
FTIR spectroscopy of chronic venous leg ulcer exudates: an approach to spectral healing marker identification
Nicolas
Cerusico
 a,
Juan P.
Aybar
a,
Silvana
Lopez
b,
Silvia G.
Molina
b,
Romina
Chavez Jara
a,
Maria Eugenia
Sesto Cabral
a,
Juan C.
Valdez
c,
Aida
Ben Altabef
d and
Alberto N.
Ramos
*a
a,
Juan P.
Aybar
a,
Silvana
Lopez
b,
Silvia G.
Molina
b,
Romina
Chavez Jara
a,
Maria Eugenia
Sesto Cabral
a,
Juan C.
Valdez
c,
Aida
Ben Altabef
d and
Alberto N.
Ramos
*a
aLaboratorio de Estudios Farmacéuticos y Biotecnología Farmacéutica, Instituto de Biotecnología Farmacéutica y Alimentaria (INBIOFAL), San Miguel de Tucumán, Tucumán, Argentina. E-mail: alnirave@gmail.com; Tel: +54381 4856596
bServicio de Dermatología, Hospital de Clínicas Presidente Nicolás Avellaneda, San Miguel de Tucumán, Tucumán, Argentina
cInstituto de Microbiología, Facultad de Bioquímica, Química y Farmacia, Universidad Nacional de Tucumán, San Miguel de Tucumán, Tucumán, Argentina
dINQUINOA-CONICET, Instituto de Química Física, Facultad de Bioquímica, Química y Farmacia, Universidad Nacional de Tucumán, San Miguel de Tucumán, Tucumán, Argentina
First published on 13th February 2018
Abstract
Chronic venous leg ulcer (CVLU) arises as a chronic venous insufficiency complication and is a major cause of morbidity throughout the world. Our hypothesis is that the CVLU exudate composition is a biochemical representation of the wound clinical state. Then, Fourier Transform Infrared (FTIR) spectroscopy could be a useful and less-invasive technique to study the clinical state of the ulcer. For this, the aim of this work was to perform a spectral characterization of the exudate from CVLU using FTIR spectroscopy to identify potential healing markers. 45 exudate samples from CVLU, 95% of the strains isolated from CVLU in planktonic and biofilm phenotypes and other related biological samples such as human plasma, serum, urine, blood cells, urea, creatinine, glucose and albumin were studied by FTIR spectroscopy. According to the vibration frequency of biomolecules’ (lipids, proteins, nucleic acids and carbohydrates) characteristic bonds in the infrared region, different spectral windows were selected and spectral areas of each window were measured. Besides, Savitzky–Golay second derivatives were obtained for all spectra and peaks from each standardized window were detected. FTIR spectroscopy allowed identification of sample types (exudate, plasma, serum, urine) as each one presents a unique relative composition and ratios range. Also, this technique could be useful to identify bacteria in the phenotypic-ulcer state and allows differentiation of whether bacteria are in the biofilm or planktonic form which is unlikely by conventional methods. In this work we found some spectral markers (areas, peaks) that allow identification of several parameters in the exudate such as (a) total cellularity, (b) inflammatory cell load, (c) bacterial load, (d) fibrin amount, and (e) inflammatory proteins. Because the measured areas or founded peaks are concentration-dependent this method could also serve to measure them. Therefore, FTIR spectroscopy could be useful to evaluate patient evolution as all these exudate parameters represent critical negative markers for wound healing.
Introduction
Fourier Transform Infrared (FTIR) spectroscopy is being increasingly used in biomedical applications with high degrees of success.1–6 Molecular bonds with an electric dipole moment that can change by atomic displacement owing to natural vibrations are IR active.7 These vibrational modes are quantitatively measurable by IR spectroscopy, providing a unique, label-free tool for studying the molecular composition and dynamics without perturbing the sample.7 FT-IR spectroscopy is a non-destructive method for the analysis of cells, tissue and fluids.1–8 However, there are no reported studies of wound fluids from Chronic Venous Leg Ulcers (CVLUs) by FT-IR spectroscopy.CVLU arises as a chronic venous insufficiency complication and is a major cause of morbidity throughout the world,9–11 with an overall prevalence ranging up to 2% in the general population13 and median ulcer durations that range from six – eight months to decades.13
Several factors are involved in the CVLU delayed healing process: venous insufficiency degree,14 infection,9,15 inflammatory molecules,9etc. Due to all of these factors involved in the CVLU development, the correct diagnostic, prognosis and its treatment are difficult, leading CVLUs to a long non-healing state.16 Wound fluid or ulcer exudate may be used as a clinical state indicator, because its complex composition is reflex of the biochemical processes that occur on the wound bed and of its chronicity.17,18 Exudate formation results from a plasma ultrafiltrate as a local inflammation consequence influenced by the wound healing process.19 When the tissue is injured, the inflammatory process begins along with the wound-healing process.20 This promotes the affluence of inflammatory cells, such as polymorphonuclears (PMN), lymphocytes, and macrophages that are key to the removal of contaminating microorganisms and infection.17,20 The exudate has high viscosity and a high protein amount (>30 g L−1)18 and several components from serum like glucose,18 urea,18,21 creatinine,18,21 lactate and salts,18,21 and tissue inflammatory molecules such as cytokines, serine proteinase, cysteine proteinase, aspartic proteinase and matrix metalloproteinases (MMPs).12,17,19 Also, the exudate contains bacteria and biofilm components such as the extracellular polysaccharide matrix (EPS)22,23 and DNA.24 Therefore, the exudate might be considered as a negative healing factor in chronic wounds because the excessively proteolytic environment will continually degrade key growth promoting agents and thus will not allow normal wound healing to occur.17 Chronic wound exudate has higher MMP levels than acute exudate which causes tissue digestion.17 There is a correlation between the elevated levels of MMPs and delayed healing.12,25 Also, MMP may cause inhibition of endothelial cell proliferation and angiogenesis.26 Finally, exudate is a physical barrier for cell displacement in the re-epithelization process.26
In summary, exudate from a chronic wound contains plasma components, inflammatory cells, proteins from the inflammatory response, bacteria and components from the bacterial biofilm matrix (Fig. 1). Our hypothesis is that the exudate composition is a biochemical representation of the clinical state of a chronic wound. Therefore, FTIR spectroscopy associated with other clinical parameters could be a useful technique that provides a less-invasive and simple way to represent the clinical state of ulcers and that allows the identification of prognosis/diagnostic markers. For this, the aim of this work was to perform the spectral characterization of exudate from CVLU using FTIR spectroscopy to identify potential healing markers.
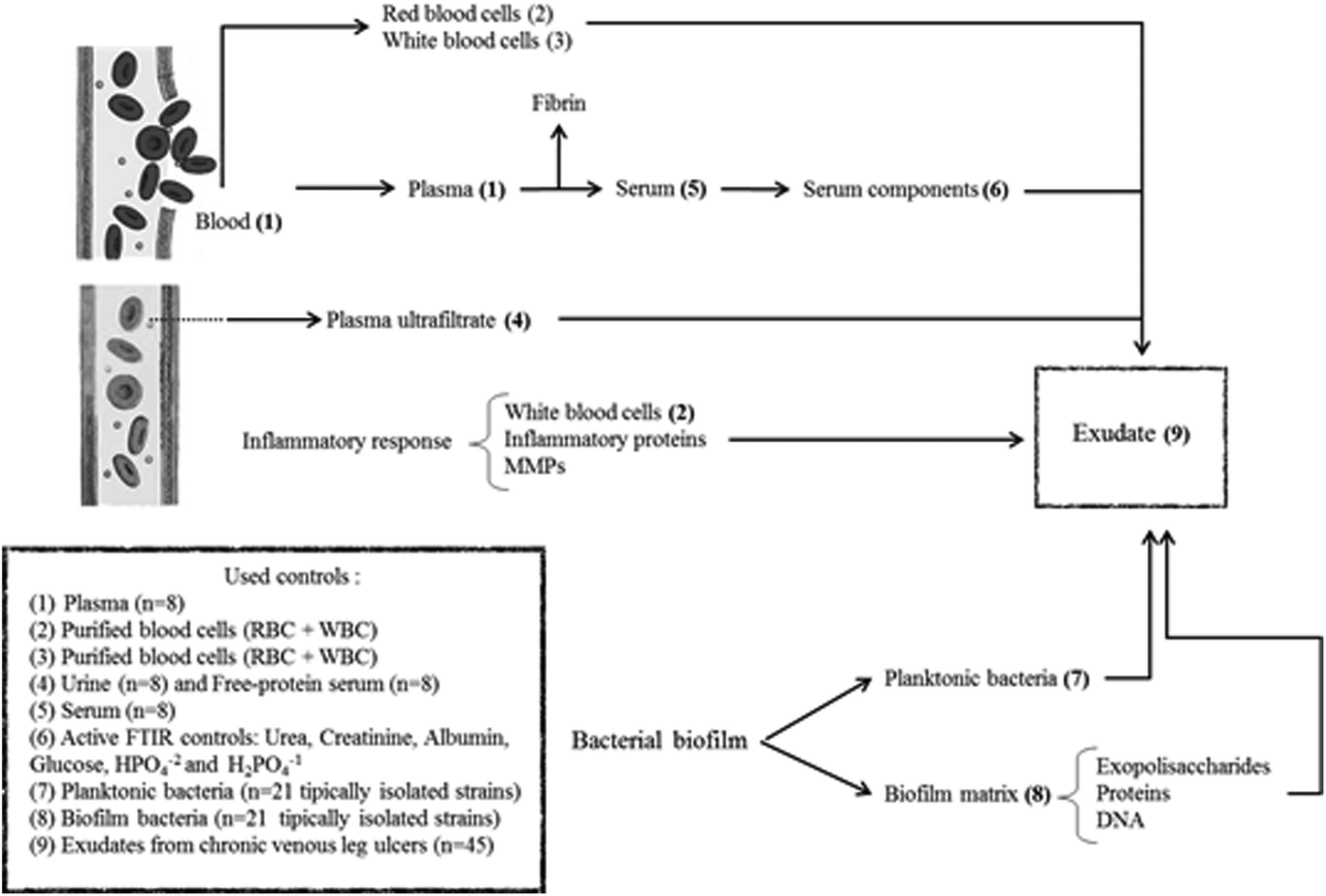 | ||
| Fig. 1 The production of wound exudate occurs as a result of vasodilation during the inflammatory early stage of healing under the influence of inflammatory mediators. As it could see in the scheme, an exudate from a chronic wound has components from plasma ultrafiltrate. For this, we use as control urine and free-protein serum. In some cases, exudate might receive blood components as plasma compounds, red blood cells (RBC) and white blood cells (WBC) which were used as controls (plasma, RBC + WBC, urea, creatinine, glucose, albumin, phosphates). Once in the wound, plasma coagulate forming a fibrin layer over de wound. To evaluate the contribution of fibrin on exudate ftir spectra, human serum was used. The difference between plasma and serum spectra in Amide I and II windows could show some information about fibrin. Finally, exudate receives contribution from planktonic and biofilm bacterial components. For this, 95% of CVLU isolated bacteria in both phenotypes were used as controls. | ||
Experimental
Ethics
This research protocol was approved and monitored by Independent Medic Ethic Committee from Argentinian Norwest (CIEM-NOA). Patients under observation signed an informed consent before being included in this protocol.Patients
For the mentioned protocol, 45 patients from Dermatology Service of Nicolas Avellaneda Hospital (San Miguel de Tucuman, Tucuman – Argentina) were selected. Patients with CVLU were diagnosed by venous doppler and clinical criteria.Inclusion criteria: (a) Ulcer location: lower-third of lower limbs. (b) Both sexes. (c) Age: between 40–80 years. (d) Ulcer size: 20 to 150 cm2. (e) Ulcer evolution time: 1 to 3 years.
Exclusion criteria: Patients with a background of (1) systemic infection, (2) cancer and/or under chemotherapy treatment, (3) autoimmune disease, and (4) drugs abuse were excluded from this study.
As relevant clinical information, patient's clinical association pathologies and ulcer evolution time were analyzed from its clinical records.
Exudate samples
Exudate was obtained by gentle aspiration with a syringe (without a needle and avoiding causing pain and bleeding) from 6 different points of the CVLU and stored at −20 °C until processing.Spectral contribution controls
In order to evaluate spectral contributions to exudate that come from the plasma ultrafiltrate, the following lyophilized controls were used (Fig. 1):Serum (n = 8): Obtained by whole blood extraction from random patients. Sera were left to clot for 15 minutes and then centrifuged for 10 min at 3000 rpm.
Plasma (n = 8): Obtained by whole blood extraction from random patients on sodium citrate 1.2% w/v (ratio: 9/1) and centrifuged for 10 min at 3000 rpm.
Free-protein serum (n = 8): This control is useful to find if there are protein contributions to exudate spectra that did not come from plasma, analyzing specifically in the protein spectral regions. Polson et al. protocol was followed to obtain this free-protein serum.27 A serum aliquot was separated and then treated with absolute ethanol 99.5% v/v (Cicarelli) (1/0.5), incubated at −20 °C for 12 h and then centrifuged at 8000 rpm for 20 minutes. This process was repeated twice to ensure serum deproteinization and corroborated with UV spectroscopy (200–400 nm)28 and the Bradford method.29
Urine (n = 8): Urine is a plasma ultrafiltrate and for this reason it could be a useful control for exudate study as a free-protein control (Fig. 1). Urine samples were taken after day-first urine from 8 volunteer human subjects in order to reduce the amount of filtered proteins to the maximum. Urine samples come from 8 different human volunteers between 30–40 years old with no kidney disease history, hepatic disease or use of chronic medication to ensure the correct glomerular function. Free-cell urines were obtained by centrifugation at 3000 rpm for 10 min.
Blood cells control (n = 8) was used to analyze the cellular contribution from inflammatory response (white blood cells – WBC) and bleeding (red blood cells – RBC) to exudate (Fig. 1). To obtain this control, a whole blood anticoagulated (citrate 1.2% w/v) aliquot was separated and centrifuged at 3000 rpm for 10 min, then plasma was separated and the remaining cells (WBC + RBC) were washed with saline three times. Finally, the cells were re-suspended in saline and stored at 4 °C until their processing.
Other serum controls: Different controls were carried out to analyze the individual contributions of most important seric molecules (Fig. 1). Glucose (Cicarelli-Argentina), urea (Cicarelli-Argentina), creatinine (Anedra-Argentina) and inorganic H2PO41−/HPO42− mix (Cicarelli-Argentina), and human albumin (Sigma-Aldrich-USA) were used as individual drug controls.
To corroborate the protein amount and confirm the origin of the protein contributions to spectra, two assays for protein determination were performed over albumin, urea, creatinine, urine, free-protein serum and serum controls: UV spectra obtained at 200–400 nm (ref. 26) and the Bradford method.27
Bacteria
To find bacterial contributions to the exudate FTIR spectra (Fig. 1), strains isolated from CVLU were studied in their planktonic and biofilm forms. Selected strains were isolated from CVLU exudate samples by conventional methods and represent 95% of the aerobic isolations.16,30–32 Isolated Gram-positive bacteria were Staphylococcus aureus, methicillin resistant S. aureus (MRSA), S. haemolyticus, coagulase negative Staphylococcus (CNS), beta-hemolytic Streptococcus and Enterococcus faecalis. Isolated Gram-negative bacteria were: Pseudomonas aeruginosa, Pseudomonas sp, Escherichia coli, Serratia marscecens, Proteus mirabilis, Enterobacter aerogenes, Enterobacter sp, Klebsiella pneumoniae, Burkholderia cepacia, Providencia sp and Citrobacter sp. All bacteria were stored at −20 °C in BHI media + glycerol (30%).Planktonic form: Each strain was activated at room temperature for 30 min, cultured in BHI broth and then incubated for 6 h at 37 °C. Cultures were centrifuged at 8000 rpm for 10 min and planktonic cell pellets were washed 3 times (saline) to remove the culture medium. Planktonic pellets were lyophilized before their spectroscopic study.
Biofilm formation: Each strain was activated at room temperature for 30 min, cultured in BHI broth or BHI broth ((plus 5% v/v) human serum for nutritionally highly demanding bacteria) (1/10 v/v) and then incubated at 37 °C until the biofilm formation (12 to 24 h depending on the strain). In some cases, bacteria were stressed to allow biofilm formation (nutritional stress, UV radiation, thermic stress). Cultures were centrifuged at 3000 rpm for 10 min (to obtain mainly a biofilm pellet). Biofilm pellets were washed 3 times (saline) to remove the culture medium and planktonic bacteria. Biofilm pellets were lyophilized before their spectroscopic study.
FTIR spectroscopy
To collect FTIR spectra, a PerkinElmer GX 1 spectrophotometer was used. Exudate samples and controls were processed as liquid samples, 5 μl of exudate sample and controls were dried under a N2 flow and vacuum over AgCl circular optical windows. Each planktonic and biofilm bacteria was processed as solid samples twice on KBr pellets of spectroscopic grade (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20). Spectra were collected with 64 scans and 4 cm−1 of resolution in the range of mid-infrared 4000 cm−1–400 cm−1. For spectral pre-processing smoothing, baseline correction and normalization with amide I band were used. Pre-processing is useful to compensate for differences in the sample quantity or a different optical pathlength.7,32
:20). Spectra were collected with 64 scans and 4 cm−1 of resolution in the range of mid-infrared 4000 cm−1–400 cm−1. For spectral pre-processing smoothing, baseline correction and normalization with amide I band were used. Pre-processing is useful to compensate for differences in the sample quantity or a different optical pathlength.7,32
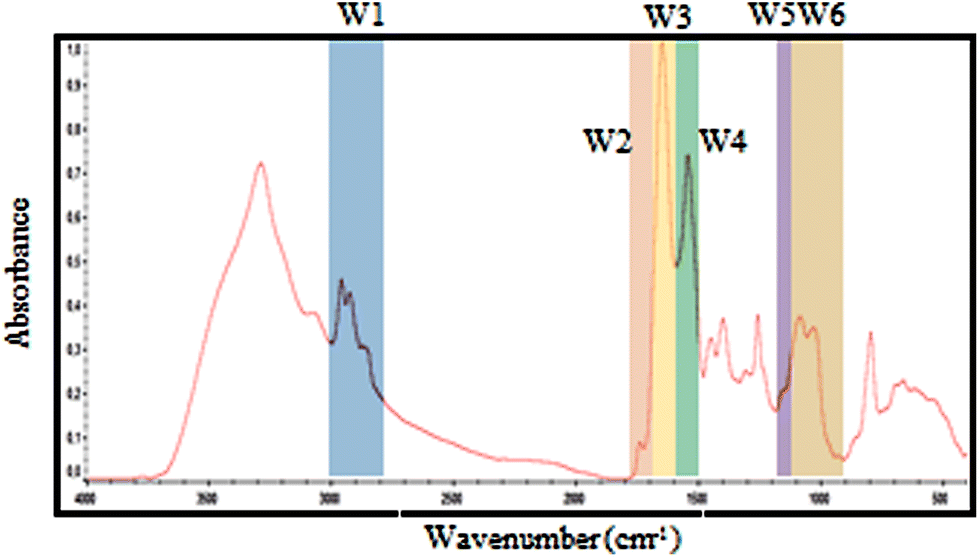 | ||
| Fig. 2 Selected spectral windows for each biomolecule marked on a typical exudate spectrum. The same windows were used for spectral contribution controls analysis. | ||
| Window | Denomination | Wavenumber (cm−1) | Proposed vibrational mode | Proposed primary source |
|---|---|---|---|---|
| W1 | CH3; CH2 | 3000–2800 | C–H asymmetric and symmetric stretch of >CH2 and CH3 present on fatty acids and lipids | Lipids, membrane phospolipids. |
| W2 | Ester bonds | 1770–1720 | >C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch of ester bonds in fatty acids O stretch of ester bonds in fatty acids |
Lipids, membrane phospolipids. |
| W3 | Amide I | 1715–1600 | >CO stretch, C–N stretch, CCN deformation in peptide bonds |
Proteins and peptides |
| W4 | Amide II | 1600–1480 | NH bend, C–N stretch, CO bend, N–C stretch | Proteins and peptides |
| W5 | Phosphate bonds | 1270–1200 | >PO2− stretch in RNA/DNA or NH bend, C–C stretch, C–N stretch, CO bend (Amide III) | Nucleic acids and proteins. |
| W6 | Carbohydrates bonds | 1190–900 | C–O, C–C stretch, C–O–H, C–O–C deformation of carbohydrates or >PO2−sym. stretch of phosphodiester group in nucleic acids | Carbohydrates, polysaccharides and nucleic acids |
Statistics
Statistical significance was evaluated using the Mann–Whitney–Wilcoxon U test for non-parametrical variables. Data analysis was performed with GraphPad Prism version 6.0 (GraphPad Software, La Jolla, CA).Results and discussion
Exudate, plasma, serum, free-protein serum and urine spectral areas
In this study, the relative composition of each biological polymer in exudates and controls (serum, plasma, free-protein serum, urine, blood cells and other related controls) (Fig. 3a–l) was studied by measurement of spectral areas and ratios between them. Also it was found that each analyzed sample by FTIR (exudates and controls) shows a unique relative composition and ratio. This could be used to identify sample types (exudates, plasmas, serum, urine) (Fig. 4). Table 2 shows the relative composition of each exudate.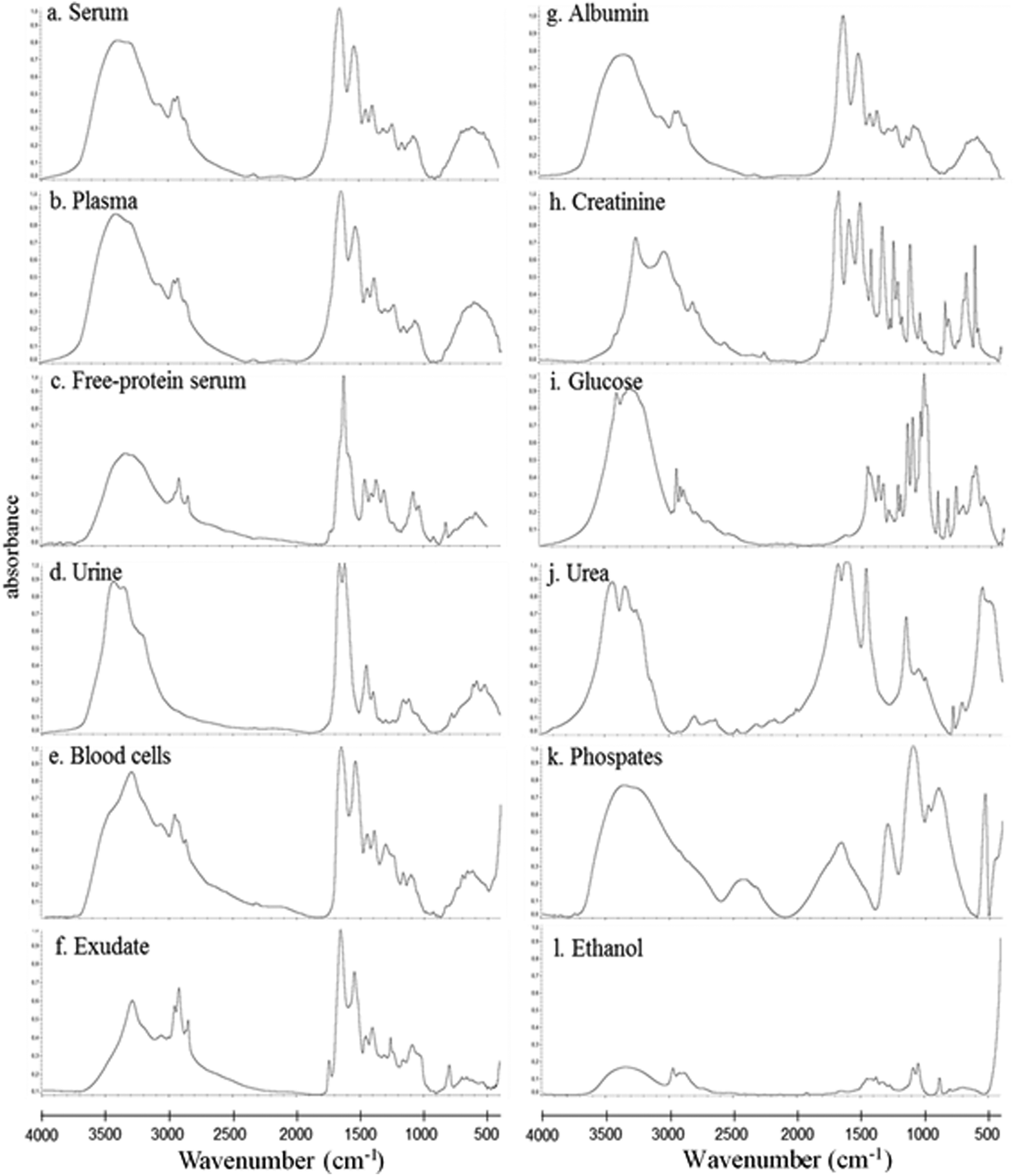 | ||
| Fig. 3 Typical FTIR spectra for different samples and controls at mid-infrared frequencies (4000 cm−1–400 cm−1). a. Serum; b. plasma; c. free-protein serum; d. urine; e. cells (WBC + RBC) control; f. exudate; g. albumin; h. creatinine; i. glucose; j. urea; k. phosphate mixture; l. absolute ethanol. | ||
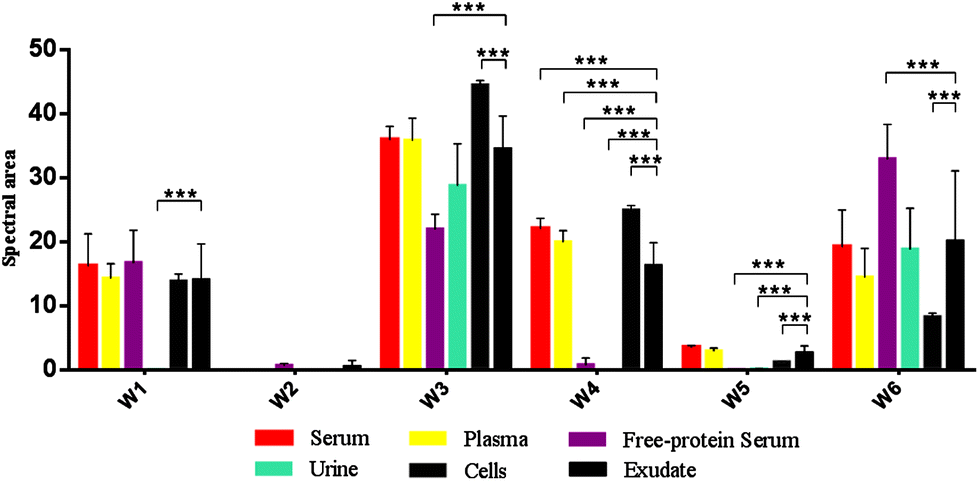 | ||
| Fig. 4 Mean FTIR spectral areas measured in each window for different samples and controls analyzed. Frequency regions for each window were determined according specific biomolecules bonds (Table 1). | ||
| Exudate | W1 | W2 | W3 | W4 | W5 | W6 |
|---|---|---|---|---|---|---|
| 001 | 11.21 | 0.58 | 22.18 | 12.19 | 2.29 | 14.81 |
| 002 | 13.50 | 0.00 | 23.56 | 11.67 | 3.37 | 13.63 |
| 003 | 11.70 | 0.00 | 27.12 | 10.00 | 1.50 | 16.27 |
| 004 | 12.97 | 0.00 | 32.90 | 15.00 | 3.31 | 18.16 |
| 005 | 19.66 | 1.75 | 28.02 | 16.17 | 2.90 | 17.82 |
| 006 | 16.12 | 0.83 | 29.76 | 15.24 | 3.71 | 24.66 |
| 007 | 20.07 | 1.76 | 34.26 | 14.82 | 3.95 | 28.06 |
| 008 | 18.60 | 1.53 | 34.36 | 14.15 | 4.13 | 29.37 |
| 009 | 9.59 | 0.00 | 36.95 | 17.42 | 1.85 | 5.68 |
| 010 | 8.61 | 0.00 | 35.14 | 15.83 | 1.54 | 4.50 |
| 011 | 14.88 | 0.23 | 36.02 | 17.70 | 2.61 | 16.84 |
| 012 | 32.68 | 4.81 | 36.60 | 17.43 | 4.37 | 25.49 |
| 013 | 16.99 | 0.00 | 37.22 | 19.65 | 2.39 | 13.34 |
| 014 | 11.78 | 0.02 | 38.12 | 16.93 | 3.31 | 22.36 |
| 015 | 19.69 | 0.64 | 28.68 | 14.63 | 2.78 | 17.02 |
| 016 | 26.54 | 3.50 | 33.72 | 18.72 | 2.76 | 12.71 |
| 017 | 10.37 | 0.00 | 49.91 | 22.36 | 1.83 | 4.88 |
| 018 | 7.77 | 0.04 | 29.77 | 16.01 | 1.61 | 9.07 |
| 019 | 19.84 | 0.15 | 32.69 | 15.91 | 5.90 | 42.69 |
| 020 | 14.25 | 0.00 | 33.23 | 17.01 | 3.61 | 22.10 |
| 021 | 17.36 | 0.36 | 36.55 | 19.44 | 2.99 | 21.10 |
| 022 | 17.92 | 0.16 | 39.36 | 18.69 | 3.92 | 20.08 |
| 023 | 12.84 | 0.01 | 36.39 | 17.13 | 2.87 | 16.66 |
| 024 | 12.01 | 0.37 | 26.85 | 11.67 | 0.95 | 14.60 |
| 025 | 7.97 | 0.12 | 33.30 | 16.95 | 1.59 | 8.09 |
| 026 | 15.97 | 0.15 | 38.86 | 20.65 | 2.96 | 21.05 |
| 027 | 10.74 | 0.00 | 35.82 | 16.84 | 2.28 | 26.92 |
| 028 | 9.75 | 0.05 | 35.51 | 16.51 | 1.35 | 17.98 |
| 029 | 18.99 | 0.55 | 37.06 | 15.54 | 5.11 | 44.04 |
| 030 | 12.19 | 0.00 | 39.63 | 18.19 | 1.86 | 10.47 |
| 031 | 10.21 | 0.00 | 39.11 | 17.10 | 1.22 | 9.64 |
| 032 | 8.41 | 0.00 | 40.35 | 18.98 | 1.96 | 38.19 |
| 033 | 20.10 | 0.00 | 34.04 | 11.59 | 2.42 | 49.04 |
| 034 | 9.26 | 0.00 | 27.40 | 13.17 | 0.97 | 10.98 |
| 035 | 10.89 | 0.05 | 35.85 | 17.67 | 2.85 | 15.27 |
| 036 | 6.67 | 0.00 | 35.11 | 17.32 | 1.32 | 6.38 |
| 037 | 8.60 | 0.09 | 35.45 | 13.34 | 2.09 | 15.94 |
| 038 | 10.57 | 0.05 | 32.84 | 12.97 | 1.17 | 24.31 |
| 039 | 10.14 | 0.00 | 35.38 | 17.76 | 2.21 | 27.06 |
| 040 | 7.64 | 0.00 | 38.39 | 21.18 | 1.75 | 5.09 |
| 041 | 9.45 | 0.02 | 42.22 | 20.88 | 2.91 | 13.17 |
| 042 | 17.02 | 0.33 | 36.76 | 17.06 | 3.78 | 29.22 |
| 043 | 9.44 | 0.09 | 36.24 | 16.67 | 1.62 | 22.28 |
| 044 | 26.60 | 2.41 | 26.04 | 7.17 | 1.97 | 42.22 |
| 045 | 14.62 | 0.82 | 38.72 | 18.44 | 4.77 | 35.42 |
| Mean | 14.05 | 0.48 | 34.52 | 16.26 | 2.63 | 20.10 |
| SD | 5.64 | 0.98 | 5.13 | 3.05 | 1.15 | 11.06 |
| MIN | 6.67 | 0.00 | 22.18 | 7.17 | 0.95 | 4.50 |
| MAX | 32.68 | 4.81 | 49.91 | 22.36 | 5.90 | 49.04 |
Oppositely, albumin shows the C–H asymmetric and symmetric stretching of >CH2 and CH3 from its hydrocarbon chain.
O stretching of ester bonds in fatty acids (Table 1). Exudates and free-protein serum were the only samples that showed this spectral band (Fig. 4). In the case of exudates, the presence of this band (and the associated peaks in the second derivative) could represent the membrane phospholipid concentration in the sample. Moreover, free-protein serum controls exhibit absorbance, which may be due to ester bond formation by the alcohol used during the deproteinization method.
O stretching of the peptidic bond of proteins and peptides, followed by C–N swinging and other vibrations of secondary protein structure components. Amide II areas are mainly determined by vibrations of the NH bend, C–N stretch, CO bend and N–C stretch (Table 1).
The protein absence of free-protein controls, like free-protein serum and urine, was demonstrated by UV spectra (Fig. 5) and the Bradford method (data not shown). These samples show lower amide I areas with respect to exudate, serum and plasma (ρ < 0.001). However, despite being free-protein samples, they still have absorbance in the amide I window (Fig. 4). This might be caused by the contribution of the CO stretching and N–H stretching and the deformation of urea,36 strong CO stretching, C–C–N bending of creatinine bonds37 as urea and creatinine spectra are also shown (Fig. 3h and j).
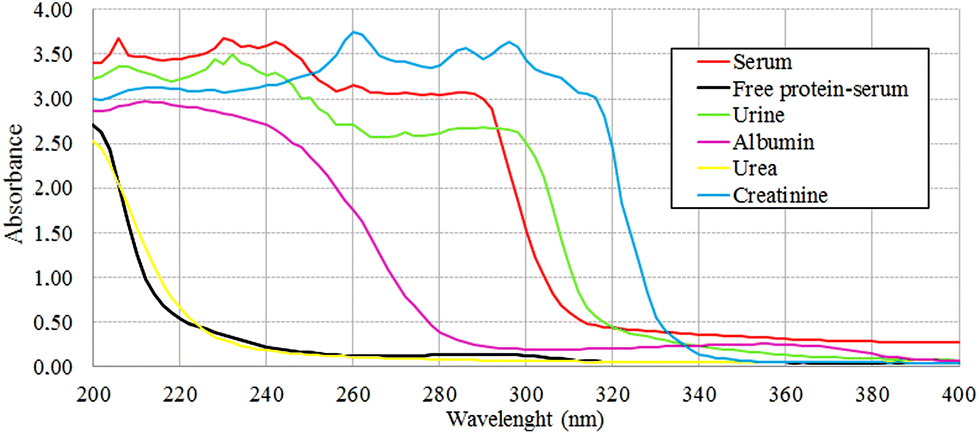 | ||
| Fig. 5 UV spectra obtained from several samples to analyze protein concentration at 280 nm. Null absorbance at 280 nm for free-protein serum was observed. Despite that urine does not have protein concentration, absorbance at 280 nm could be explained by absorbance of creatinine at this frequency. | ||
It was previously demonstrated that the wound fluid protein concentration (measured by biochemical methods) is lower than the serum protein concentration.18,21 However, there were no significant differences between amide I areas of exudate, serum and plasma (Fig. 4). As all these samples have similar concentrations of urea and creatinine,18,21 this would indicate also a similar protein concentration which is not correct. This may be because in the amide I region the contributions of the inflammatory, bacterial and serum proteins all together to exudates were detected. This compensates for the difference detected by biochemical methods that only measure inflammatory and seric proteins.18,21
Amide II areas represent the real protein content of the samples, because free-protein serum and urine show null or minimum absorbance and serum and plasma were significantly higher than exudate (ρ < 0.001) as expected (Fig. 4).
Urine and free-protein serum show null absorbance in this region, which is logical as both samples are DNA/RNA/protein free (Fig. 4 – phosphate bonds). In contrast, areas observed in serum and plasma could be caused by proteins while the observed areas in cells and exudates could be a result of the sum of nucleic acids and proteins. Taking into account that: (1) the exudate protein content is lower than the serum protein content (Fig. 4 – Amide II);18,21 (2) there is no significant difference between phosphate bonds as in serum and exudate, and (3) phosphate areas in cells are significantly lower than phosphate areas in exudates (ρ < 0.001); we could assume that the difference between the phosphate areas from exudates and cells is mainly determined by nucleic acids indirectly related to the exudate cellularity (Fig. 4 – Phosphate bonds).
In serum and plasma, area values probably came from the vibrational modes of glycoproteins, glucose and other sugars. Cells presented absorbance because of the membrane glycoproteins’ presence. Albumin has absorbance in this window because of the C–OH stretching and vibrational modes of serine, threonine and tyrosine.2 In exudates, area values probably came from the vibrational modes of cellular glycoproteins, glucose, seric glycoproteins, lipopolysaccharides from planktonic bacteria and exopolysaccharides from the bacterial biofilm matrix. There is an important variability in the polysaccharide concentration among all samples which is demonstrated by the elevated standard deviation (SD) in the polysaccharide area from exudate samples (Fig. 4 – Carbohydrate bonds). Taking into account that all controls present low SD we could assume that the elevated SD in exudates may be due to different biofilm matrix exopolysaccharides’ contribution from infecting bacteria.23,33 Because of this polysaccharide areas in exudates could indirectly represent its biofilm load.
Free-protein serum carbohydrate areas were significantly higher than exudate areas (ρ < 0.001). This may be due to spectral contributions of the remnant alcohol (C–OH) from the deproteinization method (Fig. 3l and 4).
Spectral areas of bacteria
We analyzed 95% of the aerobic clinical isolates16 from CVLU exudates. Each strain showed a unique relative composition for planktonic and biofilm phenotypes (Table 3). This could be useful to identify bacteria in the phenotypic-ulcer state. This also allows differentiation of whether bacteria are in biofilm or planktonic phenotype, which is unlikely by conventional methods. Besides, as mentioned above, a broad variability among polysaccharide areas for all bacteria spectra was observed. This could be as a result of the different exopolysaccharide composition in each case.| Bacteria | W1 | W3 | W4 | W5 | W6 | 2nd derivative peaks | |
|---|---|---|---|---|---|---|---|
| Planktonic phenotype | Staphylococcus haemolyticcus | 10.22 | 65.80 | 1.35 | 17.12 | 43.73 | 1750, 1095 |
| Staphylococcus aureus | 22.30 | 42.22 | 6.77 | 21.52 | 97.23 | 1259, 1239, 1090 | |
| MRSA | 17.11 | 55.06 | 9.02 | 11.66 | 54.63 | 968 | |
| Enterococcus faecalis | 12.76 | 48.41 | 14.02 | 18.00 | 40.53 | 1719, 1614, 1546, 1074 | |
| Enterococcus faecalis | 25.52 | 56.20 | 7.09 | 11.27 | 65.84 | 1719, 1090 | |
| Beta-hemolytic Streptococcus | 7.06 | 45.64 | 10.08 | 3.70 | 42.39 | 1695, 1545, 1230, 1078, 1015, 970 | |
| Proteus mirabilis | 13.69 | 47.82 | 12.77 | 8.09 | 30.82 | 1060 | |
| Proteus mirabilis | 12.97 | 49.84 | 16.28 | 6.56 | 30.02 | — | |
| Enterobacter sp. | 12.45 | 58.33 | 16.7 | 8.34 | 31.88 | — | |
| Enterobacter aerogenes | 16.43 | 56.51 | 10.65 | 8.60 | 56.45 | — | |
| Pseudomona sp. | 14.93 | 53.93 | 13.28 | 9.77 | 53.02 | 1731, 1227, 1177, 1127 | |
| Pseudomona sp. | 12.82 | 50.22 | 10.18 | 4.70 | 37.13 | 1731, 1227, 1097 | |
| Pseudomona. aeruginosa | 25.50 | 50.20 | 3.00 | 3.90 | 44.70 | 1665 | |
| Providencia sp. | 8.91 | 45.65 | 9.96 | 4.68 | 20.31 | — | |
| Citrobacter sp. | 13.75 | 49.88 | 13.85 | 5.63 | 36.25 | 1716 | |
| Klebsiella pneumoniae | 13.45 | 45.30 | 12.02 | 5.53 | 40.96 | 1641 | |
| Klebsiella pneumoniae | 16.74 | 53.97 | 8.92 | 6.90 | 59.65 | 1641 | |
| Serratia marcescens | 17.29 | 55.37 | 15.08 | 8.90 | 36.78 | 2874 | |
| Escherichia coli | 9.79 | 48.46 | 15.66 | 4.89 | 23.08 | 1236, 1120 | |
| Escherichia coli | 14.98 | 33.81 | 14.99 | 5.45 | 36.82 | 1236, 1120 | |
| Burkholderia cepacia | 15.26 | 56.20 | 18.16 | 7.97 | 29.43 | — | |
| Biofilm phenotype | Staphylococcus haemolyticcus | 12.95 | 56.15 | 17.60 | 8.44 | 33.67 | — |
| Staphylococcus aureus | 15.09 | 56.79 | 15.04 | 9.36 | 47.67 | 2744, 984 | |
| Beta-hemolytic Streptococcus | 14.05 | 61.75 | 13.69 | 7.44 | 52.79 | 969 | |
| Beta-hemolytic Streptococcus | 11.12 | 34.2 | 13.25 | 6.57 | 45.63 | 969 | |
| Enterococcus faecalis | 13.08 | 45.26 | 12.02 | 7.10 | 41.70 | 1634, 1212 | |
| Enterococcus faecalis | 20.31 | 49.39 | 5.45 | 10.92 | 95.41 | 1212 | |
| MRSA | 13.78 | 59.48 | 12.17 | 8.01 | 50.41 | — | |
| MR-CNS | 9.90 | 34.04 | 11.36 | 5.63 | 44.37 | 1075 | |
| CNS | 10.04 | 61.39 | 8.58 | 11.83 | 41.58 | 971 | |
| Proteus mirabilis | 13.08 | 58.88 | 15.17 | 8.00 | 30.20 | 1511, 1637, 1619, 1238, 1089 | |
| Proteus mirabilis | 13.07 | 47.27 | 14.23 | 4.415 | 27.34 | 1511, 1637, 1619, 1238, 922 | |
| Enterobacter sp | 14.89 | 46.87 | 16.23 | 8.23 | 24.29 | — | |
| Enterobacter aerogenes | 16.47 | 48.02 | 13.50 | 11.14 | 77.33 | — | |
| Pseudomona aeruginosa | 17.00 | 43.80 | 11.30 | 6.90 | 27.20 | 1085 | |
| Pseudomona sp. | 14.33 | 45.83 | 16.60 | 5.74 | 28.29 | 1223 | |
| Pseudomona sp. | 16.55 | 55.84 | 17.32 | 7.77 | 34.7 | — | |
| Citrobacter sp. | 10.24 | 47.99 | 10.77 | 5.20 | 26.76 | — | |
| Klebsiella penumoniae | 15.48 | 47.55 | 12.19 | 8.39 | 49.68 | 2873, 1163, 1104, 1068, 990 | |
| Klebsiella penumoniae | 16.97 | 49.61 | 11.50 | 7.70 | 57.43 | 2873 | |
| Escherichia coli | 11.55 | 50.5 | 16.40 | 6.54 | 30.43 | — | |
| Burkholderia cepacia | 12.53 | 47.76 | 15.5 | 5.39 | 25.25 | — | |
Peaks
A deep study of the peaks founded in 2nd derivative spectra from exudate samples, controls and bacteria in both phenotypes was performed. Here we only show the typical sample peaks that could have clinical significance.In the CH3/CH2 region (W1) a characteristic peak at ∼2933 cm−1 was found in exudate spectra (100%) and in blood cell spectra (100%). This peak could represent the presence of cellular membrane phospholipids from inflammatory cells since this peak is absent in bacteria and controls.
In the ester bond region (W2) a peak between 1716–1713 cm−1 was found in exudates (75%) and bacteria in both biofilm (100%) and planktonic (83%) phenotypes. Since this peak is absent in blood cells spectra and other controls, it could represent membrane phospolipids from bacterial cells.
In the amide I region (W3), 100% of plasma and exudate samples showed a peak at 1690 cm−1. Furthermore, 100% of serum samples present a peak at 1695 cm−1 with lower absorbance. This displacement and lower absorbance could be owing to fibrinogen that is the only proteic difference between plasma and serum (Fig. 6). Therefore, this peak could be useful to measure fibrin amounts in exudates.
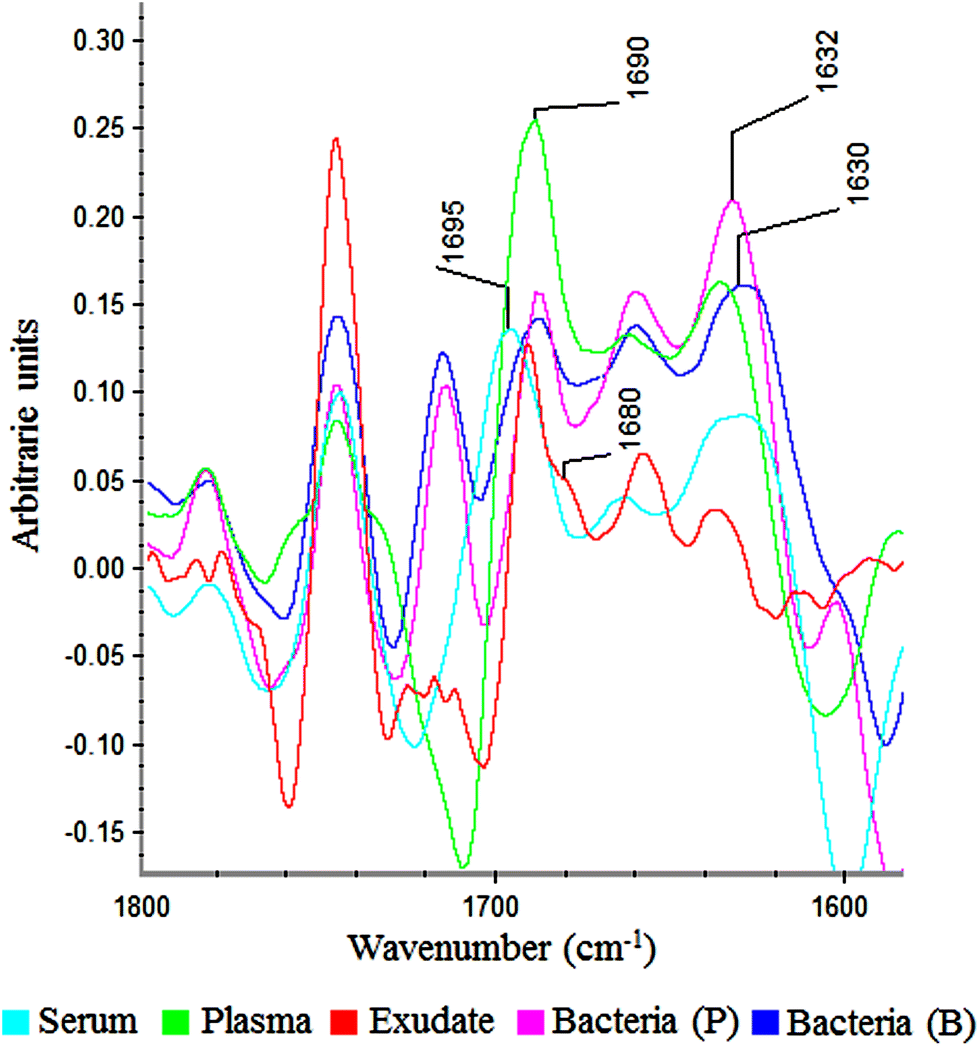 | ||
| Fig. 6 Savitzky–Golay 2nd derivative from spectra of serum, plasma, exudate and bacteria in planktonic (p) and biofilm (b) phenotypes between 1800–1600 cm−1. | ||
In all bacteria spectra (planktonic and biofilm), peaks between 1633–1629 cm−1 and 1623–1616 cm−1 were the ones with higher absorbance than other amide I peaks (Fig. 6). Besides, a specific peak between 1682 and 1680 cm−1 was founded only in exudate samples (100%). Because this peak is absent in plasma, serum and bacteria, it would represent tissue pro-inflammatory proteins. Among them we can find proteases as serine proteinase, cysteine proteinase, aspartic proteinase and matrix metalloproteinases (MMPs).12,17,19 If it is demonstrated that this peak belongs to exudate proteases, it would be extremely useful for the ulcers prognosis, as there is a correlation between elevated levels of proteases and delayed healing.12,25,26
In the amide II region (W4) a peak at 1497 cm−1 was found in 100% of exudate and serum controls although we couldn't find a possible assignment for it.
In the phosphate bonds region (W5), a specific peak between 1262 and 1260 cm−1 only in 100% of exudates (with an important absorbance) was found. Hence, it could be another representative peak for proteases as was previously assigned to Amide III vibrations (Table 1). Planktonic bacteria present a peak between 1244 and 1242 cm−1 and eukaryotic cells present a peak between 1236 and 1234 cm−1. These peaks represent a DNA A-form marker for antisymmetric PO2− stretch.37 Therefore, these peaks could represent prokaryotic/eukaryotic load in the sample as both are present in exudates.
In the carbohydrates bonds region (W6) an extraordinary variability of peaks was founded. There are only a few peaks that were sample-characteristic as 1171–1174 cm−1 for exudate (100%) and 1097–1093 cm−1 for biofilm and planktonic bacteria (100%). The rest of the founded peaks might represent the variability produced by biofilm matrix exopolysaccharides and glycoproteins in exudates and glycoproteins in plasma and serum.
Conclusion
FTIR spectroscopy allows us to identify sample types (exudates, plasmas, serum, urine, planktonic bacteria, biofilm bacteria) as each one presents a unique relative composition and ratios range. Also, this technique could be useful to identify bacteria in the phenotypic-ulcer state and allows us to differentiate if bacteria are in the biofilm or planktonic form which is unlikely by conventional methods.Because the measured areas or the located peaks are concentration-dependent, this method could serve to study several parameters in exudate as follows:
(1) Exudate cellularity.
(a) Total cellularity could be estimated from the CH3/CH2 window area.
(b) Inflammatory cells load could be estimated from the ester bond window area or by measuring 2933 cm−1 and/or 1236–1234 cm−1 peak areas from the 2nd derivative (SG).
(c) Bacterial load could be estimated by measuring 1716–1713 cm−1 and/or 1244–1242 cm−1 peak areas from the 2nd derivative (SG).
(2) Exudate total protein content
(a) In complex human fluid samples like exudates, urine, serum or plasma is advisable to use amide II areas to estimate the total protein content.
(d) The fibrin amount could be estimated by measuring the 1690 cm−1 peak area from the 2nd derivative (SG).
(b) Inflammatory proteins could be estimated by measuring 1682–1680 cm−1 and/or 1262–1260 cm−1 peak areas from the 2nd derivative (SG).
(3) Exudate biofilm load could be indirectly estimated by measuring the carbohydrate bond area.
All of these exudate parameters could be useful to evaluate patient evolution as cells and proteins from inflammatory response, fibrin and planktonic or biofilm bacterial load represent critical negative markers for wound healing. Hence, FTIR spectroscopy could be a useful technique that provides a less-invasive and simple way to represent the clinical state of the wound.
In the future, the use of other spectral contribution controls could allow the identification of more specific markers in exudate. For example, hemoglobin as a bleeding marker, purified specific phospholipids from eukaryotic membranes as an inflammatory cellularity marker, lipopolysaccharides and peptidoglycan as a bacterial cellularity marker, matrix metalloproteinases (MMP-2, MMP-8, MMP-9) as protease activity markers and different exopolysaccharides from the bacterial biofilm matrix (i.e. alginate) as specific biofilm infection markers.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by grants: BID 3664 PICT 2014, BID 2530 PICT 2014 and D-TEC 0022/2013 from the National Agency for Scientific and Technological Promotion, Argentina.References
- K. Gajjar, J. Trevisan, G. Owens, P. J. Keating, N. J. Wood, H. F. Stringfellow, P. L. MartinHirschb and F. L. Martin, Analyst, 2013, 138, 3917–3926 RSC.
- M. J. Baker, E. Gazi, M. D. Brown, J. H. Shanks, P. Gardner and M. E. Sesto Cabral, Br. J. Cancer, 2008, 99(11), 1859–1866 CrossRef CAS PubMed.
- B. Singh, R. Gautam, S. Kumar, B. N. Vinay Kumar, U. Nongthomba, D. Nandi, G. Mukherjee, V. Santosh, K. Somasundaram and S. Umapathy, Curr. Sci., 2012, 102(2), 232–244 CAS.
- C. Petibois and B. Desbat, Trends Biotechnol., 2010, 28(10), 495–500 CrossRef CAS PubMed.
- P. D. Lewis, K. E. Lewis, R. Ghosal, S. Bayliss, A. J. Lloyd, J. Wills, R. Godfrey, P. Kloer and L. Aj Mur, BMC Cancer, 2010, 10(1), 640–670 CrossRef CAS PubMed.
- J. Ollesch, S. L. Drees, H. M. Heise, T. Behrens, T. Brüning and K. Gerwert, Analyst, 2013, 138(14), 4092–4102 RSC.
- M. J. Baker, J. Trevisan, P. Bassan, R. Bhargava, H. J. Butler, K. M. Dorling, P. R. Fielden, S. W. Fogarty, N. J. Fullwood, K. A. Heys, C. Hughes, P. Lasch, P. L. Martin-Hirsch, B. Obinaju, G. D. Sockalingum, J. Sulé-Suso, R. J. Strong, M. J. Walsh, B. R. Wood, P. Gardner and F. L. Martin, Nat. Protoc., 2014, 9(8), 1771–1791 CrossRef CAS PubMed.
- R. K. Sahu and S. Mordechai, Future Oncol., 2005, 1(5), 635–647 CrossRef CAS PubMed.
- R. Serra, R. Grande, G. Buffone, V. Molinari, P. Perri, A. Perri, B. Amato, M. Colosimo and S. de Franciscis, Int. Wound J., 2016, 13(1), 53–58 CrossRef PubMed.
- G. A. Rahman, I. A. Adigun and A. Fadeyi, Ann. Afr. Med., 2010, 9, 1–4 CrossRef CAS PubMed.
- G. Lazarus, M. Fran Valle, M. Malas, U. Qazi, N. M. Maruthur, D. Doggett, O. A. Fawole, E. B. Bass and J. Zenilman, Wound Repair Regen., 2014, 22(1), 34–42 CrossRef PubMed.
- R. Serra, G. Buffone, D. Falcone, V. Molinari, M. Scaramuzzino, L. Gallelli and S. de Franciscis, Wound Repair Regen., 2013, 21(3), 395–401 CrossRef PubMed.
- H. Edwards, K. Finlayson, H. Skerman, K. Alexander, C. Miaskowski, B. Aouizerat and M Gibb, Identification of symptom clusters in patients with chronic venous leg ulcers, J. Pain Symptom Manage., 2014, 47(5), 867–75 CrossRef PubMed.
- R. T. Emalberhardt and J. D. Raffetto, Circulation, 2005, 111(18), 2398–2409 CrossRef PubMed.
- K. Gjødsbøl, J. J. Christensen, T. Karlsmark, B. Jørgensen, B. M. Klein and K. Krogfelt, Int. Wound J., 2006, 3(3), 225–231 CrossRef PubMed.
- S. V. Agale, Ulcers, 2013, 2013, 1–9 CrossRef.
- A. D. Widgerow, Wound Repair Regen., 2011, 19(3), 287–291 CrossRef PubMed.
- N. J. Trengove, S. R. Langton and M. C. Stacey, Wound Repair Regen., 1996, 4(2), 234–239 CAS.
- K. F. Cutting, Br. J. Community Nurs., 2003, 8(3), 4–9 CrossRef.
- A. J. Signer and R. A. Clark, N. Engl. J. Med., 1999, 341, 738–746 CrossRef PubMed.
- T. J. James, M. Hughes, G. W. Cherry and R. P. Taylor, Wound Repair Regen., 2000, 8(4), 264–269 CrossRef CAS PubMed.
- D. Metcalf and P. Bowler, Burns Trauma, 2013, 1(1), 5–12 CrossRef PubMed.
- I. W. Sutherland, Microbiology, 2001, 147(1), 3–9 CrossRef CAS PubMed.
- V. Papayannopoulos and A. Zychlinsky, Trends Immunol., 2009, 30(11), 513–521 CrossRef CAS PubMed.
- J. C. McDaniel, S. Roy and T. Wilgus, Wound Repair Regen., 2013, 21(3), 339–351 CrossRef PubMed.
- B. Amato, G. Coretti, R. Compagna, M. Amato, G. Buffone, D. Gigliotti, R. Grande, R. Serra and S. de Franciscis, Int. Wound J., 2015, 12(6), 641–645 CrossRef PubMed.
- C. Polson, P. Sarkar, B. Incledon, V. Raguvaran and R. Grant, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2003, 785(2), 263–275 CrossRef CAS.
- C. N. Pace, F. Vajdos, L. Fee, G. Grimsley and T. Gray, Protein Sci., 1995, 4(11), 2411–2423 CrossRef CAS PubMed.
- W. Gebauer, ELS, 2002, 2–4 Search PubMed.
- K. Kirketerp-Møller, P. Ø. Jensen, M. Fazli, K. G. Madsen, J. Pedersen, C. Moser, T. Tolker-Nielsen, N. Høiby, M. Givskov and T. Bjarnsholt, J. Clin. Microbiol., 2008, 46(8), 2717–2722 CrossRef PubMed.
- D. D. Rhoads, R. D. Wolcott, Y. Sun and S. E. Dowd, Int. J. Mol. Sci., 2012, 13(3), 2535–2550 CrossRef CAS PubMed.
- P. Lasch, Chemom. Intell. Lab. Syst., 2012, 117, 100–114 CrossRef CAS.
- A. Bosch, D. Serra, C. Prieto, J. Schmitt, D. Naumann and O. Yantorno, Appl. Microbiol. Biotechnol., 2006, 71(5), 736–747 CrossRef CAS PubMed.
- D. Naumann and P. Lasch, Biomedical Spectroscopy, Encyclopedia of analytical chemistry, Wiley J & Sons, 2015 Search PubMed.
- K. Liu, R. A. Shaw, A. Man, T. C. Dembinski and H. H. Mantsch, Clin. Chem., 2002, 48(3), 499–506 CAS.
- M. Khanmohammadi, K. Ghasemi, A. B. Garmarudi and M. Ramin, Spectrochim. Acta, Part A, 2014 Search PubMed.
- M. Banyay, M. Sarkar and A. Graslund, Biophys. Chem., 2003, 104, 477–488 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |