
A divergent and concise total synthesis of (−)-lycoposerramine R and (+)-lycopladine A†
Sheng
Chen
ab,
Jinming
Wang
ab and
Fayang G.
Qiu
 *ab
*ab
aGuangzhou Institute of Biomedicine and Health, The Chinese Academy of Sciences, 190 Kaiyuan Ave., The Science Park of Guangzhou, Guangdong 510530, China. E-mail: qiu_fayang@gibh.ac.cn
bThe University of The Chinese Academy of Sciences, Beijing, 100049, China
First published on 9th March 2018
Abstract
A concise, asymmetric and divergent synthesis of lycoposerramine R and lycopladine A is presented. The synthesis features the palladium-catalyzed cycloalkenylation of a silyl enol ether for assembling the 5/6-hydrindane system and generating a quaternary carbon center in one step.
Club mosses, such as Lycopodium complanatum and Lycopodium carinatum, are a rich source of structurally complex and biologically active alkaloids (Fig. 1).1–3 Lycoposerramine-R (1), isolated by Takayama and co-workers in 2009, was characterized to have a previously unknown skeleton consisting of a fused tetracyclic ring system with four chiral centers, a pyridone ring, and cis-fused hydrindane.4 Its simplified pyridine congener lycopladine A (2) was isolated from L. complanatum in 2006 and showed modest cytotoxicity against murine lymphoma cells.5 During the past decade, owing to their compact structures as well as their biological activities, these alkaloids have aroused the interest of a large number of research groups, whose studies have culminated in the completion of several elegant total syntheses of some lycopodium alkaloids and some new synthetic methodologies for assembling their core structures.6 To date, 4 total syntheses have been reported for lycoposerramine R (1)6h–k and 7 for lycopladine A (2), respectively.6l–r
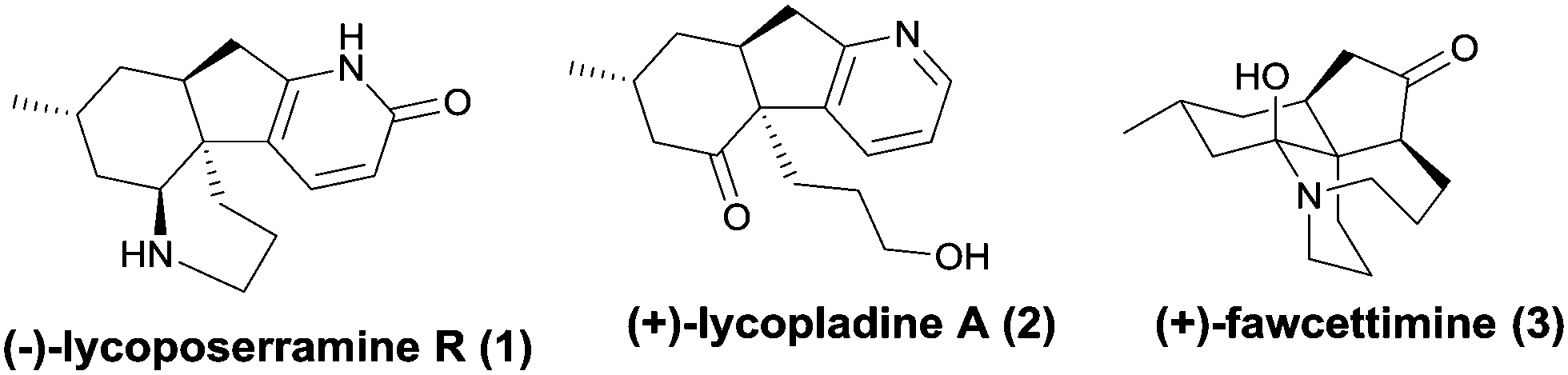 | ||
| Fig. 1 The structures of lycoposerramine R, lycopladine A, and fawcettimine. | ||
In this communication, we report a facile, alternative entry to these alkaloids that involves some novel chemistry involving the palladium-catalyzed cycloalkenylation of a silyl enol ether,7 a reaction that we believe will have general utility. As shown in the retrosynthetic analysis (Scheme 1), we reasoned that both lycoposerramine R (1) and lycopladine A (2) might be constructed from the common intermediate RS-1 through several different transformations. Intermediate RS-1 in turn might be accessed from silyl enol ether RS-2via a sequence of palladium-catalyzed cycloalkenylation of silyl enol ether followed by SeO2/TBHP oxidation. Silyl enol ether RS-2 might be obtained from the stereoselective conjugate addition of a Grignard reagent RS-4 prepared from commercial 4-bromo-1-butene8 to an α,β-unsaturated carbonyl compound RS-3, followed by trapping the enolate with TMSCl, while RS-3 could be derived from the readily accessible phenylsulfide 19via the introduction of a C3 unit.
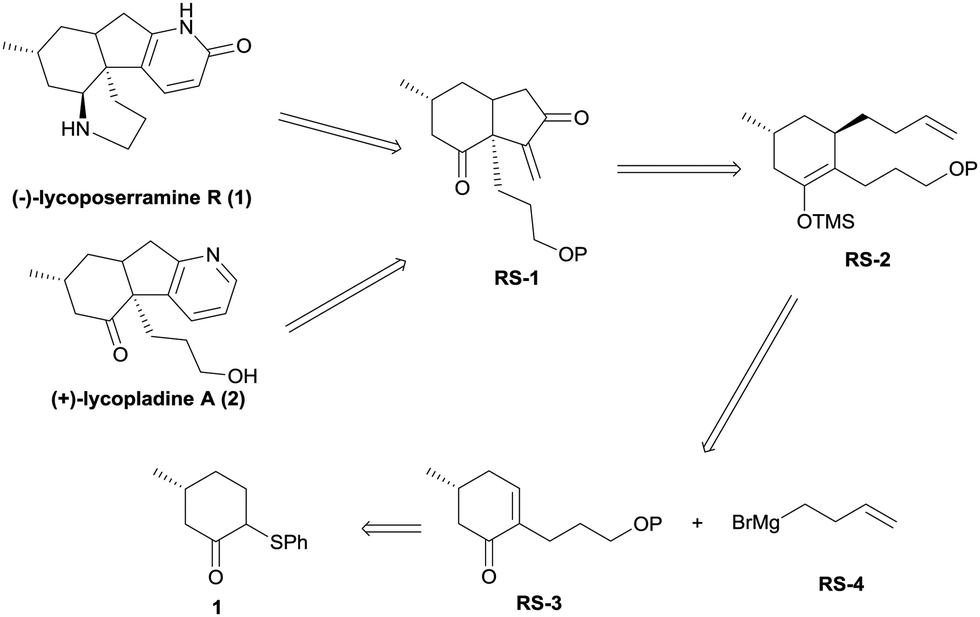 | ||
| Scheme 1 Retrosynthetic analysis of (−)-lycoposerramine R (1) and (+)-lycopladine A (2). | ||
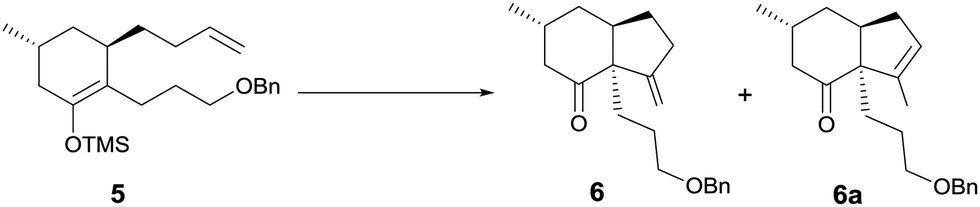
Based on the above analysis, the synthetic strategy seemed feasible. Thus, alkylation of enolate of 1 (Scheme 2) with iodide 210 afforded phenylsulfenyl ketone 3 as a diastereomeric mixture (dr = 2.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in 65% yield, oxidation of which with m-CPBA at −78 °C followed by warming to room temperature afforded enone 4.6q After the copper(I)-mediated conjugate addition of the Grignard reagent freshly prepared from 4-bromo-1-butene to enone 4 to generate an enolate, TMSCl was added at −20 °C to yield silyl enol ether 5 in 85% overall yield.
:1) in 65% yield, oxidation of which with m-CPBA at −78 °C followed by warming to room temperature afforded enone 4.6q After the copper(I)-mediated conjugate addition of the Grignard reagent freshly prepared from 4-bromo-1-butene to enone 4 to generate an enolate, TMSCl was added at −20 °C to yield silyl enol ether 5 in 85% overall yield.
 | ||
| Scheme 2 Synthesis of silyl enol ether 5. | ||
At this stage, we began to investigate the key cycloalkenylation (Table 1). Surprisingly, treatment of the silyl enol ether 5 with stoichiometric amounts of palladium acetate in dry THF yielded exo-olefin 6 along with endo-olefin 6a in 35% and 17% yields, respectively. After many unfruitful attempts, it was found that when treated with 10 mol% of palladium acetate in dry DMSO under a balloon pressure of oxygen at 45 °C, silyl enol ether 5 underwent cycloalkenylation and exo-olefin 6 was obtained in 48% yield together with endo-olefin 6a in 26% yield. Allylic oxidation of 6 using SeO2/TBHP, followed by Dess–Martin oxidation yielded the desired key intermediate 7 in 63% yield. Treatment of the endo-olefin 6a with m-CPBA, followed by Al(Oi-Pr)3 and oxidation by the Dess–Martin reagent yielded 7 in 58% yield (Scheme 3).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst (equiv.) | Solvent | Temp. | Additives (equiv.) | 6 (%) | 6a (%) |
| 1 | PdCl2(PPh3)2 (1.0) | THF | RT | — | 0 | 0 |
| 2 | Pd(CF2COOH)2 (1.0) | THF | RT | — | Trace | Trace |
| 3 | PdCl2 (1.0) | THF | RT | — | 23 | 10 |
| 4 | Pd(OAc)2 (1.0) | THF | RT | — | 35 | 17 |
| 5 | Pd(OAc)2 (0.1) | THF | RT | Cu(OAc)2·H2O (1.0) | 6 | 2 |
| 6 | Pd(OAc)2 (0.1) | THF | RT | Ag2CO3 (1.0) | 6 | 2 |
| 7 | Pd(OAc)2 (0.1) | THF | RT | Benzoquinone (1.0) | 6 | 2 |
| 8 | Pd(OAc)2 (0.1) | DMSO | RT | O2 | 33 | 16 |
| 9 | Pd(OAc)2 (0.1) | DMSO | 45 °C | O2 | 48 | 26 |
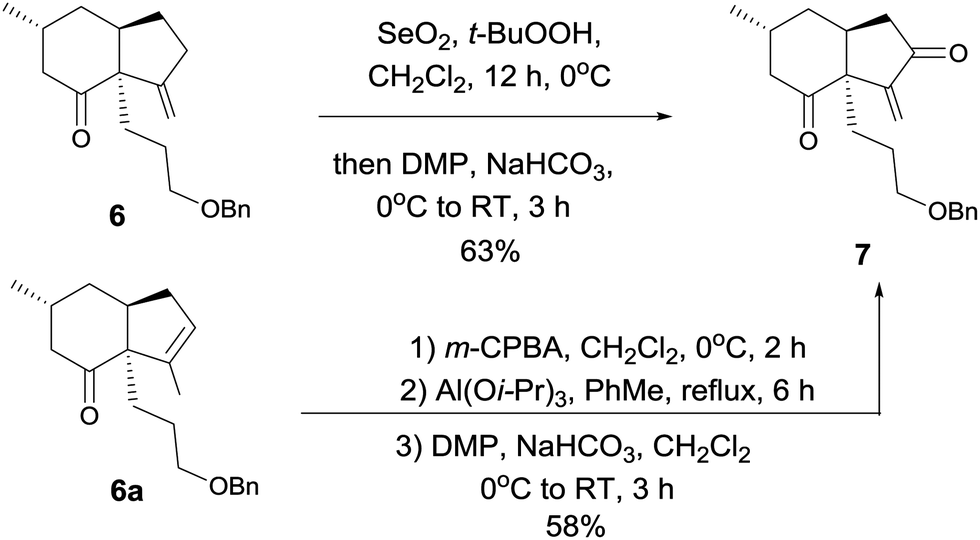 | ||
| Scheme 3 Synthesis of key intermediate 7. | ||
Addition of 2-(phenylsulfonyl)acetamide11 to intermediate 7 in the presence of sodium hydride, followed by treatment with methanolic hydrogen chloride, resulted in the formation of intermediate 8 in 62% yield (Scheme 4). Removal of the benzyl group by treatment with 10% Pd/C in EtOH under a hydrogen atmosphere gave intermediate 9 (85%). Dess–Martin oxidation of this alcohol yielded ketoaldehyde 10 (92%), which when treated with ammonium acetate in the presence of NaBH3CN in methanol at room temperature for 24 h afforded (−)-lycoposerramine R (1) in 65% yield. Synthetic (−)-lycoposerramine R (1) was identical in all respects to the natural product.
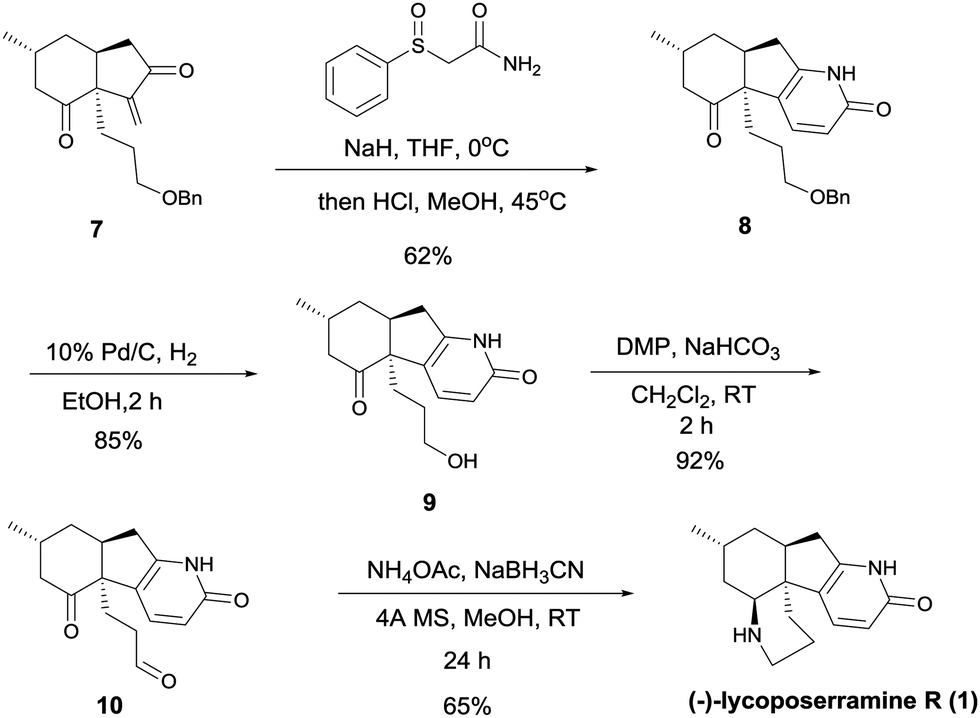 | ||
| Scheme 4 Total synthesis of (−)-lycoposerramine R (1). | ||
With intermediate 7 in hand, the synthesis of (+)-lycopladine A (2) was investigated (Scheme 5). When treated with (N-vinylimino)phosphorene12 in dry benzene at 90 °C in a sealed tube, intermediate 7 underwent cyclization to afford intermediate 11 in 65% yield. Finally, removal of the benzyl group in 11 gave (+)-lycopladine A (2) (70%). The synthetic (+)-lycopladine A (2) showed identical spectroscopic properties in all respects to the natural product.
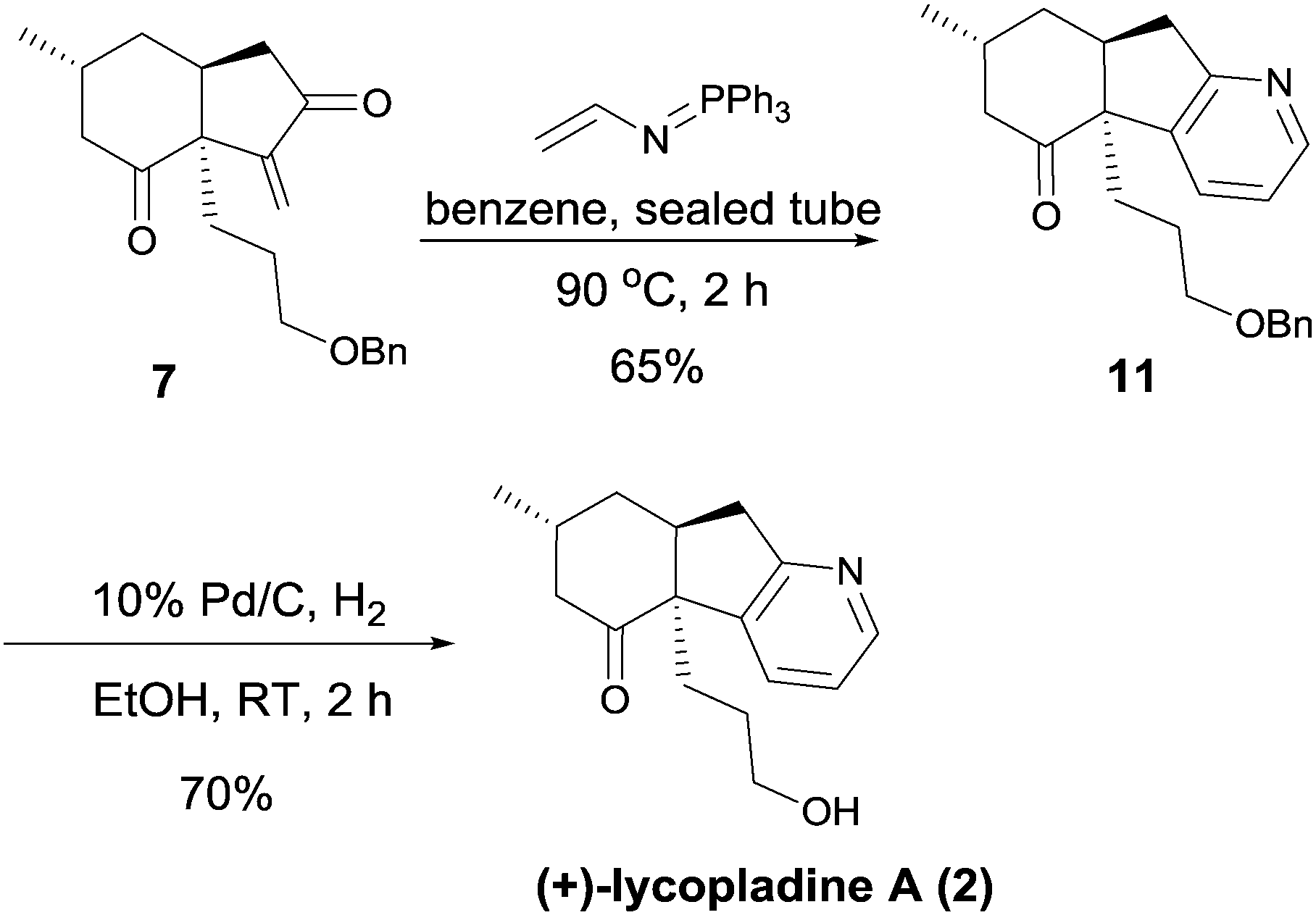 | ||
| Scheme 5 Total synthesis of (+)-lycopladine A (2). | ||
In summary, by using a divergent strategy we have developed a concise, asymmetric total synthesis of both (−)-lycoposerramine-R (1) and (+)-lycopladine A (2) from known phenylsulfide 1 in 9 and 7 steps, respectively. The key features of the current synthesis include a palladium-catalyzed cycloalkenylation of silyl enol ether 5 for assembling the 6,5-fused hydrindane and generating a quaternary carbon center in one step. The application of these synthetic studies to an enantioselective synthesis of the related fawcettimine-type alkaloid 3 will be reported in due course.
We are grateful to the National Natural Science Foundation of China for the financial support of this work (Grant #21372221 and #21572228).
Conflicts of interest
There are no conflicts to declare.Notes and references
- X. Ma and D. R. Gang, Nat. Prod. Rep., 2004, 21, 752 RSC.
- Y. Hirasawa, J. Kobayashi and H. Morita, Heterocycles, 2009, 77, 679 CrossRef CAS.
- P. Siengalewicz, J. Mulzer and U. Rinner, in the Alkaloids: Chemistry and Biology, ed. K. Hans-Joachim, Academic Press, New York, 2013; vol. 72, p 1 Search PubMed.
- K. Katakawa, N. Kogure, M. Kitajima and H. Takayama, Helv. Chim. Acta, 2009, 92, 445 CrossRef CAS.
- K. I. Ishiuchi, T. Kubota, H. Morita and J. I. Kobayashi, Tetrahedron Lett., 2006, 47, 3287 CrossRef CAS.
- For selected recent examples of the total synthesis of lycopodium alkaloids, see: (a) B. K. Hong, H. H. Li, J. B. Wu, J. Zhang and X. G. Lei, Angew. Chem., Int. Ed., 2015, 54, 1011 CrossRef CAS PubMed; (b) P. S. Chauhan, J. R. Sacher and S. M. Weinreb, Org. Lett., 2015, 17, 806 CrossRef CAS PubMed; (c) K. W. Lin, B. Ananthan, S. F. Tseng and T. H. Yan, Org. Lett., 2015, 17, 3938 CrossRef CAS PubMed; (d) C. Bosch, B. Fiser, E. Gomez-Bengoa, B. Bradshaw and J. Bonjoch, Org. Lett., 2015, 17, 5084 CrossRef CAS PubMed; (e) R. A. Samame, C. M. Owens and S. D. Rychnovsky, Chem. Sci., 2016, 7, 188 RSC; (f) B. M. Williams and D. Trauner, Angew. Chem., Int. Ed., 2016, 55, 2191 CrossRef CAS PubMed; (g) Y. Ochi, S. Yokoshima and T. Fukuyama, Org. Lett., 2016, 18, 1494 CrossRef CAS PubMed; (h) V. Bisai and R. Sarpong, Org. Lett., 2010, 12, 2552 CrossRef PubMed; (i) H. Ishida, S. Kimura, N. Kogure, M. Kitajima and H. Takayama, Tetrahedron, 2015, 71, 51 CrossRef CAS; (j) H. Ishida, S. Kimura, N. Kogure, M. Kitajima and H. Takayama, Org. Biomol. Chem., 2015, 13, 7762 RSC; (k) F. W. W. Hartrampf, T. Furukawa and D. Trauner, Angew. Chem., Int. Ed., 2017, 56, 893 CrossRef CAS PubMed; (l) F. W. W. Hartrampf and D. Trauner, J. Org. Chem., 2017, 82, 8206 CrossRef CAS PubMed; (m) L. Meng, J. Org. Chem., 2016, 81, 7784 CrossRef CAS PubMed; (n) K. Hiroya, Y. Suwa, Y. Ichihashi, K. Inamoto and T. Doi, J. Org. Chem., 2011, 76, 4522 CrossRef CAS PubMed; (o) X. J. Liu and S. L. You, Angew. Chem., Int. Ed., 2017, 56, 1 CrossRef; (p) J. E. DeLorbe, M. D. Lotz and S. F. Martin, Org. Lett., 2010, 12, 1576 CrossRef CAS PubMed; (q) S. T. Staben, J. J. Kennedy-Smith, D. Huang, B. K. Corkey, R. L. LaLonde and F. D. Toste, Angew. Chem., Int. Ed., 2006, 45, 5991 CrossRef CAS PubMed; (r) T. Xu, X. L. Luo and Y. Yang, Tetrahedron Lett., 2013, 54, 2858 CrossRef CAS.
- (a) M. Toyota, A. llangovan, R. Okamoto, T. Masaki, M. Arakawa and M. Ihara, Org. Lett., 2002, 4, 4293 CrossRef CAS PubMed; (b) A. S. Kende, B. Roth, P. J. Sanfilippo and T. J. Blacklock, J. Am. Chem. Soc., 1982, 104, 5808 CrossRef CAS; (c) A. S. Kende, B. Roth and P. J. Sanfilippo, J. Am. Chem. Soc., 1982, 104, 1784 CrossRef CAS.
- S. Kumar, P. D. Thornton, T. O. Painter, P. Jain, J. Downard, J. T. Douglas and C. Santini, J. Org. Chem., 2013, 78, 6529 CrossRef CAS PubMed.
- D. Caine, K. Proter and R. A. Cassell, J. Org. Chem., 1984, 49, 2647 CrossRef CAS.
- B. Guillaume, St. O. Miguel and B. C. Andre, Org. Lett., 2008, 10, 5497 CrossRef PubMed.
- T. Nishimura, A. K. Unni, S. Yokoshima and T. Fukuyama, J. Am. Chem. Soc., 2013, 135, 3243 CrossRef CAS PubMed.
- R. K. Alan, M. Roman, V. S. Christian and F. G. Mikhail, J. Org. Chem., 1994, 59, 2740 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc01626g |
| This journal is © The Royal Society of Chemistry 2018 |