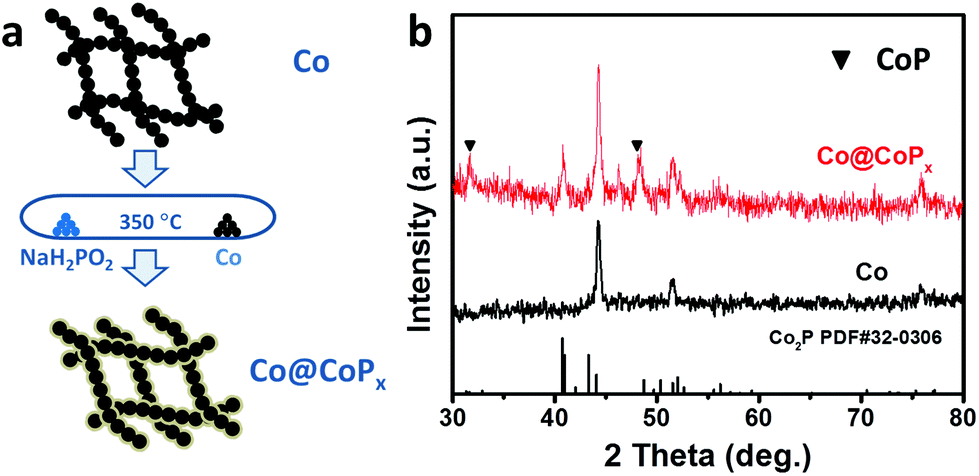 | ||
| Fig. 1 (a) Schematic illustration of the synthesis of the Co@CoPx nanochains. (b) XRD patterns of Co and Co@CoPx nanochains (prepared under 1 h). | ||
SEM and TEM were used to study the detailed morphology of the samples. Co@CoPx exhibits a nanochain structure (Fig. 2a), which is similar to Co (Fig. S1†), indicating the retention of the structural integrity after phosphorization. The Co@CoPx nanochains exhibit a diameter of 50–60 nm and length of up to several micrometers. The TEM image (Fig. 2c) also indicates that the Co@CoPx nanochains possess a core–shell structure with shell thickness of about 10 nm. The shell of Co@CoPx does not show distinct lattice fringes because of the poor crystallinity of CoPx.
 | ||
| Fig. 2 (a) SEM image of the Co@CoPx nanochains (prepared under 1 h). (b–c) TEM and (d) HRTEM images of Co@CoPx nanochains (prepared under 1 h). | ||
Co@CoPx nanochains with phosphorization time of 15 min, 0.5 h, 1.5 h and 2 h were also prepared. The diffraction peak intensity and shell thickness of CoPx increase as the phosphorization time increased from 15 min to 2 h (Fig. S2†). The perfect core–shell nanochains were acquired at preparation times ranging from 15 min to 1 h (Fig. S3 and S4†). On extending the phosphorization time, some pores appeared, and the connection between the Co nanospheres was severed because of the consumption of Co, which may influence the conductivity of the nanochains and their application in electrocatalysis.
XPS was performed to analyze the composition and valence state of Co@CoPx. The Co 2p spectrum can be deconvoluted into three spin–orbit doublets and two shakeup satellites (denoted as Sat.). The peaks located at 778.7 eV and 793.8 eV are assigned to the 2p3/2 and 2p1/2 states of Co0, respectively. The doublets located at 781.9 and 797.8 eV can be attributed to the 2p3/2 and 2p1/2 states of Co bonded to P, while the doublets appearing at 783.55 and 799.6 eV can be ascribed to the 2p3/2 and 2p1/2 states of Co bonded to O, respectively.39–42 The satellite peaks appear at 786.0 and 803.7 eV. Fig. 3b shows the P 2p XPS spectrum, which can be deconvoluted into three peaks. The peaks at 129.6 and 132.7 eV can be ascribed to the 2p3/2 and 2p1/2 core levels of P in CoPx, respectively. The peak at 134.55 eV can be assigned to the oxidized P since the catalyst was exposed to air.
