
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pharmaco-genomic investigations of organo-iridium anticancer complexes reveal novel mechanism of action†
Jessica M.
Hearn‡
ab,
George M.
Hughes‡
 ac,
Isolda
Romero-Canelón
ad,
Alison F.
Munro
e,
Belén
Rubio-Ruiz
e,
Zhe
Liu
af,
Neil O.
Carragher
*e and
Peter J.
Sadler
*a
ac,
Isolda
Romero-Canelón
ad,
Alison F.
Munro
e,
Belén
Rubio-Ruiz
e,
Zhe
Liu
af,
Neil O.
Carragher
*e and
Peter J.
Sadler
*a
aDepartment of Chemistry, University of Warwick, UK. E-mail: p.j.sadler@warwick.ac.uk; Fax: +44 (0)24 7652 3819; Tel: +44 (0)24 7652 3818
bWarwick Systems Biology Centre, University of Warwick, UK
cMOAC DTC, University of Warwick, UK
dSchool of Pharmacy, University of Birmingham, UK
eCancer Research UK Edinburgh Centre, Institute of Genetics and Molecular Medicine, University of Edinburgh, UK
fDepartment of Chemistry and Chemical Engineering, Qufu Normal University, China
First published on 13th November 2017
Abstract
Resistance to platinum drugs (used in >50% of cancer chemotherapies) is a clinical problem. Other precious metal complexes with distinct mechanisms of action might overcome this. Half-sandwich organometallic complexes containing arene or cyclopentadienyl (Cp) ligands show promise. We screened two iridium(III) complexes [Ir(CpXbiph)(ppy)Cl] (ZL49, 1, ppy = phenylpyridine) and [Ir(CpXph)(azpyNMe2)Cl]PF6 (ZL109, 2, azpyNMe2 = N,N-dimethylphenylazopyridine) in 916 cancer cell lines from 28 tissue types. On average, complex 2 was 78× more potent than 1, 36× more active than cisplatin (CDDP), and strongly active (nanomolar) in patient-derived ovarian cancer cell lines. RNA sequencing of A2780 ovarian cells revealed upregulation of antioxidant responses (NRF2, AP-1) consistent with observed induction of reactive oxygen species (ROS). Protein microarrays, high content imaging and cell cycle analysis showed S/G2 arrest, and late-stage DNA damage response without p53 requirement. The triple-negative breast cancer cell line OCUB-M was highly sensitive to 2 as were cell lines with KIT mutations. Complex 2 exhibits a markedly different pattern of antiproliferative activity compared to the 253 drugs in the Sanger Cancer Genome database, but is most similar to osmium(II) arene complexes which share the same azopyridine ligand. Redox modulation and DNA damage can provide a multi-targeting strategy, allowing compounds such as 2 to overcome cellular resistance to platinum anticancer drugs.
Significance to metallomicsHalf-sandwich organoiridium complexes show promising activity, with novel mechanisms of action that may combat platinum resistance and side effects seen commonly in current anticancer chemotherapy regimes. The field of systems pharmacology provides methods to elucidate the often multitargeted mechanisms of metallodrugs. Here, we explore the link between transcriptomic and phenotypic data in ovarian cancer cells, using state-of-the-art high-throughput methods, together with data collected from Sanger's Genomics of Drug Sensitivity in Cancer screen and primary tumour cell lines. Our studies ultimately reveal not only the subtle effects of ligands on complex activity, but unique mechanisms shared only with osmium arene complexes. |
Introduction
The square-planar platinum drugs cisplatin (CDDP), carboplatin and oxaliplatin are now used in over 50% of cancer chemotherapies, but intrinsic and acquired resistance represent major clinical problems across cancer indications. CDDP has a primary mechanism of action (MoA) which involves DNA targeting, induction of DNA bending, and apoptosis.1 Other precious metal complexes might be able to overcome resistance if they have different MoAs. This appears to be the case for platinum drug oxaliplatin and candidate drug phenanthriplatin, which are not cross-resistant with CDDP. Although oxaliplatin is classified as a DNA alkylating agent like CDDP, recent evidence now points primarily to ribosome biogenesis stress, rather than DNA damage.2Pseudo-octahedral ‘half-sandwich’ organometallic arene and cyclopentadienyl complexes also show promise for overcoming platinum resistance. Osmium(II) arene complexes and iridium(III) cyclopentadienyl (CpX) complexes [M(arene/CpX)(N,N)(X)]n+ can target DNA when N,N is a chelated diamine ligand.3,4 In contrast, when N,N is an azopyridine ligand, the complexes can cause redox stress in cancer cells and target mitochondrial metabolism.5–8 Azopyridine ligands are strong π-acceptors and withdraw electron density from the metal making the M–C bond to the monodentate ligand X (e.g. Cl or I) much less reactive.
We have discovered potent antiproliferative activity for organo-Os(II) and organo-Ir(III) complexes by phenotypic screening and have shown that screening in the National Cancer Institute (NCI-60) and Sanger 800+ panel of cancer cell lines when combined with transcriptomics, reactive oxygen species (ROS), protein micro-arrays, high content imaging and other assays can provide valuable insight into their MoAs.9,10
Here we report the results of phenotypic screening and in-depth MoA profiling of two organo-iridium(III) complexes (Fig. 1) [Ir(η5-CpXbiph)(ppy)Cl] (ZL49, 1, ppy = phenylpyridine) and [Ir(η5-CpXph)(azpyNMe2)Cl]PF6 (ZL109, 2, azpyNMe2 = N,N-dimethylphenylazopyridine) in the Sanger Cancer Genome project panel of 916 cancer cell lines with known gene sequences from 28 tissue types. Time-dependent up- and down-regulation of genes has been studied by RNA sequencing, together with the detection of selected proteins and quantitative analysis of cell cycle, and apoptosis phenotypes.
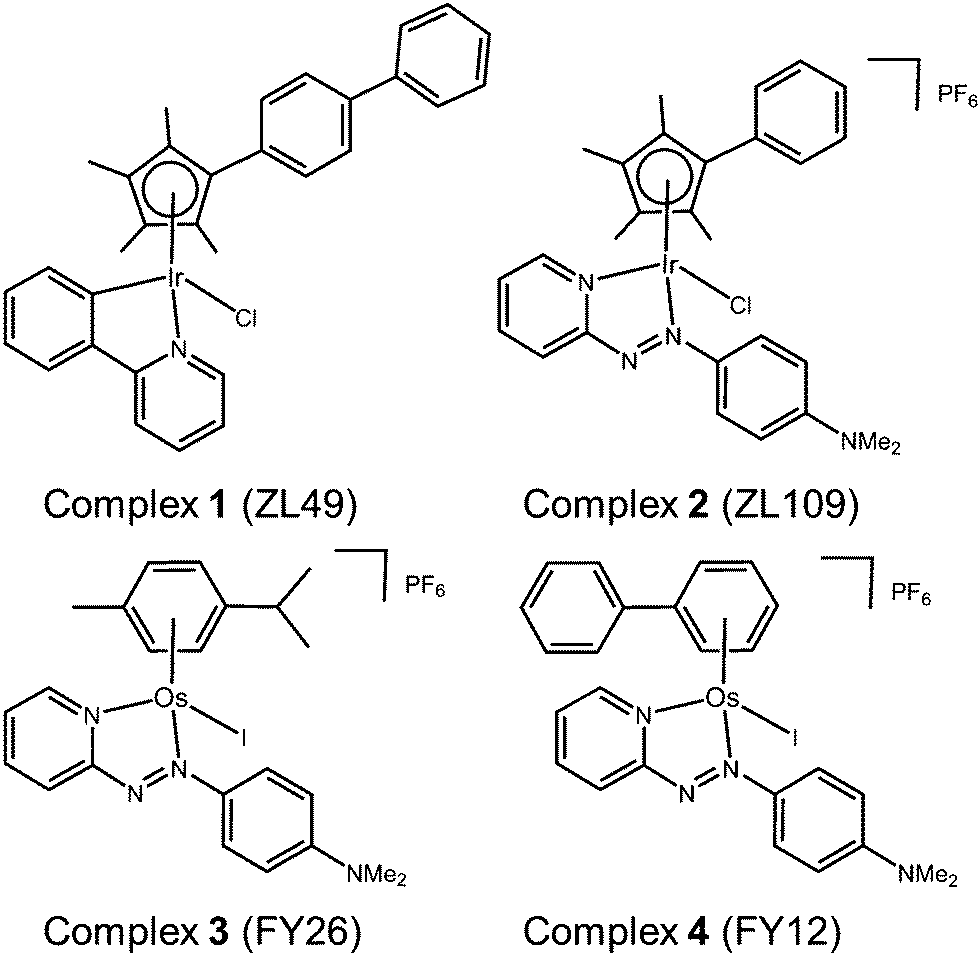 | ||
| Fig. 1 Organo-Ir(III) complexes 1 and 2 and organo-Os(II) complexes 3 and 4 referenced in this work. | ||
These studies point to a unique MoA for organo-iridium complexes with little in common with previously-screened agents, except organo-osmium complexes.
Results
Antiproliferative activity
Initially, we screened organo-iridium complexes 1 ([Ir(η5-CpXbiph)(ppy)Cl]) and 2 ([Ir(η5-CpXph)(azpy-NMe2)Cl])PF6 in the NCI-60 screen.8,11 Complex 2 was more active than 1, however both were potent with mean IC50 values of <1 μM, more potent than the clinical drug CDDP. Both compounds were particularly active in colorectal cell line COLO205 and breast cell line MDA-MB-468, with renal cell lines showing resistance (much lower sensitivity) to both.The higher activity of complexes 1 and 2 towards COLO205 and MDA-MB-468 can be attributed to redox deficiencies in these cell lines. COLO205 colorectal cells have reduced levels of glutathione-S-transferase P1 (GSTP1), an enzyme which detoxifies drugs by conjugating them to glutathione (GSH) often limiting the efficacy of anticancer agents.12 Without this enzyme, there may be less conjugation and inactivation by GSH, which could increase the sensitivity of cells towards anticancer compounds.
The breast cell line, MDA-MB-468 showed a consistently high sensitivity in the NCI-60 screen (IC50 values 17 nM to 1.8 μM). This cell line has a glucose 6-phosphate dehydrogenase (G6PD) A phenotype, meaning it lacks the proper functioning of this enzyme in the pentose phosphate pathway.13 G6PD is the rate-limiting enzyme in this pathway; its deficiency blocks the conversion of glucose-6-phosphate to 6-phosphoglucono-D-lactone and in the process prevents the conversion of NADPH to NADP+, a required step in GSH synthesis.14 This suggests that a GSH deficiency may play an important role in potentiating the activity, something previously demonstrated when A2780 cells were co-incubated with low levels of the GSH inhibitor, L-buthionine sulfoximine. Multiple studies have shown that this deficiency prevents the cells from mediating oxidative stress, and often results in apoptosis and necrosis of cells in patients.15
Comparison of the patterns of selectivity for both complexes with those of >40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 synthetic compounds and natural products in the NCI database and drugs that showed a Pearson's correlation coefficient (r) > 0.5, revealed only 3 drugs with similar patterns of selectivity to that of 1 and 2, highlighting their novelty.
000 synthetic compounds and natural products in the NCI database and drugs that showed a Pearson's correlation coefficient (r) > 0.5, revealed only 3 drugs with similar patterns of selectivity to that of 1 and 2, highlighting their novelty.
Now we have screened complexes 1 and 2 in 916 cell lines from 28 tissue types in the Sanger Cancer Genome project panel and compared the patterns of antiproliferative activity with the 256 compounds in the ‘Genomics of Drug Sensitivity in Cancer’ database.16
In Fig. 2, the IC50 values for 1 and 2 and CDDP are compared for 809 cell lines in the screen using box and whisker plots. On average across all cell lines, complex 2 is 78× more potent than complex 1 and 36× more potent than CDDP.
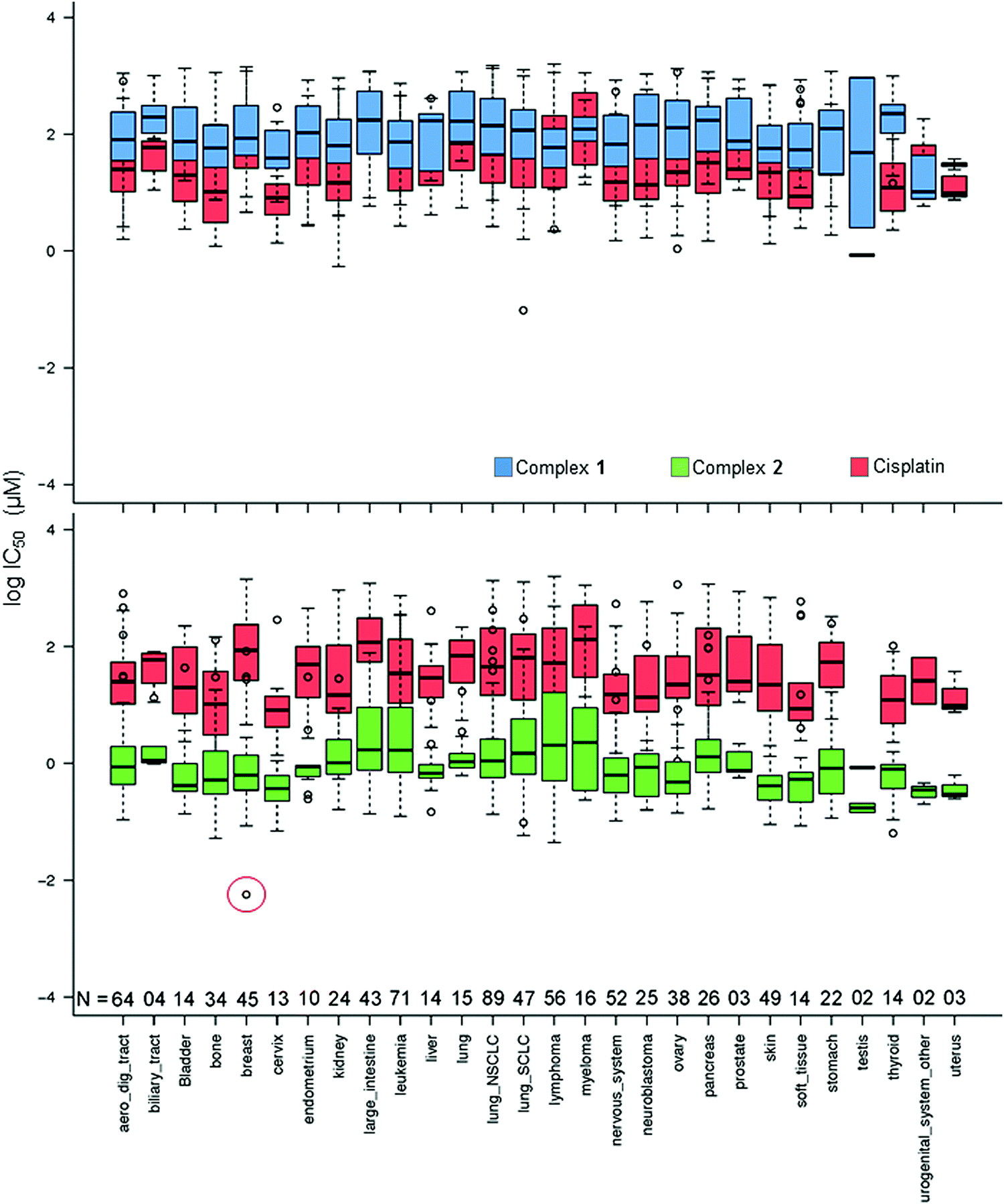 | ||
| Fig. 2 Box and whisker plots for log IC50 (μM) values, complex 1 (blue), 2 (green) and CDDP (red) in the Sanger screen, separated by tissue type. N denotes the number of cell lines of each tissue type in the screen. The red circle denotes the IC50 value for the breast cancer line OCUB-M that exhibits extreme sensitivity to 2 and, in contrast, low sensitivity to both 1 and CDDP. Total number of cell lines included = 809 (to match the cell lines screened against CDDP). IC50 values which fall outside the experimental screening concentrations were calculated using a curve-fitting algorithm, and as such have a large confidence interval.16 | ||
Complex 2 exhibits its highest mean activity in cancers of testicular, urogenital, cervical and uterine tissues, although the number of cell lines derived from these tissues is considerably lower than for many of the other tissue types. Complex 1 shows its highest activity in the SU-DHL-5 lymphoma cell line; however, this potency is lower than that of 2 and CDDP.
Complex 2 displays its highest activity in the OCUB-M breast cancer cell line, and notably is ca. 5 orders of magnitude more active than CDDP. OCUB-M is a triple negative breast cancer cell line with high expression of E-cadherin and laminin receptors.17 There are 10 genetic mutations in this cell line, one of which is a missense mutation in p53. As mentioned above, in previous work it was also shown that MDA-MB-468, another triple negative cell line, had significant susceptibility to 2.8 Triple-negative breast cancer is particularly difficult to treat; therefore further studies of 2 in this area would seem warranted.
Complex 2 and the organo-osmium complex FY12 (complex 4 in Fig. 1), which notably contains the same chelated azopyridine ligand, are amongst the 15 most potent drugs screened against OCUB-M in the Sanger panel. Five others are microtubule-targeting drugs, constituting all the drugs in this category in the screen. The successful treatment of OCUB-M with microtubule inhibitors may be linked to the over-expression of TUBG1 (tubulin SF) and MAP7 (microtubule-associated protein) and the down-regulation of MAP1LC3B (microtubule associated protein 1 light chain 3 beta), MICAL-1 (microtubule associated monooxygenase, calponin and LIM domain containing 1) and TTBK2 (Tau tubulin kinase). The over-expressed genes mediate microtubule formation and stabilisation. The down-regulated genes can cause destabilisation and depolymerisation of tubulin and actin, respectively.
Although sensitive to complex 2, OCUB-M is highly insensitive to CDDP. This might be linked to mutations in its p53 protein, which has been repeatedly correlated with CDDP resistance.18 Fig. S1 (ESI†) shows the distribution of IC50 values for 1, 2 and CDDP, where boxes show the median, upper and lower quartiles. The red lines mark the threshold for lower sensitivity compared to complex 2. Complex 2 shows considerably higher activity than complex 1 (by 78×) and CDDP (by 36×), as well as being significantly more potent when compared to the mean activity over all ca. 200 drugs included in the screen (Table S4, ESI†). The IC50 values for 46 of the 916 cell lines lie above the upper whisker threshold for 2. This population of less sensitive cell lines falls within the activity ranges of 1 and CDDP. The occurrence of so many cell lines far outside the interquartile range for 2 might be due to a common resistance feature, although may also reflect the tighter spread amongst values for the more sensitive cell lines.
Table 1 lists the 10 cell lines exhibiting the lowest sensitivity to complex 2. The tissue types displaying the highest percentages are lymphoma, myeloma and leukemia, each with >10% of their population less sensitive to 2 (Fig. S2, ESI†). These particular tissues make up the blood-derived cancers. Although some factor specific to tissues derived from the embryonic mesoderm may contribute to the low sensitivity, other mesodermally-derived tissues, such as bone, urogenital and kidney tissue do not exhibit a large proportion of resistant cells, making it more likely that some other feature of blood-derived cancers confers resistance. Cancers of ectodermally-derived tissues such as the skin and nervous system show little or no cell lines in the resistant population.
| Subtype | Cell line | Resistance drug | Drug target |
|---|---|---|---|
| Lung | LU-65 | Vorinostat and piperlongumine | HDAC |
| NCI-H2081 | Piperlongumine | Increases ROS levels | |
| NCI-H2135 | Piperlongumine | Increases ROS levels | |
| Lymphoma | WSU-DLCL2 | LAQ824 | HDAC |
| A4-Fuk | N/A | N/A | |
| Aerodigestive | ESO51 | Piperlongumine | Increases ROS levels |
| OACM5-1 | TW 37 | BCL-2, BCL-XL | |
| Leukemia | PF-382 | Piperlongumine | Increases ROS levels |
| Myeloma | U-266 | NU-7441 | DNAPK |
| Stomach | NCI-SNU-1 | BEZ235 | PI3K, mTORC1/2 |
It is apparent that resistance to piperlongumine treatment is common for these cell lines: 19 out of 46 cell lines least sensitive to complex 2 can also be classified as resistant to piperlongumine, an organic natural product isolated from the fruit of the pepper plant Piper longum. Piperlongumine has been evaluated as a novel senolytic agent, selectively inducing death of senescent cells,19 killing cancer cells by targeting the stress response to ROS.20
The level of similarity between 1 and 2 and other drugs in the screen was evaluated from differences in their IC50 values. To account for the fact that large positive and large negative differences could cancel each other out, the standard deviations of these differences were also calculated. A drug was considered similar to the index drug if the total mean difference in IC50 was close to zero and the SD was low (denoted by the red circle in Fig. 3).
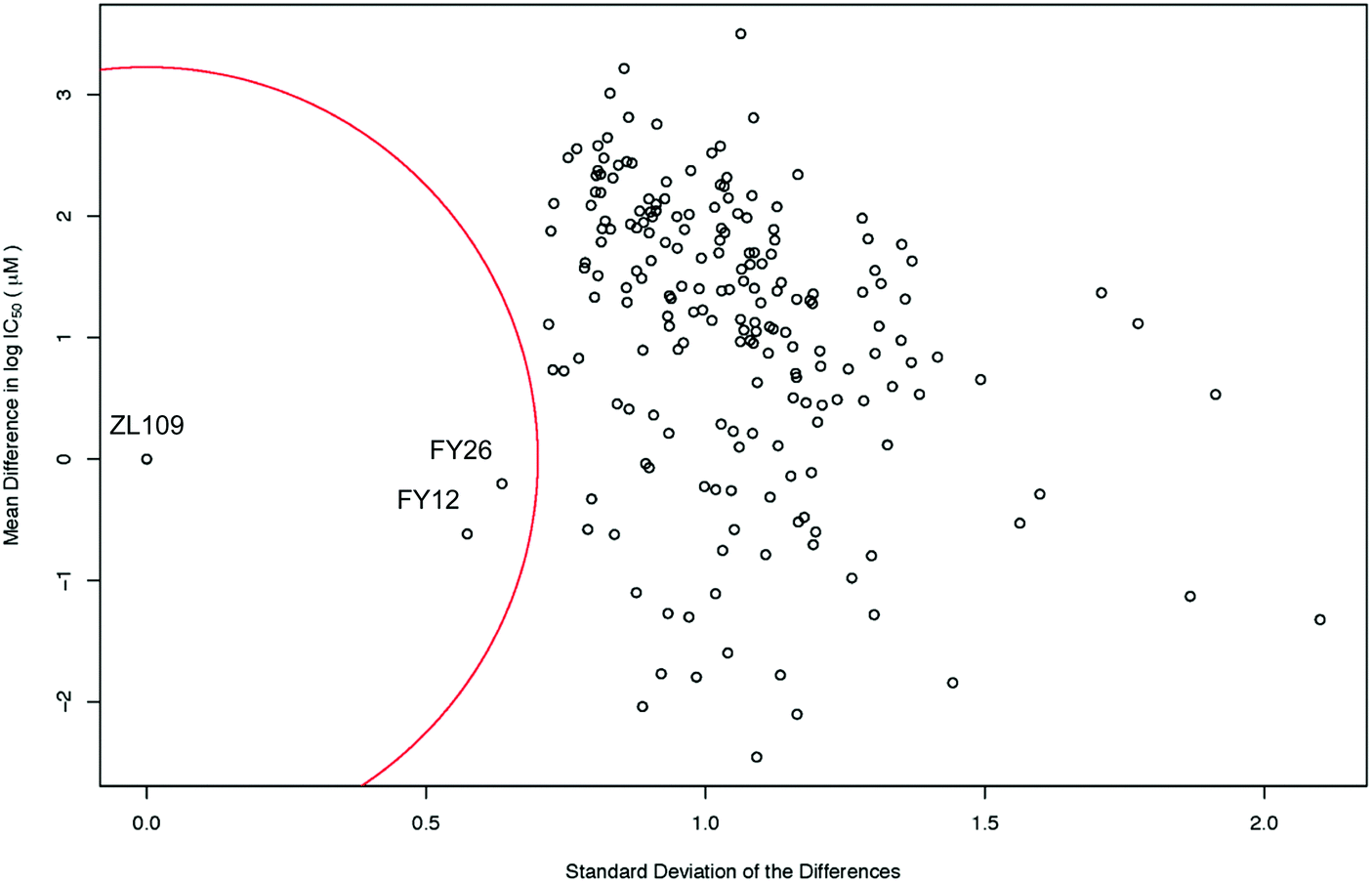 | ||
| Fig. 3 Scatter plot of the mean difference in log IC50 values (μM) between all screened drugs and complex 2 (ZL109) vs. the SD of these differences. The red circle of radius 0.7 standard deviations denotes a ‘region of significant similarity where the mean difference and standard deviation are low. The similarity with two osmium arene complexes which share the same chelated azopyridine ligand (complexes 3 (FY26) and 4 (FY12), Fig. 1) is evident. | ||
Intriguingly, the data for iridium complex 2 matched most closely those of FY12 ([Os(η6-bip)(azpy-NMe2)I]+) and FY26 ([Os(η6-p-cym)(azpy-NMe2)I]+), (complexes 4 and 3 respectively, in Fig. 1) two organo-osmium compounds studied previously in our laboratory.21 FY12/FY26 and 2 share the same bidentate azopyridine ligand, but have different C-bound π-donor ligands (neutral arene versus negatively-charged cyclopentadienyl), different monodentate halido ligands (chloride versus iodide) as well as metal centres (Os(II) versus Ir(III)). The much higher similarity of these two matches to complex 2 compared to other drugs tested in the screen is evident, demonstrating novelty in the MoA of complex 2 (Fig. 3).
To extract significant correlations between activity and oncogenic behaviour of compounds from the Sanger screen, an analysis of variance (ANOVA) test can be used. The dependent variable is presented as an n × 2 matrix for each compound, containing IC50 and b (slope) values, and therefore, a multivariate ANOVA (MANOVA) is used for the analysis. For each gene, a linear model explains these activity observables with ‘mutation status’ as the predictive variable. Only genes mutated in >2 cell lines were included in the analysis, and a Benjamin–Hochberg multiple testing correction was used to identify significant results (FDR < 0.20). This quantitative assessment of pharmacogenomics uses an effect size parameter, calculated between wild type and mutant populations. If the effect size is >1, the difference in mean value between the mutated and wild-type populations is greater than the variance in at least one population, and the effect is therefore large.
It is evident that when the CASP8 gene is mutated, cells become sensitized to 1 (Table 2). This gene encodes caspase-8, which is involved in apoptosis and signalling cascades through TNF and NFkB. The only other drug in the screen for which CASP8 coding mutation confers sensitivity is ABT-888 (veliparib), a PARP inhibitor. The PARP family of proteins are involved primarily in DNA repair and induction of programmed cell death. Where the DNA damage is beyond repair, PARP is inhibited so as to preserve cell ATP. The cell then undergoes programmed cell death.
| Complex | Mutated gene | No. cell lines | Effect size | FDR |
|---|---|---|---|---|
| 1 | CASP8 | 17 | 0.57 | 0.18 |
| 2 | KIT | 4 | 0.83 | 0.08 |
The KIT gene coding mutation is associated with the sensitivity of four NSCLC cell lines to complex 2, and encodes the human homolog of the proto-oncogene C-KIT, a type-III transmembrane receptor for the stem cell growth factor. Once bound to growth factor, this protein activates signal transduction through the cell, affecting cell survival and differentiation.
Matched pairs of patient-derived platinum sensitive and resistant ovarian cancer cell models
Matched pairs of ovarian cancer cells directly isolated from patients before and after relapse to platinum therapy are likely to better represent clinical drug resistance and relapse mechanisms relative to drug resistant cell clones generated following in vitro drug exposure. Complexes 1 and 2 were tested in the matched patient-derived ovarian cancer cell lines PE01 (patient prior to treatment), PE04 (patient after taxane/platinum treatment)22 and also PE01-CDDP (PE01 cells exposed to low levels of CDDP in vitro to create Pt resistance), Table 3.| Complex | Cell line | IC50 (μM) |
|---|---|---|
| 1 | PE01 | 22.4 |
| PE04 | 64.3 | |
| PE01-CDDP | 110.3 | |
| 2 | PE01 | 1.21 |
| PE04 | 1.65 | |
| PE01-CDDP | 1.56 | |
It can be seen from Table 3 that complex 2 maintains high activity in all 3 patient-derived cell lines. Conversely, 1 has poor activity in PE01 and loses activity in cells from patients treated with taxane/CDDP and even more so in CDDP-resistant cells. This shows that the activity of 2 is unaffected by CDDP and/or taxane resistance in these ovarian cell lines, unlike 1, suggesting a lack of cross resistance due to its different MoA. Given the rise in occurrence of clinical platinum resistance, this is a promising finding for complex 2.
Gene expression analysis
To investigate the MoA of the more active complex 2, A2780 ovarian cancer cells were exposed to the IC50 concentration (0.40 μM) and the differential gene expression was explored using RNA sequencing across a 48 h time series; measuring gene expression after 4, 12, 24 and 48 h exposure. Approximately 30 million 50 bp, paired-end reads were generated per sample, and sequence reads were mapped to the hg19 (GRCh37) human genome (accession E-MTAB-5991). Table S1 (ESI†) gives the sequencing statistics for the experiment.Fig. S3 (ESI†) shows multidimensional scaling (MDS) plots for complex 2- and control-exposed samples across the time series. These plots are a means of visualising the level of similarity in sample sets, by placing each of them in N-dimensional space, where between – object distances are preserved.
The MDS plots show natural separation of the samples into clusters, and good agreement between the triplicate measurements. The biggest source of variation is by time point, followed by exposure-status, i.e. whether they are exposed to a control or to complex 2. Fig. S3B (ESI†) provides clear evidence that the samples exposed for 48 h are significantly different from those exposed for shorter time periods, both in the control and in the compound-exposed samples.
Fig. S4A (ESI†) shows the number of differentially expressed genes (DEGs) at 4, 24 and 48 h, and highlights where differential expression overlapped. Fig. S4B (ESI†) shows the number of those genes that were up- or down-regulated across the series. The highest number of differentially expressed genes was found after 12 h exposure (746) and 24 h (719), with significant differential expression (DE) after just 4 h exposure (349). Fig. S4B (ESI†) shows that at 12 h, there is significantly more down-regulation than up-regulation, almost a 2:1 ratio, compared to 24 h, where there is ca. 1:1 ratio of up- and down-regulation.
We carried out pathway analysis with Ingenuity Pathway Analysis software which uses an extensive literature database (Ingenuity Knowledge Base) to integrate biological and chemical pathway perturbation information.23 DEGs with 1.0 < LogFC > 1.0 and FDR < 0.05 were assigned to biological pathways. Table S2 (ESI†) shows the top five upstream regulators of these pathways, across the time series. The tables show the p value as calculated by a hypergeometric test of over representation, and the z-score that describes the extent of activation/inhibition of each pathway. One of the hurdles with using high-content assays is that a drug's MoA can be swamped by larger cellular responses, like cell survival/death and stress signalling which control often overlapping downstream processes. This makes it difficult to pinpoint the MoA, however, these results show that cells activate stress response pathways even after short times of exposure to organo-iridium complex 2.
Since previous studies on complex 2 suggested that oxidative stress may be a part of its MoA, we examined the DE of oxidative stress response markers (Fig. 4).8 The NRF2 transcription factor (encoded by the NFE2L2 gene) is involved in an important antioxidant response pathway, binding to the hARE (human antioxidant response element) cis-element to transactivate detoxifying/antioxidant genes. AP-1 complexes C-FOS/C-JUN and FRA-1/c-JUN, encoded by FOS, FOSL1 and JUN genes, compete with NRF2 for binding to hARE to active antioxidant genes. Two domains of NRF2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription.24
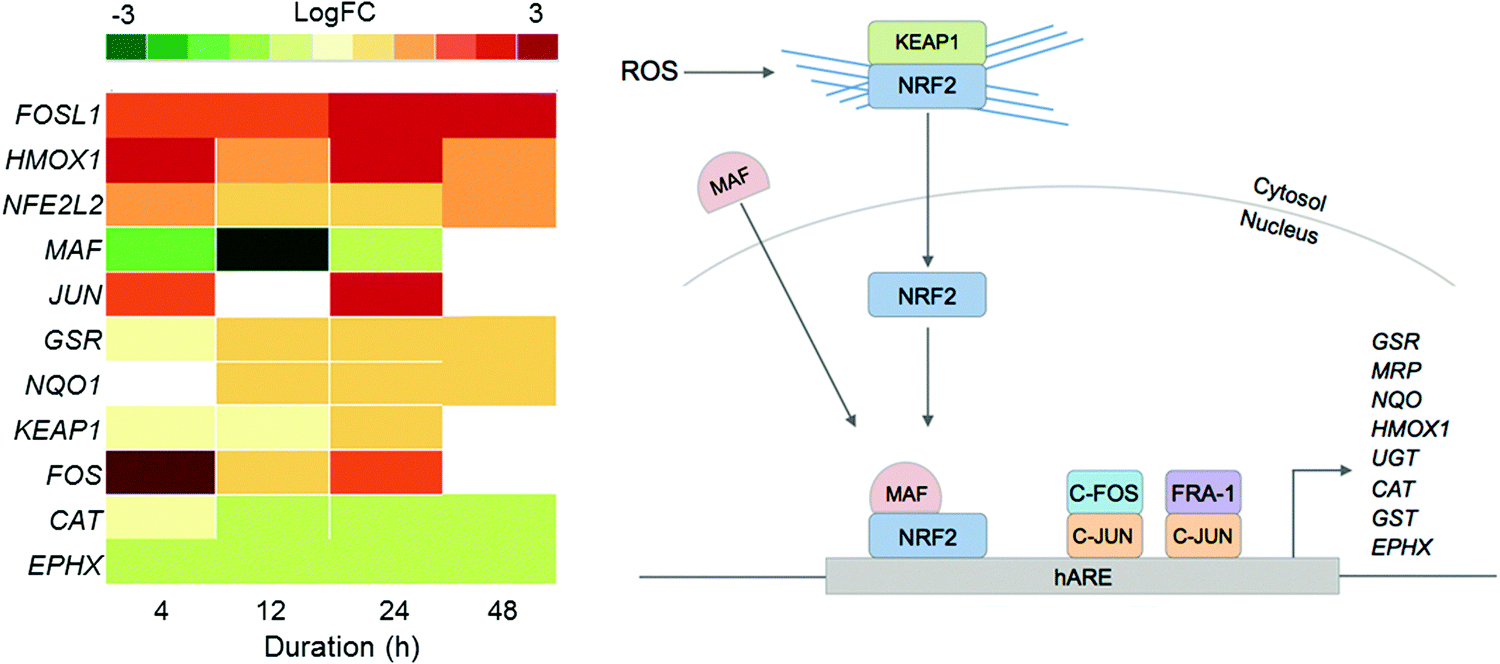 | ||
| Fig. 4 Heat map of DEGs in the oxidative stress response pathway over the 48 h exposure of A2780 human ovarian cancer cells to complex 2. Only DEGs with FDR < 0.10 are included. | ||
Even though NFE2L2 is up-regulated throughout the time series in response to 2, the MAF co-factor is significantly down-regulated. Of the two AP-1 complexes, FRA-1 (FOSL1) is up-regulated throughout the time series, showing significant DE after just 4 h and highest expression after 24 h. The second AP-1 complex, with C-FOS (FOS) is also up-regulated for 2, with the biggest response at 4 h, suggesting that the AP-1 complex may play a role during early exposure, and the FRA-1 complex a role in later exposure. Collectively this shows that all three transcription factors for oxidative stress response, NRF2 and the two AP-1 complexes, play a role in the cellular response to 2.
Only a selection of antioxidant genes downstream of the transcriptional activators are up-regulated, and those that are up-regulated are not drastically so. Antioxidant genes CAT (catalase) and EPHX (epoxide hydrolase), are both down-regulated. GSR (glutathione reductase) and NQO (NADP(H) quinone oxidoreductase) are both up-regulated. The low-level expression of downstream effectors may suggest that A2780 ovarian cancer cells have a poor mechanism for responding to ROS.
Using flow cytometry, measurements were taken to assess the capability of 2 to generate ROS and superoxide (SO) in A2780 cells after 24 h exposure at IC50 concentration at 310 K (Fig. S5, ESI†). Table S3 (ESI†) highlights the results and shows a significant production of total ROS and SO in 84% of cells exposed to complex 2, with the remaining 16% still showing significant levels of total ROS alone.
Given the similarity of iridium cyclopentadienyl complex 2 and osmium arene complex 3 (FY26) in the Sanger screen, the regulation of the ROS response pathway, as seen by RNA analysis, was also compared. In previous work, the same cell lines were exposed to FY26 at its IC50 concentration with the same protocol used here; DEGs were analysed across a 48 h time series (Fig. S6, ESI†).
Interestingly, although both compounds induce an oxidative stress response, there are differences in the components of the pathway that are up- and down-regulated. For example, FOS is up-regulated in response to iridium complex 2, but down-regulated in response to osmium FY26. This suggests that the oxidative stress response to complex 2 activates both AP-1 complexes, but activates only one in response to FY26.
Comparison of flow cytometry data for ROS measurements also supports a difference in the ROS levels in cells responding to 2 compared to FY26.6 For example, when exposed to FY26, 95% of cells showed high total ROS and high SO levels, compared to the 84% in response to 2. Yet, the levels of total ROS alone were drastically reduced in response to FY26, with only 2.5% of cells in this state, compared to 16% in response to 2.
The apoptotic cell response in Fig. S7 (ESI†) shows significant down-regulation of effector caspases after 12 h (CASP-9, -6, -2, -3 and -7). Genes for inhibitors of apoptosis proteins (IAP), which inhibit the activation of caspase proteins, such as BIRC2 and more significantly BIRC3, are up-regulated after 4 h. BIRC5, which codes for survivin, another IAP, was down-regulated throughout the time series, suggesting specific modulation of apoptotic signals through caspase inhibition.
Protein microarrays
Zeptosens reverse phase protein microarrays (RPPA) were used to study the cellular levels of key proteins in 2-exposed cells versus controls. RPPA measures the abundance of total protein levels and phosphorylated proteins using epitope-specific antibodies. The Relative Fluorescence Intensity (RFI) values obtained for each protein, following secondary fluorescent antibody detection, were normalized to a house-keeping protein in each sample (prohibitin) before the ratios of RFI between 2-exposed and control samples were determined across the time-series (4, 24, 48 and 72 h), to quantify the abundance of total protein and phospho-epitopes relative to vehicle (DMSO) controls. Given the similarity in the Sanger screen between iridium complex 2 and the osmium complex 3 (FY26), we compared our previously published RPPA data to those of 2 to explore the DNA damage response.During cellular response to DNA damage, ATM, among other proteins, is mobilised and auto-phosphorylated to ATM*, which in turn phosphorylates CHK2 to CHK2*. At this point, the response pathway branches into a rapid response via CDC25A, and a delayed response via p53 and p21. Fig. 5 shows that the protein response reaches a peak at 24 and 72 h exposure, for both complex 2 and FY26.
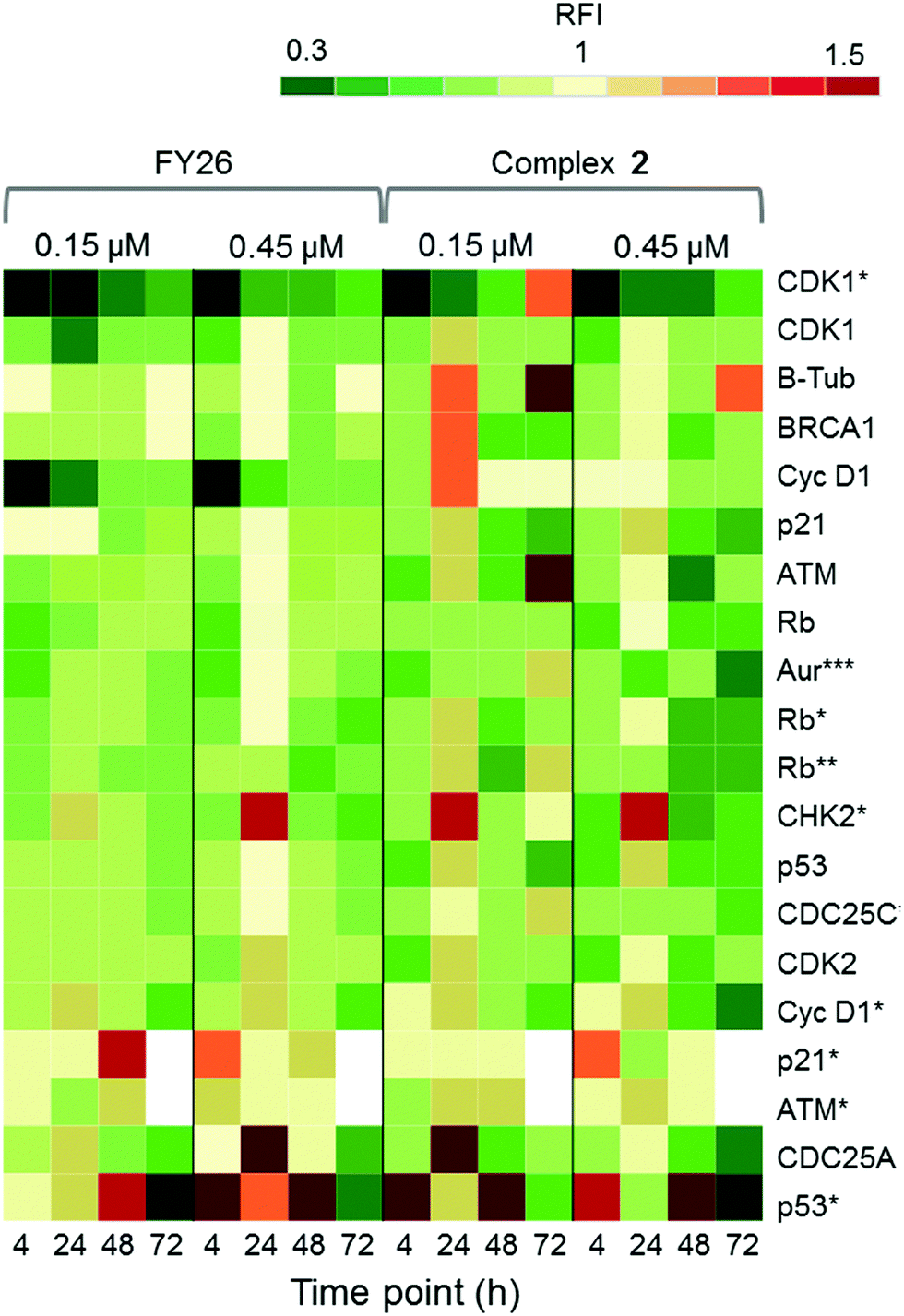 | ||
| Fig. 5 DDR/cell cycle investigations. Heat map showing the relative fluorescence intensity (RFI) values for a selection of DNA damage response (DDR) proteins in cells exposed to 2 across a 72 h time series. Data previously published for osmium complex FY26 are also included for comparison.6 RFI values <1 indicate a drug-induced decrease in protein levels, RFI = 1 indicates no-drug effect and RFI > 1 indicates drug-induced increases in protein levels. | ||
CHK2*, CDC25A and p53* all show increased levels in response to both compounds. However, only 2 appears to mobilize a BRCA1 DNA damage response after 24 h, and primarily at a low concentration.
Additionally, after exposure to 2, M-phase markers are detected, something not seen for FY26. For example, there is a high level of β-tubulin in response to 0.15 μM 2 at both 24 and 72 h, with an additional increase in the levels of CDK1. This suggests that cells still have the proteins available to allow progression from G2 to M phase of the cell cycle. At the higher dose of 2, both tubulin and CDK1 levels do not change from the control at 24 h. The higher levels of Rb* protein at 24 h suggest that cells have passed, or are at least able to pass, the G1/S transition point. This is not seen for FY26, for which levels of these proteins are lower in compound-exposed cells. Therefore, in response to 2, cells may be held in late S phase, or G2 phase of the cell cycle, whereas in response to FY26 cells are more likely held in G1 phase.25
Flow cytometry experiments were performed on A2780 cells exposed to complex 2 and compared to those reported for FY26, to detect the populations of cells in various stages of the cell cycle (Fig. 6). The red fluorescence of propidium iodide (PI) dye, which intercalates into DNA, correlates with the DNA content of the cells, and is twice as high in G2/M as in G0/G1 cells.
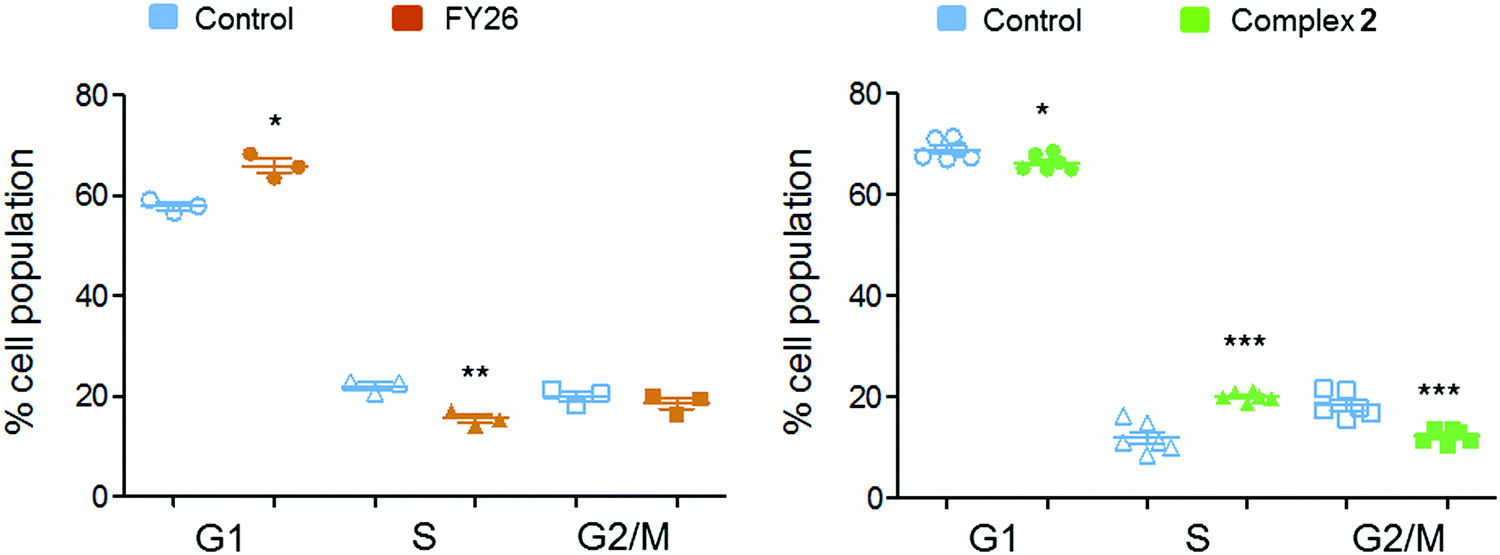 | ||
| Fig. 6 Comparison of the effects of iridium complex 2 and osmium complex 3 on the population of the various phases of the A2780 cell cycle. Cell populations for control samples are shown in blue and those for 24 h 2-exposed samples in green and 3-exposed in brown. * Indicates p < 0.10, ** indicates p < 0.05 and *** indicates p < 0.01 after a Welch two sample t-test between control- and compound-exposed samples in each stage of the cell cycle. | ||
After 24 h exposure to 2, the population of cells in the S phase of the cell cycle had increased, with subsequent G1 and G2/M populations decreasing. Conversely, exposure to 3 (FY26) caused G1 arrest, with lower levels of cells populating the S-phase. S-phase arrest is often associated with compounds that cause DNA damage, suggesting this could be a more prominent part of the MoA for complex 2.
RPPA was also used to study further the differential levels of eighteen apoptotic proteins; five pro-survival proteins and thirteen pro-apoptotic proteins, in cells responding to 2 across a 72 h time series. This was compared to data previously reported for FY26 (complex 3).
Fig. 7 shows that complex 2 induced high levels of IAP proteins, survivin and XIAP. Osmium complex 3 (FY26) also induced these same proteins, however, to a much lower level. BCL-X, which is a pro-survival protein located in the mitochondria, was down-regulated during most of the time series, but up-regulated at 24 h, suggesting specific modulation of the apoptotic pathway. Of the pro-apoptotic proteins, most were down-regulated, however, BCL-2 and BAK, which are initiator proteins, were up-regulated at 24 h. In addition, PARP cleaved by CASP-3 (PARP*), was present in higher levels after 24 h, particularly at the lower concentration of administered compound.
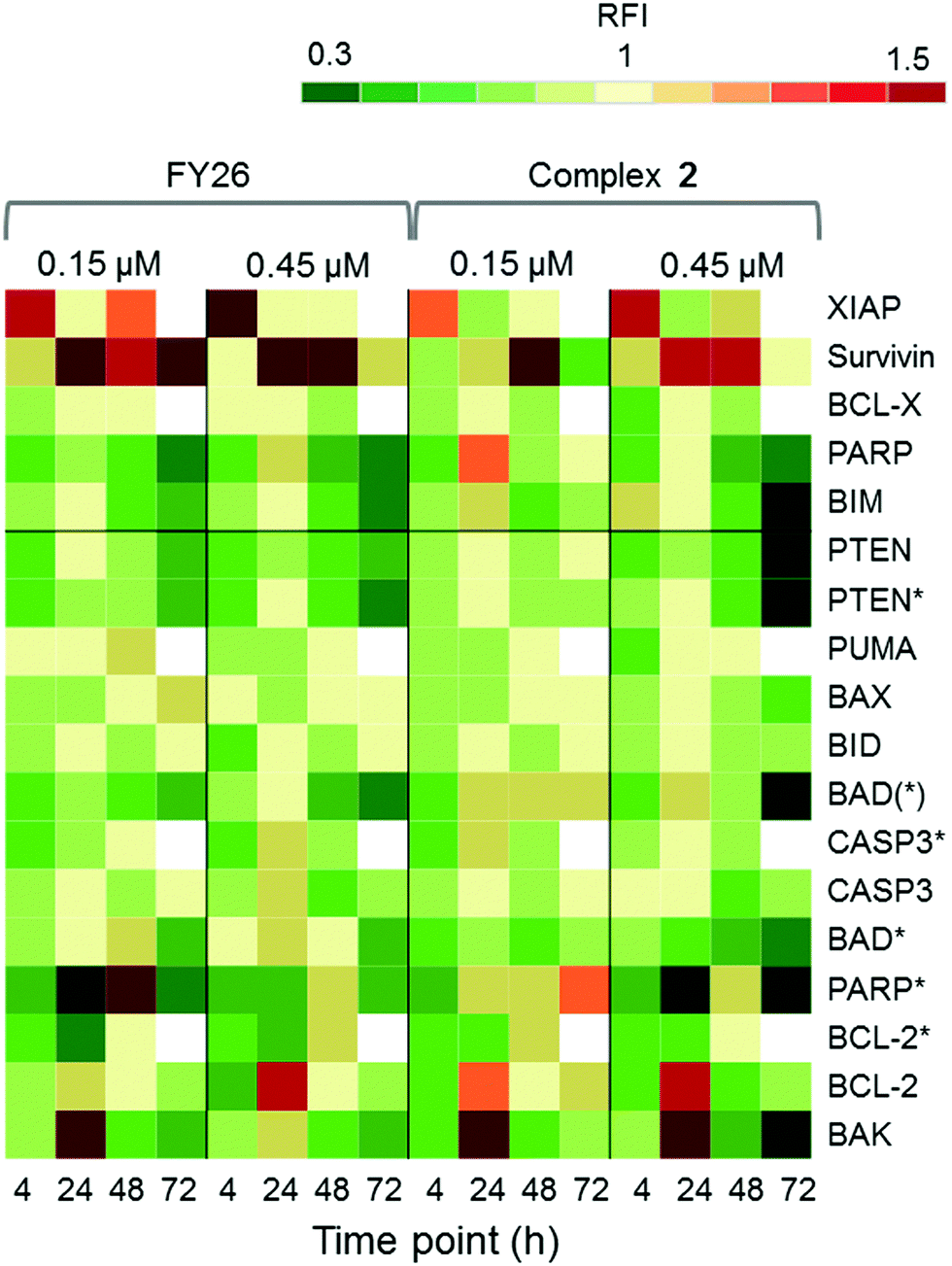 | ||
| Fig. 7 Apoptosis investigations. Heat map showing the relative fluorescence intensity (RFI) values for a selection of apoptotic proteins in A2780 cells exposed to 2 across a 72 h time series. Data reported for 3 (FY26) are presented for comparison. | ||
Flow cytometry studies were performed to determine the level of apoptotic cells after exposure to 2. These experiments measure the fluorescence of annexin V and propidium iodide dyes, where the former identifies early apoptotic cells and the latter identifies late-apoptotic and/or necrotic cells. During early apoptosis, membrane-bound phosphatidylserine proteins, which ordinarily face into the cytoplasm, translocate to the outside of the cell membrane and allow annexin V binding.26 A cell in early apoptosis, will still maintain its membrane integrity, therefore the DNA intercalator dye, propidium iodide, cannot pass through. Once the cell has lost viability, its membrane becomes permeable to propidium iodide, which fluoresces upon binding to DNA.
Fig. 8 shows that there are different levels of apoptosis after 24 and 48 h exposure to 2. After 24 h exposure, there was a slight decrease in cell viability, with a more significant decrease when exposed to a higher dose of 2 (94.2%) compared to the lower dose (95.1%). There was a higher population of cells in early apoptosis at the higher dose compared to the lower (2.0% and 1.3%, respectively), but a lower number of late apoptotic (1.1% and 1.5%) and necrotic cells (2.2% and 2.3%).
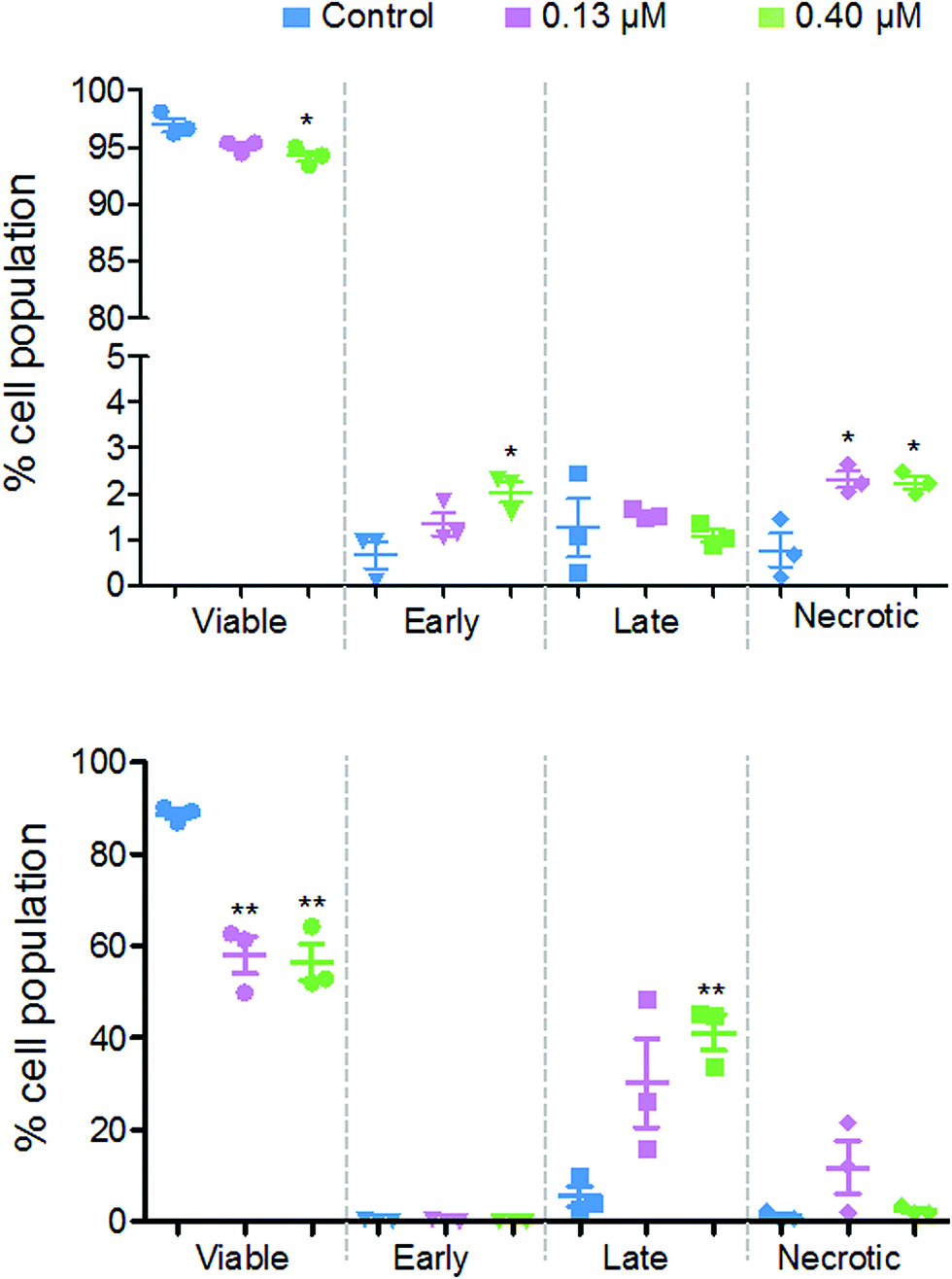 | ||
| Fig. 8 The populations (%) of the various stages of apoptosis/cell death after exposure of A2780 human ovarian cancer cells to 0.13 μM (purple) or 0.40 μM (green) organo-iridium complex 2 for 24 h (top) and 48 h (bottom). | ||
After 48 h there was a more significant drop in cell viability, now down to 58.0% at 0.13 μM and 56.4% at 0.40 μM complex 2. There were on average, a larger population of cells in late apoptosis at the higher dose of 2 compared to the lower dose, 41.2% and 30.2%, respectively. However, there were more necrotic cells present after exposure to the lower dose of 2, 11.8% and 2.5%.
Imaging
High content screening was used to further explore the mechanism of cell death in A2780 and OVCAR-3 ovarian cancer cells after 48 h exposure to various concentrations of 1 and 2. After this period, adherent cells were stained with DAPI, a blue stain for nuclear DNA, and Nucview™488, a caspase 3 biosensor that becomes activated and fluorescent upon endogenous caspase activation inside the cell. Fig. 9 shows representative microscopy images of caspase 3 positive apoptotic cells following exposure to 2.5 μM 1 and 2 for 48 h.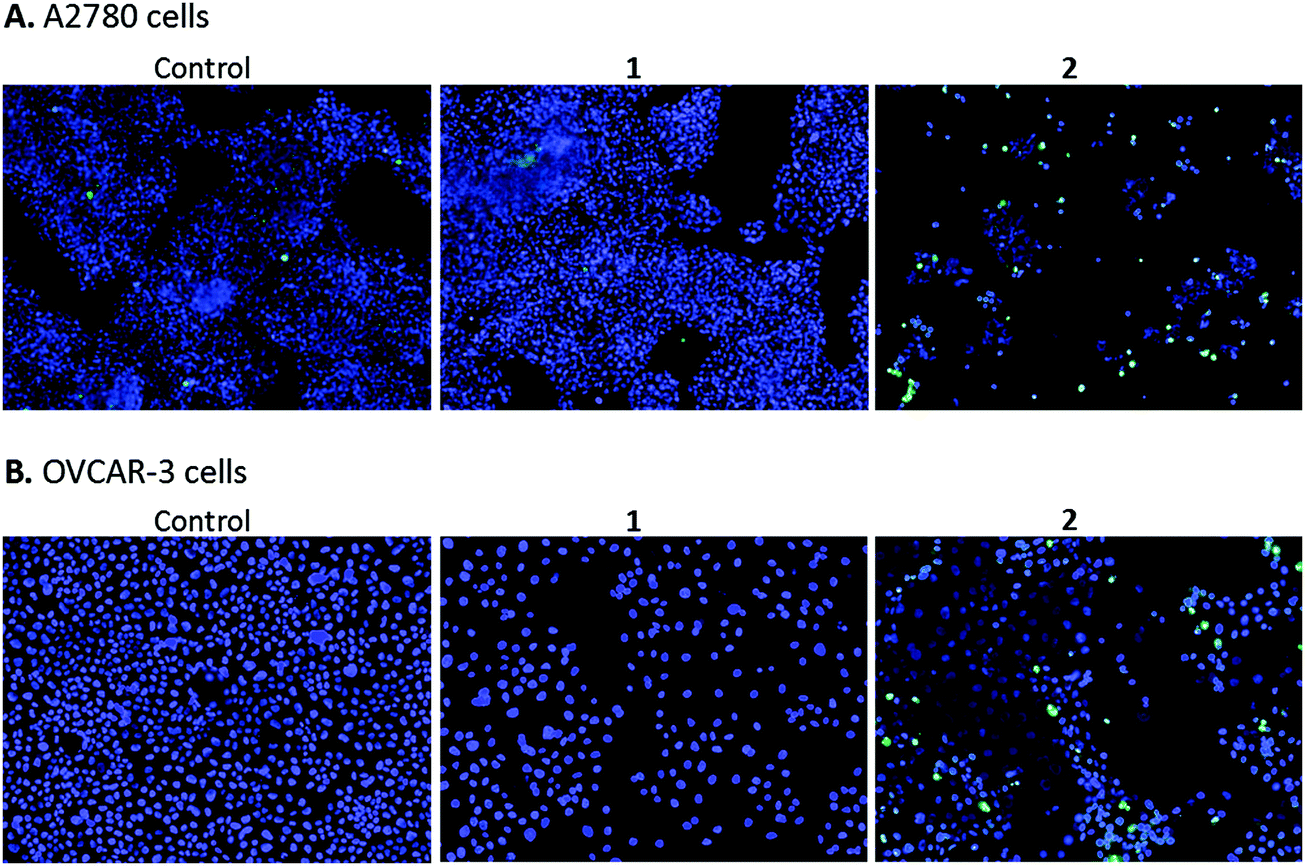 | ||
| Fig. 9 Fluorescence images showing caspase positive apoptotic A2780 (A) and OVCAR-3 (B) human ovarian cancer cells 48 h after exposure to no added compound (control), 2.5 μM 1 and 2.5 μM 2. DAPI stains nuclei of cells (blue), and NucView™ reagent stains caspase activity in apoptotic cells (green). | ||
A2780 cells exposed to 1 and 2 show very different levels of apoptosis, with 1 inducing very little cell death compared to the DMSO control. Complex 2 shows significantly higher activity in both ovarian cell lines compared to 1, particularly at the higher concentration of 2.5 μM (representative images in Fig. 9). Comparing cell lines provided interesting results and highlights the difference in cell selectivity between 1 and 2. In A2780 cells, 1 was inactive and 2 was highly active in reducing cell count, however, in OVCAR-3 cells the cell count reduction was more comparable (results not shown). In general, the activity of complexes at the single cell level27 depends on the metal itself, its oxidation state, the types of metal and numbers of bound ligand atoms, and the coordination geometry.28,29 Some complexes can be relatively inert and might reach the target site intact (as administered), but in general metal complexes are pro-drugs and may undergo metal- or ligand-based redox reactions as well as ligand substitution reactions before they reach targets.30,31
Discussion
Since we have adopted a phenotypic screening approach, attempting to optimise the design of complexes based on their chemical properties and their activity, the next stage after discovery of active complexes is to elucidate the nature of their MoA to guide further chemical optimization and/or patient selection strategies. In the present case our approach assessed correlations between anticancer activity and cell line gene mutations comparing patterns of activity with a range of previously screened compounds with known target sites. Comparing just the patterns of activity has already been fruitful on the smaller scale of the NCI-60 cell line screens.8 In general, metallodrugs are likely to be multi-targeted, attack several biochemical pathways simultaneously, and require a systems pharmacology approach for analysis of activity.32 We also compare these iridium complexes with two organo-osmium complexes for which we also have Sanger screening data. This comparison highlights the remarkable influence of the azopyridine ligand on activity.In the present case, the major difference between complexes 1 and 2 is the change in the chelated ligand from a negatively-charged C,N-bound phenylpyridine (1) to a neutral N,N-bound N-dimethylphenylazopyridine (2). Hence the overall charge on the complex changes from 0 (neutral) to +1. However, we previously reported that replacing only one atom from the N,N-chelating ligand 2,2′-bipyridine in the Ir(III) pentamethylcyclopentadienyl complexes to a C,N-chelating ligand phenylpyridine leads to a significant increase in anticancer activity.28,33 Therefore, the change in charge on the complex seems not to play a vital role in the control of their anticancer activity. In addition, we found that potency toward cancer cells increased with additional phenyl substitution on the Cp* ring: CpXbiph > CpXph > Cp* for the cyclopentadienyl Ir(III) complexes.10 Complex 1 also has an additional phenyl substituent on the cyclopentadienyl ring, which not only enhances lipophilicity but can also intercalate between DNA bases if that is an attack site. However, interestingly, the CpXph complex 2 displayed a more potent antiproliferative activity than the CpXbiph complex 1. It seems that the azopyridine ligand plays a major role in the anticancer activity.8 The azopyridine ligand is a strong π-acceptor, withdrawing electron density from the metal. This makes complex 2 (as well as 3 and 4) more inert towards hydrolysis than complex 1, which more readily undergoes aquation (replacement of monodentate chloride with water) in aqueous media and leads to deactivation before reaching its biological targets.30
We have also shown previously for Os complexes 3 and 4, that the additional phenyl group in 4 increases activity, but does not play a key role in the MoA. It is likely to increase cell uptake, resulting in a more potent compound.34 This has also been shown for Ir complexes.9
Our previous work has shown the difference in activity between neutral complex 1 and a charged analogue, [Ir(η5-CpXbiph)(ppy)py]+, in which the Cl ligand is substituted by pyridine. The charged complex has a higher potency, however, this was linked to the slower hydrolysis of the Ir-py bond, which affected the downstream MoA. However, in contrast, the Ir–Cl bond in complex 2 does not readily undergo hydrolysis. Thus even though previous work has demonstrated the importance in the Ir–Cl bond, the bidentate ligand seems to have a dominant effect, in line with the high activity of the Os-azopyridine complexes 3 and 4.21
Pharmacogenomic screening
The antiproliferative potency of organo-iridium complexes 1 and 2 compared to CDDP towards the wide variety of cancer cell lines in both the NCI-60 and Sanger – ca. 900 cell lines follows the order: 2 > CDDP > 1. Both the lipophilicity and overall charge on a complex can have a major effect on its biological activity. This trend is also apparent for patient-derived cancer cell lines which display distinct sensitivities to platinum agents (Table 3), where 1 has poor activity in all 3 cell lines, particularly in CDDP-resistant cell lines with an IC50 value >100 μM. These results show that complex 2 and not 1 would therefore be a candidate for combating platinum-resistant cancers and suggest that complex 2 might have a non-DNA-based MoA, unlike CDDP.The Sanger screen was used to identify cell lines of interest and to correlate activity with similarities in cell line genetics to explore the MoA. It is apparent that 2 has a novel pattern of activity compared to other drugs in the Sanger screen for which data are available (Table 2). It is the only drug to have a sensitivity association with mutations in the KIT gene,34,35 primarily found in NSCLC. KIT encodes C-KIT, an important cytokine receptor commonly associated with blood and bone cancers.36 Once the receptor is activated it controls signalling for cell survival, proliferation and differentiation. KIT mutations have been associated with an increased sensitivity to imatinib and has been proposed as an important biomarker in imatinib treatment.37 Functional C-KIT often confers chemoresistance by augmenting the expression of DNA repair genes. The mutated KIT gene may therefore render cells more sensitive to DNA damage, consistent with DNA attack being partly responsible for the activity of 2, as discussed further below.38
Interestingly, the Sanger screen highlighted a significant similarity between 2 ([Ir(η5-CpXph)(azpy-NMe2)Cl]+) and two previously studied Os complexes: FY12 ([Os(η6-bip)(azpy-NMe2)I]+) (4) and FY26 ([Os(η6-p-cym)(azpy-NMe2)I]+), (3), Fig. 1, suggesting that their activity is closely linked to the azpy-NMe2 ligand, which is conserved across all three compounds. Moreover since Ir(III) and Os(II) are isoelectronic (outer shell electronic configuration 5d6) and the complexes have similar 3D structures, global charges and probably log p values, it might be expected that they have similar profiles of biological activity. Although the activity of 2 does not correlate with that of any other drugs screened, those of organo-osmium complexes FY12 and FY26 do. Particularly noteworthy is the similarity in activity between FY26, bleomycin and SN-38 (the active metabolite of irinotecan, Camptosar), where cell lines with R184 amplifications are more resistant to all three compounds. Bleomycin, used in the treatment of testicular, cervical, lymphoma and cancers of the head and neck, causes both single- and double-DNA strand breaks in cancer cells.39 It can chelate iron, bind to DNA and react with oxygen to produce ROS, which damage DNA.
Topoisomerase enzymes are required for the unwinding of supercoiled DNA and the cutting and re-ligation of DNA during replication and protein synthesis. The active metabolite of irinotecan, SN-38, is a topoisomerase I (TOP1) inhibitor.40 TOP1 relaxes supercoiled DNA during replication and transcription, and generates and ligates cleaved single strands. Inhibitors of TOP1 prevent the ligation of cleaved strands, maintaining DNA breaks and activating cell death mechanisms in cancer cells.
Oxidative stress
The mitochondrion is the biggest source of ROS production in the cell, and mutations in OXPHOS machinery in A2780 cells perpetuate ROS production.41 NRF2 is up-regulated in response to 2, and we have shown by flow cytometry that this compound induces a significant ROS response. SO is normally converted to oxygen and H2O2 by superoxide dismutase (SOD), which is then broken down by catalase (CAT) to water and oxygen.42 Neither CAT nor SOD genes were up-regulated after exposure to 2. In fact, CAT was expressed in higher levels in the control samples, highlighting the oxidatively-stressed nature of A2780 cells, and that no response to H2O2 was mobilised after compound exposure. Disruption of CAT and SOD function has been shown to have a knock-on effect on the function of other antioxidant enzymes, potentiating the damage caused by ROS.43 The down-regulation of key response genes suggests that 2 induces ROS production and also reduces the ability of A2780 cells to respond to excessive ROS.The production of ROS from mitochondria has a cyclic effect on the function of mitochondria, potentiating the production of higher levels of ROS.44 These ROS can damage mtDNA and proteins within the mitochondria, in addition to disrupting the polarisation of the mitochondrial membrane.45 Mitocans, which target the bioenergetics of cancer cells and increase ROS, are becoming a focus of anticancer drug development.46 ROS generation by 2 may also occur by other mechanisms, as well as through mitochondria, ROS production during cell stress is well documented, and occurs indirectly in response to many drugs, including CDDP.47
Complex 2 shared similarity in its stress response to the organo-osmium complex FY26, 3. The Sanger screen points to close similarities in their MoAs. Previous experiments on FY26 have likewise demonstrated induction of ROS in A2780 cells.6 It seems clear that the azpy ligand plays a central role in inducing ROS by both these organo-iridium and organo-osmium complexes, however, the differences between the two structures causes a difference in ROS responses at the gene and the cellular level, with complex 2 resulting in high levels of total ROS but reduced levels of SO compared to FY26, and subsequently different transcriptional responses being activated.
Recently we have shown that although organo-osmium complex 3 is relatively inert in aqueous chemical solutions, it is activated in cancer cells and the iodide ligand is rapidly pumped out, and studies by nanofocussed X-ray fluorescence spectroscopy show that osmium from complex 3 localises in the mitochondria of human ovarian cancer cells.48,49
DNA damage and cell cycle arrest
Cells are equipped with mechanisms to repair DNA damage, however, ovarian cancers often lack the proper tools for DNA damage repair (DDR).50 Resistance to conventional platinum therapy often originates from the ability of cancer cells to repair DNA lesions caused by CDDP, even in cell lines with DDR deficiencies.51 DNA damage caused by CDDP is often not selective for cancer cells, and can be just as damaging to normal cells, especially with high-dose CDDP. In contrast, it is also possible to damage DNA through indirect mechanisms, and DNA is a well-known target for ROS, particularly guanine bases.52 DNA damage caused indirectly by anticancer compounds, through selective ROS generation, could provide a more targeted approach to achieving this MoA.There was measurable protein activation of DDR through the ATM-CHK2-p53-p21 pathway, however, there was also activation of the BRCA1 protein. RPPA suggested either S or G2/M phase arrest was possible in cells exposed to 2, with S and G2/M phase protein markers up-regulated at 24 and at 72 h. Flow cytometry experiments confirmed S phase arrest after 24 h exposure (Fig. 6). BRCA1 is an important protein in the S phase of the cell cycle during DNA damage and is required for homologous recombination and DNA damage-induced S and G2/M phase arrest.53 It could therefore follow that 2 does not require functional p53 for its MoA, as it does not require cells to be held in G1.
Collectively this could suggest that organo-iridium complex 2 has a more directed effect on DNA by exerting its activity either during DNA replication (S phase) or after the DNA content has been doubled (G2 phase). Organo-osmium complex FY26 appears to be most effective prior to DNA replication (G1 phase), and therefore may not have a DNA-focussed MoA.
Apoptosis
Modes of cell death can often be distinguished using whole cell imaging techniques.54 However, specific protein and gene markers can also be studied. For example, apoptosis is normally characterised by caspase production, and by distinct budding of cell membrane structures into so-called apoptotic bodies. In contrast, oncosis involves swelling and not shrinking of the cell and is caused by non-specific cell injury.55 Necrosis is commonly identified as its own mode of cell death, but in fact necrosis is the last stage in any cell death pathway, i.e. both oncosis and apoptosis lead to necrosis and cell death.Most anticancer agents aim to activate apoptosis in cancer cells, simply because this is the most controlled form of cell death, involving phagocytosis in the final stages. However, cancer cells often develop mechanisms to evade apoptotic cell death which serves to highlight the merits of alternative mechanisms, like pyroptosis, an immune-induced mechanism of cell death.56 RNAseq data suggested poor transcriptional activation of apoptotic caspase markers by 2, but some transcription of apoptotic initiators. RPPA further supported this conclusion, with waves of apoptotic initiation towards the middle of the time course, followed by recovery towards the end (Fig. 7). By comparison, FY26 showed even less up-regulation of apoptotic components.
Late apoptosis was detectable by flow cytometry for 2-exposed cells at the 48 h time point. However, by 72 h, the cell population had recovered viability. This supports an occurrence of a wave of cell death. Using high content screening (HCS) to detect levels of caspase-3, the effector-phase of apoptosis was explored. HCS proved to be a useful method in visualising cell death for multiple compounds; it was possible to extract compound-based trends and make predictions about the level of apoptosis at higher concentrations, where there was a significant increase in apoptotic cells at higher concentrations for 2 compared to 1. ROS release has been shown to modulate apoptosis and may therefore play a central role in delaying apoptosis.57
Conclusions
Screening of two organo-iridium complexes in the Sanger screen of 916 cancer cell lines revealed a remarkably high potency for the phenylazopyridine complex 2 compared to the phenylpyridine organo-iridium complex 1 and the clinical platinum drug cisplatin (CDDP).The screen also revealed a particularly high susceptibility of triple negative breast cell lines to 2, and potential correlations to ROS-inducing drugs. Notably the pattern of activity of 2 towards the wide range of cancer cell lines was different to 253 previously screened drugs suggesting an unusual MoA, but remarkably similar to organo-osmium complex FY26 which shares the same N,N-chelated phenylazopyridine ligand.
The MoA for both iridium complex 2 and osmium complex 3 appears to involve the rapid generation of ROS in cells, and can achieve selectivity for cancer versus normal cells since cancer cells have defective mitochondria, the usual source of ROS. However, more interesting is the difference in the exact nature of the ROS response triggered; with complex 2 inducing less of a superoxide response. This difference in the induction of ROS caused cells to arrest in S/G2 phase in response to 2, whereas for 3 cells arrested in G1. Apoptosis seemed to be apparent after longer exposure periods, with interesting pro- and anti-apoptotic protein dynamics measured across the time series.
Ultimately this study provides a clear example of the benefits of combining different theoretical and experimental methods to explore novel and complex MoAs. In particular the combination of such methods can allow the separation of intricate differences in activity between compounds to interpret phenotypic response at molecular and cellular levels, which inform further preclinical development and clinical translation.
Materials and methods
Compound synthesis and characterisation
All compounds were synthesised and characterised as described previously.8,21,33Antiproliferation assays
Complexes 1 and 2 were screened under the ‘Genomics of Drug Sensitivity in Cancer’ project at the Sanger Institute (Cambridge, UK).Briefly, cells were seeded in 96 well plates at ca. 15% confluency and left to incubate for 24 h at 310 K, 5% CO2, 95% air and 100% relative humidity. Cells were treated with nine concentrations of each compound and returned to the incubator for 72 h. For suspension cell lines, cells were treated with compound immediately following plating, and returned to the incubator. Cells were stained and quantitation of fluorescent signal intensity was performed using a plate reader. IC50 values were returned to The University of Warwick for downstream analysis. MANOVA analysis was performed by the Sanger Bioinformatics Institute.
Primary cancer cell screening
Primary patient cell lines were seeded in 96 well plates with ca. 5000 cells per well in RPMI-1640 with 1% (v/v) 2 mM glutamine. The plates were incubated at 310 K for 48 h. Stock solutions of each compound were prepared and cells exposed across a dose range for a further 48 h. Cells were extracted and the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay (Life Technologies) was used to determine cell viability.RNA sequencing
TruSeq kits (Illumina) were used to prepare samples for sequencing as per the manufacturer's guidelines. From total RNA, mRNA samples were purified, fragmented and reverse transcribed to cDNA. Sequencing libraries were produced, with incorporated barcodes to allow multiplexing. Samples were sequenced on an Illumina Hiseq2000 instrument across 5 lanes (6 samples per lane). 50 base pair, paired-end reads were recorded producing ca. 60 million reads in total, per sample.A2780 cells were seeded at 3 × 106 cells per 100 mm petri dish. Plates were incubated for 24 h at 310 K, 5% CO2, 95% air and 100% relative humidity. Complex 2 (400 nM) and vehicle control solutions were added and after 4, 12, 24 and 48 h cells were collected and whole-cell RNA extracted (RNeasy plus mini kit, Qiagen). Samples were run on a NanoDrop 1000 spectrophotometer machine and the absorbance at 230, 260 and 280 nm recorded to calculate the 260/230 and 260/280 ratios. Samples with A260/230 > 2.0 and A260/280 > 1.9 were passed. The concentration of RNA in each solution was also estimated using the NanoDrop and was verified using a 2100 Agilent Bioanalyzer and an RNA 6000 Nano Kit (Agilent), and the Qubit assay (Life Technologies). All samples had a RNA integrity number (RIN) > 9.50. A minimum of 1 μg RNA for each sample was transferred to Oxford Genomics Centre (Wellcome Trust Centre for Human Genetics) in a total of 30 μL RNase free water in skirted 96 well plates.
Data deposition: the sequencing data reported in this paper have been deposited in ArrayExpress (accession E-MTAB-5991).
Data analysis
Preliminary filtering was performed on the data to remove all samples with quality scores (Q) < 20. FASTQ files for the forward and reverse reads were integrated and aligned to the hg19 (GRCh37) human genome using TopHat2. Files were returned to The University of Warwick in BAM format. Samtools (version 0.1.19-44428cd) was used to explore the raw BAM files, extracting chromosomal reads and summary statistics. BAM files were sorted by read name and then converted to SAM format. HTSeq (version 0.5.4p5) was used to map the read locations to genomic regions using the hg19 genome construct with the intersection-nonempty program option. Resulting mapped files, were then analysed for differential gene expression using the edgeR package (version 3.1.10) in the R statistical programme (version 3.0.2). A list of differentially expressed genes was obtained for each time point (4, 12, 24 and 48 h) and used together with Ingenuity Pathway Analysis software to assign significantly expressed genes to biological pathways.Reverse phase protein microarrays (RPPA)
4 × 105 A2780 cells were seeded per well and cells were pre-incubated in drug-free media for 48 h. After this, cells were treated at 150 nM and 450 nM for 4, 24, 48 and 72 h. Control samples were treated with media containing 0.1% DMSO. Cell lysates were collected and deposited onto a Zeptosens chip. Chips were blocked and washed prior to performing a dual antibody immunoassay comprising of a 24 h incubation of primary antibodies followed by 2.5 h incubation with secondary Alexa-Fluor 647 conjugated antibody detection reagent. The immunostained arrays were imaged using the ZeptoREADER instrument and analysed using ZeptoView 3.1 software. Local normalisation of sample signal to a reference BSA grid was used to compensate for any intra- or inter-array/chip variation. RFI values were further normalised to a house keeping protein (prohibitin) and to the negative control, to provide the final RFI to represent the relative abundance of total, phosphorylated and cleaved proteins in compound-treated samples relative to the DMSO control for each time point.Apoptosis assays
A2780 cells were seeded at 1 × 106 cells per well in 6-well plates. Cells were preincubated in drug-free media at 310 K for 24 h in a 5% CO2 humidified atmosphere. Drug/compound solutions were added and the cells left to incubate for a further 24, 48 or 72 h. Following exposure, the drug-containing medium was removed, and cells were washed, harvested and stained with Annexin V FITC and propidium iodide (Biovision, Annexin V-FITC Apoptosis Kit) according to the manufacturer's instructions. Control samples stained with just propidium iodide or Annexin V FITC were also included for compensation purposes. The samples were analysed using Beckton Dickinson FACScan with fluorescence detection running Cell Quest software (20000 events were collected from each sample). Data were processed using Flowjo software (version 7.2.5).
Cell cycle analysis
A2780 cells were seeded at a density of 1 × 106 cells per well in a 6-well plate and pre-incubated in drug-free media at 310 K for 24 h in a 5% CO2 humidified atmosphere. Drug solutions were added and the cells left to incubate for a further 24 h. Cells were collected and washed twice with PBS. Cells were fixed with 70% ethanol and stored at −293 K for 24 h. For analysis, cells were transferred into PBS, incubated with RNase (0.2 mg mL−1) and propidium iodide (0.05 mg mL−1) for 40 min at 310 K and then analysed by flow cytometry using a Beckton Dickinson FACScan with fluorescence detection. The resulting DNA histograms were quantified using the FlowJo software (version 7.2.5).High content screening (HCS)
Compounds 1 and 2 were imaged by HCS at The Edinburgh Cancer Research Centre. Cells were seeded in a 96-well plate at 5000 cells per well and incubated for 48 h before treatment with each compound for a further 48 h. Prior to image acquisition, cells were incubated with 4 μg mL−1 DAPI (Sigma D8417), and 1 μM NucView™ (Biotium) reagent for 0.5 h. Microscopic images of DAPI and NucView™ stained cells were acquired with a 10× objective and appropriate optical filters using the Olympus ScanR high-content imaging microscope. Merged images of DAPI and Nucview™ cells were created using the Olympus ScanR imaging software Cell-IR™.Author contributions
JMH, IRC, AFM, NOC, and PJS designed the research. JMH, IRC, AFM, BRR, ZL and NOC performed the research. JMH, GMH, IRC, AFM, NOC, and PJS analysed data; and JMH, GMH, IRC, NOC and PJS wrote the paper. All authors contributed to the final version.Conflicts of interest
We have no competing interests.Acknowledgements
We thank the European Research Council (grant 247450), the Wellcome Trust (grants 086357, 102696 and 107691) the Biotechnology and Biological Sciences Research Council (grant 324594, Systems Biology studentship for J. M. H.), Engineering and Physical Sciences Research Council (grant EP/F500378/1, MOAC Studentship for G. M. H.), Drs Ultan McDermott and Matthew Garnett (Sanger Institute) for their help and advice with the Sanger screening studies, and Professor David Wild (Warwick University) and EU COST Action CM1105 for helpful discussions.References
- D. Wang and S. J. Lippard, Cellular processing of platinum anticancer drugs, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- P. M. Bruno, Y. Liu, G. Y. Park, J. Murai, C. E. Koch, T. J. Eisen, J. R. Pritchard, Y. Pommier, S. J. Lippard and M. T. Hemann, A subset of platinum-containing chemotherapeutic agents kills cells by inducing ribosome biogenesis stress, Nat. Med., 2017, 23, 1–13 CrossRef PubMed.
- H. Kostrhunova, J. Florian, O. Novakova, A. F. A. Peacock, P. J. Sadler and V. Brabec, DNA interactions of monofunctional organometallic osmium(II) antitumor complexes in cell-free media, J. Med. Chem., 2008, 51, 3635–3643 CrossRef CAS PubMed.
- V. Novohradsky, Z. Liu, M. Vojtiskova, P. J. Sadler, V. Brabec and J. Kasparkova, Mechanism of cellular accumulation of an iridium(III) pentamethylcyclopentadienyl anticancer complex containing a C,N-chelating ligand, Metallomics, 2014, 6, 682–690 RSC.
- Y. Fu, A. Habtemariam, A. M. Basri, D. Braddick, G. J. Clarkson and P. J. Sadler, Structure–activity relationships for organometallic osmium arene phenylazopyridine complexes with potent anticancer activity, Dalton Trans., 2011, 40, 10553 RSC.
- J. M. Hearn, I. Romero-Canelon, A. F. Munro, Y. Fu, A. M. Pizarro, M. J. Garnett, U. McDermott, N. O. Carragher and P. J. Sadler, Potent organo-osmium compound shifts metabolism in epithelial ovarian cancer cells, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E3800–E3805 CrossRef CAS PubMed.
- S. H. van Rijt, I. Romero-Canelón, Y. Fu, S. D. Shnyder and P. J. Sadler, Potent organometallic osmium compounds induce mitochondria-mediated apoptosis and S-phase cell cycle arrest in A549 non-small cell lung cancer cells, Metallomics, 2014, 6, 1014–1022 RSC.
- J. M. Hearn, I. Romero-Canelón, B. Qamar, Z. Liu, I. Hands-Portman and P. J. Sadler, Organometallic iridium(III) anticancer complexes with new mechanisms of action: NCI-60 screening, mitochondrial targeting, and apoptosis, ACS Chem. Biol., 2013, 8, 1335–1343 CrossRef CAS PubMed.
- Z. Liu, A. Habtemariam, A. M. Pizarro, S. A. Fletcher, A. Kisova, O. Vrana, L. Salassa, P. C. A. Bruijnincx, G. J. Clarkson, V. Brabec and P. J. Sadler, Organometallic half-sandwich iridium anticancer complexes, J. Med. Chem., 2011, 54, 3011–3026 CrossRef CAS PubMed.
- Z. Liu and P. J. Sadler, Organoiridium complexes: anticancer agents and catalysts, Acc. Chem. Res., 2014, 47, 1174–1185 CrossRef CAS PubMed.
- K. D. Paull, R. H. Shoemaker, L. Hodes, A. Monks, D. A. Scudiero, L. Rubinstein, J. Plowman and M. R. Boyd, Display and analysis of patterns of differential activity of drugs against human tumor cell lines: development of mean graph and COMPARE algorithm, J. Natl. Cancer Inst., 1989, 81, 1088–1092 CrossRef CAS PubMed.
- D. Sidler, A. Brockmann, J. Mueller, U. Nachbur, N. Corazza, P. Renzulli, A. Hemphill and T. Brunner, Thiazolide-induced apoptosis in colorectal cancer cells is mediated via the Jun kinase-Bim axis and reveals glutathione-S-transferase P1 as Achilles/’ heel, Oncogene, 2012, 31, 4095–4106 CrossRef CAS PubMed.
- Y.-P. Wang, L.-S. Zhou, Y.-Z. Zhao, S.-W. Wang, L.-L. Chen, L.-X. Liu, Z.-Q. Ling, F.-J. Hu, Y.-P. Sun, J.-Y. Zhang, C. Yang, Y. Yang, Y. Xiong, K.-L. Guan and D. Ye, Regulation of G6PD acetylation by SIRT2 and KAT9 modulates NADPH homeostasis and cell survival during oxidative stress, EMBO J., 2014, 33, 1304–1320 CAS.
- J. E. Frank, Diagnosis and management of G6PD deficiency, Am. Fam. Physician, 2005, 72, 1277–1282 Search PubMed.
- T. Efferth, U. Fabry, P. Glatte and R. Osieka, Increased induction of apoptosis in mononuclear cells of a glucose-6-phosphate dehydrogenase deficient patient, J. Mol. Med., 1995, 73, 47–49 CrossRef CAS PubMed.
- W. Yang, J. Soares, P. Greninger, E. J. Edelman, H. Lightfoot, S. Forbes, N. Bindal, D. Beare, J. A. Smith, I. R. Thompson, S. Ramaswamy, P. A. Futreal, D. A. Haber, M. R. Stratton, C. Benes, U. McDermott and M. J. Garnett, Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells, Nucleic Acids Res., 2013, 41, 955–961 CrossRef PubMed.
- T. Sawada, Y. S. Chung, B. Nakata, T. Kubo, Y. Kondo, T. Sogabe, N. Onoda, Y. Ogawa, N. Yamada and M. Sowa, Establishment and characterization of a human breast cancer cell line, OCUB-1, Hum. Cell, 1994, 7, 138–144 CAS.
- T. Aas, A.-L. Børresen, S. Geisler, B. Smith-Sørensen, H. Johnsen, J. E. Varhaug, L. A. Akslen and P. E. Lønning, Specific P53 mutations are associated with de novo resistance to doxorubicin in breast cancer patients, Nat. Med., 1996, 2, 811–814 CrossRef CAS PubMed.
- Y. Wang, J. Chang, X. Liu, X. Zhang, S. Zhang, X. Zhang, D. Zhou and G. Zheng, Discovery of piperlongumine as a potential novel lead for the development of senolytic agents, Aging, 2016, 8, 2915–2926 Search PubMed.
- L. Raj, T. Ide, A. U. Gurkar, M. Foley, M. Schenone, X. Li, N. J. Tolliday, T. R. Golub, S. A. Carr, A. F. Shamji, A. M. Stern, A. Mandinova, S. L. Schreiber and S. W. Lee, Selective killing of cancer cells with a small molecule targeting stress response to ROS, Nature, 2012, 475, 231–234 CrossRef PubMed.
- Y. Fu, A. Habtemariam, A. M. Pizarro, S. H. Van Rijt, D. J. Healey, P. A. Cooper, S. D. Shnyder, G. J. Clarkson and P. J. Sadler, Organometallic osmium arene complexes with potent cancer cell cytotoxicity, J. Med. Chem., 2010, 53, 8192–8196 CrossRef CAS PubMed.
- S. P. Langdon, S. S. Lawrie, F. G. Hay, M. M. Hawkes, A. McDonald, I. P. Hayward, D. J. Schol, J. Hilgers, R. C. F. Leonard and J. F. Smyth, Characterization and properties of nine human ovarian adenocarcinoma cell lines, Cancer Res., 1988, 48, 6166–6172 CAS.
- Ingenuity Systems, Ingenuity Pathway Analysis.
- Y. Katoh, K. Itoh, E. Yoshida, M. Miyagishi, A. Fukamizu and M. Yamamoto, Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription, Genes Cells, 2001, 6, 857–868 CrossRef CAS PubMed.
- I. Romero-Canelon, M. Mos and P. J. Sadler, Enhancement of Selectivity of an Organometallic Anticancer Agent by Redox Modulation, J. Med. Chem., 2015, 58, 7874–7880 CrossRef CAS PubMed.
- I. Vermes, C. Haanen, H. Steffens-Nakken and C. P. Reutelingsperger, A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V, J. Immunol. Methods, 1995, 184, 39–51 CrossRef CAS PubMed.
- P. Horvath, N. Aulner, M. Bickle, A. M. Davies, E. Del Nery, D. Ebner, M. C. Montoya, P. Ostling, V. Pietiainen, L. S. Price, S. L. Shorte, G. Turcatti, C. von Schantz and N. O. Carragher, Screening out irrelevant cell-based models of disease, Nat. Rev. Drug Discovery, 2016, 15, 751–769 CrossRef CAS PubMed.
- Z. Liu, L. Salassa, A. Habtemariam, A. M. Pizarro, G. J. Clarkson and P. J. Sadler, Contrasting reactivity and cancer cell cytotoxicity of isoelectronic organometallic iridium(III) complexes, Inorg. Chem., 2011, 50, 5777–5783 CrossRef CAS.
- N. P. E. Barry and P. J. Sadler, Exploration of the medical periodic table: towards new targets, Chem. Commun., 2013, 49, 5106–5131 RSC.
- Z. Liu, I. Romero-Canelon, B. Qamar, J. M. Hearn, A. Habtemariam, N. P. E. Barry, A. M. Pizarro, G. J. Clarkson and P. J. Sadler, The potent oxidant anticancer activity of organoiridium catalysts, Angew. Chem., Int. Ed., 2014, 53, 3941–3946 CrossRef CAS PubMed.
- C. Wang, J. Liu, Z. Tian, M. Tian, L. Tian, W. Zhao and Z. Liu, Half-Sandwich Iridium N-Heterocyclic Carbene Anticancer Complexes, Dalton Trans., 2017, 6870–6883 RSC.
- J. Liu and M. Chen, Systems Pharmacology for the Study of Anticancer Drugs: Promises and Challenges, Clin. Pharmacol. Biopharm., 2015, 4, 140–142 Search PubMed.
- Z. Liu, A. Habtemariam, A. M. Pizarro, G. J. Clarkson and P. J. Sadler, Organometallic iridium(III) cyclopentadienyl anticancer complexes containing C,N-chelating ligands, Organometallics, 2011, 30, 4702–4710 CrossRef CAS.
- C. Andre, A. Hampe, P. Lachaume, E. Martin, X. P. Wang, V. Manus, W. X. Hu and F. Galibert, Sequence analysis of two genomic regions containing the KIT and the FMS receptor tyrosine kinase genes, Genomics, 1997, 39, 216–226 CrossRef CAS PubMed.
- C. E. Edling and B. Hallberg, c-Kit—a hematopoietic cell essential receptor tyrosine kinase, Int. J. Biochem. Cell Biol., 2007, 39, 1995–1998 CrossRef CAS PubMed.
- A. McIntyre, B. Summersgill, B. Grygalewicz, A. J. Gillis, J. Stoop, R. J. van Gurp, N. Dennis, C. Fisher, R. Huddart, C. Cooper, J. Clark, J. W. Oosterhuis, L. H. Looijenga and J. Shipley, Amplification and overexpression of the KIT gene is associated with progression in the seminoma subtype of testicular germ cell tumors of adolescents and adults, Cancer Res., 2005, 65, 8085–8089 CrossRef CAS PubMed.
- F. S. Hodi, C. L. Corless, A. Giobbie-Hurder, J. A. Fletcher, M. Zhu, A. Marino-Enriquez, P. Friedlander, R. Gonzalez, J. S. Weber, T. F. Gajewski, S. J. O'Day, K. B. Kim, D. Lawrence, K. T. Flaherty, J. J. Luke, F. A. Collichio, M. S. Ernstoff, M. C. Heinrich, C. Beadling, K. A. Zukotynski, J. T. Yap, A. D. Van Den Abbeele, G. D. Demetri and D. E. Fisher, Imatinib for melanomas harboring mutationally activated or amplified kit arising on mucosal, acral, and chronically sun-damaged skin, J. Clin. Oncol., 2013, 31, 3182–3190 CrossRef CAS PubMed.
- C. Wichmann, I. Quagliano-Lo Coco, Ö. Yildiz, L. Chen-Wichmann, H. Weber, T. Syzonenko, C. Döring, C. Brendel, K. Ponnusamy, A. Kinner, C. Brandts, R. Henschler and M. Grez, Activating c-KIT mutations confer oncogenic cooperativity and rescue RUNX1/ETO-induced DNA damage and apoptosis in human primary CD34+ hematopoietic progenitors, Leukemia, 2015, 29, 279–289 CrossRef CAS PubMed.
- S. M. Hecht, Bleomycin: New Perspectives on the Mechanism of Action, J. Nat. Prod., 2000, 1, 158–168 CrossRef.
- R. Mullangi, P. Ahlawat and N. R. Srinivas, Irinotecan and its active metabolite, SN-38: Review of bioanalytical methods and recent update from clinical pharmacology perspectives, Biomed. Chromatogr., 2010, 24, 104–123 CrossRef CAS PubMed.
- S. I. Grivennikov and M. Karin, Inflammatory cytokines in cancer: tumour necrosis factor and interleukin 6 take the stage, Ann. Rheum. Dis., 2011, 70, i104–i108 CrossRef CAS PubMed.
- M. P. Murphy, How mitochondria produce reactive oxygen species, Biochem. J., 2009, 417, 1–13 CrossRef CAS PubMed.
- S. Turkseven, A. Kruger, C. J. Mingone, P. Kaminski, M. Inaba, L. F. Rodella, S. Ikehara, M. S. Wolin and N. G. Abraham, Antioxidant mechanism of heme oxygenase-1 involves an increase in superoxide dismutase and catalase in experimental diabetes, Am. J. Physiol.: Heart Circ. Physiol., 2005, 289, H701–H707 CrossRef CAS PubMed.
- C. Henchcliffe and M. F. Beal, Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis, Nat. Clin. Pract. Neurol., 2008, 4, 600–609 CrossRef CAS PubMed.
- F. S. Hosnijeh, Q. Lan, N. Rothman, C. S. Liu, W. Cheng, A. Nieters, P. Guldberg, A. Tjønneland, D. Campa, A. Martino, H. Boeing, A. Trichopoulou, G. Masala, E. Weiderpass, M. Dorronsoro, J. Ram, B. Melin, A. S. Johansson, J. Malm, S. Borgquist, P. H. Peeters, H. B. Bueno-de-mesquita and N. Wareham, Mitochondrial DNA copy number and future risk of B-cell lymphoma in a nested case-control study in the prospective EPIC cohort, Blood, 2014, 124, 530–535 CrossRef CAS PubMed.
- J. Neuzil, L. F. Dong, J. Rohlena, J. Truksa and S. J. Ralph, Classification of mitocans, anti-cancer drugs acting on mitochondria, Mitochondrion, 2013, 13, 199–208 CrossRef CAS PubMed.
- Y. Chen, P. Jungsuwadee, M. Vore, D. A. Butterfield and D. K. St. Clair, Collateral Damage in Cancer Chemotherapy: Oxidative Stress in Nontargeted Tissues, Mol. Interventions, 2007, 7, 147–156 CrossRef CAS PubMed.
- C. Sanchez-Cano, I. Romero-Canelón, Y. Yang, I. J. Hands-Portman, S. Bohic, P. Cloetens and P. J. Sadler, Synchrotron X-Ray Fluorescence Nanoprobe Reveals Target Sites for Organo-Osmium Complex in Human Ovarian Cancer Cells, Chem. – Eur. J., 2017, 1–6 Search PubMed.
- R. J. Needham, C. Sanchez-Cano, X. Zhang, I. Romero-Canelón, A. Habtemariam, M. S. Cooper, L. Meszaros, G. J. Clarkson, P. J. Blower and P. J. Sadler, In-Cell Activation of Organo-Osmium(II) Anticancer Complexes, Angew. Chem., Int. Ed., 2016, 1017–1020 Search PubMed.
- J. Assis, D. Pereira and R. Medeiros, Ovarian cancer and DNA repair: DNA ligase IV as a potential key, World J. Clin. Oncol., 2013, 4, 14–24 CrossRef PubMed.
- L. Galluzzi, L. Senovilla, I. Vitale, J. Michels, I. Martins, O. Kepp, M. Castedo and G. Kroemer, Molecular mechanisms of cisplatin resistance, Oncogene, 2012, 31, 1869–1883 CrossRef CAS PubMed.
- N. R. Jena, DNA damage by reactive species: mechanisms, mutation and repair, J. Biosci., 2012, 37, 503–507 CrossRef CAS PubMed.
- N. Foray, D. Marot, A. Gabriel, V. Randrianarison, A. M. Carr, M. Perricaudet, A. Ashworth and P. Jeggo, A subset of ATM- and ATR-dependent phosphorylation events requires the BRCA1 protein, EMBO J., 2003, 22, 2860–2871 CrossRef CAS PubMed.
- G. Kroemer, L. Galluzzi, P. Vandenabeele, J. Abrams, E. S. Alnemri, E. H. Baehrecke, M. V. Blagosklonny, W. S. El-Deiry, P. Golstein, D. R. Green, M. Hengartner, R. A. Knight, S. Kumar, S. A. Lipton, W. Malorni, G. Nuñez, M. Peter, J. Tschopp, J. Yuan, M. Piacentini, B. Zhivotovsky and G. Melino, Classification of Cell Death, Cell Death Differ., 2009, 16, 3–11 CrossRef CAS PubMed.
- G. Majno and I. Joris, Apoptosis, oncosis, and necrosis. An overview of cell death, Am. J. Pathol., 1995, 146, 3–15 CAS.
- T. Bergsbaken, S. L. Fink and B. T. Cookson, Pyroptosis: host cell death and inflammation, Nat. Rev. Microbiol., 2010, 7, 99–109 CrossRef PubMed.
- H.-U. Simon, A. Haj-Yehia and F. Levi-Schaffer, Role of reactive oxygen species (ROS) in apoptosis induction, Apoptosis, 2000, 5, 415–418 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7mt00242d |
| ‡ These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |