
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolydisperse emulsion digital assay to enhance time to detection and extend dynamic range in bacterial cultures enabled by a statistical framework†
Samantha A.
Byrnes
 ,
Elizabeth A.
Phillips
,
Toan
Huynh
,
Bernhard H.
Weigl
and
Kevin P.
Nichols
*
,
Elizabeth A.
Phillips
,
Toan
Huynh
,
Bernhard H.
Weigl
and
Kevin P.
Nichols
*
Intellectual Ventures Laboratory, 14360 SE Eastgate Way, Bellevue, WA 98007, USA. E-mail: knichols@intven.com
First published on 21st May 2018
Abstract
Microbiological culture remains the most sensitive method for detecting viable and infectious bacteria, but these methods often require at least 24 hours to visibly identify bacterial growth. Lab-on-a-chip applications have utilized methods to isolate bacteria in picoliter-sized reaction vessels, resulting in digitized signals that offer improved time-to-detection and improved quantification. Although a great improvement, these approaches typically require expensive and specialized equipment, trained laboratory personnel, and maximum addressable volumes that can be orders of magnitude less than needed for clinically relevant limits of detection. To address these limitations, we have developed a simple method for preparing and semi-quantitatively analyzing small-volume droplets for performing digital culture, allowing for the detection of bacteria. This work includes a description of the method, characterization of resulting droplet sizes, comparison to traditional culture, and a statistical framework to quantify results. Though polydisperse, the droplet size distribution was consistent over different experiments, and there was a correlation between the observed number of positive droplets and the bulk concentration that can serve as a calibration curve for samples with unknown droplet size distributions. This statistical framework enables the simplification of droplet preparation and allows for accurate quantification even with polydisperse droplet sizes. The application of this method can also be extended to a variety of settings for the detection or quantification of bacteria in complex samples.
Introduction
The most common method for detection of viable bacteria in a sample is microbiological culture. In liquid bulk culture, any resulting signal from growth – such as a fluorescent reporter – is diluted in the bulk matrix, reducing the signal-to-noise ratio (SNR) and limit of detection. The development of micro-cultures using isolated picoliter-sized (pL-sized) volumes helps overcome the effects of diffusion and leads to an improved SNR. However, the total addressable volume in typical microfluidic systems is often orders of magnitude lower than clinically or environmentally relevant sample sizes, where limits of detection on the order of single cells per 10s or 100s of mL are common.1Methods have been developed to identify single bacteria in a sample such as volume reduction through filtration2 and target isolation through bead-based capture.3,4 Although effective, these methods can be complex requiring multiple user steps or lack sensitivity due limited ability to sample large volumes.3
The basis of the method reported here is the digitalization of individual bacteria into isolated droplets, or culture vessels. While many droplets contain no target bacteria, those with one or more bacteria will have bacterial and reagent concentrations higher than that of the bulk sample, leading to faster culture and higher SNRs, especially when combined with more sensitive reporter systems such as fluorescence.5–8 Water-in-oil (w/o) emulsions have been reported to create stable, isolated, pL-sized culture vessels.9 These emulsion systems contain a dispersed or aqueous phase, a continuous or oil phase, and an emulsifying agent, which is often a surfactant.10 The choice of the emulsifying agent can be critical for culture applications because aging can lead to the coalescence of droplets if the selected emulsifying agent does not maintain long-term droplet stability.11 The droplet volume is also an important factor. Droplets should be as small as possible (but not smaller than the target bacteria), so that the concentrations of targets in them are high and the detection time is shortened. Therefore, the required droplet sizes usually range from 10s–100s μm in diameter (<1–100s pL). In addition to reducing the effects of diffusion, a non-aqueous continuous phase encapsulates single bacterium into droplet vessels for isolated culture.
There are two main categories of emulsion preparation: bulk solution approaches and microfluidic approaches. Bulk emulsions are generated by mixing two immiscible fluids and applying force to breakdown larger micelles into smaller droplets. Forces can be applied by vigorous shaking by hand or through the use of an active homogenizer.12 Ushikubo et al. used a commercially available homogenizer to prepare droplets that ranged in size from 2.8–35 μm (<1–30 pL) depending on the forces applied.13 In these systems, median droplet size is dictated by the Weber number at a given shear strain and fluid viscosity.14 These approaches are effective, but tend to lead to droplets that are polydisperse. During emulsion formation, polydispersity or the droplet size range depends on the viscosity ratio between the dispersed and continuous phases.15 This ratio is influenced by multiple factors including the composition of the oil and emulsifier, interfacial tension, and shear rate of emulsification.13,15 According to Boxall et al., water-in-oil emulsion formation is dominated by viscous forces during turbulent mixing. These viscous forces can be inhomogenous resulting in varied droplet sizes controlled by locally turbulent flows.16,17 Traditionally, this polydispersity has been a challenge for digitization due to the lack of uniformity and certainty of equal distribution of reagents. Due to these constraints, microfluidic systems were developed to produce more controlled, uniform droplets.
Microfluidic droplet generation approaches rely on interfacial tension and viscous shear stresses caused by the geometry of microfluidic chips and fluid flow rates.18,19 Resulting droplet diameters range from 1–50 μm (1s–100s pL). Additionally, a considerable amount of research has identified appropriate surfactants9,20 and other stabilizers21,22 to help maintain droplet integrity in-chip. This approach was first demonstrated in capillary microfluidic systems,23 and has since been modified for a variety of pL-sized droplets24–26 and used for in-chip bacterial culture.27,28 There are also commercially available systems such as SlipChip29–31 and Fluidigim.32 Although effective, all of these systems still require specialized chips, expensive equipment, and trained technicians for operation, limiting their potential use to laboratory-settings and locations with consistent electricity. Additionally, most of these systems have maximum addressable volumes that can be orders of magnitude less than clinically relevant sample sizes.33
To overcome the limitations of bacterial detection using microfluidic droplet generation, volume reduction through filtration, or bead-based target isolation, we have demonstrated a simple, low-cost method for droplet generation and in-droplet bacterial culture. Droplet preparation is based on a known method of bulk emulsion formation, which relies on agitation. Our system uses off-the-shelf components and is compatible with a wide range of oils and input sample types. Also, this system can function with a range of volumes and uses the entire input sample, therefore increasing total addressable sample volume. Our method can use simple hand-mixing to prepare droplets, removing the constraints of laboratory equipment, electricity, and trained personnel making it a good fit across a range of settings. However, it is not limited to hand-made droplets, and can be used with a simple laboratory vortexer if it is available.
We characterized the droplet volumes made by different methods and compared bacterial growth in bulk and hand-made droplets. We also performed a series of emulsion experiments to obtain a calibration curve, and compared the observed results with the calculated expected outputs34 using a fluorescent growth reporter for improved sensitivity.35
Materials and methods
Reagent preparation
MI broth was purchased from Hach (Loveland, CO, USA), PicoSurf 1 was purchased from Dolomite Microfluidics (Royston, UK), MI agar for preparing plated cultures was purchased from Fisher Scientific (Waltham, MA, USA), and MUG supplement, Lysogeny broth (LB), resazurin sodium salt were purchased from Sigma Aldrich (St Louis, MO, USA).E. coli cell culture
Escherichia coli (E. coli, strain BL-21, purchased from ATCC, Manassas, VA, USA) was cultured at 37 °C with shaking (250 rpm) overnight in LB. When ready for use, cultures were diluted to the desired working concentration in MI media with an additional 0.5 mM of MUG supplement. After dilution, 10 μL aliquots were plated on MI agar plates for overnight incubation and colony forming unit (CFU) counting. MI agar plates were prepared as suggested by the manufacturer and stored at 4 °C until use.Bulk bacterial growth measured in a plate reader
Bulk bacterial growth was monitored using the commercially-available fluorogenic substrate MUG. During growth, E. coli produces the enzyme β-glucuronidase which cleaves MUG resulting in a fluorescent compound.35 Samples with varying concentrations of E. coli were prepared in MI media supplemented with 0.5 mM MUG. The samples were aliquoted into flat-bottom 96- and 384-well plates. A range of sample volumes from 10–100 μL were prepared with equal CFUs inputted at each volume, to verify that time-to-detection was independent of culture volume. Well plates were incubated at 37 °C with orbital shaking in a Synergy H1 plate reader (BioTek, Winooski, VT, USA) and fluorescent signal was monitored every 30 minutes for 9 hours (excitation: 360 nm; emission: 450 nm; gain = 50). Aliquots were also plated on MI agar culture plates and incubated overnight at 37 °C to obtain CFU counts.Droplet preparation using a microfluidic chip
Monodisperse droplets were prepared using the commercially available droplet generating system from Dolomite Microfluidics (Royston, UK). Briefly, the set-up included the pressured-based droplet starter kit, two mitos p-pump basics, a 3-way vessel holder kit, and small quartz droplet chips with hydrophobic channel coating from Dolomite Microfluidics (Royston, UK). The p-pumps were controlled using Flow Control software from Dolomite Microfluidics (Royston, UK). The system was pressurized using an air compressor (part #2010A, California air Tools, San Diego, CA, USA) and droplet formation was monitored using an LCD Digital Microscope (model #44341, Celestron, Torrance, CA, USA).The dispersed phase was composed of aqueous droplets containing MUG-supplemental MI media and bacterial cells (between 0–103 CFU μL−1) while the continuous phase was composed of PicoSurf 1. Droplets were prepared by pressuring the system between 300–600 psi, most often with the dispersed phase 0–100 psi higher than the continuous phase. After droplets were prepared, the chip ports were sealed using microseal “B” plate sealing film (BioRad, Hercules, CA, USA). After an experiment was complete, chips were thoroughly washed (at least 10× chip volume) with DI water followed by acetone. Chips were then pumped dried with air and autoclaved to sterilize prior to reuse. Chips were not cleaned with bleach to preserve the hydrophobic coatings on the channels.
Simplified droplet preparation
Polydisperse droplets were prepared using our in-house method to compare with aforementioned monodisperse droplets and bulk culture. Equal volumes of MUG-supplemented MI media with cells (between 102–105 CFU per 100 μL) and PicoSurf 1 were added to a 1.7 mL Eppendorf tube. Droplets were formed using two methods: inverting the tubes 10× times or vortexing for 5 seconds at the maximum speed. The emulsions formed were ready for use without any additional preparation.To monitor the stability of droplets prepared with our simplified method, emulsions with the varying input E. coli CFUs described above were incubated at 37 °C for up to 144 hours. At regular intervals, bacterial growth from each sample was quantified and compared to a standard plate count (at 24 hours). Droplet sizes over time were also monitored to ensure they remained consistent – aging of unstable emulsions usually results in increased droplet size over time.36 Droplets and bacterial growth were monitored using a Nikon Ti2 microscope as described below.
Imaging droplets and bacterial growth using a microscope
Droplets prepared using our simplified method were imaged using a Nikon Eclipse Ti2 inverted microscope at 100× magnification – 10× objective with a 10× eyepiece – (Nikon Instruments Inc., Melville, NY, USA). An 18 μL (10 μL to fill chamber and extra to minimize air bubbles when sealed) aliquot of the emulsion was loaded to each chamber of a Countess cell counting chamber slide (Fisher Scientific, Waltham, MA, USA) and sealed with “B” plate sealing film (BioRad, Hercules, CA, USA) for imaging. Bright field and fluorescent images were automatically captured from twenty-five 0.339 mm2 regions of each chamber.Droplet size was measured using bright field images with automatic exposure and gain. Bacterial growth was monitored in the dark using a DAPI filter cube (Nikon Instruments Inc., Melville, NY, USA) with 1 second exposure and 1× gain. A SOLA light engine was used as the fluorescent light source (Nikon Instruments Inc., Melville, NY, USA). The same fluorescent substrate, MUG, was detected in droplets using the same excitation and emission wavelengths as bulk bacterial growth measured in a plate reader. During growth experiments, loaded Countess slides were heated at 37 °C in an incubation chamber.
Calculations of the expected positive number of droplets using measured volumes
In a particular experiment, the expected number of positive droplets was calculated using the binomial distribution. The probability a particular droplet is positive was calculated using the input concentration (via the Poisson distribution). To account for droplet volume polydispersity, we added the new step that utilizes the measured volume distribution (in comparison to traditional digital assays with monodisperse droplets).Image analysis
The brightness and contrast was held constant across all images taken with the microscope. Droplet size was measured using MATLAB's Image Processing Toolbox (MATLAB R2017a, The MathWorks, Inc., Boston, MA, USA), see the ESI, Fig. S1 and S2,† for example code and images. Positive droplets were manually counted after background subtraction from a zero CFU input sample.Results and discussion
Comparing methods for droplet formation
In this work, we aimed to demonstrate a simple, low-cost method for droplet generation and in-droplet bacterial culture that didn't rely on complex droplet generating equipment. To characterize our method, we used the principles of bulk-based emulsion formation coupled with off-the-shelf components. Our method can be used in a variety of settings by either hand-mixing or vortexing to prepare droplets. We compared our method to the commercially available Dolomite Droplet formation system. The Dolomite system was able to reproducibly prepare droplets on the order of 30 μm (15 pL), Fig. 1A, while our simplified method produced droplets ranging from 20–150 μm (1–1000 pL), Fig. 1B.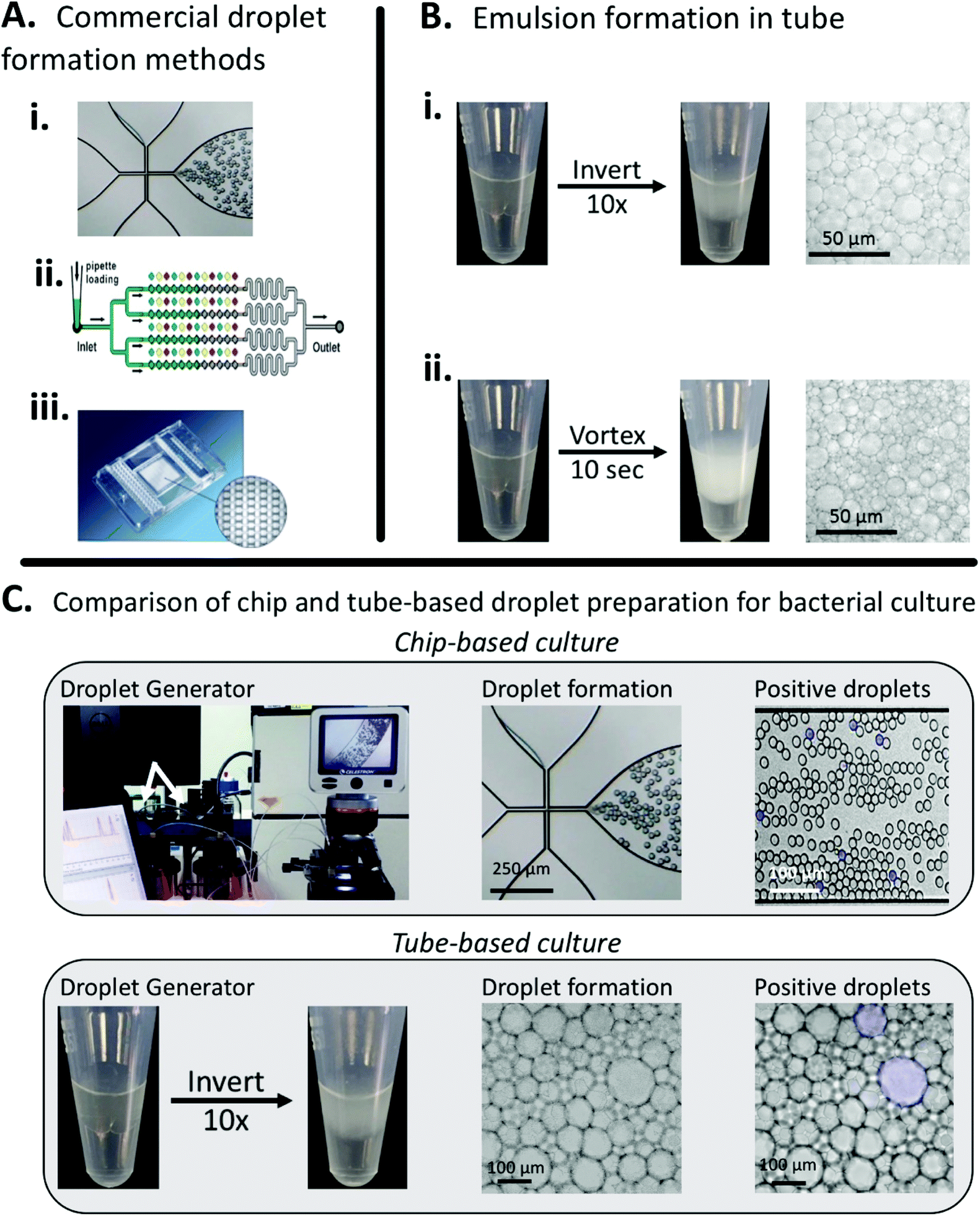 | ||
| Fig. 1 Comparing conventional droplet-forming methods to our easy, low-cost, minimally instrumented method. A. Commercial, instrumented methods for droplet formation from (i) Dolomite,37 (ii) SlipChip38 (reproduced from ref. 31 with permission from the Royal Society of Chemistry), and (iii) Fluidigim39 (from ref. 32 reprinted with permission from AAAS). B. Simplified droplet formation by hand-mixing and vortexing. C. Both the commercial Dolomite system and our method can be used for in-droplet cell culture and quantification. Images show droplets containing actively growing E. coli. By counting the number of fluorescent droplets, inputted bacterial concentration can be quantified. | ||
Both methods create stable droplets suitable for multi-hour experiments, Fig. S3,† but our method has multiple advantages over the chip-based system, principally a larger total addressable volume, but also including rapid droplet formation (seconds vs. minutes) and ease of use (no equipment vs. specialized pumps and chips) Additionally, we've demonstrated that our method for droplet preparation is compatible with in-droplet bacterial culture, Fig. 1C.
Monitoring bacterial growth in bulk and in droplets
First, we focused on comparing time-to-detection for bulk culture and droplets (both mono- and polydisperse). Previous work has shown a decreased time-to-detection for in-droplet culture due to reduced effects of nutrient diffusion and increased signal-to-noise ratio.8,27,28 We demonstrated that emulsions prepared with our method show reduced times-to-detection compared to bulk, well-plate culture and enable direct quantification of bacterial count, Fig. 2.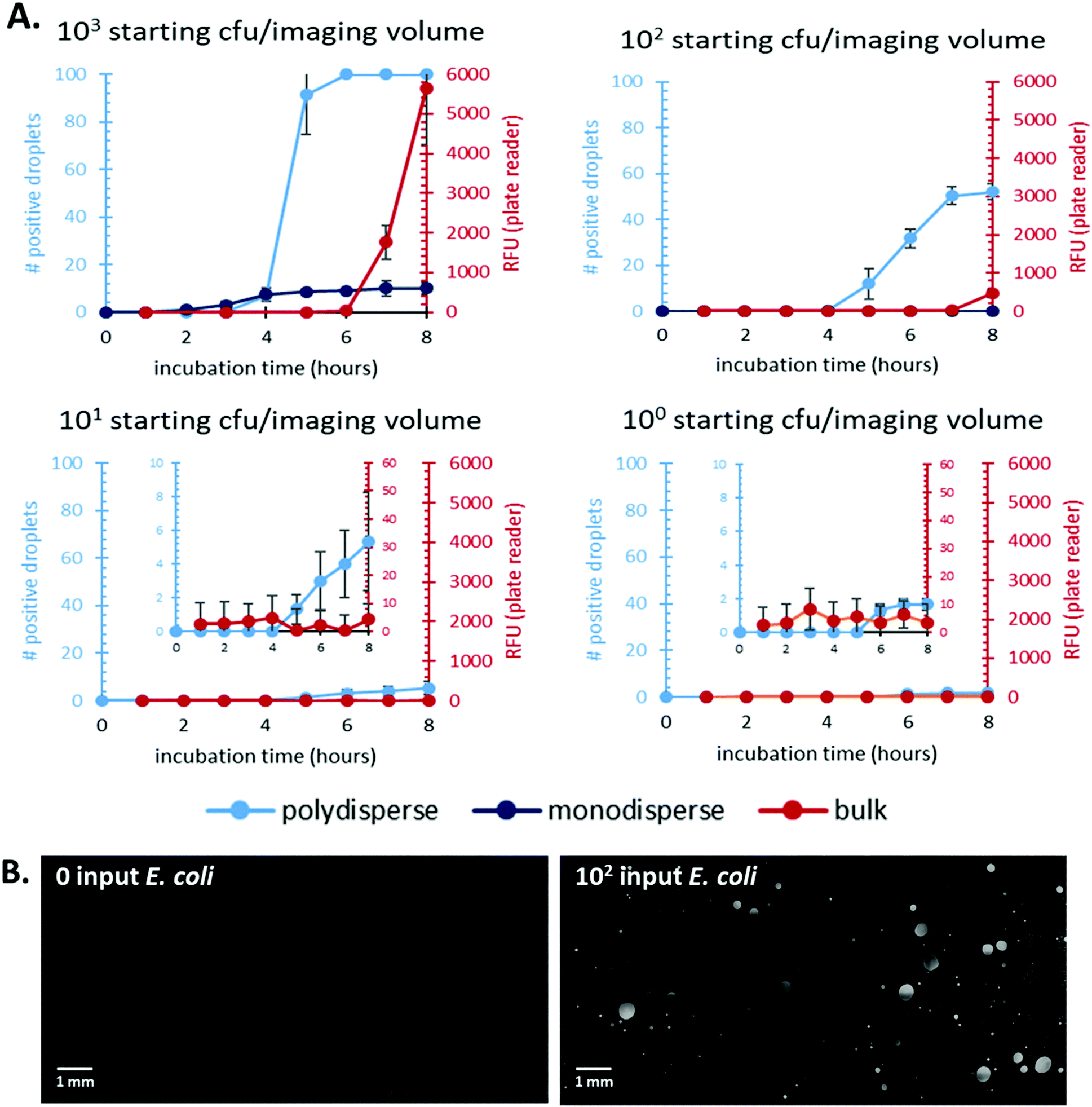 | ||
| Fig. 2 Comparing E. coli cells cultured in droplets made using a droplet-forming chip (monodisperse) or our simplified method (polydisperse) to cells cultured in bulk (in well-plates). A. Time-to-detection and quantification for inputs ranging from 100–103 starting cfu E. coli per imaged volume. Averages of N = 3 (for polydisperse and bulk) and N = 2 (for monodisperse) are reported with error bars corresponding to ±one standard error. The droplet-based samples (polydisperse and monodisperse) were quantified by counting the number of fluorescent droplets. B. Images post-culture of 0 and 102 input per 10 μL for polydisperse droplet samples. Images were collected and stitched together using the Nikon Ti2 software and analysed via ImageJ. | ||
Additionally, our polydisperse droplet distribution showed the same time-to-detection as the monodisperse droplet distribution for higher input CFU samples. We were also able to show a higher dynamic range for the polydisperse system, likely due to the higher volume of sample this system processed. The statistical correction overcomes any bias as a result of polydisperse droplet populations, and the simplicity of a shaken emulsion preparation compared to monodisperse droplet creation removes the need for expensive microfluidic equipment and trained personnel for droplet formation.
The lowest bacterial count the polydisperse system quantified was 101 CFU. This limit is driven by the amount of sample that can be processed and not the polydispersity of the system. Future work will explore methods to increase the volume processed by the system and should further reduced the detection limit.
We compared the total number of bacteria in each system's imaging volume in order to quantitatively compare experimental results to known inputs. We verified that varying volumes of bulk culture did not affect time-to-detection using the plate reader, Fig. S4.† Each bulk culture volume contained the same total number of starting bacteria. We chose to use data from the 10 μL bulk sample (well-plate dimensions constrained us from testing lower volumes) to compare to droplet-based cultures as it provided the closest to volumetric parity. The total imaged volume of the polydisperse emulsion was ∼10 μL. We determined this value by measuring droplet size and adjusting for the droplets’ aqueous fraction of the emulsion imaged. Fig. 2B shows representative images of bacterial culture in polydisperse droplets for a 0 and 102 input samples.
The time-to-detection and signal over background was shown for a range of input E. coli amounts from 100–103 bacteria. Detection in bulk culture lagged significantly behind in-droplet culture for all inputs tested. Additionally, the polydisperse droplet cultures showed a wider dynamic range and at least an order of magnitude lower limit of detection compared to bulk cultures. These results are summarized in Table 1.
| Input CFU | Bulk – well plate | Simplified droplet method |
|---|---|---|
| ND = not detected over the course of an 8-hour experiment. | ||
| 103 | 420 | 240 |
| 102 | 480 | 300 |
| 101 | ND | 360 |
| 100 | ND | ND |
In addition to time-to-detection, we studied the stability of droplets made with our simplified method. Initially, we tracked droplet size during the course of an eight-hour experiment at 37 °C. The tracked droplets ranged in size from ∼30–400 pL and remained stable during the 8 hours, Fig. S3.†
We also tracked the number of droplets with positive bacterial growth over time. Droplets were prepared with varying input amounts of E. coli ranging from 102–105 bacteria per 100 μL droplets. All samples were then incubated at 37 °C for up to 144 hours. At regular intervals, aliquots of each emulsion were imaged and bacterial concentration was quantified based on the number of positive droplets. Stable emulsions should plateau to a constant number of positive droplets, indicating that each droplet remains an isolated system. Fig. S5† shows that the emulsions do remain stable over the course of the 144-hour experiment for each input tested. Additionally, these results indicate that each droplet remains an isolated environment with no fluorescent substrate diffusing between droplets because the number of positive droplets remains stable over the 144 hours.
Another advantage of bacterial detection using the MUG substrate is its previous demonstration for E. coli detection in real world samples. Work has been published that uses MUG to identify bacteria in water40 and soil.41 This compatibility indicates that future work in our system should accommodate testing of complex samples.
Droplet size distribution and quantification
A larger droplet is more likely to contain one or more target bacteria, and thus, has a larger probability of turning “on”, i.e. becoming a positive droplet. A wide range of volumes means a wide range of probabilities that a droplet turns on, making the statistical understanding of this approach non-trivial.Therefore, we developed a statistical procedure to calculate the expected number of positive droplets. The bulk concentration is called λ. In an experiment with n droplets, the number of positive droplets (those with 1 or more bacteria) is called a. The probability a droplet with known volume v turns positive is P0(λ, v) = 1 − e−λv. The probability a randomly chosen droplet (among droplets of polydisperse sizes) turns positive is,
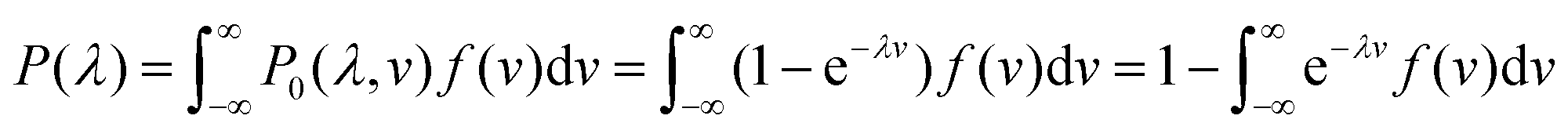
 . The experiment is now similar to flipping a biased coin, so the number of positive droplets follows a binomial distribution with mean of n × Pon and standard deviation of
. The experiment is now similar to flipping a biased coin, so the number of positive droplets follows a binomial distribution with mean of n × Pon and standard deviation of 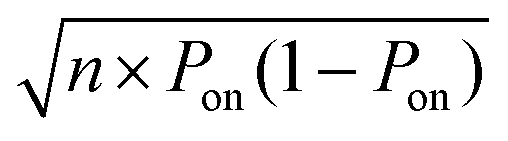 .
.
Consequently, the assay result (number of positive droplets) depends on the volume distribution. Our simplified method results in polydisperse droplets that can range from 10s to 100s μm in diameter (1–1000 pL), while droplets made in chip are monodisperse, Fig. 3A. With such a wide range of droplet volumes, it is important to verify that their distribution is consistent over multiple experiments using the same method of preparation including a consistent agitation frequency and duration. We measured droplet volumes across experiments with varying bulk concentrations of E. coli (1, 10, 100, 1000 bacteria per μL, each in 3 replicates) and found the resulting distributions to be similar, even though they span orders of magnitude, Fig. 3B. This consistency of droplet size distribution across experiments and the statistical framework described above allows for accurate quantification of bacteria even with polydisperse droplet sizes.
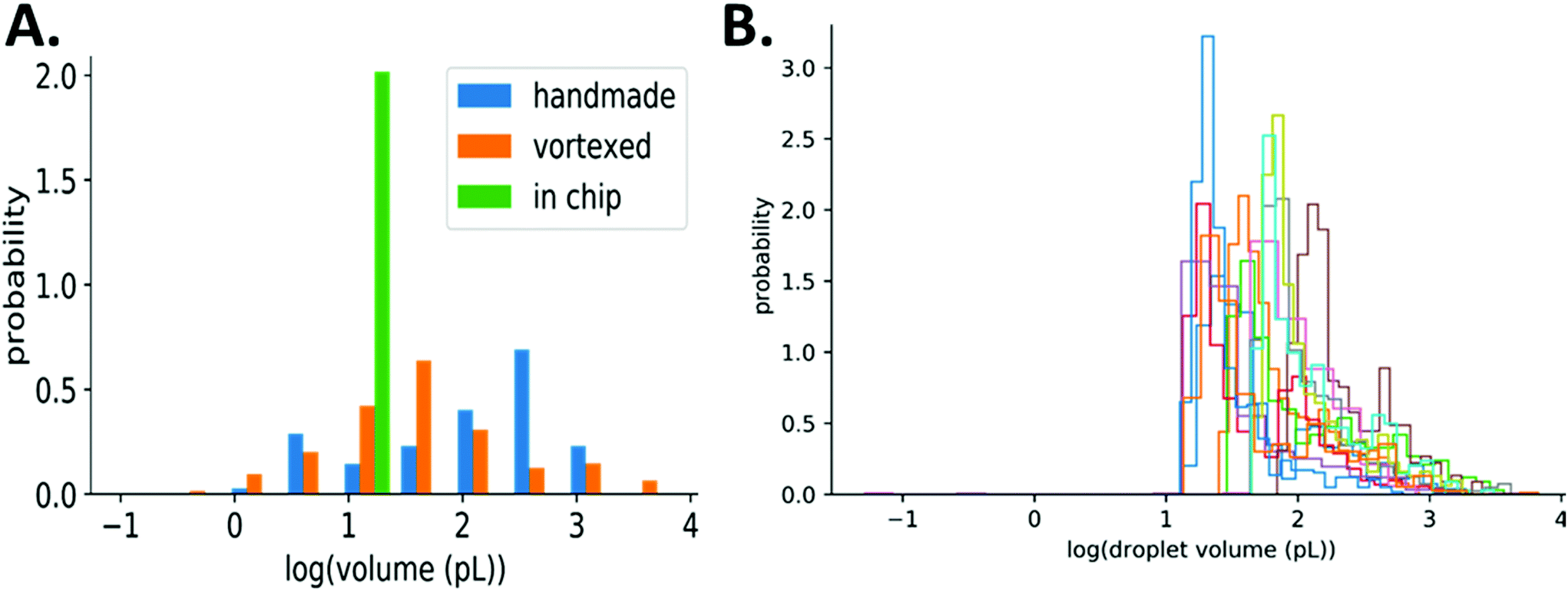 | ||
| Fig. 3 Droplet size distribution and quantification. A. Comparing droplet sizes made by manual inversion (handmade), vortexing, or using the Dolomite chip. B. Histograms of measured droplet volumes from twelve separate experiments using the same method of preparation including a consistent agitation frequency and duration. Overall, the range of droplet volumes is consistent across experiments. | ||
Our experimentally derived data shows an expected linear relationship between the input cell count and the number or positive droplets, Fig. 4A. Using the measured droplet volumes and statistics described above, we can compare our experimental data to the statistically predicted number of positive droplets for each sample, Fig. 4B. For low input cell concentrations, the experimental counts fall within the 95% confidence intervals predicted by the statistics. At higher bacterial input, the number of positive droplets began to saturate the ability to accurately count, resulting in a more varied distribution (Fig. S6†) and a dynamic range of 0–103 CFU per imaged area. Because this variation falls at the higher input concentrations, it is less vital for applications where this technology would be impactful, such as counting very rare events like bacteria in drinking water. However, to overcome this limitation, future iterations of this work will include processing larger sample volumes and incorporating an automated counting mechanism to help more accurately count high numbers of positive droplets and therefore expand the dynamic range.
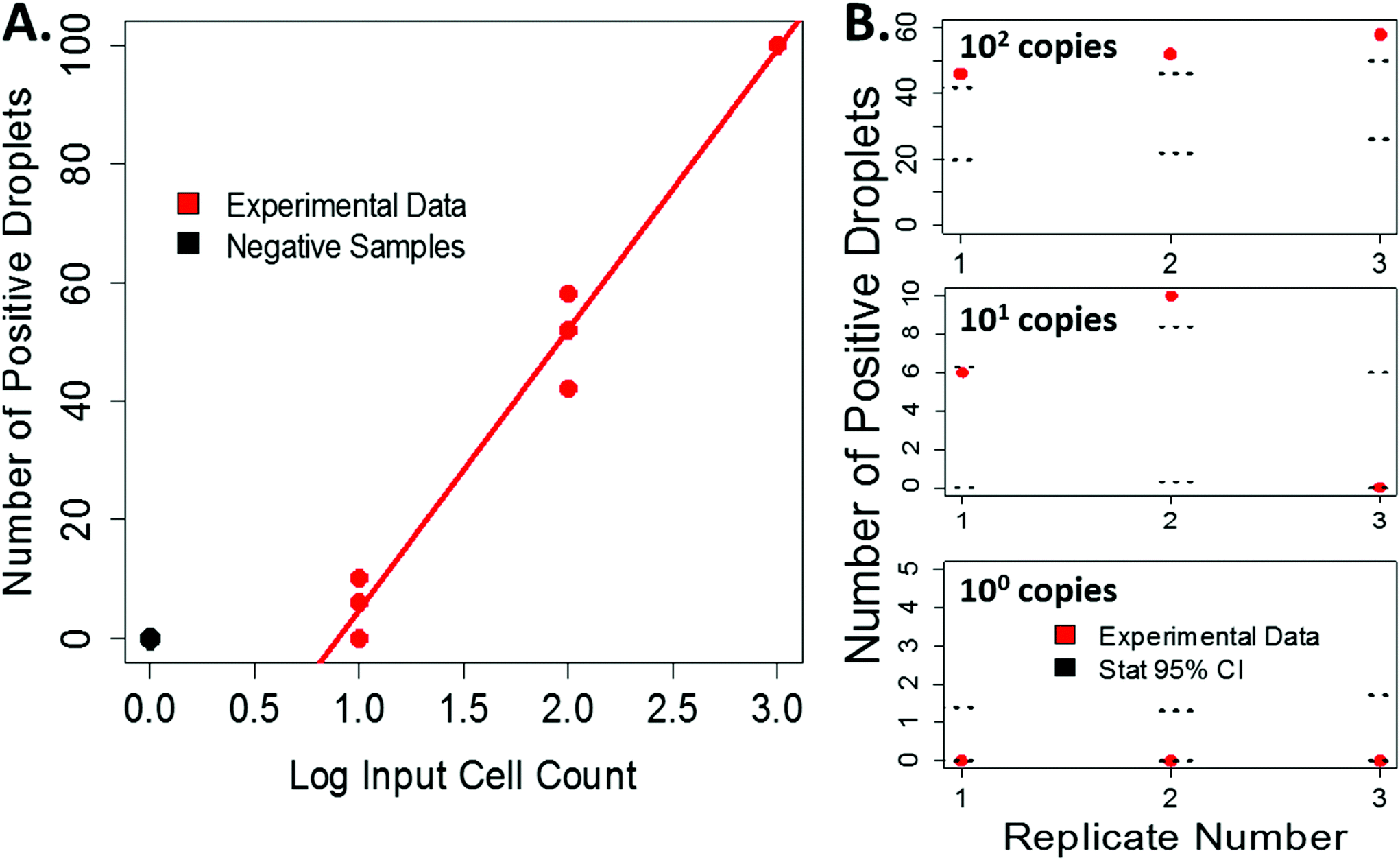 | ||
| Fig. 4 Comparing experimental data to statically derived droplet counts. A. Experimentally counted positive droplets follows an expected linear relationship (note the y-axis is linear while the x-axis is log-scale); as input cell count increases, so does the number of positive droplets, N = 3 per input cell count (linear regression model: y = 47.33x − 42.67) B. For low input cell concentrations, the statistically derived 95% CI for the predicted number of positive droplets aligns well with most of the experimental data. For higher input cell counts, number of positive droplets began to saturate the ability to accurately count, resulting in a more varied distribution. The black dashed lines denote the 95% confidence interval (95% CI) from the statistical predictions for each individual replicate based on the distribution of droplet sizes observed in each sample. | ||
Conclusions
Here we have demonstrated a simple-to-use, low cost method for preparing small-volume droplets for performing digital culture. First, the system was shown to accurately quantify input bacteria counts. Our simplified method requires significantly less equipment and time to execute compared to droplet-forming microchips. Additionally, our polydisperse droplet preparation method showed a similar time-to-detection to monodisperse droplets and an improved time-to-detection over more traditional bulk-based culture methods. The polydisperse droplets prepared with this system also remained stable for up to 144 hours, indicating that these methods could be compatible with pathogens that require longer incubation times, such as mycobacterium.We have also characterized the range of droplet sizes created in our system and presented statistical methods to quantify the number of bacteria in samples with polydisperse droplet sizes. Importantly, this statistical approach also enables the quantification of bacteria in a sample without having to measure the actual distribution of droplet sizes, even in very polydisperse samples. The combination of rapid droplet preparation and this statistical framework leads the way for implementing this method in a variety of locations including limited resource settings. The samples in this manuscript were characterized using a traditional laboratory microscope; but for future applications, a reader, could be used in conjunction with the statistical method to quantify rare events from large volumes such as water samples. Future work will aim to demonstrate this improved and affordable reader technology with realistic samples.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding was provided by Intellectual Ventures’ Global Good Fund. The authors also gratefully acknowledge Bill Gates and Nathan Myhrvold for their support and supervision.We thank David Bell from Global Good, Tim Chang from Intellectual Ventures Laboratory for help with imaging, and Shivani Gupta from Global Good's Virtual Research Analyst Network for background information gathering which helped guide experimental design. We thank our colleague Akos Somoskovi who provided valuable discussion and feedback on experimental design and analysis.
Notes and references
- UNICEF, UNICEF Target Product Profile Rapid E. Coli Detection, 2017, https://www.unicef.org/supply/files/UNICEF_TPP_Rapid_E._coli_Detection_v2.2_Final_2017.08.04.pdf Search PubMed.
- M. D. Sobsey, 9 Methods to Identify and Detect Microbial Contaminants in Drinking Water, in Identifying Future Drinking Water Contaminants, The National Academies Press, 1999, pp. 173–260 Search PubMed.
- R. T. Noble and S. B. Weisberg, A review of technologies for rapid detection of bacteria in recreational waters, J. Water Health, 2005, 3(4), 381–392, DOI:10.2166/wh.2005.051.
- V. Tanchou, Review of Methods for the Rapid Identification of Pathogens in Water Samples, 2014, DOI:10.2788/18775.
- S. Patiño, L. Alamo and M. Cimino, et al., Autofluorescence of Mycobacteria as a Tool for Detection of Mycobacterium tuberculosis, J. Clin. Microbiol., 2008, 46(10), 3296–3302, DOI:10.1128/JCM.02183-07.
- S. Sakakihara, S. Araki and H. Noji, A single-molecule enzymatic assay in a directly accessible femtoliter droplet array, Lab Chip, 2010, 10, 3355–3362, 10.1039/c0lc00062k.
- R. Ghodbane, D. Raoult and M. Drancourt, Dramatic reduction of culture time of Mycobacterium tuberculosis, Sci Rep, 2014, 4, 4236, DOI:10.1038/srep04236.
- F. Lyu, M. Xu, Y. Cheng, M. Kim, J. Rao and S. K. Y. Tang, Rapid Detection of Blac Using Droplet-Based Microfluidics for Use in the Diagnosis and Drug Susceptibility Testing of Tuberculosis, Biomicrofluidics, 2015, 44120, 903–905 Search PubMed.
- C. Holtze, A. C. Rowat and J. J. Agresti, et al., Biocompatible surfactants for water-in-fluorocarbon emulsions, Lab Chip, 2008, 8(10), 1632–1639, 10.1039/B806706F.
- J. Bibette, L. Leal Calderon and P. Poulin, Emulsions: basic principles, Rep. Prog. Phys., 1999, 62(6), 969 CrossRef.
- F. Macritchie, Barriers to coalescence in stablizied emulsions, Nature, 1967, 215, 1159–1160 CrossRef.
- A. Gupta, H. B. Eral, T. A. Hatton and P. S. Doyle, Nanoemulsions: formation, properties and applications, Soft Matter, 2016, 12(11), 2826–2841, 10.1039/C5SM02958A.
- F. Y. Ushikubo and R. L. Cunha, Stability mechanisms of liquid water-in-oil emulsions, Food Hydrocolloids, 2014, 34, 145–153 CrossRef.
- G. I. Taylor, The formation of emulsions in definable fields of flow, Proc. R. Soc. London, Ser. A, 1934, 146(858), 501–523 CrossRef.
- J. Weiss and G. Muschiolik, Factors affecting the droplet size of water-in-oil emulsions (W/O) and the oil globule size in Water-in-oil-in-water emulsions (W/O/W), J. Dispersion Sci. Technol., 2007, 28(5), 703–716, DOI:10.1080/01932690701341819.
- J. A. Boxall, C. A. Koh, E. D. Sloan, A. K. Sum and D. T. Wu, Droplet size scaling of water-in-oil emulsions under turbulent flow, Langmuir, 2012, 28(1), 104–110, DOI:10.1021/la202293t.
- J. A. Boxall, C. A. Koh, E. D. Sloan, A. K. Sum and D. T. Wu, Measurement and calibration of droplet size distributions in water-in-Oil emulsions by particle video microscope and a focused beam reflectance method, Ind. Eng. Chem. Res., 2010, 49(3), 1412–1418, DOI:10.1021/ie901228e.
- A. R. Abate and D. A. Weitz, Syringe-vacuum microfluidics: A portable technique to create monodisperse emulsions, Biomicrofluidics, 2011, 5(1), 014107 CrossRef PubMed.
- T. S. Kaminski, O. Scheler and P. Garstecki, Droplet microfluidics for microbiology: techniques, applications and challenges, Lab Chip, 2016, 16, 2168–2187, 10.1039/C6LC00367B.
- J.-C. Baret, Surfactants in droplet-based microfluidics, Lab Chip, 2012, 12(3), 422–433, 10.1039/C1LC20582J.
- M. Pan, L. Rosenfeld and M. Kim, et al., Fluorinated pickering emulsions impede interfacial transport and form rigid interface for the growth of anchorage-dependent cells, ACS Appl. Mater. Interfaces, 2014, 6(23), 21446–21453, DOI:10.1021/am506443e.
- P. Gruner, B. Riechers and L. Andreina, et al., Stabilisers for water-influorinated-oil dispersions: Key properties for micro fluidic applications, Curr. Opin. Colloid Interface Sci., 2015, 20, 183–191, DOI:10.1016/j.cocis.2015.07.005.
- P. B. Umbanhowar, V. Prasad and D. A. Weitz, Monodisperse Emulsion Generation via Drop Break Off in a Coflowing Stream, Langmuir, 2000, 16(2), 347–351 CrossRef.
- C. Li, M. Boban and A. Tuteja, Open-channel, Water-in-oil Emulsification in Paper-Based Microfluidic Devices, Lab Chip, 2017, 17(8), 1436–1441 RSC.
- S. Arai, M. Okochi, T. Hanai and H. Honda, Micro-compartmentalized cultivation of cyanobacteria for mutant screening using glass slides with highly water-repellent mark, Biotechnol. Rep., 2014, 4(1), 151–155, DOI:10.1016/j.btre.2014.10.003.
- S. Mashaghi and A. M. van Oijen, External control of reactions in microdroplets, Sci. Rep., 2015, 5, 11837, DOI:10.1038/srep11837.
- D. Kang, M. M. Ali and K. Zhang, et al., Rapid detection of single bacteria in unprocessed Digital Detection, Nat. Commun., 2014, 5, 1–10, DOI:10.1038/ncomms6427.
- E Tumarkin, L Tzadu and E. Csaszar, et al., High-throughput combinatorial cell co-culture using microfluidics, Integr. Biol., 2011, 3(6), 653–662, 10.1039/c1ib00002k.
- S. Begolo, F. Shen and R. F. Ismagilov, A microfluidic device for dry sample preservation in remote settings, Lab Chip, 2013, 13(22), 4331–4342, 10.1039/c3lc50747e.
- B. Sun, F. Shen, S. E. McCalla, J. E. Kreutz, M. A. Karymov and R. F. Ismagilov, Mechanistic evaluation of the pros and cons of digital RT-LAMP for HIV-1 viral load quantification on a microfluidic device and improved efficiency via a two-step digital protocol, Anal. Chem., 2013, 85(3), 1540–1546, DOI:10.1021/ac3037206.
- SlipChip, https://careers.smartrecruiters.com/SlipChip.
- Fluidigm, https://www.fluidigm.com/.
- Z. Chen, P. Liao, F. Zhang, M. Jiang, Y. Zhu and Y. Huang, Centrifugal micro-channel array droplet generation for highly parallel digital PCR, Lab Chip, 2017, 17, 235–240, 10.1039/C6LC01305H.
- J. E. Kreutz, T. Munson, T. Huynh, F. Shen, W. Du and R. F. Ismagilov, Multiplexed quantification of nucleic acids with large dynamic range using multivolume digital RT-PCR on a rotational SlipChip tested with HIV and hepatitis C viral load, Anal. Chem., 2011, 83, 8158–8168, DOI:10.1021/ja2060116.
- A. H. Farnleitner, L. Hocke and C. Beiwl, et al., Rapid enzymatic detection of Escherichia coli contamination in polluted river water, Lett. Appl. Microbiol., 2001, 33(3), 246–250, DOI:10.1046/j.1472-765X.2001.00990.x.
- F. MacRitchie, Barrier to coalescence in stabilized emulsions, Nature, 1967, 1159–1160 CrossRef.
- Dolomite Microfluidics, http://www.dolomite-microfluidics.com/.
- W. Du, L. Li, K. P. Nichols and R. F. Ismagilov, SlipChip, Lab Chip, 2009, 2286–2292, 10.1039/B908978K.
- Science, 2009, vol. 324, issue 5924, p. 280 DOI:10.1126/science.324.5924.280a .
- T. Garcia-Armisen, P. Lebaron and P. Servais, B-D-glucuronidase activity assay to assess viable Escherichia coli abundance in freshwaters, Lett. Appl. Microbiol., 2005, 40, 278–282 CrossRef PubMed.
- A. Unc, J. Gardner and S. Springthorpe, Recovery of Escherichia coli from Soil after Addition of Sterile Organic Wastes, Appl. Environ. Microbiol., 2006, 72(3), 2287–2289 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8an00029h |
| This journal is © The Royal Society of Chemistry 2018 |