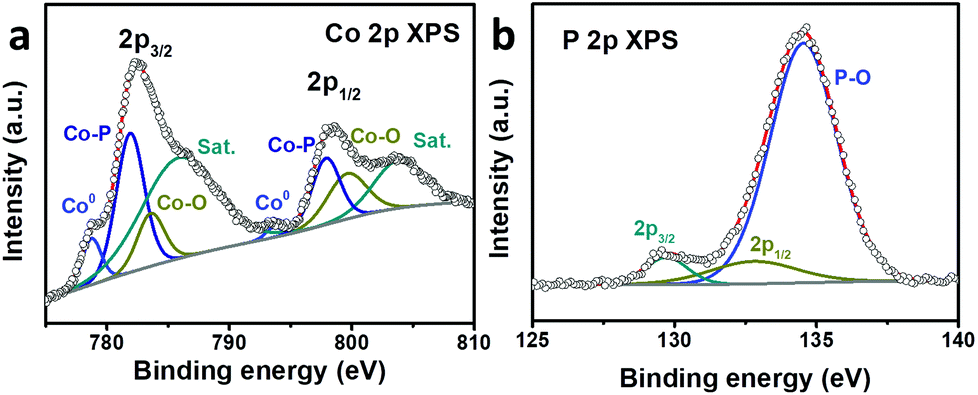 | ||
| Fig. 3 (a) Co 2p and (b) P 2p high resolution XPS spectra of Co@CoPx. | ||
The OER catalytic performance of the Co@CoPx nanochains catalyst was studied using a three-electrode system in 0.1 M KOH, and the iR-corrected polarization curves were recorded. The CV curve of Co@CoPx displays distinct redox peaks before oxygen evolution (Fig. S6a†). The first redox couple (OI/RI) can be assigned to Co3+/Co2+, and the second redox couple (OII/RII) can be assigned to Co4+/Co3+. For comparison, the performances of CoPx, Co and RuO2 were also acquired in the same way. As shown in Fig. 4a, the Co@CoPx nanochains show superior performance to the Co nanochains and CoPx with higher current density and lower onset potential. When the current increases to 10 mA cm−2, the Co@CoPx nanochains present an overpotential of 312 mV, which is much better than that of Co (408 mV), CoPx (310 mV) and also slightly superior to that of the RuO2 catalyst (317 mV). The Co@CoPx nanochains exhibit a Tafel slope of 81.7 mV dec−1, which is lower than that of Co (120 mV dec−1) and CoPx (87.0 mV dec−1), indicating its high OER kinetics. Since the catalytic process involves a valence state change in Co, the intense redox peak of Co@CoPx at 1.2 V also indicates that Co@CoPx has more catalytic sites than Co and CoPx. The excellent OER performance of the Co@CoPx nanochains can be attributed to the synergistic effect between Co and CoPx. CoPx is the catalytically active phase for OER, while the Co nanochains act as an electrical conductive channel, which promotes efficient electron exchange between the surface CoPx and the electrolyte. In addition to the synergistic effect, the electron injection process between Co and CoPx may also improve the OER performance of CoPx (Fig. 4c and d). The Fermi level of Co is higher than that of CoPx; thus, Co can inject electrons into CoPx and build up an internal electric field in their interface until they achieve the same Fermi level. This process manipulates the work function of CoPx and reduces the overpotential needed to drive OER. The electron injection-promoted oxygen evolution process has been proven in our previous studies on metal–oxide and metal–sulfide heterostructures.28,29 DFT calculations have proven that coupling with metals can change the adsorption energy of the reaction intermediates on oxides or sulfides, and thus change the rate determining step of the oxygen evolution process.28,29
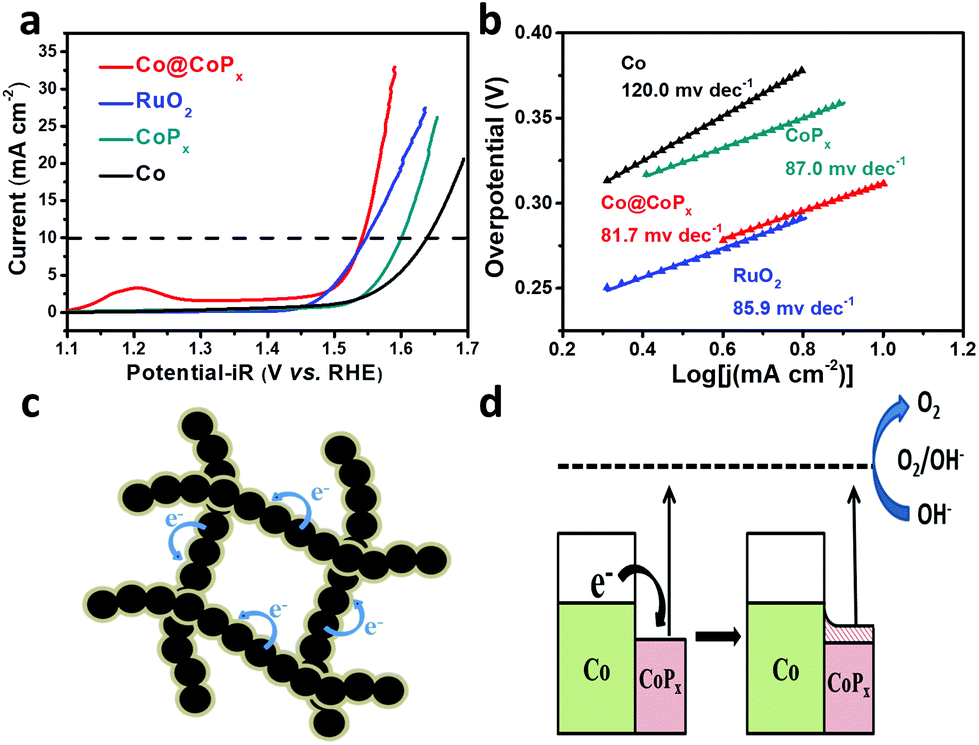 | ||
| Fig. 4 (a) Polarization curves and (b) Tafel plots of Co, CoPx, Co@CoPx and RuO2. (c), (d) schematic illustration of the electron injection process between Co and CoPx. | ||
To estimate the exposure of ion-accessible sites, the electrochemically active surface area (ECSA) was measured. The ECSA is linearly proportional to the electrochemical double layer capacitance (Cdl), which can be calculated from the cyclic voltammograms recorded in the region where no redox reaction occurs.36,37 The CV curves of Co@CoPx are presented in Fig. 5a and the CV curves of Co and CoPx are presented in Fig. S6.†Cdl was acquired by plotting j (ja–jc) at 0.94 V (vs. RHE) against the scan rate, the slope of which is twice the Cdl. The result indicates that Co@CoPx has much larger ECSA than CoPx. CoPx in Co@CoPx is supported by Co nanochains, which can prevent CoPx from aggregation during the reaction. A high ECSA means more catalytic active sites, which also contribute to the excellent OER performance of Co@CoPx.
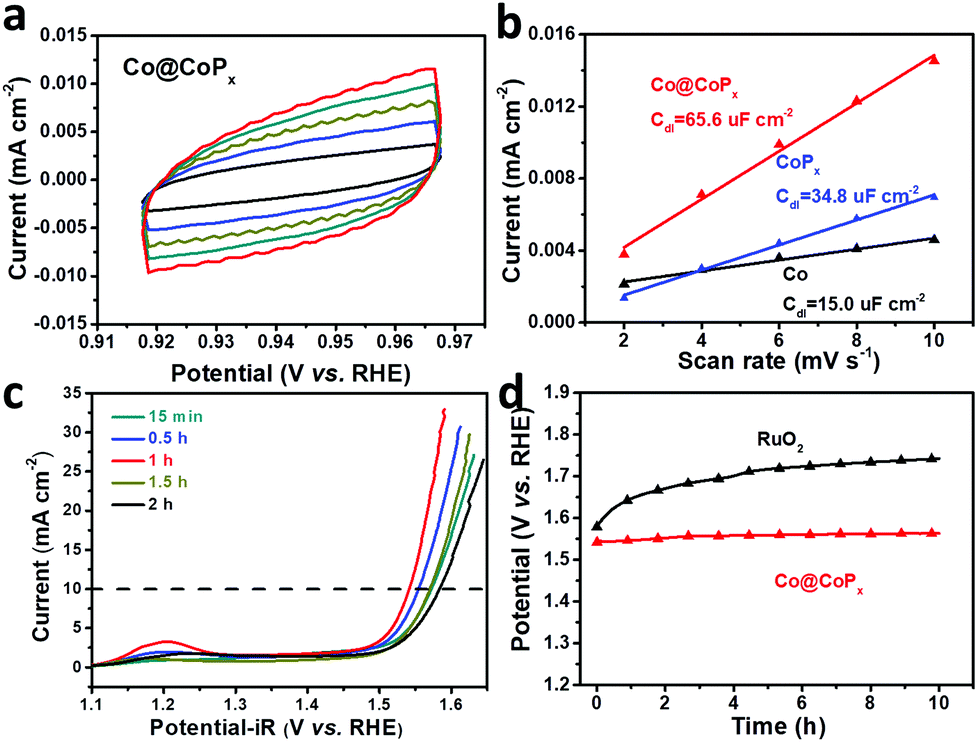 | ||
| Fig. 5 (a) Cyclic voltammetry curves of Co@CoPx in the region where no redox reaction occurs; the scan rates are 10, 8, 6, 4, and 2 mV s−1. (b) Current density as a function of the scan rate for the Co nanochains, CoPx and Co@CoPx nanochains. (c) Polarization curves of the samples prepared under different phosphorization times. (d) Chronopotentiometry curves of Co@CoPx and RuO2. | ||
The effect of the CoPx shell thickness on the OER activity of Co@CoPx was also studied. The polarization curves of Co@CoPx prepared with different phosphorization times are shown in Fig. 5c. CoPx is the catalytically active phase in Co@CoPx; thus, the OER performance initially improved with an increase in the amount of CoPx and the sample prepared for 1 h shows the best catalytic activity. When the reaction time was further extended, the thickness of CoPx increased and the interaction between Co and CoPx decayed rapidly. The sample with long phosphorization time also show some pores in the Co nanochains (Fig. S4†). The electron transfer path was blocked by these pores, which reduced the electrical conductivity of the Co@CoPx nanochains. Consequently, the catalytic performance of the samples prepared with longer reaction time decreased. The sample prepared for 1 h has the optimal structure and exhibits the best OER performance. Apart from its high catalytic activity, the Co@CoPx composite also shows excellent durability for the OER. Fig. 5d shows the long-term chronopotentiometry test of Co@CoPx and RuO2. The performance of Co@CoPx shows only a slight decay, while the overpotential of RuO2 increased significantly since the surface RuO2 is easily oxidized into water soluble RuO42−.43,44 The polarization curves and SEM image of Co@CoPx after the stability test also indicate that the morphology of the sample changes only slightly during the oxygen evolution process (Fig. S8 and S9†).
Conclusions
In summary, Co@CoPx core–shell nanochains were synthesized via an arc-discharge method and subsequent phosphorization process. The thickness of the CoPx shell can be altered by controlling the phosphorization time. The sample prepared under 1 h exhibits optimal OER performance with an overpotential of 310 mV at the current density of 10 mA cm−2, which is better than that of CoPx (370 mV), Co (408 mV), and RuO2 (317 mV). The high catalytic activity of Co@CoPx can be attributed to the synergistic effect between Co and CoPx. The highly conductive Co nanochains improve the kinetics of the OER on CoPx, and the electron injection process from Co to CoPx reduces the overpotential of OER. Apart from its high OER catalytic performance, the Co@CoPx nanochains also exhibited good durability during the long-term test.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by National key R&D Program of China (Grant 2016YFB0901600), Science and Technology Commission of Shanghai (Grant 16JC1401700), the Key Research Program of Chinese Academy of Sciences (Grants No. QYZDJ-SSW-JSC013 and KGZD-EW-T06).Notes and references
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef PubMed.
- M. Gao, W. Sheng, Z. Zhuang, Q. Fang, S. Gu, J. Jiang and Y. Yan, J. Am. Chem. Soc., 2014, 136, 7077–7084 CrossRef PubMed.
- Z.-L. Wang, D. Xu, J.-J. Xu and X.-B. Zhang, Chem. Soc. Rev., 2014, 43, 7746–7786 RSC.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef PubMed.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef.
- S. W. Lee, C. Carlton, M. Risch, Y. Surendranath, S. Chen, S. Furutsuki, A. Yamada, D. G. Nocera and Y. Shao-Horn, J. Am. Chem. Soc., 2012, 134, 16959–16962 CrossRef PubMed.
- J. Wang, H. x. Zhong, Y. l. Qin and X. b. Zhang, Angew. Chem., Int. Ed., 2013, 52, 5248–5253 CrossRef PubMed.
- F. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172–2192 RSC.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef PubMed.
- Z. Chen, D. Cummins, B. N. Reinecke, E. Clark, M. K. Sunkara and T. F. Jaramillo, Nano Lett., 2011, 11, 4168–4175 CrossRef PubMed.
- M. R. Gao, Y. F. Xu, J. Jiang, Y. R. Zheng and S. H. Yu, J. Am. Chem. Soc., 2012, 134, 2930–2933 CrossRef PubMed.
- X.-Y. Yu, Y. Feng, B. Guan, X. W. Lou and U. Paik, Energy Environ. Sci., 2016, 9, 1246–1250 Search PubMed.
- A. Grimaud, K. J. May, C. E. Carlton, Y. L. Lee, M. Risch, W. T. Hong, J. Zhou and Y. Shao-Horn, Nat. Commun., 2013, 4, 2439 CrossRef PubMed.
- L. Han, S. Dong and E. Wang, Adv. Mater., 2016, 28, 9266–9291 CrossRef PubMed.
- J. Wang, W. Cui, Q. Liu, Z. Xing, A. M. Asiri and X. Sun, Adv. Mater., 2016, 28, 215–230 CrossRef PubMed.
- B. M. Hunter, H. B. Gray and A. M. Muller, Chem. Rev., 2016, 116, 14120–14136 CrossRef PubMed.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1 Search PubMed.
- S. Anantharaj, S. R. Ede, K. Sakthikumar, K. Karthick, S. Mishra and S. Kundu, ACS Catal., 2016, 6, 8069–8097 CrossRef.
- J. Ryu, N. Jung, J. H. Jang, H.-J. Kim and S. J. Yoo, ACS Catal., 2015, 5, 4066–4074 CrossRef.
- Y.-P. Zhu, Y.-P. Liu, T.-Z. Ren and Z.-Y. Yuan, Adv. Funct. Mater., 2015, 25, 7337–7347 CrossRef.
- L. Jiao, Y.-X. Zhou and H.-L. Jiang, Chem. Sci., 2016, 7, 1690–1695 RSC.
- H. Yang, Y. Zhang, F. Hu and Q. Wang, Nano Lett., 2015, 15, 7616–7620 CrossRef PubMed.
- D. Das and K. K. Nanda, Nano Energy, 2016, 30, 303–311 CrossRef.
- Z. H. Xue, H. Su, Q. Y. Yu, B. Zhang, H. H. Wang, X. H. Li and J. S. Chen, Adv. Energy Mater., 2017, 7, 7 Search PubMed.
- X. B. Yu, S. Zhang, C. Y. Li, C. L. Zhu, Y. J. Chen, P. Gao, L. H. Qi and X. T. Zhang, Nanoscale, 2016, 8, 10902–10907 RSC.
- P. Wang, F. Song, R. Amal, Y. H. Ng and X. L. Hu, ChemSusChem, 2016, 9, 472–477 CrossRef PubMed.
- X. Yuan, J. Yin, Z. Liu, X. Wang, C. Dong, W. Dong, M. S. Riaz, Z. Zhang, M. Chen and F. Huang, ACS Appl. Mater. Interfaces, 2018, 10, 11565–11571 Search PubMed.
- X. Wang, X. Liu, C.-J. Tong, X. Yuan, W. Dong, T. Lin, L.-M. Liu and F. Huang, J. Mater. Chem. A, 2016, 4, 7762–7771 Search PubMed.
- X. Yuan, M. S. Riaz, X. Wang, C. Dong, Z. Zhang and F. Huang, Chem. – Eur. J., 2018, 24, 3707–3711 CrossRef PubMed.
- X. Yuan, H. Ge, X. Wang, C. Dong, W. Dong, M. S. Riaz, Z. Xu, J. Zhang and F. Huang, ACS Energy Lett., 2017, 2, 1208–1213 CrossRef.
- Z. Xiong and X. S. Zhao, J. Am. Chem. Soc., 2012, 134, 5754–5757 CrossRef PubMed.
- C. Sun, H. Li and L. Chen, Energy Environ. Sci., 2012, 5, 8475 Search PubMed.
- J. Tian, Y. Sang, Z. Zhao, W. Zhou, D. Wang, X. Kang, H. Liu, J. Wang, S. Chen and H. Cai, Small, 2013, 9, 3864–3872 CrossRef PubMed.
- X. Yuan, H. Ge, X. Liu, X. Wang, W. Chen, W. Dong and F. Huang, J. Alloys Compd., 2016, 688(Part A), 613–618 Search PubMed.
- J. Bao, X. Zhang, B. Fan, J. Zhang, M. Zhou, W. Yang, X. Hu, H. Wang, B. Pan and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 7399–7404 CrossRef PubMed.
- J. Xie, X. Zhang, H. Zhang, J. Zhang, S. Li, R. Wang, B. Pan and Y. Xie, Adv. Mater., 2017, 29, 1604765 CrossRef PubMed.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef PubMed.
- Y. Bai, H. Zhang, Y. Feng, L. Fang and Y. Wang, J. Mater. Chem. A, 2016, 4, 9072–9079 Search PubMed.
- Y. Pan, Y. Lin, Y. Liu and C. Liu, Catal. Sci. Technol., 2016, 6, 1611–1615 Search PubMed.
- L. Ai, Z. Niu and J. Jiang, Electrochim. Acta, 2017, 242, 355–363 CrossRef.
- T. Liu, K. Wang, G. Du, A. M. Asiri and X. Sun, J. Mater. Chem. A, 2016, 4, 13053–13057 Search PubMed.
- E. J. M. Osullivan and J. R. White, J. Electrochem. Soc., 1989, 136, 2576–2583 CrossRef.
- S. Zhuiykov, Proc. Int. Symp. Olfaction Electron. Noses, 2009, 1137, 34–37 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: XRD pattern, TEM images of Co@CoPx nanochains, electrocatalytic properties of Co@CoPx nanochains. See DOI: 10.1039/c8qi00428e |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2018 |