
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding solid-state photoswitching in [Re(OMe2-bpy)(CO)3(η1-NO2)] crystals via in situ photocrystallography†
Lauren E.
Hatcher
 *
*
Department of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: l.e.hatcher@bath.ac.uk
First published on 19th June 2018
Abstract
Single-crystal-to-single-crystal nitro → nitrito isomerism is reported for the novel rhenium(I)-bipyridine complex [Re(OMe2-bpy)(CO)3(η1-NO2)], achieving a maximum conversion of 66% to a photoinduced nitrito-(η1-![[O with combining low line]](https://www.rsc.org/images/entities/char_004f_0332.gif) NO) isomer under continuous illumination. The 3D X-ray structure of the photoinduced isomer is determined by steady-state and psuedo-steady-state photocrystallographic methods, providing insight into the structural changes required to accommodate photoswitching. Photocrystallographic kinetic studies follow the progress of photoswitching with time, determining a reaction rate constant of k = 0.38(2) min−1 at 150 K. Linkage isomerism is fully-reversible on warming, and pseudo-steady-state experiments confirm that the photoexcited state is retained, at measurable occupancy, up to a temperature of 240 K. These results confirm the validity of combining photoactive linkage isomer and Re(I) photocatalyst chemistries, and the detailed determination of the photoexcited state structure will facilitate the future design of new photoactive Re(I) crystals for a range of applications.
NO) isomer under continuous illumination. The 3D X-ray structure of the photoinduced isomer is determined by steady-state and psuedo-steady-state photocrystallographic methods, providing insight into the structural changes required to accommodate photoswitching. Photocrystallographic kinetic studies follow the progress of photoswitching with time, determining a reaction rate constant of k = 0.38(2) min−1 at 150 K. Linkage isomerism is fully-reversible on warming, and pseudo-steady-state experiments confirm that the photoexcited state is retained, at measurable occupancy, up to a temperature of 240 K. These results confirm the validity of combining photoactive linkage isomer and Re(I) photocatalyst chemistries, and the detailed determination of the photoexcited state structure will facilitate the future design of new photoactive Re(I) crystals for a range of applications.
Introduction
Molecular photoswitches that display a fast, useful change in their physical properties in response to irradiation, are an increasingly important area of photochemistry research. They are broadly applicable across a range of disciplines, from the design of smart materials and optoelectronics,1 to biomedical applications such as photodynamic therapy2,3 and fluorescent tracers for in vivo imaging,4 and to environmental solutions such as waste water treatment5,6 and solar energy.7Sunlight is our most abundant and, as yet, under-exploited energy source, delivering more energy to the earth's surface in one hour than is used by humanity in an entire year,7 and can produce electricity via far cleaner routes than fossil fuels. Though advances continue in this area, solar energy alternatives remain expensive.7 As such, there is a continued need to design new, more efficient materials for solar harvesting, to achieve the ultimate goal of providing cheap and clean energy for all.
Taking inspiration from nature, much research is devoted to the design of biomimetic materials for artificial photosynthesis. Molecular switches find application here as photosensitizers and photocatalysts, accelerating key chemical reactions that transform light into other useful forms of energy.8 Photoactive organometallics are a key focus area, particularly in the design of homogeneous photocatalysts in solution. Among the most well studied materials are Re(I) systems of the general formula [Re(R2-bpy)(CO)3(X)],9–12 and this popularity is due in part to the fact that they are one of the few systems that combine both photosensitizer and photocatalytic centre within the same molecule,13 promoting efficient charge transfer. Despite this interest, there remain some key issues for Re(I) photocatalysts. They suffer from problems common to other homogeneous catalysts, including issues with separation and recycling, as well as concerns over their long-term stability in solution. As such, there is significant interest in immobilising Re(I) systems into solid state scaffolds, enabling heterogeneous functionality.14–16 Current Re(I) complexes also do not absorb across a broad enough range of the solar spectrum,17 and key aspects of their photocatalytic mechanism are also not well understood. This is because of difficulties distinguishing between many structurally similar potential intermediates via solution spectroscopy.9,11 It is here that dynamic structural chemistry methods, such as Photocrystallography,18 can now make important contributions.
Since the late 1990s, photocrystallographic methods have been used to follow dynamic photoswitching in a variety of crystalline materials in situ and in real time.19 Among the earliest systems to be studied are solid state linkage isomers, in which a long lived, metastable photoexcited isomer is produced at low temperature.20 Building on seminal work by Coppens on metal-nitrosyl compounds,21 a variety of linkage isomer systems have been studied including nitrosyl,22–25 sulfur dioxide,26–29 dinitrogen30 and nitrite species.20,31–38 In particular, nitro → nitrito isomerism is well studied by photocrystallographic methods, with the first 100% conversion in a single crystal reported in 2009 for [Ni(dppe)(Cl)(η1-NO2)].35 Several other crystals capable of 100% conversion have since been designed, and there is also interest in systems capable of room temperature photoswitching for real world applications.36 Our recent work has demonstrated that the photoinduced nitrito-(η1-NO) isomer is strongly temperature dependent, enabling tuning of the excited state lifetime across a range of timescales.39 This high degree of facile control makes these materials ideal test systems for the design of new photocrystallographic methods and equipment.
A handful of previous reports highlight an interest in combining photocatalytic Re(I) species with linkage isomer chemistry. Work by Dominguez et al. in 2014 reports the preparation of pure crystals of both [Re(bpy)(CO3)(η1-NO2)] and [Re(Me2-bpy)(CO3)(η1-ONO)] from the reaction mixture. While the authors were able to demonstrate changes in solution state UV/vis and IR spectra on irradiation that may be indicative of photoinduced linkage isomer switching, no single-crystal-to-single-crystal transformation is reported.40 In 2016, Kia and Safari also reported the synthesis of [Re(Me2-phen)(CO3)(η1-ONO)], but again no evidence of solid state switching is found.41 There remains scope for detailed, in situ photocrystallographic investigations with [Re(R2-bpy)(CO3)(X)] systems, delivering key structural insight into how these systems respond to irradiation.
This work reports the synthesis, crystallisation and in situ photocrystallographic study of the novel complex [Re(OMe-bpy)(CO3)(η1-NO2)]. The 3D single crystal X-ray structure of a metastable state (MS) photoexcited nitrito-(η1-NO) isomer is determined, and the progress of the nitro → nitrito process is followed in real time by photocrystallographic kinetic methods. Further manipulation of the maximum excited state conversion level is also achieved under continuous illumination, via pseudo-steady-state photocrystallographic techniques.
Experimental
Synthetic procedures
Rhenium pentacarbonyl chloride [Re(CO)5Cl], 4,4′-dimethoxy-2,2′-bipyridine (OMe2-bpy), silver triflate (AgOTf) and potassium nitrite (KNO2) were all purchased from Sigma Aldrich and used as received. All solvents were reagent grade and used as purchased without further purification.CSD entry = YIYMAW. P21/c, a = 8.725 Å, b = 15.160 Å, c = 14.750 Å, β = 91.82°, V = 1948.732 Å3 (at 100 K).42
Experimental. P21/c, a = 8.701 Å, b = 15.184 Å, c = 14.710 Å, β = 91.70°, V = 1942.5 Å3 (at 150 K).
Single crystal X-ray crystallography
Single crystal X-ray data were recorded on a dual source Rigaku-Oxford Diffraction Gemini A Ultra diffractometer, equipped with an Atlas CCD detector and an Oxford Cryosystems Cryojet-XL liquid nitrogen flow device for temperature control. Data collection, indexing and integration were performed using the Rigaku-Oxford Diffraction software CrysAlisPro.43 Structures were solved by dual-space methods in SHELXT,44 or by direct methods in SHELXS,45 and refined by full matrix least-squares on F2 in SHELXL.46 Hydrogen atoms were positioned geometrically and refined using a riding model. The hydrogen atom isotropic displacement parameters were fixed to Uiso(H) = 1.5 × (for CH3) or Uiso(H) = 1.2 × (for CH) the Ueq of the parent atom.Photocrystallography
For photocrystallographic studies, the standard single crystal X-ray diffraction set-up was modified to incorporate a purpose-built LED ring array,47 which positions five LEDs (405 nm: Bivar UV5TZ-405-15, λp = 405 ± 2.5 nm, 3.4 V, 15 mA, 40 mW) at a distance of approximately 1 cm from the crystal in a uniform arc. This set-up helps to ensure even irradiation on all sides of the crystal, and enables the collection of complete single-crystal X-ray data while the array remains in place. To further ensure even illumination, for steady-state experiments the crystal was also continuously rotated about the phi axis during irradiation.Results and discussion
Ground state single crystal X-ray structure
A crystal of 1 was flash frozen at 150 K and the GS single crystal X-ray structure was determined in the dark. 1 crystallises in the tetragonal space group I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) , with one Re complex molecule in the asymmetric unit. The Re-centre adopts a distorted octahedral geometry and is bound to three carbonyl ligands, one bidentate OMe2-bpy ligand and one monodentate nitrite group, which is coordinated solely as the nitro-(η1-
, with one Re complex molecule in the asymmetric unit. The Re-centre adopts a distorted octahedral geometry and is bound to three carbonyl ligands, one bidentate OMe2-bpy ligand and one monodentate nitrite group, which is coordinated solely as the nitro-(η1-![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif) O2) isomer. Fig. 1a shows the full atomic connectivity in the asymmetric unit, and selected X-ray data are provided in Table 1.
O2) isomer. Fig. 1a shows the full atomic connectivity in the asymmetric unit, and selected X-ray data are provided in Table 1.
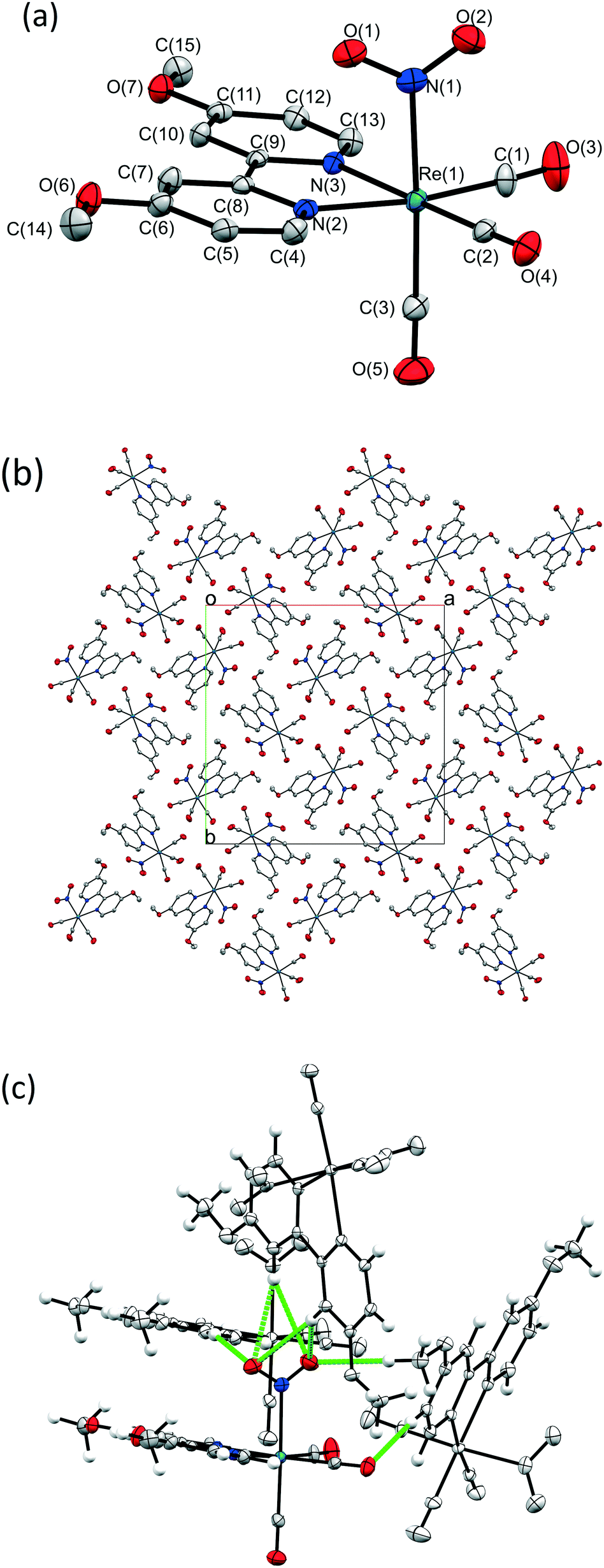 | ||
| Fig. 1 Single-crystal X-ray structure of the GS of 1 at 150 K. (a) Atomic connectivity in the asymmetric unit, ellipsoids at 50% probability, hydrogen atoms removed for clarity; (b) crystal packing arrangement; (c) C–H⋯O interactions involving the nitro-NO2 ligand. | ||
| Ground state (GS) | Metastable state (MS)a | |
|---|---|---|
| a The MS structure at 150 K is recorded in the 180K_405nm_150K.cif crystallographic information file, see ESI. | ||
| Photoconversion | 0% | 58% |
| Irradiation time (total) | 0 min | 180 min |
| Temperature | 150(1) K | 150(1) K |
| Wavelength | 0.71073 Å | 0.71073 Å |
| Empirical formula | C15H12N3O7Re1 | C15H12N3O7Re1 |
| Formula weight | 532.48 g mol−1 | 532.48 g mol−1 |
| Crystal system | Tetragonal | Tetragonal |
| Space group |
I |
I |
| Unit cell parameters (constrained) | a = 22.2989(5) Å | a = 22.6482(5) Å |
| c = 6.6471(2) Å | c = 6.5591(2) Å | |
| Volume | 3305.2(2) Å3 | 3364.4(2) Å3 |
| Z | 8 | 8 |
| Density (calculated) | 2.140 g cm−1 | 2.102 g cm−1 |
| Absorption coefficient μ | 7.398 mm−1 | 7.268 mm−1 |
| F(000) | 2032 | 2032 |
| R(int) | 0.0362 | 0.0748 |
| Completeness (to θ = 25.00°) | 0.987 | 0.997 |
| R 1 (observed data I > 2σ(I)) | 0.0275 | 0.0453 |
| wR2 (all data) | 0.0554 | 0.1121 |
| Reflections (independent) | 6711 (3356) | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 493 (3157) 493 (3157) |
| Flack parameter | −0.031(11) | −0.048(11) |
Fig. 1b shows the crystal packing arrangement in 1. The [Re(OMe2-bpy)(CO)3(NO2)] molecules appear closely packed, however a contact surface void space calculation in the CCDC software Mercury48 (probe radius 1.2 Å, grid spacing 0.1 Å) reveals small pockets of space at points along the 4-fold rotation axes, comprising a total of 65.65 Å3, or 2% of the unit cell volume. These voids suggest inefficient packing between the carbonyl ligands oriented towards each other in this region (see Fig. S1, ESI†). Any void space in the crystal lattice is significant as it could potentially assist the atomic movements necessary for linkage isomerism to occur. Due to the lack of traditional hydrogen bond donors in 1 there are no strong hydrogen bonds between molecules. However, there are a number of intermolecular CH⋯O contacts, including several to the potentially photoactive nitro-(η1-O2) group (see Fig. 1c, Table S1 ESI†). These intermolecular interactions are significant because, for linkage isomerism to occur, these contacts must be broken and potentially re-formed. As such, they present a likely barrier to the progress of nitro → nitrito photoswitching in 1.
Steady-state photocrystallography with 1 at 150 K
The same crystal was held at 150 K and irradiated, in situ on the diffractometer, using the LED ring array set-up outlined above. A wavelength of 405 nm, towards the tail of the absorption maxima, was selected by reference to the solid state absorption spectrum of 1 (Fig. S2/S3, ESI†). This choice reflects the results of previous investigations, in which wavelengths in the tail of the main absorption band are found to promote solid state photoswitching, helping to maximise the penetration of the excitation light through the crystal.49In light of previous photocrystallographic experiments with metal-nitrite crystals,20,31,34,36,50 a long-lived metastable isomer is expected on irradiation of 1 at 150 K. Thus, using the steady-state photocrystallographic method,20 after an illumination period of 1 min the LEDs were switched off and a second X-ray dataset was collected. This revealed that photoactivation had been achieved, with 29% of the crystal now converted to an endo-nitrito-(η1-NO) isomer (Fig. 2a). The steady-state experiment was then continued, with irradiation paused at regular intervals to allow the collection of a complete X-ray diffraction dataset. These results were then used to plot the photoexcitation profile for 1 with respect to irradiation time at 150 K (Fig. 2b). The photoreaction initially progresses quickly, but the conversion rate rapidly slows with irradiation time, producing a sigmoidal-shaped profile. A maximum conversion of 58% to the endo-nitrito isomer was determined after a total of 3 h illumination. Further irradiation caused no appreciable change, confirming that a photostationary state had been achieved at this temperature. This excitation level was retained when the crystal was held at 150 K in the dark for a further 1 h, proving that the excited state isomer is metastable at this temperature. A significant reduction in the quality of the diffraction data was seen on irradiation, which is reflected in the increased R-indices for the MS structure (Table 1, S3†).
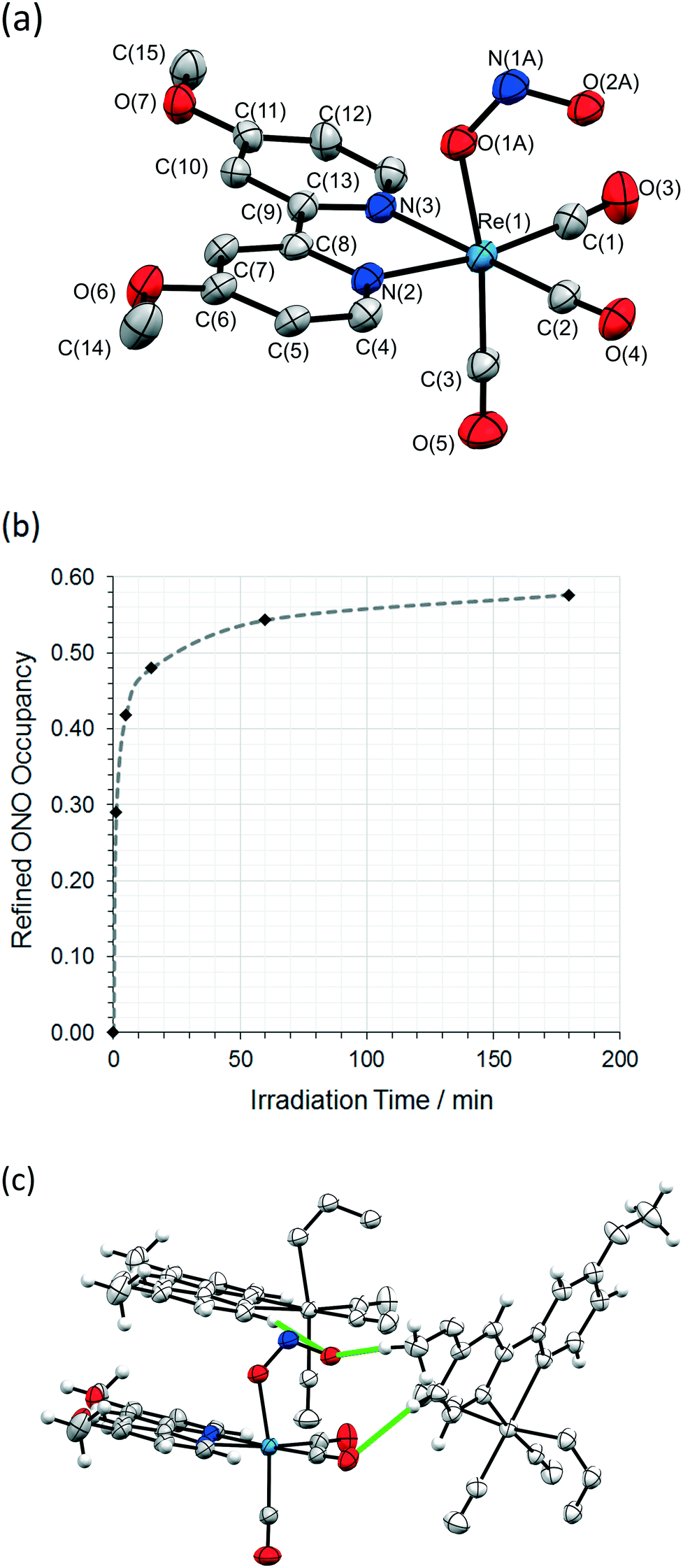 | ||
| Fig. 2 Photo-induced metastable state (MS) structure and refined occupancy levels for 1 at λ = 405 nm. (a) Single-crystal X-ray structure showing the atomic connectivity for the MS endo-nitrito-(η1-ONO) isomer at 150 K. (b) Graph of photoconversion level vs. irradiation time in a single-crystal of 1 at 150 K. (c) C–H⋯O interactions involving the photoinduced nitrito-ONO ligand. | ||
Analysis of the photoconversion profile in Fig. 2b can provide kinetic information on the progress of nitro → nitrito photo-switching in 1. As shown previously in the literature, the JMAK theory for solid state kinetics provides a good fit for single-crystal-to-single-crystal photoreactions,36,39,50–53 and can be adapted for both incomplete excitation and decay.39,51 The general equation for JMAK analysis is given as:
| α(t) = 1 − e−ktn | (1) |
| D = n − 1 | (2) |
Here, as the photoreaction in 1 remains incomplete at 150 K, the data were fitted with a modified version of eqn (1),51 which allows the initial and final conversion fractions to be parameterised:
| α(t) = α(f) − [α(f) − α(0)]e−ktn | (3) |
A least squares regression analysis was performed on the data points in Fig. 3 using eqn (3), producing a good fit to the data with a value of 0.996 for R2. The initial and final conversion fractions were fixed to their experimental values of α(0) = 0 and α(f) = 0.576, while the parameters k and n were allowed to refine freely. A reaction rate constant of k = 0.38(2) min−1 was determined, which is comparable to those reported previously for nitro → nitrito conversion processes in the single crystal at low temperature.36,39,50 For the Avrami exponent, a refined value of n = 0.35(2) was determined. While a value of n ≈ 1 (D = 0) is indicative of zero-dimensional, or homogeneous, growth of the MS phase through the crystal, a value of n < 1 suggests a negative dimensionality for the production of the MS phase, which not physically realistic. This result indicates that eqn (3) is insufficient to accurately model photoconversion in 1, and there is likely to be some other physical processes limiting the progress of the nitro → nitrito conversion through the crystal that are not accounted for by simple JMAK theory. Possible limiting factors could include incomplete penetration of excitation light through the crystal bulk, or steric influences resulting from strain in the crystal lattice on photoswitching (see the comparison of GS/MS discussion below).
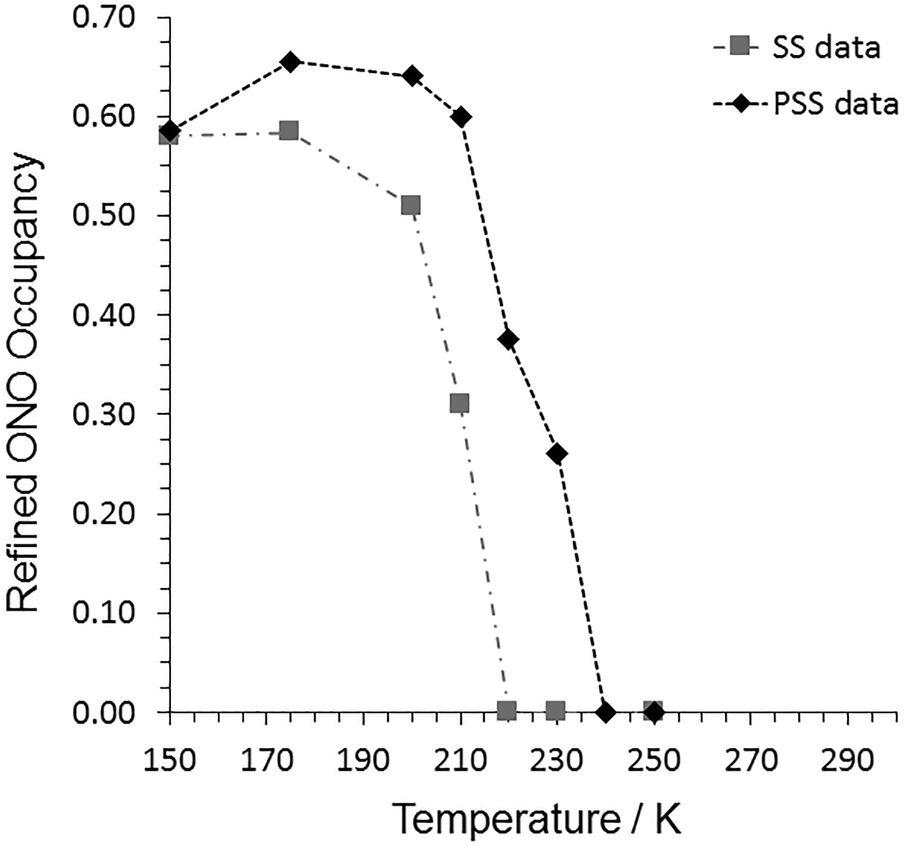 | ||
| Fig. 3 Refined nitro:nitrito isomer occupancies from variable temperature parametric studies with 1, recorded under both steady-state (SS) and pseudo-steady-state (PSS) conditions. | ||
The final step of the steady-state photocrystallographic experiment is to determine the temperature range over which the system is metastable. The crystal in its photoinduced MS was next subject to a variable temperature study between 150 and 300 K (Fig. 3, Tables S2/S4†). The MS occupancy remained constant on heating to 175 K, above which temperature the crystal then began to decay back towards the GS, indicating an approximate temperature for the apparent “metastable limit” for this crystal system.391 then finally reverts back to a 100% GS nitro-(η1-O2) isomer by 220 K.
Comparison of GS and MS structures of 1 at 150 K
The crystal packing arrangement in 1 is largely unchanged on irradiation, excepting the change in nitrite coordination mode (see Fig. S4/S5, ESI†). However, a comparison of Fig. 1c and 2c shows that there is a significant rearrangement in the intermolecular C–H⋯O contacts involving the isomerising ligand. For nitro → nitrito isomerism to occur, six C–H⋯O contacts to the nitro-(η1-O2) ligand need to be broken, with an average C⋯O distance of 3.52(2) Å. In the MS, there are just two C–H⋯O contacts formed by the nitrito-(η1-NO) ligand, with a longer average C⋯O distance of 3.57(4) Å. Full details of the C–H⋯O distances in the MS are given in Table S5, ESI.† Overall, the intermolecular interactions involving the photoinduced isomer are longer, and therefore likely to be weaker, than those influencing the GS isomer, suggesting that conversion to the MS nitrito-ONO arrangement will not be energetically favourable.
A comparison of the unit cell dimensions in Table 1 for the GS and MS structures reveals that there is a significant increase of 59.2(3) Å3 (or 1.79%) in the unit cell volume on photoactivation. This is a reasonably large change in unit cell volume in comparison to those reported previously, and may point to an explanation for the incomplete level of photoactivation in 1. Typically, metal-nitrite crystals displaying high levels of photoactivation, up to 100%, tend to produce a volume change of <1%.34–36 This larger unit cell change may suggest a significant amount of strain is induced in the crystal as a result of the photoisomerisation, which in turn limits the overall conversion fraction that can be accommodated while maintaining single crystal integrity.
In addition to an assessment of overall changes in the unit cell, the “reaction cavity” concept can provide insight into the local structure changes in the region of the isomerising nitrite group, helping to rationalise how or why the photoreaction proceeds.57,58 The reaction cavity is defined as the volume encapsulating the photoactive group and, in particular, the change in the reaction cavity volume ΔVc is a useful measure of how much the local structure must change in order to accommodate nitro → nitrito rearrangement. Table 2 provides reaction cavity analyses performed on the GS and MS structures of 1 using the CCDC software Mercury.48 The reaction cavity volume more than doubles on irradiation of the crystal, with a value for ΔVc of 10.2 Å3 per molecule, equating to a 101.5% increase. In addition, a qualitative visual assessment of the reaction cavities reveals that their shape is also significantly altered on excitation (see Fig. S6, ESI†). Both of these factors indicate that there is a significant change required in the local crystal structure to accommodate the isomerisation, which is expected to induce considerable strain on the surrounding crystalline array. This provides a likely explanation not only for the incomplete photoconversion level in 1, but also for the significant reduction in data quality on irradiation, and for the low value of the Avrami exponent n in the JMAK kinetic analysis above.
| V c per unit cell/Å3 | V c per molecule/Å3 | ΔVc (MS-GS)/Å3 | ΔVc (MS-GS)/% | |
|---|---|---|---|---|
| GS | 80.26 | 10.03 | 10.19 | 101.5 |
| MS | 161.76 | 20.22 |
Steady-state photocrystallography at 170 K
In our recent work with Pd and Pt-nitrite linkage isomer crystals that also show incomplete conversion at low temperature, we found that irradiation at higher temperatures, just below the “metastable limit” for the system, can help to increase the overall conversion level.31 As such, a second steady-state photocrystallographic experiment was also conducted with 1 at 170 K. Following a GS X-ray structure determination at 170 K, a crystal was then irradiated for a period of 3 h, before the LEDs were switched off and a second X-ray dataset collected (see Table S6, ESI†). A maximum nitrito-ONO occupancy of 60% was determined from these data, which is comparable, within error, to the photostationary state achieved at 150 K. As such, in the case of the Re(I)-complex 1, it is apparent that raising the temperature does not have a marked effect on the overall level of photoactivation that can be achieved under steady-state photocrystallographic conditions.Pseudo-steady-state photocrystallography with 1
A crystal of 1 was next subjected to a pseudo-steady-state photocrystallographic investigation to determine if the nitro:nitrito ratio can be further manipulated under continuous illumination. Previous work has shown that a photostationary equilibrium is induced between the forwards photoexcitation and the reverse thermal decay processes with continuous irradiation,32,36,39 and these conditions are easily achieved using the LED ring array set-up. The crystal was first mounted at 150 K and irradiated for a period of 3 h to ensure that an equilibrium nitro:nitrito occupancy had been reached at this temperature. A complete X-ray dataset was then collected, with the LEDs remaining on and in place, to reveal the pseudo-steady-state photoconversion level. Irradiation was then continued while the temperature was raised, pausing at regular intervals to enable further pseudo-steady-state experiments to be collected.
The refined nitro:nitrito occupancy levels from these data are shown in Fig. 3 and Table S7.† At 150 K, the pseudo-steady-state conversion fraction is the same, within error, as that obtained after 3 h illumination in the steady-state experiments. However, on warming the crystal to 175 K, continuous illumination induces an increase in the maximum conversion level in 1, achieving a maximum nitrito-(η1-NO) occupancy of 66%. Selected X-ray crystal data and structure information for the pseudo-steady state data at 150 and 175 K are provided in Table 3. At higher temperatures the equilibrium excited state isomer occupancy gradually decreases, finally returning to the 100% GS nitro-(η1-NO2) isomer by 240 K.
| PSS 150 K | PSS 175 K | |
|---|---|---|
| Photoconversion | 59% | 66% |
| Temperature | 150(2) K | 175(2) K |
| Wavelength | 0.71073 Å | 0.71073 Å |
| Empirical formula | C15H12N3O7Re1 | C15H12N3O7Re1 |
| Formula weight | 532.48 g mol−1 | 532.48 g mol−1 |
| Crystal system | Tetragonal | Tetragonal |
| Space group |
I |
I |
| Unit cell parameters (constrained) | a = 22.719(2) Å | a = 22.739(2) Å |
| c = 6.5611(8) Å | c = 6.5506(8) Å | |
| Volume | 3386.7(7) Å3 | 3387.2(7) Å3 |
| Z | 8 | 8 |
| Density (calculated) | 2.089 g cm−1 | 2.088 g cm−1 |
| Absorption coefficient μ | 7.220 mm−1 | 7.219 mm−1 |
| F(000) | 2032 | 2032 |
| R(int) | 0.0618 | 0.0652 |
| Completeness (to θ = 25.00°) | 0.996 | 0.996 |
| R 1 (observed data I > 2σ(I)) | 0.0430 | 0.0443 |
| wR2 (all data) | 0.0975 | 0.0966 |
| Reflections (independent) | 6030 (2903) | 6047 (2900) |
| Flack parameter | −0.016(18) | −0.01(2) |
This result is interesting as it suggests that, under continuous illumination conditions, raising the temperature helps overcome some of the barriers limiting the overall photoconversion level. To assess this more quantitatively, a direct comparison may be made between the pseudo-steady-state structures at 150 and 175 K. Table 3 shows that there is a negligible 0.5(10) Å3 (or 0.014%) increase in unit cell volume on warming under continuous illumination, and there is similarly no significant increase in the void space in the crystal, using a contact surface calculation in Mercury (probe radius 1.2 Å, grid spacing 0.1 Å). Interestingly, this observation is at odds with the results of the steady-state studies and tends not to support the theory of steric strain as a barrier to photoconversion, as it would be expected that an increase in excitation level should correlate with an observed increase in free space in the overall crystal structure. Similarly, an assessment of the reaction cavity volumes at 150 and 175 K under continuous irradiation shows no significant change (see Table S8†). However, as irradiation continues throughout the pseudo-steady-state experiment, the data quality is observed to decrease considerably with a significant increase in R-indices, reduction of I/σ and loss of high angle diffraction data (see Table S9†). As such, it is possible that damage to the crystal may also influence the overall photoconversion fraction observed in these studies (e.g. due to loss of crystallinity at the crystal surface and/or crystal fracturing during the experiment), and so care should be taken when drawing any more detailed conclusions from these data.
Conclusions
This study delivers the first 3D structure confirmation of photoinduced nitro → nitrito conversion in a Re(I)-bpy single crystal, providing a detailed determination of the photoinduced [Re(OMe2-bpy)(CO)3(η1-NO)] excited state structure. The combination of photoactive linkage isomer chemistry with that of Re(I)-bpy complexes broadens the scope of dynamic structural studies on linkage isomers and also relates the photoactive properties of this and other Re(I) molecules to photocatalytic studies on related systems. The detailed, 3D understanding of the structural changes that occur on photoactivation at the atomic-scale in 1 will benefit the design of new and more efficient Re(I) compounds, in both the solid and solution state, which can be further optimised towards specific photoswitching applications.
The successful use of 405 nm light to induce a considerable (66%) photoactivation level in the system is of particular importance, as many Re(I)-photocatalyst systems are limited to absorption at shorter wavelengths in the UV region.17 By broadening the range of the UV/vis absorption spectrum in the solid state, these Re(I)-nitrite crystal systems can absorb a wider range of the solar spectrum and therefore have the potential to increase solar conversion efficiency.
The incomplete photoconversion level achieved in 1, even under continuous illumination, and the considerable reduction in X-ray data quality on irradiation are both considerable limitations for the development of single crystals of 1 for real photoswitchable device solutions. Given the indication of significant steric strain in the crystal, and its potential impact on the photoactivation levels observed, future studies will focus on the development of new Re(I) crystal systems containing bulkier auxiliary fragments (ligands and/or counter-ion moieties) – a crystal engineering route that we have previously shown can successfully facilitate higher levels of nitro → nitrito conversion by controlling the reaction cavity volume and minimising ΔVc.34,36
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
LEH would like to that the UK Engineering and Physical Sciences Research Council (EPSRC) for financial support. This work is supported by EPSRC Programme Grant EP/K004956/1 and EP/M010481/1.Notes and references
- O. Sato, Nat. Chem., 2016, 8, 644–656 CrossRef PubMed.
- Y.-L. Luo, Y.-S. Shiao and Y.-F. Huang, ACS Nano, 2011, 5, 7796–7804 CrossRef PubMed.
- J. Liu, L. Zhang, J. Lei, H. Shen and H. Ju, ACS Appl. Mater. Interfaces, 2017, 9, 2150–2158 CrossRef PubMed.
- H. Hyun, M. H. Park, E. A. Owens, H. Wada, M. Henary, H. J. M. Handgraaf, A. L. Vahrmeijer, J. V. Frangioni and H. S. Choi, Nat. Med., 2015, 21, 192 CrossRef PubMed.
- J. Shen, R. Steinbach, J. M. Tobin, M. Mouro Nakata, M. Bower, M. R. S. McCoustra, H. Bridle, V. Arrighi and F. Vilela, Appl. Catal., B, 2016, 193, 226–233 CrossRef.
- Y. Wang, H. Liu, M. Zhang, W. Duan and B. Liu, RSC Adv., 2017, 7, 16232–16237 RSC.
- N. S. Lewis, Science, 2016, 351(6271), aad1920 CrossRef PubMed.
- F. E. Osterloh, ACS Energy Lett., 2017, 2, 445–453 CrossRef.
- Y. Kou, Y. Nabetani, D. Masui, T. Shimada, S. Takagi, H. Tachibana and H. Inoue, J. Am. Chem. Soc., 2014, 136, 6021–6030 CrossRef PubMed.
- L. A. Worl, R. Duesing, P. Chen, L. D. Ciana and T. J. Meyer, J. Chem. Soc., Dalton Trans., 1991, 849–858 RSC.
- J. Shakeri, H. Farrokhpour, H. Hadadzadeh and M. Joshaghani, RSC Adv., 2015, 5, 41125–41134 RSC.
- H. Takeda, K. Koike, H. Inoue and O. Ishitani, J. Am. Chem. Soc., 2008, 130, 2023–2031 CrossRef PubMed.
- X. Liu, S. Inagaki and J. Gong, Angew. Chem., Int. Ed., 2016, 55, 14924–14950 CrossRef PubMed.
- C. Gao, J. Wang, H. Xu and Y. Xiong, Chem. Soc. Rev., 2017, 46, 2799–2823 RSC.
- A. Dhakshinamoorthy, A. M. Asiri and H. García, Angew. Chem., Int. Ed., 2016, 55, 5414–5445 CrossRef PubMed.
- T. L. Easun, J. Jia, J. A. Calladine, D. L. Blackmore, C. S. Stapleton, K. Q. Vuong, N. R. Champness and M. W. George, Inorg. Chem., 2014, 53, 2606–2612 CrossRef PubMed.
- A. Genoni, D. N. Chirdon, M. Boniolo, A. Sartorel, S. Bernhard and M. Bonchio, ACS Catal., 2017, 7, 154–160 CrossRef.
- P. Coppens, Angew. Chem., Int. Ed., 2009, 48, 4280–4281 CrossRef PubMed.
- L. E. Hatcher and P. R. Raithby, Coord. Chem. Rev., 2014, 277–278, 69–79 CrossRef.
- L. E. Hatcher and P. R. Raithby, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2013, 69, 1448–1456 CrossRef PubMed.
- M. D. Carducci, M. R. Pressprich and P. Coppens, J. Am. Chem. Soc., 1997, 119, 2669–2678 CrossRef.
- N. Casaretto, D. Schaniel, P. Alle, E. Wenger, P. Parois, B. Fournier, E.-E. Bendeif, C. Palin and S. Pillet, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2017, 73, 696–707 CrossRef PubMed.
- B. Cormary, S. Ladeira, K. Jacob, P. G. Lacroix, T. Woike, D. Schaniel and I. Malfant, Inorg. Chem., 2012, 51, 7492–7501 CrossRef PubMed.
- P. Coppens, I. Novozhilova and A. Kovalevsky, Chem. Rev., 2002, 102, 861–884 CrossRef PubMed.
- D. V. Fomitchev, P. Coppens, T. Li, K. A. Bagley, L. Chen and G. B. Richter-Addo, Chem. Commun., 1999, 2013–2014 RSC.
- J. M. Cole, K. Y. M. Yeung, G. Pace, S. O. Sylvester, D. Mersch and R. H. Friend, CrystEngComm, 2015, 17, 5026–5031 RSC.
- S. O. Sylvester and J. M. Cole, J. Phys. Chem. Lett., 2013, 4, 3221–3226 CrossRef.
- K. F. Bowes, J. M. Cole, S. L. G. Husheer, P. R. Raithby, T. L. Savarese, H. A. Sparkes, S. J. Teat and J. E. Warren, Chem. Commun., 2006, 2448–2450 RSC.
- A. Y. Kovalevsky, K. A. Bagley, J. M. Cole and P. Coppens, Inorg. Chem., 2002, 42, 140–147 CrossRef PubMed.
- D. V. Fomitchev, K. A. Bagley and P. Coppens, J. Am. Chem. Soc., 2000, 122, 532–533 CrossRef.
- L. E. Hatcher and P. R. Raithby, CrystEngComm, 2017, 19, 6297–6304 RSC.
- L. E. Hatcher, J. Christensen, M. L. Hamilton, J. Trincao, D. R. Allan, M. R. Warren, I. P. Clarke, M. Towrie, D. S. Fuertes, C. C. Wilson, C. H. Woodall and P. R. Raithby, Chem. – Eur. J., 2014, 20, 3128–3134 CrossRef PubMed.
- L. E. Hatcher, E. J. Bigos, M. J. Bryant, E. M. MacCready, T. P. Robinson, L. K. Saunders, L. H. Thomas, C. M. Beavers, S. J. Teat, J. Christensen and P. R. Raithby, CrystEngComm, 2014, 16, 8263–8271 RSC.
- L. E. Hatcher, M. R. Warren, D. R. Allan, S. K. Brayshaw, A. L. Johnson, S. Fuertes, S. Schiffers, A. J. Stevenson, S. J. Teat, C. H. Woodall and P. R. Raithby, Angew. Chem., Int. Ed., 2011, 50, 8371–8374 CrossRef PubMed.
- M. Warren, S. Brayshaw, A. Johnson, S. Schiffers, P. Raithby, T. Easun, M. George, J. Warren and S. Teat, Angew. Chem., 2009, 121, 5821–5824 CrossRef.
- L. E. Hatcher, CrystEngComm, 2016, 18, 4180–4187 RSC.
- R. Kaminski, K. N. Jarzembska, S. E. Kutyla and M. Kaminski, J. Appl. Crystallogr., 2016, 49, 1383–1387 CrossRef.
- A. Y. Kovalevsky, G. King, K. A. Bagley and P. Coppens, Chem. – Eur. J., 2005, 11, 7254–7264 CrossRef PubMed.
- L. E. Hatcher, J. M. Skelton, M. R. Warren, C. Stubbs, E. L. da Silva and P. R. Raithby, Phys. Chem. Chem. Phys., 2018, 20, 5874–5886 RSC.
- S. E. Domínguez, P. Alborés and F. Fagalde, Polyhedron, 2014, 67, 471–480 CrossRef.
- R. Kia and F. Safari, Inorg. Chim. Acta, 2016, 453, 357–368 CrossRef.
- M. E. Viguri, J. Pérez and L. Riera, Chem. – Eur. J., 2014, 20, 5732–5740 CrossRef PubMed.
- CrysAlis Pro, Rigaku Oxford Diffraction Data Collection and Data Reduction GUI, Version 171.38.43 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- M. Sheldrick George, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- S. K. Brayshaw, J. W. Knight, P. R. Raithby, T. L. Savarese, S. Schiffers, S. J. Teat, J. E. Warren and M. R. Warren, J. Appl. Crystallogr., 2010, 43, 337–340 CrossRef.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. v. d. Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef.
- I. Abdelmoty, V. Buchholz, L. Di, C. Guo, K. Kowitz, V. Enkelmann, G. Wegner and B. M. Foxman, Cryst. Growth Des., 2005, 5, 2210–2217 CrossRef.
- S. E. Bajwa, T. E. Storr, L. E. Hatcher, T. J. Williams, C. G. Baumann, A. C. Whitwood, D. R. Allan, S. J. Teat, P. R. Raithby and I. J. S. Fairlamb, Chem. Sci., 2012, 3, 1656–1661 RSC.
- R. More, G. Busse, J. Hallmann, C. Paulmann, M. Scholz and S. Techert, J. Phys. Chem. C, 2010, 114, 4142–4148 CrossRef.
- D.-K. Cao, T. V. Sreevidya, M. Botoshansky, G. Golden, J. Brown Benedict and M. Kaftory, J. Phys. Chem. A, 2010, 114, 7377–7381 CrossRef PubMed.
- J. B. Benedict and P. Coppens, J. Phys. Chem. A, 2009, 113, 3116–3120 CrossRef PubMed.
- M. Avrami, J. Chem. Phys., 1941, 9, 177–184 CrossRef.
- M. Avrami, J. Chem. Phys., 1940, 8, 212–224 CrossRef.
- M. Avrami, J. Chem. Phys., 1939, 7, 1103–1112 CrossRef.
- E. V. Boldyreva, Solid State Ionics, 1997, 101–103(Part 2), 843–849 CrossRef.
- Y. Ohashi, Crystallogr. Rev., 2013, 19, 2–146 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1842772–1842793. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ce00774h |
| This journal is © The Royal Society of Chemistry 2018 |