
Carbon dots as analytical tools for sensing of thioredoxin reductase and screening of cancer cells†
Jagpreet Singh
Sidhu
a,
Ashutosh
Singh
b,
Neha
Garg
b,
Navneet
Kaur
*c and
Narinder
Singh
 *a
*a
aDepartment of Chemistry, Indian Institute of Technology Ropar, Rupnagar, Punjab 140001, India. E-mail: nsingh@iitrpr.ac.in
bSchool of Basic Sciences, Indian Institute of Technology Mandi, Kamand, Mandi, Himachal Pradesh-175005, India
cDepartment of Chemistry, Panjab University, Chandigarh 160014, India. E-mail: navneetkaur@pu.ac.in
First published on 12th February 2018
Abstract
Thioredoxin Reductase (TrxR) is a redox regulating enzyme which is predestined for the maintenance of redox homeostasis of mammalian cells. However, the elevated level of TrxR is associated with the progress of various types of tumors and therefore, this is a significant target for the detection of cancer cells. Herein, an easily engineered ‘Turn ON’ fluorescent sensor probe has been synthesized for the detection of TrxR and cell imaging using carbon dots. The emission intensity of fCDs on complexation with Cu2+ ions was drastically quenched. Subsequently, the addition of TrxR to the solution of the fCDs-Cu2+ complex leads to the cleavage of the disulfide bond of the fCDs, which acclaim the release of 3-mercaptopropionic acid. 3-Mercaptopropionic acid, being a strong bi-dentate chelating agent for Cu2+ ions, extracted Cu2+ from the coordination sphere of fCDs and restored the original fluorescence intensity of fCDs. Thus, the probe is operating with a simple process of “ON–OFF” emission switching due to Cu2+ and “OFF–ON” switching with TrxR. The probe has been successfully used for real-time application to monitor TrxR activities in the complex biological system. The fluorescence images of MCF-7 and HeLa cells after incubation with the fCDs-Cu2+ complex were recorded under a confocal laser scanning microscope (CLSM) as a function of time. Enhancement in the emission intensity of cancer cells after 2 h of treatment demonstrates the potential application of the sensor probe for the bioimaging of endogenous TrxR in living cells and screening of cancer cells. Such fluorescent probes will open the door for the development of promising clinical devices for the diagnosis of cancer cells.
Introduction
Thioredoxin Reductase (TrxR) is a selenocysteine enzyme, which along with co-factor NADPH, regulates the redox homeostasis of cells.1,2 The overexpression of TrxR under hypoxic conditions or under oxidative stress is associated with cancer progression which is the second largest fatal disease after cardiovascular disorders.3–6 Therefore, qualitative and quantitative determination of TrxR has gained considerable attention for the diagnosis of cancer cells.7,8 Conventional analytical tools are available for the detection of TrxR, however, fluorescence sensors are the most widely accepted tool for the screening of biological analytes.9,10 The extensive review of literature revealed that some of the reported fluorescent sensor probes are suffering from the interference of other biological molecules which leads to false positive results.11 There are another set of probes which are otherwise selective for sensor response to TrxR, nevertheless, they exhibit limitations as promising clinical devices for the diagnosis of cancer cells due to their tedious multi-step organic synthesis, photobleaching, cellular toxicities, and low quantum yields.12,13 Thus, facile, inexpensive, efficient, non-toxic, selective and specific probes for biological systems are substantially required.14–16 These requirements can be addressed using nanomaterial-based fluorescent sensors owing to their superior optical properties and facile synthesis route as compared to organic receptors.17,18 Carbon dots (CDs) has gained significant attentions for the detection of biological analytes and cellular imaging.19 These CDs are zero-dimensional structured nano-materials (particle size <10 nm) and possess low toxicity to living tissues.20,21 In addition to their high biocompatibility, CDs also offer high photochemical stability, impressive water solubility, and outstanding fluorescence behavior, which make them excellent fluorescent tools for cellular imaging.22–26 There is still another important issue of background signals associated with cellular imaging which usually arises from the cellular components when probes are excited at lower wavelengths. However, due to the excitation-dependent emission behavior of CDs, the background signal arising at a lower wavelength in biological samples can be eliminated by changing the excitation source to get emission at a longer wavelength. For example, if the background signal interferes with cellular imaging in the blue and green channels, the red channel can be employed to get better contrast of cellular images at a higher wavelength.27–30 Therefore, we considered all the above-mentioned parameters and employed the CDs as the basic fluorophore unit for TrxR detection and cellular imaging. For the detection of TrxR in mammalian cells, the basic core structure of the sensor probe is fabricated with a disulfide bond which acts as a substrate for TrxR.31 The theme of sensor operation lies with the fact that the reduction of the disulfide bond of the sensor probe in the presence of TrxR leads to a change in the fluorescence signal of the sensor probe. Where the design of the sensor is concerned, the CDs were prepared using the hydrothermal method and then fabricated with disulfide linker 3–3′-dithiodipropanoic acid (DTPA) to yield functionalized carbon dots (fCDs). A comparison of recognition properties revealed that the amino coated CDs did not produce any significant changes in the emission profile in response to Cu2+. However, on DTPA, the functionalization interaction of CDs was significantly increased towards Cu2+ ions and the emission of fCDs was quenched drastically. Herein, the DTPA has exhibited a dual function: (a) improve the binding affinity of the sensor towards Cu2+ and (b) act as a substrate for TrxR. Therefore, upon addition of Cu2+, the blue emission of fCDs was quenched due to the chelation properties of DTPA towards Cu2+ while in the presence of TrxR, the disulfide of DTPA was reduced to 3-mercaptopropionic acid which carries the Cu2+ ion away from the CD surface. Enhancement in blue emission in response to TrxR was easily observed with the naked eye under a UV lamp of wavelength 365 nm. These results prompted us to explore the practicability of their use in biological systems. Therefore, the fCDs-Cu2+ sensor probe was further evaluated for the endogenous imaging of TrxR in cancer cells. Fluorescence images of cancer cells upon treatment with the sensor probe were recorded using a CLSM. Furthermore, a cytotoxicity assay was also performed against MCF-7 and HeLa cells.Results and discussion
Synthesis and characterization of CDs and fCDs
The synthesis scheme of the sensor probe is shown in Scheme S1,† which was initiated with the synthesis of CDs bearing –NH2 as a surface functional group.32 The mixture of 4,7,10-trioxa-1,13-tridecane diamine (TTDDA) and citric acid in water was heated at 180 °C for 6 h in an autoclave and after cooling the reaction mixture to room temperature, the CDs were purified through a dialysis membrane (MW 3500 DA) against water. The purified CDs were characterized with TEM, Powder XRD, FTIR, DLS, fluorescence and UV-vis absorption spectroscopy. TEM images revealed that CDs were monodispersed in a solution phase and the average particle size was 4 nm with spherical morphology (Fig. 1A). The crystallinity of CDs was evaluated by recording the selected-area electron diffraction (SAED) image, which disclosed the good crystalline nature of CDs (Fig. 1B). The disorder pattern of carbon atoms of CDs was analyzed from a broad diffraction peak at 22°(2θ) of powder X-ray diffraction (XRD) spectra which is supported by the literature reports (Fig. 1C).33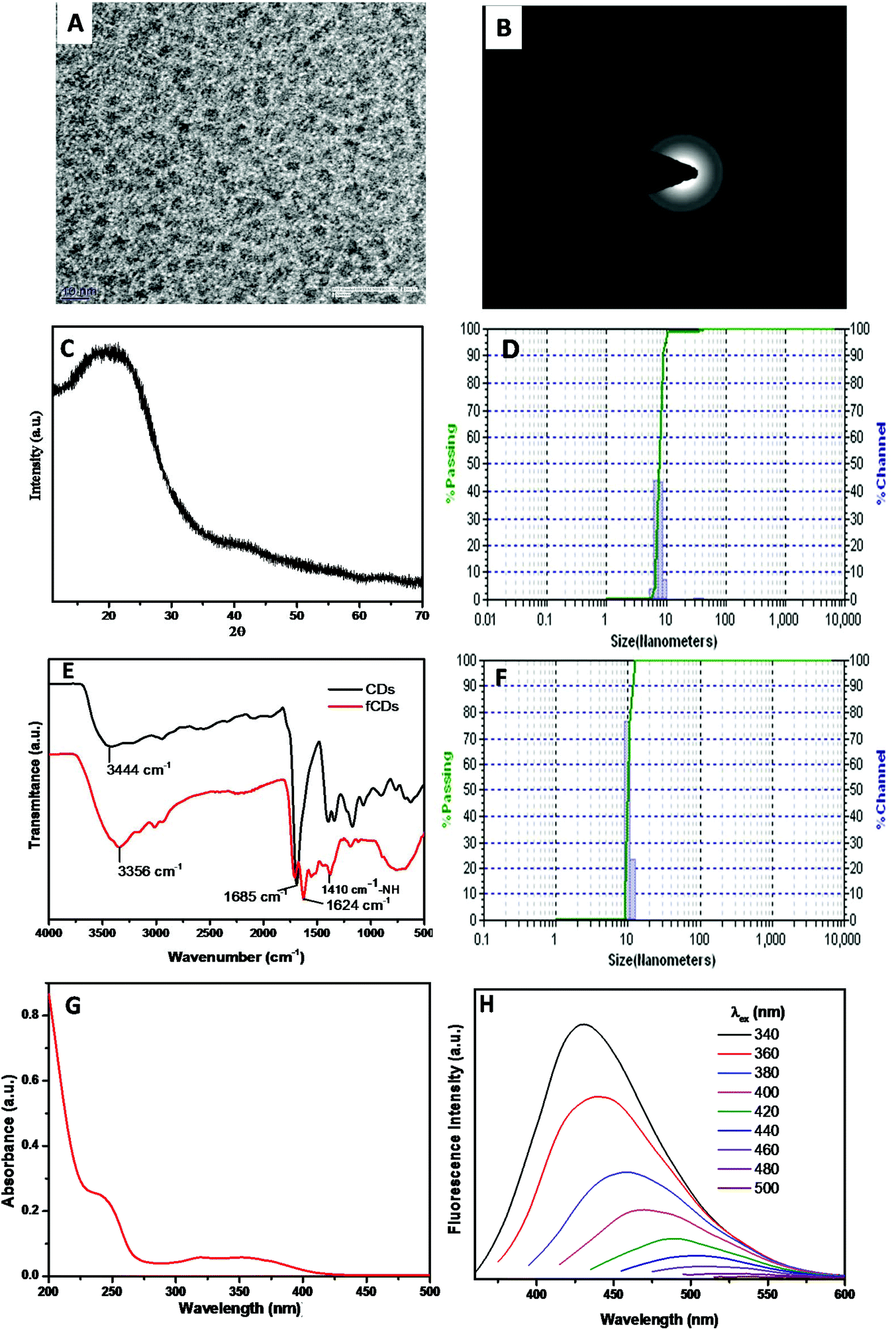 | ||
| Fig. 1 Characterization of CDs: (A) TEM image of CDs exhibiting uniform distribution of the particle with an average particle size of 4 nm. (B) Selected-area electron diffraction image of CDs signifies the crystalline nature of CDs. (C) Powder XRD pattern of CDs shows the disorder pattern of carbon atoms (D) DLS histogram of CDs showing a particle diameter of 7 nm. (E) FTIR spectra of CDs (black line) and fCDs (red line). (F) DLS histogram of fCDs showing a particle diameter of 9 nm. (G) UV-vis absorption spectra of CDs exhibiting two absorption peak at 350 nm and 247 nm corresponding to n–π and π–π* transitions, respectively. (G) Emission spectra of CDs at different excitation wavelengths varied from 340 nm to 500 nm. | ||
The diameter of particles (7 nm) in the solution phase was analyzed from the histogram of a dynamic light scattering (DLS) instrument which is somewhat greater than that estimated with TEM analysis (Fig. 1D). The higher particle size of CDs signifies the interactions of the solvated molecules with the particle surface which contributes to increasing the particle diameter in the solution phase.26 The surface elemental composition of CDs was analyzed by recording the XPS measurement (Fig. S1†). Full XPS spectra exhibited three different peaks at 284.1 eV (C 1s), 399 eV (N 1s) and 531.4 eV (O 1s) which represent the presence of carbon, nitrogen, and oxygen atoms in the CDs composition. Information of surface functional groups on CDs was obtained from FTIR spectra (Fig. 1E black line). The broad absorption band at 3300–3100 cm−1 assigned to –OH and –NH2 stretching vibrations.34 A small band at 2974 cm−1 signifies the –CH2 vibrations, while the strong vibrational band at 1685 cm−1 has been assigned to –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretches of the carbonyl group.35 The –C–O stretching band has been assigned to 1250 cm−1, whereas C–N stretching vibration corresponds to the absorption band at 1474 cm−1. Furthermore, UV-vis absorption and fluorescence studies were performed to investigate the photophysical properties of CDs. The aqueous solution of CDs possesses two absorption bands at 350 nm and 247 nm which correspond to π–π* and n–π* transitions, respectively (Fig. 1G). In daylight, an aqueous solution of CDs was a light brownish clear solution and displayed strong blue fluorescence emission under UV light (irradiation at 365 nm). Photoluminescence spectra of CDs reveal that CDs showed excitation-dependent emission behavior and displayed maximum emission intensity when excited at 340 nm. Emission intensity decreased gradually as the excitation wavelength was increased from 340 nm to 500 nm (Fig. 1H). A characteristic feature, “excitation-dependent emission” behavior, of CDs is in accordance with literature reports.36 A change in the emission profile with varying the excitation wavelength arises from different emissive states, conjugate systems and zigzag sites on CDs. Different theories such as quantum confinement, surface trap model, giant red-edge effect, edge states and heteroatom atom models have been proposed to prove this mechanism of CD emission, however, the exact mechanism is still obscure.37–39 The quantum yield of CDs was about 21% as determined by the absolute method. Furthermore, the quantity of the amino group present on the CD surface (1.30 μmol mg−1) has been determined by measuring the absorbance of Ruhemann's purple at 570 nm using alanine as a reference standard (Fig. S2†).40
O stretches of the carbonyl group.35 The –C–O stretching band has been assigned to 1250 cm−1, whereas C–N stretching vibration corresponds to the absorption band at 1474 cm−1. Furthermore, UV-vis absorption and fluorescence studies were performed to investigate the photophysical properties of CDs. The aqueous solution of CDs possesses two absorption bands at 350 nm and 247 nm which correspond to π–π* and n–π* transitions, respectively (Fig. 1G). In daylight, an aqueous solution of CDs was a light brownish clear solution and displayed strong blue fluorescence emission under UV light (irradiation at 365 nm). Photoluminescence spectra of CDs reveal that CDs showed excitation-dependent emission behavior and displayed maximum emission intensity when excited at 340 nm. Emission intensity decreased gradually as the excitation wavelength was increased from 340 nm to 500 nm (Fig. 1H). A characteristic feature, “excitation-dependent emission” behavior, of CDs is in accordance with literature reports.36 A change in the emission profile with varying the excitation wavelength arises from different emissive states, conjugate systems and zigzag sites on CDs. Different theories such as quantum confinement, surface trap model, giant red-edge effect, edge states and heteroatom atom models have been proposed to prove this mechanism of CD emission, however, the exact mechanism is still obscure.37–39 The quantum yield of CDs was about 21% as determined by the absolute method. Furthermore, the quantity of the amino group present on the CD surface (1.30 μmol mg−1) has been determined by measuring the absorbance of Ruhemann's purple at 570 nm using alanine as a reference standard (Fig. S2†).40
To utilize the CDs for TrxR detection, TrxR substrate DTPA was functionalized on the CD surface. To functionalize the CDs, the dithiodipropionic anhydride (DTDPA) form of DTAP was synthesized and then coupled using the amide bond formation reaction in the presence of EDC. The surface functionalization of CDs with DTDPA was analyzed with zeta potential measurements, FTIR spectroscopy, and by estimating the number of amino groups remained on the CD surface. As DTDPA was functionalized on CDs, a significant change in IR spectra was observed (Fig. 1E red line). The broad and intensified vibrational band at 3356 cm−1 demonstrates the stretches of –OH and –NH groups. Furthermore, the band at 1624 cm−1 suggested the carbonyl absorption stretch of –COOH of the DTAP ligand. The absorption band at 2955 cm−1 and 2875 cm−1 corresponds to –CH2 and –CH3 stretches of the alkyl chain of DTPA. The small deep band at 1410 cm−1 indicates the –NH stretch, further suggesting the amide coupling between CDs and DTDPA. For more confirmation of the coupling reaction, the zeta potential of fCDs was recorded. The zeta potential of CDs was found to be 15 mV and it was reduced to 4.5 mV for fCDs which also demonstrates the consumption of free amine groups on the CD surface (Fig. S3†). The hydrodynamic size of fCD (9 nm) was confirmed from the histogram of DLS (Fig. 1F).
Strategy for TrxR detection through the displacement assay
The development of chemosensors for the detection of biologically important molecules has gained a lot of attention, and works on the principle of metal complex ensemble displacement, hydrogen bonding interactions, supramolecular interactions and so on.41–43 The metal displacement assay, among the various sensing mechanistic approaches, has been proved as the most successful method for the detection of biological important analytes.44 In the cation displacement assay, some of the anions displace the cation from the binding site and give the corresponding changes in the signal. Here, we designed the same strategies to displace the cation from the fCD surface for the sensing of TrxR because the Cu2+ ions exhibit strong affinity towards 3-mercaptopropionic acid. Thus, we have chosen Cu2+ for the preparation of the sensor probe.45,46 Our strategy is to reduce the S–S bond of fCDs with TrxR, only then Cu2+ will displace from the fCD surface and the fluorescence of CDs will regain.37,38 As shown in Fig. 2A, a deep blue emission of fCDs was significantly quenched by the addition of Cu2+ in HEPES buffer. The mechanism of quenched fluorescence emission in response to Cu2+ refers to the coordination of Cu2+ with carboxyl and thio groups of DTPA to form the fCDs-Cu2+ complex as predicted from 13C NMR of the fCDs-Cu2+ complex. The carbon signal corresponds to the –CO group shifted downfield (δ value) compared to CDs which suggested that Cu2+ most probably bind through the oxygen atom of the carbonyl group. The downfield shifting of alkylic carbon was also observed in 13C NMR (Fig. 2C). With increase in the concentration of Cu2+ from 0–2.6 μM, the relative fluorescence intensity (F0/F) of fCDs shows good linear relationship (r2 = 0.99). Therefore, this quenching mechanism was described by the Stern–Volmer equation: F0/F = 1 + Ksv[Q] (F0 and F are the fluorescence emission intensity of fCDs in the absence and presence of Cu2+, respectively. Ksv is the association constant and Q is the concentration of Cu2+). However, above this concentration, nonlinearity in fluorescence response with respect to the Cu2+ concentration was observed which indicates the dynamic and static quenching mechanism.47 The ksv was calculated to be 2.1 × 104 (Fig. 2B and S4†). The quenching of the fluorescence intensity of fCDs may be due to the partial transfer of an electron to the empty orbital of Cu2+ ions.48 To further investigate the quenching mechanism, TCPS studies were performed. The average life-time of fCDs was decreased from 7.1 ns to 3.7 ns after reacting with Cu2+ which suggested the ultrafast transfer of electrons from fCDs to d orbital of metal ions (Fig. S5†).
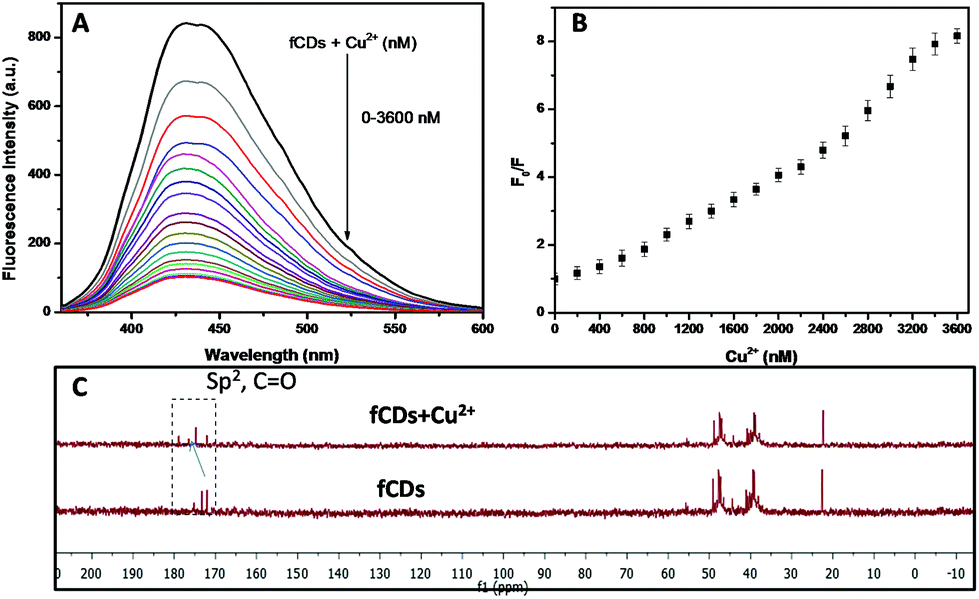 | ||
| Fig. 2 Photophysical properties and characterization of the fCD-Cu2+ complex. (A) Emission spectra (λem = 446 nm) of fCDs upon addition of different concentrations of Cu2+. (B) Relative fluorescence emission (F0/F at λem = 446 nm) upon addition of Cu2+. (C) 13C NMR of fCDs and fCDs-Cu2+. The carbon signal corresponding to carbonyl carbon shifted downfield which indicates the binding of Cu2+ with carbonyl oxygen. | ||
For the sensing studies of TrxR, 50 μg mL−1 solution of the sensor probe was prepared in HEPES buffer (pH 7.4). On addition of TrxR (100 nM), the fluorescence intensity of the sensor probe was restored in a time-dependent manner as shown in Fig. 3A and B. Enhancement in fluorescence intensity (blue emission) with the addition of TrxR was clearly observed under UV lamp radiation of 365 nm. After 100 min of incubation, a plateau was reached when no further change in fluorescence intensity was observed. Enhancement in the fluorescence intensity predicted as TrxR caused the reduction of the disulfide bond which results in the release of 3-mercaptopropionic acid from the CD surface. The 3-mercaptopropionic acid forms the bi-dentate chelate coordinate with Cu2+ and can be kept away from the CD surface. The formation of a bidentate chelate of 3-mercaptopropionic acid with Cu2+ has been confirmed by mass spectroscopy. Mass correspondence to the bidentate chelate of Cu2+ was found to be 273 (Fig. 3E) in the presence of TrxR which confirmed the reduction of a disulfide bond. Furthermore, the displacement of Cu2+ from the fCD surface was predicted by measuring the fluorescence decay time (Fig. S5†). On addition of 100 nM TrxR to sensor probe solution, the average decay time of fCDs-Cu2+ was increased from 3.7 ns to 6.7 after 100 min of incubation. The increase in fluorescence decay time suggested no transfer of an electron from fCDs to metal ions. On the basis of all these results and literature reports, the graphical representation of the detection mechanism and the Cu2+ binding mechanism has been predicted (Fig. 3E)47,49 since the fluorescence of the sensor probe is unambiguously switched on by TrxR. Furthermore, the selectivity of the sensor probe towards TrxR was evaluated using a panel of biological thiols and similar enzymes (Cysteine, Glutathione, Homocysteine, NADH and GSH reductase) and enhancement in fluorescence intensity was recorded (Fig. 3C and D). To our delight, none of these reducing molecules significantly enhanced the fluorescence of the sensor probe at 1 mM concentration. For the comparison of the sensitivity study, the sensitivity factor was calculated for the sensor probe as shown in Table S1.† These experimental results suggested that the prepared sensor probe is highly selective towards the TrxR. However, interference from GSH was observed only at a very high concentration (5 mM) with sensitivity factor 0.65. Therefore, we can say that the sensor probe is quite selective towards TrxR at 100 nM of TrxR and experiences a little influence only from the high concentration of GSH. For the quantitative applications of fluorescent probes, the kinetic behavior of enzymes was evaluated by measuring the Km value. Km is the Michaelis Menten constant which denotes the amount of substrate at which half of the maximum velocity of the enzymatic reaction is reached.50 A lower Km value indicates the higher binding affinity of the substrate and vice versa. Herein, the Km value was found to be 5.5 μg at 460 nm (Fig. S6†). With the progressive addition of TrxR in fCDs-Cu2+ solution, a linear increase in fluorescence intensity was observed at a 446 nm emission wavelength and the limit of detection was calculated to be 2.0 × 10−8 M (Fig. S7†). In order to calculate the limit of detection, a calibration curve was obtained between the fluorescence intensity at 446 nm and the concentration of TrxR. The regression equation was applied at a lower concentration and the detection limit was calculated based on the standard deviation and the slope of the curve using a formula LOD = 3 SD/k (SD: standard deviation of blank sensor solution and k: slope). Fluorescence responses of fCDs-Cu2+ to TrxR were also recorded in the presence of different concentrations of salt and pH to evaluate the effect of salt concentration over probe response. The optimum pH for the sensing study was found to be 5.5–7.5 and the salt concentration was tolerable up to 1 μM (Fig. S8A and S8B†).
 | ||
| Fig. 3 Qualitative and quantitative investigation of TrxR: (A) fluorescence emission spectra of fCDs-Cu2+ in relation to time with the addition of TrxR (100 nM). (B) Fluorescence intensity curve of fCDs-Cu2+ with a time scale from 0–120 min upon addition of TrxR (100 nM). (C) Fluorescence response of fCDs-Cu2+ (50 μg mL−1) in the presence of different disulfide reducing molecules present in the mammalian system and enzymes at different concentrations. (D) The bar graph of the fluorescence intensity of fCDs-Cu2+ in the presence of different biomolecules and fold of fluorescence increment was recorded. (E) Idealized schematic representation of TrxR detection. The Cu2+ ion binds with fCDs and causes the quenching of fluorescence intensity of fCDs. When TrxR was added to the fCDs-Cu2+ complex, 3-mercaptopropionic acid was released from the CD surface and forms the bidentate complex with Cu2+ ions which results into the emission of CDs that was recovered. All the experiments were performed in the presence of 0.1 mM of NADPH. | ||
Cytotoxicity assay and bioimaging of TrxR in cancer cells
The MTT assay was carried out to appraise the cytotoxicity of CDs, fCDs and the fCDs-Cu2+ complex against MCF-7 cell lines. It was worth describing that CDs did not exhibit any significant toxicity against MCF-7 cells. More than 87% of cells retained their viability even after the 100 μg mL−1 concentration of CDs and ≥82% cells remained viable in response to fCDs (100 μg mL−1) (Fig. S9†). Cytotoxicity results demonstrate the insignificant toxicity of CDs and fCDs towards the mammalian cells. However, fCDs-Cu2+ significantly inhibits the growth of cancer cells. At 50 μg mL−1 concentration, cell viability was reduced to 80% and it was further reduced to 67% at 100 μg mL−1 concentration in both types of cells. Retardation of the viability of cancer cells after treatment with the sensor probe ascertains the cytotoxic nature of Cu2+. Therefore, such CD metal complexes can also be used for the treatment of cancer cells in addition to their detection (Fig. S10†).Applications of the sensor probe were further expanded in bioimaging fields, especially for the cellular imaging of TrxR. For the cellular imaging of endogenous TrxR, MCF-7 and HeLa cells were employed in this study. 1 × 105 numbers of cells were grown in 6 well plates at 37 °C for 24 h. After successful washings, cells were incubated with 40 μg mL−1 of fCDs-Cu2+ for 30 min and images of cells were collected under a CLSM after 30 and 120 min of incubation with the sensor probe. Very weak emission at different excitation wavelengths was observed after 30 min (Fig. 4A and C) of incubation which suggested that fCDs existed in the complex form and no enzymatic reaction proceeded in this time. Notably, after 120 min, strong excitation dependent emission from the cytoplasm of cells was observed in both types of cells (Fig. 4B and D). Strong emission at the different excitation wavelengths (blue, green and red) after 120 min of incubation specifies the reduction of the disulfide bond owing to endogenous TrxR and Cu2+ ions have been released from the fCD surface. These results signify the permeability of the sensor through the cell wall and sensitivity towards TrxR in the cytoplasm of cells. Hence this is worth mentioning here that enhancement in the emission intensity of cells using such a sensing approach could serve as competitive tool for the detection of endogenous TrxR and related biological activities.
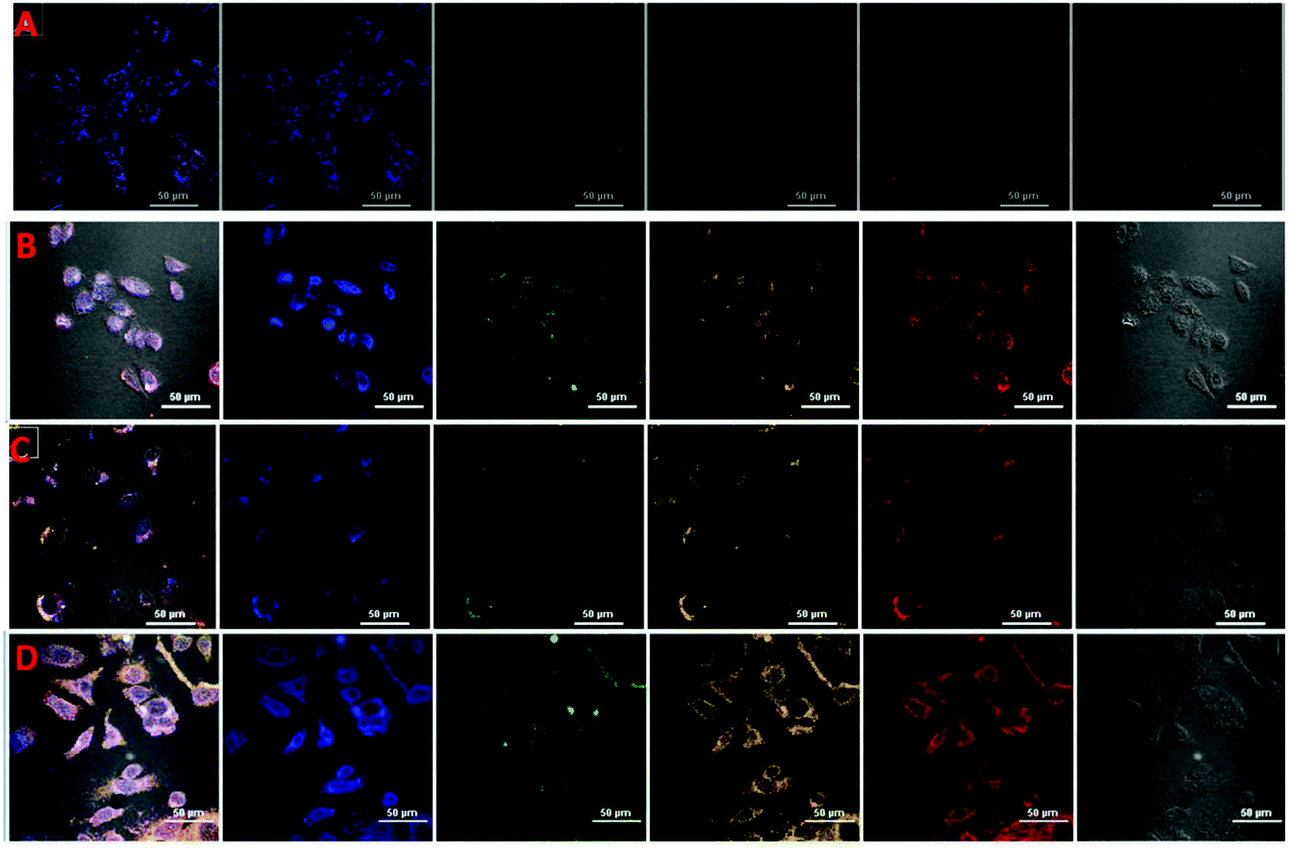 | ||
| Fig. 4 CLSM images of MCF-7 and HeLa cells. MCF-7 cells after 30 min (A and C) and 120 min (B and D) of incubation with fCDs-Cu2+. Very low emission intensity suggested the existence of the Cu2+ complex of fCDs in the cytoplasm of cells. The high fluorescence intensity of cells in (B) and (D) indicates the breakdown of the fCDs-Cu2+ complex in the cytoplasm of cells by the virtue of disulfide bond reduction by TrxR. The release of free Cu2+ from the surface of CDs results in the restoration of excitation dependent fluorescence emission of fCDs. Because of the excitation-dependent emission nature of CDs, cells emit at different wavelengths also. | ||
Conclusion
In conclusion, CDs and the Cu2+ metal ion complex as a ‘Turn On’ fluorescence sensor for TrxR have been synthesized for the detection of TrxR and live cell images were recorded to monitor the endogenous activity of TrxR. Inspired by the binding affinity of 3-mercaptopropionic acid with Cu2+ and the anticancer activity of Cu2+, CDs were covalently coupled with DTDPA and the metal complex prepared. We established a fluorescence assay for TrxR, which works on the principle of reduction of the disulfide bond of DTPA and the generation of 3-mercaptopropionic acid. 3-Mercaptopropionic acid chelated with a Cu2+ ion which takes away the Cu2+ ion from the CD surface and the fluorescence intensity of CDs was regained. The sensor probe has exhibited significant selectivity and sensitivity towards the TrxR. The fluorescence response of the sensor probe was stable at a wide range of pH (5–7.4) and salt concentration (up to 1 mM). For the real time applications, MCF-7 and HeLa cells were treated with the 3-mercaptopropionic complex and a remarkable change in the emission intensity of cells was clearly observed under a CLSM. Due to the presence of metal ions, the MTT assay was performed and found that at 100 μg mL−1 concentration, fCDs-Cu2+ reduced the cellular growth to 62% while more than 82% of cell viability was retained in CDs and fCD treated cells. Therefore, the current ‘Turn On’ fluorescent strategies could be extended to utilize their applications in the clinical field and may shed new light on the improvement of the more specific sensor for monitoring of enzymatic activities.Experimental section
Chemical and instruments
4,7,10-Trioxa-1,13-tridecanediamne (TTDDA) and citric acid were purchased from Sigma-Aldrich. 3-Mercaptopropionic acid, dimethylaminopyridine (DMAP) and acetyl chloride were procured from Avra Synthesis. Thioredoxin reductase from rat liver was obtained from Sigma-Aldrich. Fetal bovine serum (FBS), DMEM and DMSO were supplied by Hi-Media. All the chemicals were used without further purification. Milli-Q water was used in all synthetic and analytical experiments. Absorption spectra were recorded on a Shimadzu UV-2400 spectrophotometer. Fluorescence studies were performed using a PerkinElmer LS 55 fluorescence spectrophotometer. The PL quantum yield and the decay time were recorded on a PicoQuant FluoTime 300 High-Performance Fluorescence Lifetime Spectrometer using a Time-Correlated Single Photon Counting (TCSPC) technique. The single exponential function was used to measure the fitted PL decay curve. The response function of the instrument was acquired with LUDOX. The morphology of CDs was characterized by TEM (Transmission electron microscope) using a Hitachi (H-7500) instrument. The particle diameter was measured by using DLS (Dynamic Light Scattering) with an external probe of a Metrohm Microtrac Ultra Nanotrac particle size analyzer. IR spectra of solid samples were recorded by using a solid cell technique on a Bruker Tensor 27 spectrophotometer. The powder XRD pattern of CDs was obtained from an analytical X'PERT PRO instrument. A Nikon Eclipse 540 Ti−U inverted microscope was used to collect fluorescence images of cells.Synthesis of CDs and fCDs
Dithiodipropionic anhydride (DTDPA) was synthesized according to the reported procedure.51 Briefly, DTPA (2 g, 9.6 mmol) and acetyl chloride (15 mL) solution were refluxed for 7 h. The solvent was evaporated under vacuum; the residue was precipitated into excess ethyl ether to afford DTDPA. Precipitates were filtered out and dried under vacuum. Amino coated CDs were prepared according to the reported procedure with little modification.52 Briefly, 0.5 g of TTDDA was dissolved in 20 mL of glycerol and 0.5 g of citric acid was added into it. The resulting solution was stirred for 15 min in a round bottom flask under nitrogen conditions and transferred to a Teflon lined autoclave. The autoclave was tightly sealed and kept at 180 °C in a muffler furnace for 6 h. A dark brown solution was dialyzed against water for 48 h to get the CDs. Thus, the obtained CDs were freeze dried and kept at 4 °C for further use. DTDPA (20 mg, 0.10 mmol) was dissolved in 1 mL of DMSO and transferred to 5 mg solution of CDs in 3 mL of water. 2 mg of DMAP was added into the solution and let the reaction mixture to a stirrer for 20 h at room temperature. The reaction mixture was dialyzed (3500 MWCO) against water for 72 h to remove the organic impurities and freeze dried to obtain fCDs. The synthesized product was stored at 4 °C in the dark. Purified CDs were characterized by recording the UV-vis, fluorescence spectra, FTIR, and TEM analysis.Quantum yield measurement
The quantum yield was measured by the absolute method at room temperature. A quartz cuvette having a path length of 10 mm was used for all the liquid samples. The solvent without any sample in the same cuvette was used as a blank sample. 2-Aminopyridine (QY: 84%) and rhodamine 101 (QY: 99%) were chosen as a reference standard to test the accuracy of apparatus.Recognition properties of fCDs and monitoring of TrxR
For the sensing of TrxR, fCDs and the Cu2+ complex was prepared by mixing 5 mg of fCDs and 1 mg of CuNO3. The mixture was allowed to stir for 4 h followed by dialysis against water for 40 h. The aqueous solution was freeze dried and stored at 4 °C. In HEPES buffer (pH 7.4), 50 μg mL−1 solution of fCDs-Cu2+ was prepared. The 100 nM concentration of TrxR in 0.1 mM of NADPH was prepared for the sensing assay. The concentration of fCDs-Cu2+ and TrxR was kept constant and the change in the emission intensity was recorded with respect to time. Photoluminescence properties were recorded at 37 °C. Each experiment was performed in triplicate.Cellular studies for imaging and cytotoxicity assay
In DMEM media containing 10% FBS, MCF-7 and HeLa cell lines with cell density of 1 × 105 per well were grown at 37 °C in humidified 5% CO2 for 24 h. Old culture media were removed and new DMEM media containing 50 μg mL−1 concentration of dimethylaminopyridine were added. After addition of complex containing media, cells were incubated for 60 min at 37 °C in 5% CO2. The adherent cells were washed with PBS three times to remove the extracellular CDsM-Cu complex. Glass slides were taken out and analyzed under a confocal laser scanning microscope (CLSM). Cell images were acquired under the blue, green and red channels of the CLSM. The in vitro cytotoxicity of fCDs and fCDs-Cu2+ was evaluated by the MTT assay using MCF-7 and HeLa cell lines. Both the cells were cultured in DMEM media containing 10% FBS at 37 °C in 5% CO2 for 24 h. After 24 h of incubation, media were replaced with fCDs and fCDs-Cu2+ containing new DMEM medium and further incubated for 24 h. The cell without any treatment was chosen as a control. The absorbance of cells was measured after treatment with a MTT dye to evaluate the viability of cells. Cells without any treatment were selected as a control sample.Conflicts of interest
There are not conflicts to declare.Acknowledgements
We are grateful to DST-SERB India (Project No. EMR/2014/000613) for providing financial support.References
- R. Rubbiani, I. Kitanovic, H. Alborzinia, S. Can, A. Kitanovic, L. A. Onambele, M. Stefanopoulou, Y. Geldmacher, W. S. Sheldrick and G. Wolber, J. Med. Chem., 2010, 53, 8608–8618 CrossRef CAS PubMed.
- M. McNaughton, L. Engman, A. Birmingham, G. Powis and I. A. Cotgreave, J. Med. Chem., 2004, 47, 233–239 CrossRef CAS PubMed.
- S. Lin, L. M. Del Razo, M. Styblo, C. Wang, W. R. Cullen and D. J. Thomas, Chem. Res. Toxicol., 2001, 14, 305–311 CrossRef CAS PubMed.
- B. Zhang, D. Duan, C. Ge, J. Yao, Y. Liu, X. Li and J. Fang, J. Med. Chem., 2015, 58, 1795–1805 CrossRef CAS PubMed.
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2016, 66, 7–30 CrossRef PubMed.
- U. Ikoba, H. Peng, H. Li, C. Miller, C. Yu and Q. Wang, Nanoscale, 2015, 7, 4291–4305 RSC.
- Z. Luo, L. Yu, F. Yang, Z. Zhao, B. Yu, H. Lai, K.-H. Wong, S.-M. Ngai, W. Zheng and T. Chen, Metallomics, 2014, 6, 1480–1490 RSC.
- M.-H. Yoo, X.-M. Xu, B. A. Carlson, A. D. Patterson, V. N. Gladyshev and D. L. Hatfield, PLoS One, 2007, 2, e1112 Search PubMed.
- L. Huang, Y. Chen, B. Liang, B. Xing, G. Wen, S. Wang, X. Yue, C. Zhu, J. Du and X. Bu, Chem. Commun., 2014, 50, 6987–6990 RSC.
- H. Zhang, K. Wang, X. Xuan, Q. Lv, Y. Nie and H. Guo, Chem. Commun., 2016, 52, 6308–6311 RSC.
- S. Liu and Z. Tang, J. Mater. Chem. B, 2010, 20, 24–35 RSC.
- L. Zhang, D. Duan, Y. Liu, C. Ge, X. Cui, J. Sun and J. Fang, J. Am. Chem. Soc., 2014, 136, 226–233 CrossRef CAS PubMed.
- M. Culzoni, A. M. de la Pena, A. Machuca, H. Goicoechea and R. Babiano, Anal. Methods, 2013, 5, 30–49 RSC.
- A. P. Turner, Chem. Soc. Rev., 2013, 42, 3184–3196 RSC.
- K. L. Diehl and E. V. Anslyn, Chem. Soc. Rev., 2013, 42, 8596–8611 RSC.
- D. Ding, K. Li, B. Liu and B. Z. Tang, Acc. Chem. Res., 2013, 46, 2441–2453 CrossRef CAS PubMed.
- V. Biju, Chem. Soc. Rev., 2014, 43, 744–764 RSC.
- Y. Wen, F. Xing, S. He, S. Song, L. Wang, Y. Long, D. Li and C. Fan, Chem. Commun., 2010, 46, 2596–2598 RSC.
- L. Cao, X. Wang, M. J. Meziani, F. Lu, H. Wang, P. G. Luo, Y. Lin, B. A. Harruff, L. M. Veca, D. Murray, S.-Y. Xie and Y.-P. Sun, J. Am. Chem. Soc., 2007, 129, 11318–11319 CrossRef CAS PubMed.
- Y. Wang and A. Hu, J. Mater. Chem. C, 2014, 2, 6921–6939 RSC.
- Q. Xu, P. Pu, J. Zhao, C. Dong, C. Gao, Y. Chen, J. Chen, Y. Liu and H. Zhou, J. Mater. Chem. A, 2015, 3, 542–546 CAS.
- J. Wen, Y. Xu, H. Li, A. Lu and S. Sun, Chem. Commun., 2015, 51, 11346–11358 RSC.
- W. Wang, Y. Li, L. Cheng, Z. Cao and W. Liu, J. Mater. Chem. B, 2014, 2, 46–48 RSC.
- J. S. Sidhu, A. Singh, N. Garg and N. Singh, ACS Appl. Mater. Interfaces, 2017, 9, 25847–25856 CAS.
- L.-S. Wang, M.-C. Chuang and J.-a. A. Ho, Int. J. Nanomed., 2012, 7, 4679 CAS.
- H. Liu, R. S. Li, J. Zhou and C. Z. Huang, Analyst, 2017, 142, 4221–4227 RSC.
- K. Jiang, S. Sun, L. Zhang, Y. Lu, A. Wu, C. Cai and H. Lin, Angew. Chem., Int. Ed., 2015, 54, 5360–5363 CrossRef CAS PubMed.
- S. Y. Lim, W. Shen and Z. Gao, Chem. Soc. Rev., 2015, 44, 362–381 RSC.
- S. Li, L. Wang, C. C. Chusuei, V. M. Suarez, P. L. Blackwelder, M. Micic, J. Orbulescu and R. M. Leblanc, Chem. Mater., 2015, 27, 1764–1771 CrossRef CAS.
- X. Zhu, Y. Liu, P. Li, Z. Nie and J. Li, Analyst, 2016, 141, 4541–4553 RSC.
- H. Ma, J. Zhang, Z. Zhang, Y. Liu and J. Fang, Chem. Commun., 2016, 52, 12060–12063 RSC.
- R. Wang, K.-Q. Lu, Z.-R. Tang and Y.-J. Xu, J. Mater. Chem. A, 2017, 5, 3717–3734 CAS.
- J.-H. Liu, L. Cao, G. E. LeCroy, P. Wang, M. J. Meziani, Y. Dong, Y. Liu, P. G. Luo and Y.-P. Sun, ACS Appl. Mater. Interfaces, 2015, 7, 19439–19445 CAS.
- Z. Qian, X. Shan, L. Chai, J. Ma, J. Chen and H. Feng, ACS Appl. Mater. Interfaces, 2014, 6, 6797–6805 CAS.
- T. Feng, X. Ai, G. An, P. Yang and Y. Zhao, ACS Nano, 2016, 10, 4410–4420 CrossRef CAS PubMed.
- L. Wang and H. S. Zhou, Anal. Chem., 2014, 86, 8902–8905 CrossRef CAS PubMed.
- Z. X. Gan, S. J. Xiong, X. L. Wu, C. Y. He, J. C. Shen and P. K. Chu, Nano Lett., 2011, 11, 3951–3956 CrossRef CAS PubMed.
- N. Dhenadhayalan, K.-C. Lin, R. Suresh and P. Ramamurthy, J. Phys. Chem. C, 2016, 120, 1252–1261 CAS.
- A. Sharma, T. Gadly, A. Gupta, A. Ballal, S. K. Ghosh and M. Kumbhakar, J. Phys. Chem. Lett., 2016, 7, 3695–3702 CrossRef CAS PubMed.
- T. Feng, X. Ai, H. Ong and Y. Zhao, ACS Appl. Mater. Interfaces, 2016, 8, 18732–18740 CAS.
- F. Wang, L. Wang, X. Chen and J. Yoon, Chem. Soc. Rev., 2014, 43, 4312–4324 RSC.
- B. Shi, P. Zhang, T. Wei, H. Yao, Q. Lin and Y. Zhang, Chem. Commun., 2013, 49, 7812–7814 RSC.
- D. G. Smith, I. L. Topolnicki, V. E. Zwicker, K. A. Jolliffe and E. J. New, Analyst, 2017, 142, 3549–3563 RSC.
- L. Qin, L. Hou, J. Feng, J. Chao, Y. Wang and W. J. Jin, Anal. Methods, 2017, 9, 259–266 RSC.
- Y. Gou, J. Qi, J.-P. Ajayi, Y. Zhang, Z. Zhou, X. Wu, F. Yang and H. Liang, Mol. Pharmaceutics, 2015, 12, 3597–3609 CrossRef CAS PubMed.
- Y. Sikdar, R. Modak, D. Bose, S. Banerjee, D. Bieńko, W. Zierkiewicz, A. Bieńko, K. D. Saha and S. Goswami, Dalton Trans., 2015, 44, 8876–8888 RSC.
- S. Mohapatra, S. Sahu, N. Sinha and S. K. Bhutia, Analyst, 2015, 140, 1221–1228 RSC.
- J. S. Floyd, N. Haralampus-Grynaviski, T. Ye, B. Zheng, J. D. Simon and M. D. Edington, J. Phys. Chem. B, 2001, 105, 1478–1483 CrossRef CAS.
- D. Kong, F. Yan, Z. Han, J. Xu, X. Guo and L. Chen, RSC Adv., 2016, 6, 67481–67487 RSC.
- A. Lervik, S. Kjelstrup and H. Qian, Phys. Chem. Chem. Phys., 2015, 17, 1317–1324 RSC.
- C. Liu, J. Yuan, X. Luo, M. Chen, Z. Chen, Y. Zhao and X. Li, Mol. Pharmaceutics, 2014, 11, 4258–4269 CrossRef CAS PubMed.
- Y. Wang, C. Zhang, X. Chen, B. Yang, L. Yang, C. Jiang and Z. Zhang, Nanoscale, 2016, 8, 5977–5984 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7an02040f |
| This journal is © The Royal Society of Chemistry 2018 |