
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metallomic study on the metabolism of RAPTA-C and cisplatin in cell culture medium and its impact on cell accumulation†
Hannah U.
Holtkamp
,
Sanam
Movassaghi
,
Stuart J.
Morrow
,
Mario
Kubanik
and
Christian G.
Hartinger
 *
*
School of Chemical Sciences, University of Auckland, Private Bag 92019, Auckland 1142, New Zealand. E-mail: c.hartinger@auckland.ac.nz; Web: http://hartinger.auckland.ac.nz Tel: +64 9 3737 599 ext. 83220
First published on 19th February 2018
Abstract
Metal-based anticancer agent development can be improved with advanced metallomics methods that allow for quick and efficient screening of metallodrugs for their metabolites in biological media. Cellular accumulation in in vitro settings is not always correlated with cytotoxicity; and protein binding, particularly with albumin and transferrin, can have an important influence on metallodrug transportation, selectivity, and efficacy. We contrast the time-dependent cellular accumulation of both cisplatin and the pre-clinically investigated RAPTA-C in terms of cell uptake and speciation in culture medium via CE-ICP-MS analysis. Despite RAPTA-C being administered at 40-fold higher dose than cisplatin, owing to its much higher IC50 value, the accumulation over time was only 10-fold higher. An optimised CE-ICP-MS method, through the coating of the capillary to prevent protein–capillary surface interactions, resulted in superior resolution and metal–protein adduct identification. It was then used for extracellular speciation in conjunction with [tris(acetylacetonato)cobalt(III)] as an internal standard. RAPTA-C was found to be more inert to extracellular reactions than cisplatin which could be used to rationalise the observed cellular uptake patterns. While for cisplatin both transferrin and albumin were identified as the main binding partners, RAPTA-C was found to react nearly exclusively with albumin. Moreover, this behaviour was time-dependent and our results also demonstrate that cancer cells have an influence on metal species distribution in the cell culture medium over time.
Significance to metallomicsThis study shows the impact of speciation of cisplatin and the organoruthenium developmental anticancer agent RAPTA-C on the accumulation in cancer cells. The metabolism of RAPTA-C in cell culture medium leaves it to a higher degree unreacted than cisplatin but cisplatin is taken up into cells more efficiently. Notably, the presence of cancer cells influenced the species detected for both cisplatin and RAPTA-C interaction with proteins. |
1. Introduction
Metal-based drugs are essential pillars of cancer chemotherapy. A reason for the low development rate of novel anticancer metallodrugs is the limited knowledge about their modes of action beyond the DNA-targeting ability of cisplatin.1 In particular newer generations of developmental metal-based anticancer agents are designed to target proteins which are overexpressed in tumour cells as compared to healthy cells.Ruthenium complexes are some of the most widely studied non-Pt compounds and offer novel mechanisms of action, reduced toxicity, and a different spectrum of activity, which can provide alternative treatments for cisplatin-resistant cancers.2–4 Promising Ru compounds encompass the clinically-investigated RuIII coordination compounds NAMI-A and KP1019/NKP-1339 as well as organometallic RuII(η6-arene) structures, such as RAPTA and RAED compounds. The organometallic RuII(η6-arene) scaffold provides a versatile platform for drug development because the choice of ligand allows modulation for particular biological targets and desired activity.4 RAPTA compounds of the general structure [Ru(η6-arene)(PTA)X2] (PTA = 1,3,5-triaza-7-phosphaadamantane, X = halide; see Fig. 1 for RAPTA-C with arene = p-cymene) bind preferentially to proteins while the ethylene-1,2-diamine complexes RAED exhibit a preference for DNA.5
 | ||
| Fig. 1 The chemical structures of cisplatin and RAPTA-C anticancer agents used in this study. | ||
While stability in aqueous solution and characterisation of hydrolysis and degradation products is a prerequisite in the early stage of (metal-based) drug development,6 the interaction with proteins is more difficult to elucidate, especially in biological matrices. Most metal-based anticancer agents are developed for intravenous administration, and this makes the reactions with blood components of particular significance, e.g. the human serum proteins albumin [HSA] and transferrin [hTf].1 The LADME properties of metal complexes can be dramatically influenced by serum protein binding. Classically thought to remove active forms of the complexes from circulation, there is now considerable evidence that proteins contribute to tumour selective transport,1,7–11 which has been exploited in several instances to develop novel metallodrugs.10,12–18
It is well established that cisplatin largely exhibits its therapeutic effect through the binding to DNA but there have also been studies showing that Pt–HSA and Pt–hTf adducts still show anticancer activity.19,20 RAPTA-C has shown antimetastatic21 and antiangiogenic activity,22 as well as suppression of primary tumour growth in preclinical studies, and is on track to clinical investigation.23 The interesting biological properties are most probably due to protein interactions, although no selective targeting behaviour could be identified.24 Therefore, the modes of action of both compound types are very different and this is reflected in their anticancer activity profiles.4
Bioanalytical techniques to study these complexes often include inductively-coupled plasma mass spectrometry (ICP-MS) as a highly sensitive detector that quantitatively measures metal concentrations. However, determination of the extent of binding necessitates hyphenation with separation techniques to distinguish the unreacted metal complex and metal-bound protein fractions.25 Typical techniques used are based on 1D or 2D chromatographic separations,14,26–28 gel,29–31 and capillary electrophoresis (CE),32,33 often hyphenated to MS,34–36 and taking into consideration the conditions observed in living organisms. CE-MS has been widely used for the study of aqueous stability, biological target identification and characterisation of kinetics for various types of metal complexes, including cisplatin,37–39 oxaliplatin,40 platinum complexes,41,42 Ru coordination and organometallic compounds,43–47 Zn,48 Ga,49,50 and Au complexes.51
In this study, we aimed to develop and utilise a robust CE-based separation method to study the metabolism of cisplatin and RAPTA-C in cell culture medium in order to assess the impact of speciation on the accumulation in cancer cells and eventually biological activity.
2. Methods
2.1. Chemicals
RAPTA-C and [tris(acetylacetonato)cobalt(III)] [Co(acac)3] were prepared according to procedures described previously.39,52 Cisplatin was obtained from ICN Pharmaceuticals, Inc., hydrochloric acid (36.5–37.5%) from J. T. Baker, and Suprapur® nitric acid from Merck. Sodium dihydrogen phosphate (99% purity) was supplied by AK Scientific, and disodium hydrogenphosphate dihydrate from Sigma-Aldrich. Sodium chloride (reagent grade) was purchased from Scharlau, ammonium bicarbonate (analytical grade) and ammonia solution from AnalaR, BDH Chemicals Ltd. Sodium hydroxide (1.0 N) was from Agilent and ammonium acetate was obtained from ECP Ltd. The ICP-MS standards Tm, Sc, Ge, Re in 2% HNO3 (1000 μg mL−1) were products of CPI International. Tuning Solution for ICP-MS 7500cs was supplied by Agilent Technologies, hexadimethrine bromide, polyethylene glycol (average Mw 2000), poly(vinyl pyrrolidone) (average Mw 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), human transferrin (lot# BCBR1763V), human albumin (lot# 061M1583), and filtered human serum from male AB plasma (lot# SLBM3315V) were obtained from Sigma Aldrich. High purity Type 1 water was used throughout these experiments obtained from an Elga pureflex-lab system.
000), human transferrin (lot# BCBR1763V), human albumin (lot# 061M1583), and filtered human serum from male AB plasma (lot# SLBM3315V) were obtained from Sigma Aldrich. High purity Type 1 water was used throughout these experiments obtained from an Elga pureflex-lab system.
2.2. Instrumentation and analysis conditions
:1). The interaction of cisplatin and RAPTA-C with serum proteins was studied with incubation solutions consisting of 100 μM of either cisplatin or RAPTA-C, and 50 μM of either HSA, hTf or both HSA and hTf. The samples were prepared to contain a matrix of 100 mM NaCl and 10 mM phosphate buffer (pH 7.4) designed to simulate physiological conditions. The only exception was for the ESI-MS detector where Type 1 water was used. The samples were incubated on a Grant bio thermoshaker (37 °C, 260 rpm) and aliquots were collected after 0, 2, 4, 24, 48, and 96 h and were frozen until just prior to analysis.
Human serum samples were diluted 1:4 with the incubation solution before analysis. Cell medium samples collected from cellular accumulation experiment (compare 2.3) were analysed on CE-ICP-MS without additional workup, but for CE-ESI-MS samples needed to be filtered (Vivaspin® 500 Polyethersulfone, 10 kDa, Sartorius) and the low- and high-molecular weight (LMW and HMW) fractions were collected.
All samples contained [Co(acac)3] (final concentration 10 μM) as an internal standard which was added just prior to analysis.
The calibration plots for Pt and Ru were prepared using cisplatin and RAPTA-C concentrations of 1.6, 3.1, 6.1, 12.5, 25, 50 and 100 μM with 10 μM of [Co(acac)3] as the internal standard. The limits of detection (LOD) and quantification (LOQ) were calculated as described previously.39
2.3. Cell studies
HCT116 cells were supplied by ATCC, and were grown in αMEM (Life Technologies) supplemented with 5% fetal calf serum (Moregate Biotech) at 37 °C in a humidified incubator with 5% CO2. The sulforhodamine B cytotoxicity assay to determine IC50 values and the cellular uptake experiments were carried out as described previously.56 To determine the intracellular metal concentrations, HCT116 cells (4 × 105 per well) were seeded into 6-well plates and allowed to settle for 24 h time at 37 °C and 5% CO2. The compounds RAPTA-C and cisplatin were dissolved in DMSO (10000, and 250 μM, respectively) and diluted with medium to a concentration of 1% DMSO and the previously determined IC50 values of the metal complexes. Cells were left to incubate at 4, 24, 48 and 72 h exposure times. The medium was collected and analysed by both direct infusion analysis ICP-MS to quantify the total metal content, and by PVP coated CE-ICP-MS for speciation analysis. The wells were washed twice with 1 mL of ice-cold PBS buffer. The cells were lysed with 2 mL of concentrated nitric acid (containing 0.1 μL of a 1000 ± 3 μg mL−1 thulium standard as internal standard) and digested with an Ethos Up microwave digestion system (Milestone). Then the solutions were diluted with 10 mL of H2O and the metal content was determined by ICP-MS. The ICP-MS (Agilent 7700) with an ASX-500 autosampler (CETAC Technologies) in a Serie SuSi laminar flow hood (SPECTEC) was equipped with a MicroMist nebuliser and a Scott double pass spray chamber. The carrier gas flow rate was 1 mL min−1. The instrument was tuned for cerium, cobalt, lithium, magnesium, thallium, and yttrium. The calibration standards were matched to the samples with regard to HNO3 concentration and the internal standard. The reported values are the mean of at least 3 independent uptake experiments conducted with blank wells for each substance to account for unspecific binding to the plastic of the well plates.
3. Results and discussion
3.1. Interaction of RAPTA-C with human serum proteins
CE-ICP-MS analysis was used to demonstrate that the RuIII complexes KP1019 and NKP-1339/IT139 are predominantly bound to HSA in serum in vivo.45,47 A similar method was applied to compare these Ru(III) compounds and the organoruthenium(II) complex RAPTA-C in terms of their ligand exchange kinetics with water and DNA models.43 Such studies are of particular importance in the early stage of drug development,6,57 and CE-ICP-MS has significant potential in this research area. There have been a wide variety of reports elucidating the interactions between metal complexes and proteins and also on the impact of halido/aqua ligand exchange on biomolecule binding. The methods used are often based on liquid chromatography (LC) separation and several interesting results have been reported, contributing especially to the understanding of speciation and its impact on the biological activity of metal-based anticancer agents.14,26–28,58–61 LC provides low to high flow rates, which makes it suitable for hyphenation with molecular and element-specific detectors, and benefits from constant retention times for the analytes. However, separations based on solid phase interactions also suffer from the risk of shifts in the species present in the samples, especially for reactive metal complexes. Operating in purely aqueous conditions in CE reduces that risk and even allows the use of the capillary as a reactor to resemble the environment of specific biological tissue,62 while still using minute sample amounts and allowing for short analysis times.Notably, a disadvantage of CE separation with bare fused-silica capillaries is that proteins such as HSA tend to adsorb on the capillary surface. Though experimental procedures analysing serum or HSA samples on bare fused-silica capillaries have been previously published,63–65 we have observed this phenomenon in previous studies looking at the kinetics and binding behaviour of HSA with metallodrugs. This strongly interferes with the analysis and may result in capillary blocking. Lucy et al. describe the fundamentals of this problem in detail in a recent review.66
To overcome this issue, many procedures have been investigated that involve the coating of the capillary and high adsorption resistance was observed, while being even able to separate protein isoforms with high resolution.67–72 One approach of capillary coating is based on polybrene (PB) and polyethylene glycol,73 which creates a cationic surface requiring the use of an acidic background electrolyte (BGE). This procedure was modified and then successfully applied to binding studies of Ru compounds with serum proteins.44,45 The practical limitation of this approach was that the polybrene coating is unstable and dynamic, which can lead to contamination of a hyphenated MS detector. While ICP-MS is robust, it can be problematic for ESI-MS.
Therefore, an alternative approach was investigated for the application of CE-MS in the elucidation of metallodrug protein interaction in serum and cell culture medium operating the CE in capillary zone electrophoresis mode (CZE). To avoid contamination with PB, poly(vinyl pyrrolidone) (PVP) was used to covalently coat the capillary. PVP was shown to be stable, insensitive to pH, and biocompatible.53 To study the suitability of the coating, the separation performance of a PVP-coated capillary for the serum proteins HSA and hTf was evaluated and contrast with PB-modified capillaries. In PVP coated capillaries the BGE can be broadly varied for separation and it is possible to separate the principal serum proteins HSA and hTf using a physiologically compatible buffer (25 mM phosphate buffer, pH 7.4), while PB coatings require a low pH buffer (50 mM formic acid) for separation. A significantly higher signal-to-noise ratio was found with PVP, and a superior protein resolution was achieved for the HSA and hTf mixture (Fig. 2). [Co(acac)3] was added to the samples as an internal standard to a final concentration of 10 μM prior to analysis to record the position of the EOF and to correct for run-by-run variation in migration time and peak area (Fig. 2).
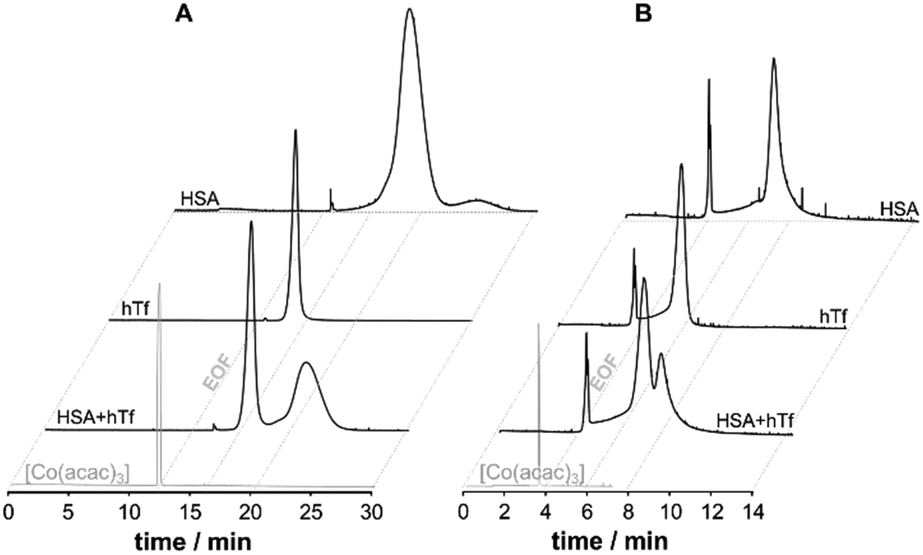 | ||
| Fig. 2 The CE-UV (214 nm) electropherograms of [Co(acac)3] (10 μM, internal standard, EOF marker) and the serum proteins (100 μM) HSA and hTf and a HSA/hTf mixture (1:1) using either (A) PVP- or (B) PB-coated capillaries. | ||
The enhanced robustness to a broad pH range allowed the use of 1 M NaOH for rinsing to regenerate the EOF, while the coating remained stable for over two weeks. There was no indication of any loss of ion sensitivity in the ESI-MS confirming the PVP is fixed firmly to the exposed silanol groups.
Given these positive results, PVP-coated capillaries were employed in CZEPVP-ICP-MS studies to evaluate the serum protein binding behaviour of the organometallic complex RAPTA-C, monitoring the Ru, Co (internal standard), Re (nebulisation stability), and P (capillary performance) traces. The total amount of measured Ru for the same sample was found to be higher with the PVP-coated capillary than when using PB (Table S2, ESI†). Although the peak area was initially more variable than with PB coating, correction with the internal standard [Co(acac)3] decreased the variability to a level comparable with PB (Fig. S2 and Table S2, ESI†). Moreover, the peak areas for the Ru-serum protein adducts measured via PVP capillaries were comparatively larger. Therefore, this improved method made it possible to identify the principal binding partners of RAPTA-C in human serum (Fig. 3). Using pure samples of HSA and hTf incubated with RAPTA-C for 96 hours as standards helped to identify the migration behaviour of the respective Ru-serum protein adducts with the hTf adduct migrating quicker than the HSA conjugate. For a 1:1 HSA/hTf mixture, approximately a 2.5:1 ratio of adduct formation with the proteins was identified, with a preference for HSA. Studies in human serum, where HSA is present in about 10-times higher concentration support this outcome and the vast majority of Ru was found attached to HSA (Fig. 3), although the peaks were broadened in these studies. Importantly, only one cobalt peak was detected in these samples, which indicated that the [Co(acac)3] not only remained intact but did not form adducts with any species present in the reaction mixture. This is another example that confirms the broad applicability of [Co(acac)3] as an internal standard in CE analyses.39
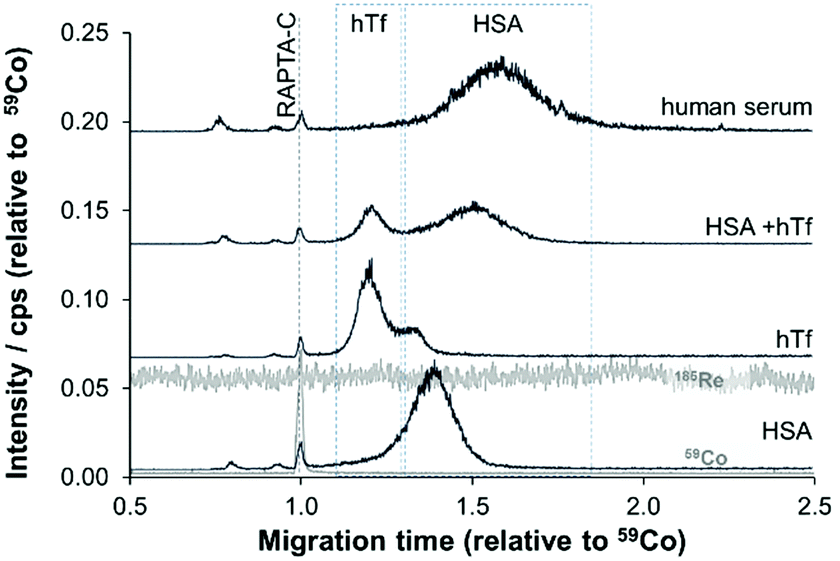 | ||
| Fig. 3 The CZE-ICP-MS electropherograms for reaction mixtures of RAPTA-C with HSA, hTf and HSA/hTf (50 μM, 1:1) in saline solution and human serum after 96 h incubation, using PVP-coated capillaries. [Co(acac)3] was added as an internal standard and used as the EOF marker. | ||
3.2. Cell uptake and speciation of RAPTA-C and cisplatin in cancer cells
The biological activity of metal-based anticancer agents is often strongly dependent on the speciation and the accumulation in cancer cells.74 Therefore, cellular uptake experiments for cisplatin and RAPTA-C were conducted in HCT116 colon cancer cells grown in αMEM supplemented with 5% fetal calf serum containing the bovine serum proteins bovine serum albumin (BSA) and transferrin (bTf). HSA has approximately the same molecular weight as BSA and they feature 76% sequence homology.75 Transferrins in general have a significant degree of conservation in their amino acid sequence (>70%),76 and hTf and bTf also have similar molecular weights. The concentrations of the complexes chosen were at or below the IC50 values for cisplatin (2.5 μM)56 and RAPTA-C (100 μM) in HCT116 cells, respectively. The cell viability was confirmed under the light microscope before cells were collected for each time point of the cell uptake analysis. Analysis with direct infusion ICP-MS demonstrated that cisplatin was accumulated more efficiently inside the HCT116 cells, while the total amount of Ru was higher despite the lower antiproliferative activity of RAPTA-C (Fig. 4). While the uptake of Ru from RAPTA-C appears to increase steadily over the time course of 72 h, the concentration of Pt detected in the cells treated with cisplatin only slightly varies between 24 and 72 h. This may be related to the differing reactivity of the complexes with proteins or other medium components.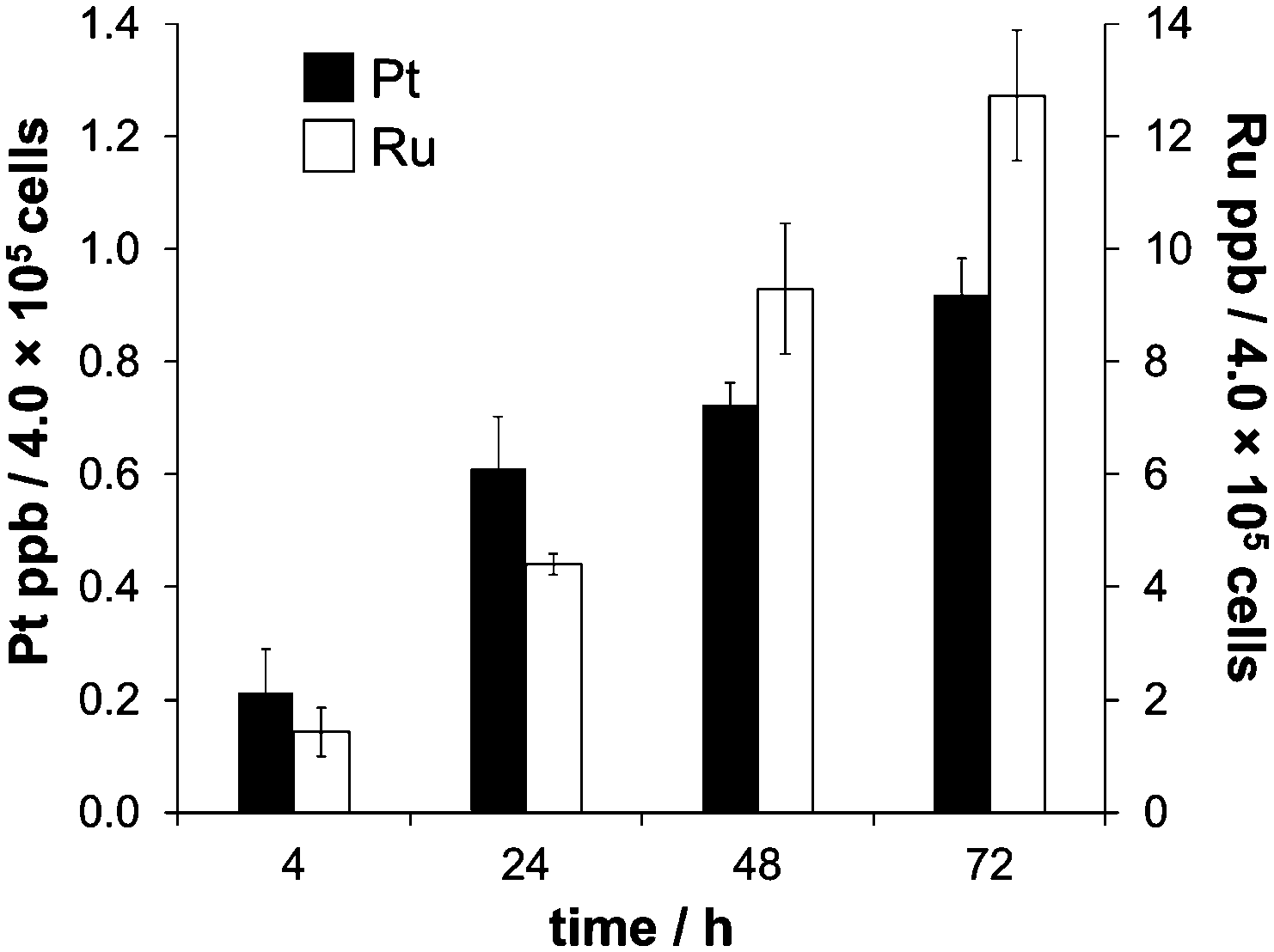 | ||
| Fig. 4 The time-dependent uptake of cisplatin (2.5 μM) and RAPTA-C (100 μM) in HCT116 colon carcinoma cells, measured at time points of 4, 24, 48 and 72 h. | ||
In order to investigate this differing behaviour further, we prepared samples of cell medium containing fetal calf serum spiked with both anticancer agents and analysed them with our newly optimised method based on PVP-coated capillaries with a particular emphasis on the influence of speciation on the uptake. Notably, analysis of the cell medium samples by CZEPVP-ICP-MS was carried out without the need for prior work up or dilution, despite the significant matrix in such samples (Fig. 5, composition listed in Tables S3 and S4, ESI†).
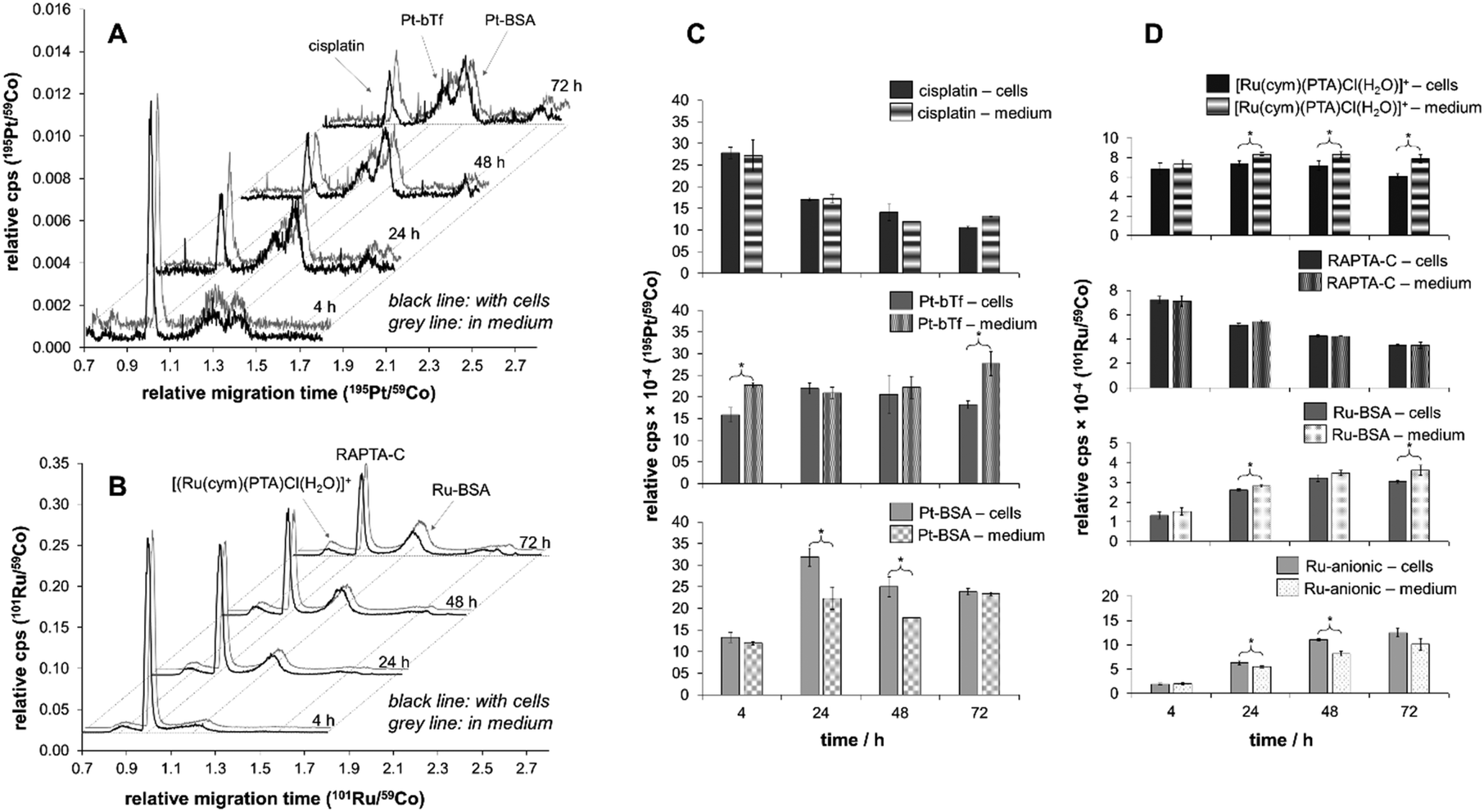 | ||
| Fig. 5 The speciation studies carried out by CZEPVP-ICP-MS. (A) 195Pt and (B) 101Ru electropherograms of cisplatin and RAPTA-C speciation over time in cell medium both in the presence of HCT116 cells (black line) and absence (grey line). The principal species of interest are identified by arrows and comparisons of cisplatin and RAPTA-C species in the presence and absence of HCT116 are made in C and D, respectively. t-Tests were carried out on the species amount in the presence of cells and in medium. Statistically significant differences at the 95% confidence level are indicated with an asterisk. | ||
For both cisplatin and RAPTA-C, the principal binding partner(s) in the cell culture medium were identified as the serum proteins. This confirms reports by Groessl et al. for cisplatin and an analogous RAPTA compound by size exclusion chromatography (SEC)-ICP-MS.3,28 It is also in line with a study by Gibson et al. who collected SEC fractions from cell extracts treated with cisplatin and determined two-thirds of the Pt content to be in molecular weight fractions of >3000 g mol−1.77 Heffeter and colleagues found large protein aggregates with high molecular weights for RuIII anticancer agents in the cytosol, which was very different to the protein adducts cisplatin formed.27
With the rising signals assigned to the BSA and bTf adducts for cisplatin and BSA for RAPTA-C, the peaks identified for the free metal complexes declined over the time of the incubation period (Fig. 5A and B). While for RAPTA-C a cationic metabolite, presumably [Ru(cym)(PTA)Cl(H2O)]+ based on previous experiments,21,43,78,79 was identified, no cationic Pt-containing peaks were observed. For both compounds, several additional Ru- or Pt-containing anionic species were detected. Attempts of using ESI-MS in a hyphenated setup did not allow identification of any adducts as the salt content of the cell medium samples was too high. Therefore, the samples were further separated with a 10 kDa cut-off filter into low (LMW) and high molecular weight (HMW) fractions. CEPVP-ESI-MS analysis of the HMW fraction successfully confirmed the identity of the principal serum protein binding partner as BSA for both cisplatin and RAPTA-C. Analysis of the LMW fraction by CZEPVP-ICP-MS clearly shows that the unknown Ru species has a molecular weight below 10 kDa (ESI,† Fig. S4). This anionic species may be formed by a reaction with phosphate or sulfate ions, or the negatively-charged amino acids aspartic and glutamic acid, all present in the cell medium solution. Adducts with some of these were detected before for cisplatin, although in absence of proteins.37
The speciation data collected by CZEPVP-ICP-MS showed a peak variability of ca. 7 and 5% for Pt and Ru after correction using [Co(acac)3] as the internal standard. This was especially beneficial for samples containing cisplatin which behaved more irregularly, probably influenced by the low concentrations of the analytes. The high reproducibility of measurements involving RAPTA-C gave LOD and LOQ values of 1.3 and 3.9 μM (ESI,† Fig. S2), while for cisplatin with this method 2.6 and 8.0 μM were obtained. Similar observations for cisplatin have been reported previously,39 and in a laser ablation (LA)-ICP-MS study contrasting the spatial distribution of cisplatin and KP1339 in animal tissue.80
Importantly, the CZEPVP method used allowed superior separation of albumin and transferrin in combination with both UV and ICP-MS detectors. This is a significant advantage as compared to PB-coated capillaries. Therefore, it was possible to use CZEPVP-ICP-MS and show significant differences in the peak areas over time in the cell medium samples that could be assigned to the presence of HCT116 cells. Overall, the presence or absence of cells did not impact the decrease of the peaks assigned to cisplatin and RAPTA-C (Fig. 5C and D) but it had a significant influence on the amount of individual species detected, probably because of different interactions between the metal complexes and the proteins present in the samples. For example, for cisplatin the amount of bTf adduct formed after 4 h was significantly higher in the sample without cells than in presence of cells (Fig. 5C). While it remains fairly constant in the presence of cells over the whole period of the experiment, in case of the cell medium samples it significantly increased after 72 h. In contrast, the amount of Pt attached to BSA was higher in the samples incubated with cells for 24 and 48 h while it was constant for the cell medium samples. Overall, the relative abundance of the BSA adduct decreased from 24 to 72 h in presence of cell. The relative distribution between original complex and protein adducts showed that RAPTA-C is present to a higher degree as unreacted complex while much of the Pt is bound to proteins and only a minor amount of unreacted cisplatin was detectable. We suggest that this is the reason for RAPTA-C to accumulate consistently over time, while the increase of Pt concentration in the cells beyond 24 h was limited.
4. Conclusions
We have introduced a new CE-based method for the analysis of metal-based anticancer agents and we have applied it in conjunction with ICP-MS detection to study the impact of speciation in cell culture medium on the cellular accumulation of cisplatin and the developmental anticancer agent RAPTA-C. The PVP coating used in these studies has been demonstrated to show considerable robustness to high salt content samples, as often found in biological environment, with no loss of peak resolution. Given these characteristics, this method has high potential for application in speciation analysis of anticancer metal complexes. Being able to generate informative results from the analysis of complex serum sample with no sample work up is an important feature of the new method as it limits the potential of alteration in speciation. With this 1D CZE method, it was possible to separate proteins such as albumin and transferrin, despite their similar molecular weights, and we obtained LOD and LOQ values that allow application of the method in biological samples. Therefore, it enabled us to quickly and confidently assess the principal protein binding partners of cisplatin and RAPTA-C with enhanced resolution. Despite using low concentrations in the cell culture experiments, especially for cisplatin, it was possible to show that the presence of cancer cells had an impact on the detected species distribution of anticancer metal complexes in terms of the amounts of protein adducts present in cell culture medium. While cisplatin was found to react with both albumin and transferrin, for RAPTA-C only an albumin adduct was detected, accompanied by a minor amount of hydrolysis product. Notably, the amount of free RAPTA-C was significantly higher than found for cisplatin, which may explain the different cellular accumulation behaviour of the two compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Auckland (University of Auckland Doctoral Scholarship to H. H. and M. K.) for the financial support. We would also like to thank Tony Chen for useful discussions and technical expertise.References
- A. R. Timerbaev, C. G. Hartinger, S. S. Aleksenko and B. K. Keppler, Chem. Rev., 2006, 106, 2224–2248 CrossRef CAS PubMed.
- R. E. Aird, J. Cummings, A. A. Ritchie, M. Muir, R. E. Morris, H. Chen, P. J. Sadler and D. I. Jodrell, Br. J. Cancer, 2002, 86, 1652–1657 CrossRef CAS PubMed.
- M. Groessl, O. Zava and P. J. Dyson, Metallomics, 2011, 3, 591–599 RSC.
- B. S. Murray, M. V. Babak, C. G. Hartinger and P. J. Dyson, Coord. Chem. Rev., 2016, 306, 86–114 CrossRef CAS.
- Z. Adhireksan, G. E. Davey, P. Campomanes, M. Groessl, C. M. Clavel, H. Yu, A. A. Nazarov, C. H. Yeo, W. H. Ang, P. Droge, U. Rothlisberger, P. J. Dyson and C. A. Davey, Nat. Commun., 2014, 5, 3462 Search PubMed.
- A. Jouyban and E. Kenndler, Electrophoresis, 2008, 29, 3531–3551 CrossRef CAS PubMed.
- H. Li and Z. M. Qian, Med. Res. Rev., 2002, 22, 225–250 CrossRef CAS PubMed.
- F. Kratz, A. Warnecke, K. Scheuermann, C. Stockmar, J. Schwab, P. Lazar, P. Drückes, N. Esser, J. Drevs, D. Rognan, C. Bissantz, C. Hinderling, G. Folkers, I. Fichtner and C. Unger, J. Med. Chem., 2002, 45, 5523–5533 CrossRef CAS PubMed.
- J. Ghuman, P. A. Zunszain, I. Petitpas, A. A. Bhattacharya, M. Otagiri and S. Curry, J. Mol. Biol., 2005, 353, 38–52 CrossRef CAS PubMed.
- F. Kratz, J. Controlled Release, 2008, 132, 171–183 CrossRef CAS PubMed.
- T. R. Daniels, T. Delgado, G. Helguera and M. L. Penichet, Clin. Immunol., 2006, 121, 159–176 CrossRef CAS PubMed.
- W. H. Ang, E. Daldini, L. Juillerat-Jeanneret and P. J. Dyson, Inorg. Chem., 2007, 46, 9048–9050 CrossRef CAS PubMed.
- V. Pichler, J. Mayr, P. Heffeter, O. Domotor, E. A. Enyedy, G. Hermann, D. Groza, G. Kollensperger, M. Galanski, W. Berger, B. K. Keppler and C. R. Kowol, Chem. Commun., 2013, 49, 2249–2251 RSC.
- M. Hanif, A. A. Nazarov, A. Legin, M. Groessl, V. B. Arion, M. A. Jakupec, Y. O. Tsybin, P. J. Dyson, B. K. Keppler and C. G. Hartinger, Chem. Commun., 2012, 48, 1475–1477 RSC.
- F. Kratz and B. Elsadek, J. Controlled Release, 2012, 161, 429–445 CrossRef CAS PubMed.
- S. Moon, M. Hanif, M. Kubanik, H. Holtkamp, T. Söhnel, S. M. F. Jamieson and C. G. Hartinger, ChemPlusChem, 2015, 80, 231–236 CrossRef CAS.
- M. Hanif, S. Moon, M. P. Sullivan, S. Movassaghi, M. Kubanik, D. C. Goldstone, T. Söhnel, S. M. F. Jamieson and C. G. Hartinger, J. Inorg. Biochem., 2016, 165, 100–107 CrossRef CAS PubMed.
- N. S. Sommerfeld, D. Strohhofer, K. Cseh, S. Theiner, M. A. Jakupec, G. Koellensperger, M. Galanski and B. K. Keppler, Eur. J. Inorg. Chem., 2017, 4049–4054 CrossRef CAS.
- J. D. Holding, W. E. Lindup, C. van Laer, G. C. Vreeburg, V. Schilling, J. A. Wilson and P. M. Stell, Br. J. Clin. Pharmacol., 1992, 33, 75–81 CrossRef CAS PubMed.
- F. Kratz, Expert Opin. Ther. Pat., 2002, 12, 433–439 CrossRef CAS.
- C. Scolaro, A. Bergamo, L. Brescacin, R. Delfino, M. Cocchietto, G. Laurenczy, T. J. Geldbach, G. Sava and P. J. Dyson, J. Med. Chem., 2005, 48, 4161–4171 CrossRef CAS PubMed.
- P. Nowak-Sliwinska, J. R. van Beijnum, A. Casini, A. A. Nazarov, G. Wagnières, H. van den Bergh, P. J. Dyson and A. W. Griffioen, J. Med. Chem., 2011, 54, 3895–3902 CrossRef CAS PubMed.
- A. Weiss, R. H. Berndsen, M. Dubois, C. Muller, R. Schibli, A. W. Griffioen, P. J. Dyson and P. Nowak-Sliwinska, Chem. Sci., 2014, 5, 4742–4748 RSC.
- M. V. Babak, S. M. Meier, K. V. M. Huber, J. Reynisson, A. A. Legin, M. A. Jakupec, A. Roller, A. Stukalov, M. Gridling, K. L. Bennett, J. Colinge, W. Berger, P. J. Dyson, G. Superti-Furga, B. K. Keppler and C. G. Hartinger, Chem. Sci., 2015, 6, 2449–2456 RSC.
- M. Groessl and C. G. Hartinger, Anal. Bioanal. Chem., 2013, 405, 1791–1808 CrossRef CAS PubMed.
- M. Sulyok, S. Hann, C. G. Hartinger, B. K. Keppler, G. Stingeder and G. Koellensperger, J. Anal. At. Spectrom., 2005, 20, 856–863 RSC.
- P. Heffeter, K. Böck, B. Atil, M. A. Reza Hoda, W. Körner, C. Bartel, U. Jungwirth, B. K. Keppler, M. Micksche, W. Berger and G. Koellensperger, J. Biol. Inorg. Chem., 2010, 15, 737–748 CrossRef CAS PubMed.
- M. Groessl, M. Terenghi, A. Casini, L. Elviri, R. Lobinski and P. J. Dyson, J. Anal. At. Spectrom., 2010, 25, 305–313 RSC.
- I. Khalaila, A. Bergamo, F. Bussy, G. Sava and P. J. Dyson, Int. J. Oncol., 2006, 29, 261–268 CAS.
- L. Hu, T. Cheng, B. He, L. Li, Y. Wang, Y.-T. Lai, G. Jiang and H. Sun, Angew. Chem., Int. Ed., 2013, 52, 4916–4920 CrossRef CAS PubMed.
- Y. Wang, L. Hu, X. Yang, Y.-Y. Chang, X. Hu, H. Li and H. Sun, Metallomics, 2015, 7, 1399–1406 RSC.
- C. G. Hartinger, M. Groessl, S. M. Meier, A. Casini and P. J. Dyson, Chem. Soc. Rev., 2013, 42, 6186–6199 RSC.
- H. U. Holtkamp and C. G. Hartinger, Drug Discovery Today: Technol., 2015, 16, 16–22 CrossRef PubMed.
- A. Prange and D. Pröfrock, Anal. Bioanal. Chem., 2005, 383, 372–389 CrossRef CAS PubMed.
- H. Cheng, P. Li, J. Liu and M. Ye, J. Anal. At. Spectrom., 2016, 31, 1780–1810 RSC.
- A. Týčová, V. Ledvina and K. Klepárník, Electrophoresis, 2017, 38, 115–134 CrossRef PubMed.
- G. Grabmann, B. Keppler and C. Hartinger, Anal. Bioanal. Chem., 2013, 405, 6417–6424 CrossRef CAS PubMed.
- T. T. T. N. Nguyen, J. Østergaard, S. Stürup and B. Gammelgaard, Anal. Bioanal. Chem., 2013, 405, 1845–1854 CrossRef CAS PubMed.
- H. U. Holtkamp, S. J. Morrow, M. Kubanik and C. G. Hartinger, J. Biol. Inorg. Chem., 2017, 1–10 Search PubMed.
- T. T. T. N. Nguyen, J. Østergaard, S. Stürup and B. Gammelgaard, Anal. Bioanal. Chem., 2012, 402, 2131–2139 CrossRef CAS PubMed.
- G. Grabmann, S. M. Meier, Y. Y. Scaffidi-Domianello, M. Galanski, B. K. Keppler and C. G. Hartinger, J. Chromatogr. A, 2012, 1267, 156–161 CrossRef CAS PubMed.
- T. T. T. N. Nguyen, J. Østergaard, S. Stürup and B. Gammelgaard, Int. J. Pharm., 2013, 449, 95–102 CrossRef CAS PubMed.
- M. Groessl, C. G. Hartinger, P. J. Dyson and B. K. Keppler, J. Inorg. Biochem., 2008, 102, 1060–1065 CrossRef CAS PubMed.
- M. Groessl, C. G. Hartinger, K. Polec-Pawlak, M. Jarosz and B. K. Keppler, Electrophoresis, 2008, 29, 2224–2232 CrossRef CAS PubMed.
- A. K. Bytzek, K. Boeck, G. Hermann, S. Hann, B. K. Keppler, C. G. Hartinger and G. Koellensperger, Metallomics, 2011, 3, 1049–1055 RSC.
- O. Dömötör, C. G. Hartinger, A. K. Bytzek, T. Kiss, B. K. Keppler and E. A. Enyedy, J. Biol. Inorg. Chem., 2013, 18, 9–17 CrossRef PubMed.
- A. K. Bytzek, G. Koellensperger, B. K. Keppler and C. G. Hartinger, J. Inorg. Biochem., 2016, 160, 250–255 CrossRef CAS PubMed.
- A. K. Bytzek, É. A. Enyedy, T. Kiss, B. K. Keppler and C. G. Hartinger, Electrophoresis, 2009, 30, 4075–4082 CrossRef CAS PubMed.
- M. Groessl, A. Bytzek and C. G. Hartinger, Electrophoresis, 2009, 30, 2720–2727 CrossRef CAS PubMed.
- É. A. Enyedy, O. Dömötör, K. Bali, A. Hetényi, T. Tuccinardi and B. K. Keppler, J. Biol. Inorg. Chem., 2015, 20, 77–88 CrossRef PubMed.
- T. T. T. N. Nguyen, J. Østergaard and B. Gammelgaard, Anal. Bioanal. Chem., 2015, 407, 8497–8503 CrossRef CAS PubMed.
- C. S. Allardyce, P. J. Dyson, D. J. Ellis and S. L. Heath, Chem. Commun., 2001, 1396–1397 RSC.
- L. Tan, X. Zheng, L. Chen and Y. Wang, J. Sep. Sci., 2014, 37, 2974–2982 CrossRef CAS PubMed.
- H. U. Holtkamp, S. J. Morrow, M. Kubanik and C. G. Hartinger, unpublished data.
- C. Artner, H. U. Holtkamp, W. Kandioller, C. G. Hartinger, S. M. Meier-Menches and B. K. Keppler, Chem. Commun., 2017, 53, 8002–8005 RSC.
- M. Kubanik, H. Holtkamp, T. Söhnel, S. M. F. Jamieson and C. G. Hartinger, Organometallics, 2015, 34, 5658–5668 CrossRef CAS.
- D. C. Crans, K. A. Woll, K. Prusinskas, M. D. Johnson and E. Norkus, Inorg. Chem., 2013, 52, 12262–12275 CrossRef CAS PubMed.
- S. Hann, A. Zenker, M. Galanski, T. L. Bereuter, G. Stingeder and B. K. Keppler, Fresenius’ J. Anal. Chem., 2001, 370, 581–586 CrossRef CAS PubMed.
- T. Falta, G. Koellensperger, A. Standler, W. Buchberger, R. M. Mader and S. Hann, J. Anal. At. Spectrom., 2009, 24, 1336–1342 RSC.
- T. T. Morris, Y. Ruan, V. A. Lewis, A. Narendran and J. Gailer, Metallomics, 2014, 6, 2034–2041 RSC.
- M. Sooriyaarachchi, G. N. George, I. J. Pickering, A. Narendran and J. Gailer, Metallomics, 2016, 8, 1170–1176 RSC.
- A. R. Timerbaev, L. S. Foteeva, K. Pawlak and M. Jarosz, Metallomics, 2011, 3, 761–764 RSC.
- A. R. Timerbaev, S. S. Aleksenko, K. Polec-Pawlak, R. Ruzik, O. Semenova, C. G. Hartinger, S. Oszwaldowski, M. Galanski, M. Jarosz and B. K. Keppler, Electrophoresis, 2004, 25, 1988–1995 CrossRef CAS PubMed.
- K. Polec-Pawlak, J. K. Abramski, O. Semenova, C. G. Hartinger, A. R. Timerbaev, B. K. Keppler and M. Jarosz, Electrophoresis, 2006, 27, 1128–1135 CrossRef CAS PubMed.
- M. Matczuk, K. Anecka, F. Scaletti, L. Messori, B. K. Keppler, A. R. Timerbaev and M. Jarosz, Metallomics, 2015, 7, 1364–1370 RSC.
- C. A. Lucy, A. M. MacDonald and M. D. Gulcev, J. Chromatogr. A, 2008, 1184, 81–105 CrossRef CAS PubMed.
- N. Iki and E. S. Yeung, J. Chromatogr. A, 1996, 731, 273–282 CrossRef CAS.
- X. Huang, L. J. Doneski and M. J. Wirth, Anal. Chem., 1998, 70, 4023–4029 CrossRef CAS PubMed.
- H. Stutz, Electrophoresis, 2009, 30, 2032–2061 CrossRef CAS PubMed.
- Y. Alahmad, N. T. Tran, I. Le Potier, E. Forest, S. Jorieux and M. Taverna, Electrophoresis, 2011, 32, 292–299 CrossRef CAS PubMed.
- R. Huang, A. R. Ferhan, L. Guo, B. Qiu, Z. Lin, D.-H. Kim and G. Chen, RSC Adv., 2014, 4, 4883–4888 RSC.
- N. N. Poulsen, J. Østergaard, N. J. Petersen, K. Daasbjerg, J. Iruthayaraj, A. Dedinaite, R. Makuska and H. Jensen, J. Sep. Sci., 2017, 40, 779–788 CrossRef CAS PubMed.
- J. F. Kelly, S. J. Locke, L. Ramaley and P. Thibault, J. Chromatogr. A, 1996, 720, 409–427 CrossRef CAS PubMed.
- A. Levina, D. C. Crans and P. A. Lay, Coord. Chem. Rev., 2017, 352, 473–498 CrossRef CAS.
- B. X. Huang, H.-Y. Kim and C. Dass, J. Am. Soc. Mass Spectrom, 2004, 15, 1237–1247 CrossRef CAS PubMed.
- M. D. Retzer, A. Kabani, L. L. Button, R.-H. Yu and A. B. Schryvers, J. Biol. Chem., 1996, 271, 1166–1173 CrossRef CAS PubMed.
- Y. Kasherman, S. Sturup and D. Gibson, J. Med. Chem., 2009, 52, 4319–4328 CrossRef CAS PubMed.
- C. Gossens, A. Dorcier, P. J. Dyson and U. Rothlisberger, Organometallics, 2007, 26, 3969–3975 CrossRef CAS.
- C. Scolaro, C. G. Hartinger, C. S. Allardyce, B. K. Keppler and P. J. Dyson, J. Inorg. Biochem., 2008, 102, 1743–1748 CrossRef CAS PubMed.
- A. E. Egger, S. Theiner, C. Kornauth, P. Heffeter, W. Berger, B. K. Keppler and C. G. Hartinger, Metallomics, 2014, 6, 1616–1625 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: ICP-MS operational values, calibration plots, components of the α-MEM cell medium and foetal calf serum and additional CE-ICP-MS data. See DOI: 10.1039/c8mt00024g |
| This journal is © The Royal Society of Chemistry 2018 |