
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembly of noble metal nanoparticles into sub-100 nm colloidosomes with collective optical and catalytic properties†
Lei
Zhang
 a,
Qikui
Fan
a,
Xiao
Sha
a,
Ping
Zhong
a,
Jie
Zhang
b,
Yadong
Yin
c and
Chuanbo
Gao
*a
a,
Qikui
Fan
a,
Xiao
Sha
a,
Ping
Zhong
a,
Jie
Zhang
b,
Yadong
Yin
c and
Chuanbo
Gao
*a
aCentre for Materials Chemistry, Frontier Institute of Science and Technology, Xi’an Jiaotong University, Xi’an, Shaanxi 710054, China. E-mail: gaochuanbo@mail.xjtu.edu.cn
bSchool of Materials Science and Engineering, Xi’an Jiaotong University, Xi’an, Shaanxi 710049, China
cDepartment of Chemistry, University of California, Riverside, California 92521, USA
First published on 16th June 2017
Abstract
Self-assembly at the nanoscale represents a powerful tool for creating materials with new structures and intriguing collective properties. Here, we report a novel strategy to synthesize nanoscale colloidosomes of noble metals by assembling primary metal nanoparticles at the interface of emulsion droplets formed by their capping agent. This strategy produces noble metal colloidosomes of unprecedentedly small sizes (<100 nm) in high yield and uniformity, which is highly desirable for practical applications. In addition, it enables the high tunability of the composition, producing a diversity of monometallic and bimetallic alloy colloidosomes. The colloidosomes exhibit interesting collective properties that are different from those of individual colloidal nanoparticles. Specifically, we demonstrate Au colloidosomes with well-controlled interparticle plasmon coupling and Au–Pd alloy colloidosomes with superior electrocatalytic performance, both thanks to the special structural features that arise from the assembly. We believe this strategy provides a general platform for producing a rich class of miniature colloidosomes that may have fascinating collective properties for a broad range of applications.
Introduction
The bottom-up self-assembly of nanoparticles represents one of the innovative tools in nanotechnology to build complex functional architectures, with the resulting assemblies showing superior collective properties over the individual nanoparticles for a broad range of applications.1–7 Among different strategies, the use of interfaces to self-assemble nanoparticles has been widely investigated,7–9 which can be achieved typically by the Langmuir–Blodgett technique,10–12 evaporation-induced self-assembly13–17 and adsorption of the nanoparticles.18,19 In particular, the self-assembly of colloidal particles at the oil/water interface of emulsion droplets can produce an interesting family of colloidosomes in the shape of hollow capsules,20,21 which may find broad use in different applications such as biosensing, imaging and biomedicine.22–27 However, conventional synthesis strategies usually give rise to micron-sized colloidosomes.28–36 The miniaturization of colloidosomes into nanoscale ones, which is a prerequisite for many applications, remains a great challenge.22,37–39 The difficulty in the miniaturization of colloidosomes mainly lies in the less favourable reduction of the Helmholtz free energy when small particles (≪50 nm) are adsorbed on the interfaces. This leads to the convenient detachment of particles from the interface,18,22 increasing the Laplace pressure between the inside and outside of the emulsion droplets, resulting in significant coalescence of the droplets.37 Therefore, although sub-micron colloidosomes have been reported in recent years, the number of reports has been very limited so far.39–51 To the best of our knowledge, little progress has been made in the synthesis of sub-100 nm colloidosomes by the interfacial assembly of particles on emulsion droplets. On the other hand, colloidosomes are usually obtained with high polydispersity from conventional synthesis.22 Therefore, it is highly desirable to establish new synthesis strategies that can afford uniform nanoscale colloidosomes with intriguing collective properties and broad applicability for different applications.In this work, we developed a novel strategy to self-assemble ultrasmall noble metal nanoparticles (typically < 4 nm) at the oil/water interface of an emulsion, which formed uniform sub-100 nm colloidosomes (with sizes as low as ∼30 nm) after a subsequent seeded growth process (Fig. 1a). We found that polyvinylpyrrolidone (PVP), as a common capping agent for the synthesis of noble metal nanoparticles, can serve dually as the oil phase and emulsifier, forming a stable emulsion under alkaline conditions and thus enabling the assembly of a variety of noble metal nanoparticles at their interfaces, resembling a Pickering emulsion.23,52 These noble metal nanoparticles were then employed as seeds for the growth of a second metal, leading to an increase in the particle size, which further minimized the Helmholtz free energy for interfacial assembly.18 On the other hand, this led to the lattice contact of the nanoparticles driven by the minimization of the surface area and thus the surface energy, which enables the structural integrity of the assembly after the removal of PVP, giving rise to colloidosomes with remarkable porosity and structural integrity. The fact that the emulsion of PVP could serve as a template for the formation of nanoparticle assemblies has rarely been reported, but is particularly interesting. This enables the vast availability and thus a flexible range of choices of seeds and second metals for the synthesis of colloidosomes in either monometallic or bimetallic alloy forms with high compositional tunability. On the other hand, this provides strong interactions between the ultrasmall noble metal nanoparticles and the oil phase, which overcomes the momentum transferred from the solvent molecules,22 making the nanoparticles strongly confined at the oil/water interface of the emulsion for the successful formation of rigid colloidosomes.37 Although micron-sized35 and sub-micron-sized noble metal colloidosomes39,42,48–51 have been reported previously, for the first time, noble metal colloidosomes have been obtained with unprecedentedly small sizes (<100 nm) in high homogeneity and yield. These colloidosomes demonstrated intriguing collective optical properties in localized surface plasmon resonance (LSPR) and collective electrocatalytic properties in the formic acid oxidation reaction (FAOR), which proved to be superior to those of the individual nanoparticles. We believe these sub-100 nm colloidosomes represent a new family of attractive nanostructures, which may open up new opportunities in pursuit of fascinating collective properties for a broad range of applications.
 | ||
| Fig. 1 (a) Formation mechanism of the colloidosomes. (b) Size distributions of the PVP-stabilized Au seeds, the emulsion after addition of DEA and the Au colloidosomes as a final product, measured by DLS. (c) TEM image of the iodine-stained PVP emulsion. (d and e) SEM and TEM images of the monometallic Au colloidosomes, respectively. | ||
Results and discussion
The interfacial self-assembly route to the colloidosomes of noble metal nanoparticles
In a typical self-assembly route to monometallic Au colloidosomes, a solution of Au seeds (<3 nm) was first prepared by chemical reduction with PVP (0.47 wt%) as the capping agent.53 Diethylamine (DEA) was added to this solution with vigorous stirring to afford a PVP emulsion. The further growth of Au on these seeds gave rise to Au nanoparticles of ∼3.7 nm, which were assembled at the oil/water interface of the PVP emulsion, forming rigid colloidosomes. Self-supported Au colloidosomes with a clean metal surface were obtained after PVP removal by repetitive washing with water and acetic acid (ESI Fig. S6 and S7†).54The formation of a PVP emulsion was critical to the synthesis of the colloidosomes, which could be monitored by dynamic light scattering (DLS, Fig. 1b). The PVP-capped Au seeds showed a narrow size distribution at ∼3.7 nm, corresponding to the hydrodynamic size of the Au nanoparticles. After the addition of DEA, the size distribution shifted to ∼57 nm, implying that large, uniform assemblies had formed. The formation of the emulsion was further confirmed by TEM imaging after staining with Lugol’s iodine (Fig. 1c), which showed discrete droplets of PVP with an average size of ∼30 nm. The droplets were irregular in shape, which may have arisen from the drying of the emulsion on a copper grid before TEM imaging. It is believed that the formation of the PVP emulsion was induced by the destabilized hydrogen bonding between PVP and water under alkaline conditions, which led to a decrease in the solubility of PVP in water and thus caused microphase separation. After further seeded growth, the size distribution of the colloid was virtually unchanged. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images reveal that the Au colloidosomes were formed in high yield and homogeneity, with an overall size of ∼31.8 nm (standard deviation of 6.5 nm), matching the size of the PVP emulsion very well (Fig. 1d and e and S1†). Meanwhile, the SEM image further verifies that the colloidosomes are three-dimensional spherical assemblies (Fig. 1d). It is worth noting that small nanoparticles (≪50 nm) do not usually easily adsorb at the oil/water interface of an emulsion due to the less favourable free energy reduction, and they are susceptible to detachment from the interface by the momentum offered by solvent molecules.18,22 In this synthesis, the oil phase consists of PVP, an excellent capping ligand for noble metals, and it may provide strong interactions with the ultrasmall noble metal nanoparticles. This interaction is favourable for overcoming thermal fluctuations and is thus favourable for the formation of colloidosomes with small primary nanoparticles.37 It is therefore reasonable that nanoparticles of ∼3.7 nm were trapped at the interface of the PVP emulsion, and highly porous, self-supported rigid colloidosomes were eventually formed due to sintering of the primary nanoparticles by lattice contact during seeded growth.
Nanoparticle self-assembly at the interfaces of the PVP emulsion represents a highly versatile strategy, which can produce a diversity of monometallic and bimetallic alloy colloidosomes with broadly tunable metal ratios, not limited to monometallic Au colloidosomes. The compositions of the colloidosomes can be conveniently tuned by choosing different metals for the seeds and the subsequent seeded growth. Remarkably, bimetallic Au–Pd (Fig. 2a and S4†), Au–Pt (Fig. 2b) and Pd–Pt alloy colloidosomes (Fig. 2c) and monometallic Pd colloidosomes (Fig. 2d) were synthesized by choosing Au, Au, Pd and Pd as the metals for the respective seeds, and Pd, Pt, Pt and Pd as the metals for the respective seeded growth. The metal compositions of these colloidosomes were confirmed by energy dispersive X-ray spectroscopy (EDS) (Fig. 2a–d).
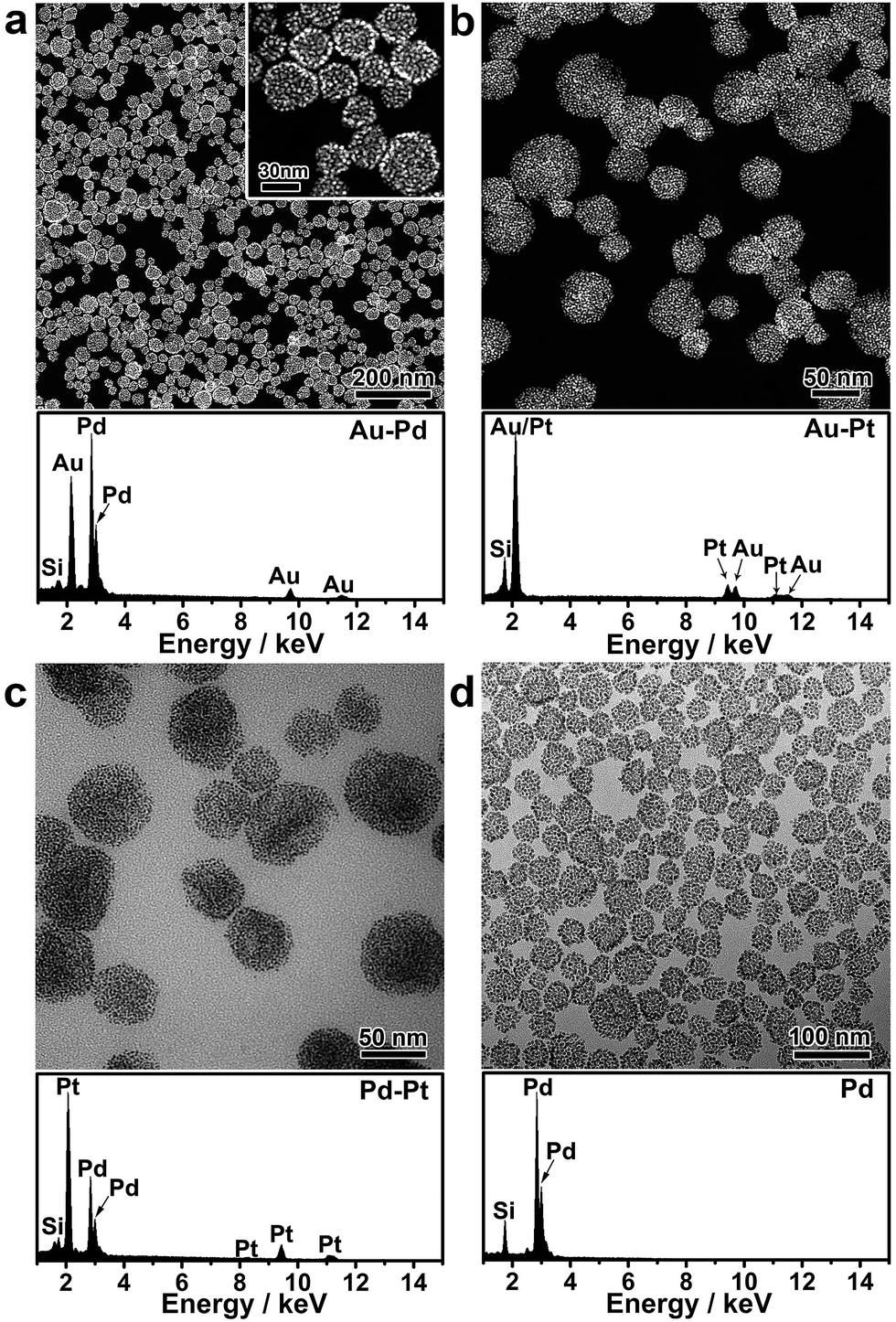 | ||
| Fig. 2 Versatility of the nanoparticle assembly approach. (a–d) Electron microscopy (HAADF-STEM or TEM) images and corresponding EDS of the bimetallic Au–Pd alloy colloidosomes (a), Au–Pt alloy colloidosomes (b), Pd–Pt alloy colloidosomes (c) and monometallic Pd colloidosomes (d). Si signals in the EDS arise from the silicon substrate. | ||
In addition, the metal ratios in the bimetallic alloy colloidosomes can be readily tuned by adjusting the ratios of the metal precursors in the synthesis. Taking Au–Pd colloidosomes as an example, the Pd/Au ratios are tunable from 0.53 to 14 by varying the amount of Na2PdCl4 in the seeded growth upon a given volume of Au seeds (Fig. 3a and Table S1†). It is worth noting that the Pd/Au ratio of the Au–Pd alloy colloidosomes is lower than that of the feeding precursor, indicating that Na2PdCl4 was partially reduced during the seeded growth. However, a clear linear relationship between the Pd/Au ratio in the final product and that in the feeding precursor can be observed, which enables the precise control of the Pd/Au ratios in the bimetallic Au–Pd alloy colloidosomes by tuning the concentrations of the precursors. The X-ray diffraction (XRD) patterns of the Au–Pd colloidosomes show a single set of reflections with their positions in between the standard peak positions of Au and Pd (Fig. 3b). Moreover, the XRD profile cannot be deconvoluted into reflections of Au and Pd, especially at high-index reflections (e.g. 220) where the respective reflections from Au and Pd are far apart from each other, confirming that an alloy of Au and Pd has been formed. With increasing Pd/Au ratios, the X-ray reflections shift continuously to high angles, implying that contraction of the face-centered cubic lattices takes place, which is in good agreement with Vagard’s law.
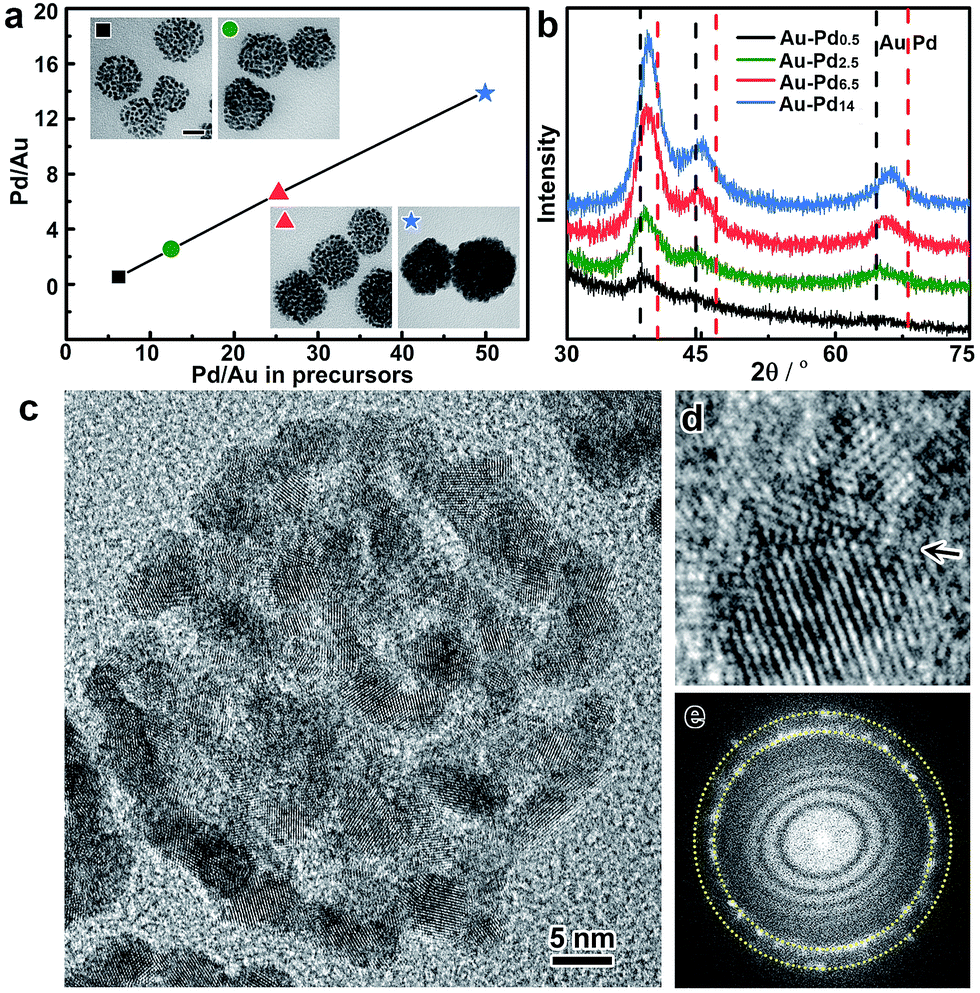 | ||
| Fig. 3 Compositional and structural analysis of the Au–Pd alloy colloidosomes. (a) The Pd/Au ratio of the Au–Pd alloy colloidosomes as a function of the ratio in the feeding precursor, measured by inductively coupled plasma mass spectrometry (ICP-MS). Inset: TEM images of the Au–Pd alloy colloidosomes with different Pd/Au ratios; scale bar: 20 nm. (b) XRD patterns of the Au–Pdx alloy colloidosomes (x = 0.5, 2.5, 6.5 and 14). The standard peak positions of Au and Pd (111, 200 and 220) are shown for reference. (c) HRTEM image of an individual colloidosome. (d) A defective boundary indicated by an arrow. (e) The corresponding Fourier diffractogram. The dotted lines indicate the {111} and {200} diffraction rings. | ||
The crystal structures of the colloidosomes were further examined, taking bimetallic Au–Pd alloy colloidosomes as an example. Uniform Au–Pd alloy colloidosomes were obtained with nanoparticle grains of ∼3.2 nm for all Pd/Au ratios investigated, which could be confirmed by high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and high-resolution TEM (HRTEM) images (Fig. 2a, 3a and c and S8†). The HRTEM images suggest that the Au–Pd colloidosomes are polycrystalline with randomly oriented nanoparticle grains, which can be attributed to the crystal growth initiated from separate seeds (Fig. 3c). Intimate contact of the interparticle lattices can be observed, forming a highly porous nanostructure and abundant crystallographic defects (Fig. 3d). The Au–Pd alloying, rather than forming a core/shell structure, can be further confirmed by the Fourier diffractogram (Fig. 3e) where only one set of the diffraction rings could be observed, without discernible diffraction spots corresponding to individual Au and Pd nanocrystals. It is assumed that the surprisingly high tendency of the metals to become an alloy may stem from the ultrasmall size of the nanoparticle grains and thus the high mobility (low melting point) of the metal atoms.55 The high mobility of the metals further favours the sintering of neighbouring particles, which accounts for the rigid nanostructure of the colloidosomes by lattice contact. In general, the colloidosomes are a family of monometallic or bimetallic alloy assemblies of nanoparticles, which are polycrystalline with high porosity and abundant crystallographic defects.
Collective optical properties of the Au colloidosomes
The noble metal nanoparticle-assembled colloidosomes show collective optical properties which are superior to those of their individual nanoparticles. The assembly of nanoparticles in the colloidosomes affords a high density of interparticle nanogaps and thus a high porosity, which may cause strong plasmon coupling at these nanogaps if the colloidosomes are composed of Au, Ag or their alloys, giving rise to peculiar optical properties and remarkable activity in surface-enhanced Raman scattering (SERS) applications. In our demonstration, Au colloidosomes were synthesized with an overall size of ∼32 nm and well-tunable interparticle nanogaps (4.6 and 1.7 nm) by changing the amount of HAuCl4 in seeded growth (Fig. 4a and b). Due to the nanoparticle assembly and thus the formation of nanogaps, the Au colloidosomes show distinct localized surface plasmon resonance (LSPR) in the visible range of the spectrum. In contrast to the individual Au nanoparticles of ∼3.7 nm, which show a broad LSPR band at ∼520 nm due to strong plasmon scattering at the particle surfaces, the Au colloidosomes with nanogaps of 4.6 and 1.7 nm display pronounced LSPR bands at ∼537 and ∼578 nm, respectively, showing a high extinction efficiency and well-defined shape of their profiles (Fig. 4c). Thus, the Au colloidosomes are wine-red and blue-violet in color, respectively, which are distinct from the colors of the individual Au nanoparticles (∼3.7 nm, light red, Fig. S9†) and the initial Au seeds for the assembly (<3 nm, light yellow) (Fig. 4c, inset). The finite-difference time-domain (FDTD) simulation implies that the pronounced LSPR band arises from nanoparticle coupling at the interparticle nanogaps, with the strongest local electromagnetic field appearing at the nanogaps, rather than in proximity of the individual nanoparticles in dipole mode resonance (Fig. 4d, inset). With a decreasing size of the interparticle nanogaps, the LSPR band of the Au colloidosomes shifts towards red, suggesting that the optical properties of the Au colloidosomes are sensitive to the size of the nanogaps and therefore can be conveniently tuned for plasmonic applications. | ||
| Fig. 4 Synthesis and optical properties of the monometallic Au colloidosomes. (a and b) TEM images of the Au colloidosomes with different gap sizes (4.6 and 1.7 nm). Inset: histograms of the size of the nanogaps measured from the TEM image. (c) Extinction spectra of the Au colloidosomes with different gap sizes, in comparison with the pristine Au seeds. Inset: corresponding digital photographs of the colloids. (d) SERS activity of the Au colloidosomes for detecting crystal violet (10−6 M) adsorbed on a silicon substrate. Au nanospheres of ∼3.7 nm and ∼30 nm were employed as control samples. Inset: FDTD simulation of the local electromagnetic field in a cross-section of an Au colloidosome under irradiation of a plane wave (633 nm). The color bar represents the amplitude ratio of E2/E02, where E and E0 are the local and incident electromagnetic fields, respectively. | ||
The intense local electromagnetic fields at the interparticle nanogaps of the Au colloidosomes represent hotspots for SERS analysis for the highly sensitive detection of the molecules of interest. We experimentally verified the SERS activity of the Au colloidosomes in comparison with Au nanospheres of ∼3.7 nm (close to the size of the nanoparticle grains, Fig. S9†) and ∼30 nm (close to the overall size of the colloidosomes, Fig. S9†). Strong and well-resolved Raman vibrational signals of crystal violet, a model analyte, have been detected from the substrate of the Au colloidosomes with nanogaps of 1.7 nm, which were ∼53 times stronger than those from the substrate of the 3.7 nm Au nanospheres, and ∼8.4 times stronger than those from the substrate of the 30 nm Au nanospheres (Fig. 4d). It is therefore clear that the Au colloidosomes are superior in fabricating highly active SERS substrates for much improved SERS activity due to the nanoparticle assembly and therefore their collective optical properties. In addition, the size of the nanogaps also plays a critical role in the SERS activity of the Au colloidosomes, with small gaps producing significantly increased Raman intensity, which enables the high designability of the Au colloidosomes for optimal activity in chemical sensing applications (Fig. 4d).
Collective catalytic properties of the Au–Pd alloy colloidosomes
On the other hand, these noble metal nanoparticle-assembled colloidosomes show collective catalytic properties, which are also superior to those of their individual nanoparticles, especially for the Pt and Pd-based colloidosomes. As crystallographic defects have been generally recognized as the active sites for many catalytic reactions, excellent catalytic activity of the colloidosomes could be expected as a result of the abundant defective boundaries arising from the intimate lattice contact of the nanoparticles in the assemblies (Fig. 3d).56–61 These defects, along with the small size of nanoparticle grains which affords a large population of unsaturated surface sites,55 in addition to the tunable alloy compositions that enable synergistic effects between the metals,60,62–65 promise excellent activity of the colloidosomes in many catalytic reactions. In addition, the assembly in the form of a self-supported nanostructure favours structural integrity and thus remarkable durability in practical catalysis.To examine the catalytic properties, monometallic Pd colloidosomes and bimetallic Au–Pd alloy colloidosomes with different Pd/Au ratios (denoted as Au–Pdx, with x representing the Pd/Au ratio: x = 2.5, 6.5 and 14) were applied in the electrochemical formic acid oxidation reaction (FAOR). For all colloidosomes investigated, the average sizes of the nanoparticle grains were ∼3.2 ± 0.2 nm, and the average overall sizes were ∼33 ± 1 nm (Fig. S8†). Commercial Pd/C (Pd 10%, size ∼3.5 nm) was employed as a benchmark, with a similar grain size to the Pd and Au–Pd colloidosomes (Fig. S10†). The electrochemically active surface areas (ECSAs) of the Au–Pd2.5, Au–Pd6.5, Au–Pd14 and Pd colloidosomes and Pd/C were 46, 41, 53, 43 and 80 m2 g−1, respectively, estimated by the underpotential deposition (UPD) of monolayer Cu (Fig. S11†). All colloidosomes were highly active in catalyzing the FOAR, with the mass activity increasing in the order of Pd/C (0.30 mA μg−1) < Pd colloidosomes (0.43 mA μg−1) < Au–Pd14 colloidosomes (0.79 mA μg−1) < Au–Pd2.5 colloidosomes (1.0 mA μg−1) < Au–Pd6.5 colloidosomes (1.3 mA μg−1) (Fig. 5a and b). Remarkably, the Au–Pd6.5 colloidosomes exhibited the highest mass activity, which was ∼4.3 times that of commercial Pd/C. This order is also valid for the specific activity of the catalysts, with the specific activity of the Au–Pd6.5 colloidosomes (3.19 mA cm−2) being ∼8.6 times that of commercial Pd/C (0.37 mA cm−2, Fig. 5b and S11†). It is surprising that compared with Pd/C, the monometallic Pd colloidosomes already showed significantly increased catalytic activity toward the FAOR, even though they were almost the same size as the primary nanoparticles, which emphasized the critical role of the nanoparticle assembly in improving the catalytic properties. It is reasonable that due to the self-assembly, the Pd nanoparticles are closely packed to afford abundant crystallographic defects, which effectively reduces the activation energy for the favorable kinetics of the FAOR.56–61 Furthermore, the alloying of Pd with Au provides an additional opportunity to improve their catalytic properties due to the bimetallic strain effect.64 The alloying with Au leads to an upward shift of the d-band centre of Pd and thus significantly improves its binding strength with formic acid for favorable reaction kinetics. Thanks to the self-assembly and the highly tunable compositions, optimal catalytic activity toward the FAOR has been achieved with the Au–Pd6.5 alloy colloidosomes as the catalyst, which shows among the highest activities of state-of-the-art Pd-based catalysts reported so far for this type of electrocatalysis (Table S2†).
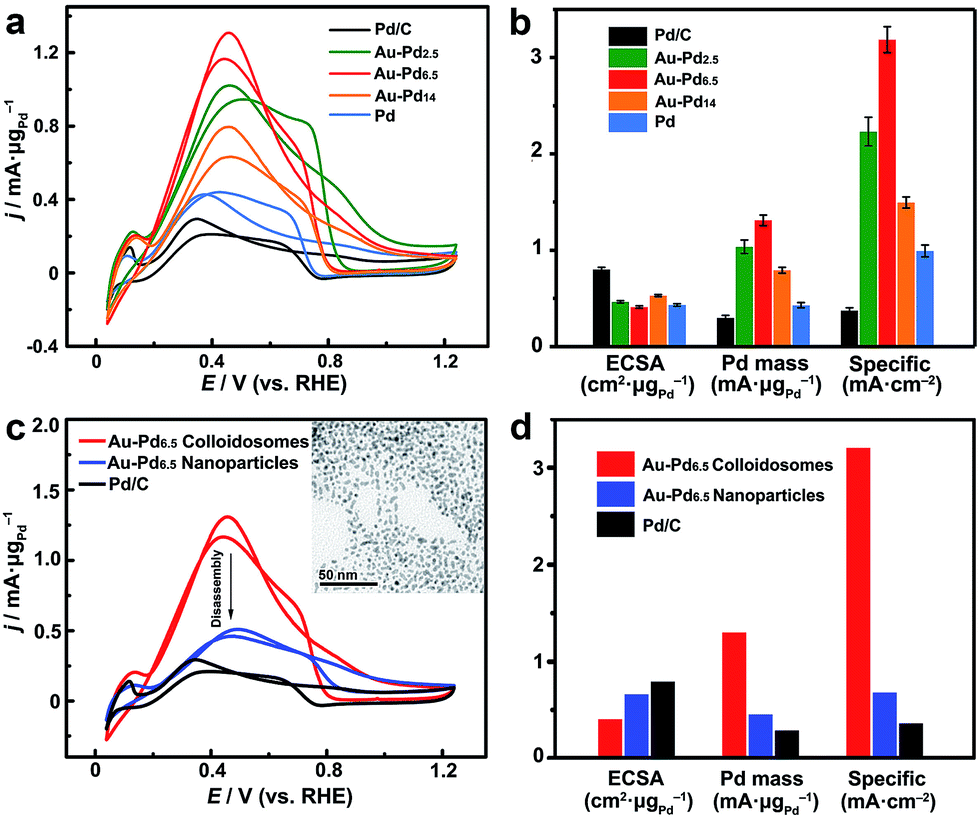 | ||
| Fig. 5 Electrocatalytic properties of Pd and Au–Pd alloy colloidosomes in the FAOR. (a) CV of the Pd and Au–Pd alloy colloidosomes with different Pd/Au ratios in N2-saturated HCOOH (0.25 M)/H2SO4 (0.5 M), in comparison with Pd/C (scan rate: 50 mV s−1). The catalytic activities were normalized to the mass of Pd. (b) Comparison of the ECSAs (measured by the UPD of Cu), mass and specific activities of the catalysts. Error bars indicate the standard deviations of the values from 3 independent measurements. (c) CV of the Au–Pd alloy nanoparticles after disassembly, in comparison with the Au–Pd alloy colloidosomes and Pd/C. Inset: TEM image of the Au–Pd alloy nanoparticles obtained by disassembly. (d) Comparison of the ECSAs (measured by the UPD of Cu), mass and specific activities of the Au–Pd alloy nanoparticles after disassembly with those of the Au–Pd alloy colloidosomes and Pd/C. | ||
In order to further reveal the collective catalytic properties of the Au–Pd alloy colloidosomes, the colloidosomes were disassembled into individual nanoparticles (inset of Fig. 5c, S12 and S13†), which were subjected to FAOR measurements (Fig. 5c and d and S14†). The average size of the resulting Au–Pd alloy nanoparticles was ∼3.3 ± 0.2 nm (Fig. S13†), similar to that of the primary nanoparticles in the Au–Pd alloy colloidosomes (∼3.2 ± 0.3 nm). After disassembly, the ECSA of the Au–Pd alloy nanoparticles increased to 1.6 times that of the colloidosomes, which can be attributed to the exposure of interparticle contact areas in the original assembly. However, the specific activity of the nanoparticles dropped to ∼21% of that of the colloidosomes, due to a loss of crystallographic defects in between the primary nanoparticles of the colloidosomes. As a result, the Au–Pd alloy nanoparticles showed much decreased mass activity after the disassembly. This illustrates that the nanoparticles are better electrocatalysts when they are self-assembled into hierarchical colloidosomes. In other words, although the surface area was compromised after the nanoparticle self-assembly, the specific activity of the colloidosomes was significantly increased due to the formation of abundant crystallographic defects, which not only compensated for the loss in surface area, but also gave rise to the much increased overall mass activity of the colloidosomes. It is worth noting that the catalytic activity of the Au–Pd nanoparticles was already much higher than that of commercial Pd/C, which can be rationalized by the synergistic effect rendered by the effective alloying of Pd with Au.
The self-assembly of the nanoparticles leads to the formation of a self-supported nanostructure, which is favorable for retaining the structural integrity and the ECSA for much enhanced stability in practical catalytic applications. The long-term durability of the catalysts for catalyzing the FAOR was thus investigated (Fig. 6). After 500 cycles of cyclic voltammetry (CV), the Au–Pd alloy colloidosomes retained ∼94% of their original catalytic activity, showing the excellent durability of the catalyst. TEM imaging confirmed the structural integrity of the colloidosomes during the cycling process. For comparison, the commercial Pd/C lost the majority of its activity after 500 cycles of electrocatalytic catalysis. Substantial agglomeration of the Pd nanoparticles can be observed by TEM after the cycling process, which accounts for the loss of the ECSA and thus the electrocatalytic activity.
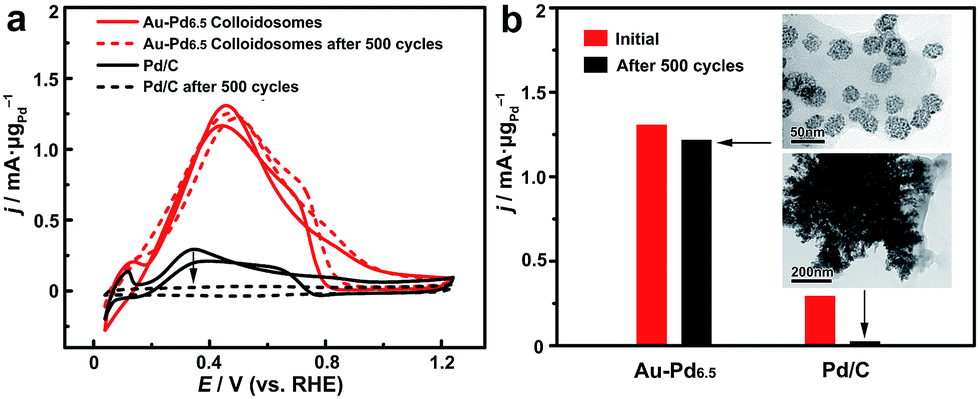 | ||
| Fig. 6 Catalytic stability of the Au–Pd alloy colloidosomes in comparison with commercial Pd/C. (a) CV of the Au–Pd alloy colloidosomes and Pd/C before and after potential sweeps for 500 cycles. (b) Comparison of the mass activities of the Au–Pd alloy colloidosomes and Pd/C before and after the potential sweeps. Inset: TEM images of the Au–Pd alloy colloidosomes (up) and Pd/C (bottom) after the cycling process. | ||
Conclusion
In summary, we have presented a unique route to self-assemble ultrasmall noble metal nanoparticles (<4 nm) at the interface of a PVP emulsion under alkaline conditions, which affords a novel family of colloidosomes with unprecedentedly small sizes (<100 nm). The advantages of this self-assembly strategy include the small size, high homogeneity and high yield of the colloidosomes that can be obtained, the generality in producing colloidosomes with broadly tunable compositions in either monometallic or bimetallic alloy forms, the versatility in tuning the porosity, the presence of abundant crystallographic defects and the formation of self-supported colloidosomes with remarkable integrity. As a result, the collective properties of the assemblies can be observed in optics and catalysis, arising from the high-density interparticle nanogaps and the abundant crystallographic defects formed by the nanoparticle self-assembly. We believe the nanoscale colloidosomes represent a new family of attractive nanostructures (see Fig. S17–S20† for colloidosomes with other metal compositions), which may open up new opportunities in chemical sensing, biomedicine, catalysis and many other applications.Experimental
Synthesis of PVP-capped Au seeds and Pd seeds
In a typical synthesis of PVP-capped Au seeds, 5 mL of PVP (Mw 29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 5 wt% in H2O) and 10 μL of HAuCl4 (0.25 M) were dissolved in 5 mL of H2O. With vigorous stirring, 600 μL of ice-cold NaBH4 (0.1 M) was injected into the solution, producing a yellowish solution with Au nanoparticles of <3 nm. After stirring for 1 h, the solution was collected as a stock solution without further purification. The synthesis of PVP-capped Pd seeds followed the same procedure except for the use of 80 μL of Na2PdCl4 (0.0625 M) in place of HAuCl4.
000, 5 wt% in H2O) and 10 μL of HAuCl4 (0.25 M) were dissolved in 5 mL of H2O. With vigorous stirring, 600 μL of ice-cold NaBH4 (0.1 M) was injected into the solution, producing a yellowish solution with Au nanoparticles of <3 nm. After stirring for 1 h, the solution was collected as a stock solution without further purification. The synthesis of PVP-capped Pd seeds followed the same procedure except for the use of 80 μL of Na2PdCl4 (0.0625 M) in place of HAuCl4.
Synthesis of Au colloidosomes
In a typical synthesis of Au colloidosomes, 1.6 mL of the stock solution of Au seeds, 40 μL of diethylamine (DEA), 80 μL of HAuCl4 (0.025 M) and 1.6 mL of ascorbic acid (AA, 10 mM) were added to 16 mL of H2O in order with vigorous stirring at room temperature (25 °C). The reaction was allowed to proceed for 20 min. The resulting Au colloidosomes (gap size: ∼4.6 nm) were collected by centrifugation and washed with H2O 5 times. Au colloidosomes with a gap size of ∼1.7 nm were synthesized following a similar procedure except for the addition of 180 μL of HAuCl4 (0.025 M) instead of 80 μL of HAuCl4 (0.025 M).Synthesis of Pd colloidosomes
In a typical synthesis of Pd colloidosomes, 9.6 mL of the stock solution of Pd seeds, 960 μL of DEA, 480 μL of Na2PdCl4 (0.0625 M) and 9.6 mL of AA (10 mM) were added to 96 mL of H2O in order with vigorous stirring at 50 °C. The reaction was allowed to proceed for 24 h. The resulting Pd colloidosomes were then collected by centrifugation and washed with H2O 5 times.Synthesis of Au–Pd alloy colloidosomes
In a typical synthesis of Au–Pd colloidosomes, 9.6 mL of the stock solution of Au seeds, 960 μL of DEA, 480 μL of Na2PdCl4 (0.0625 M) and 9.6 mL of AA (10 mM) were added to 96 mL of H2O in order with vigorous stirring at 50 °C. The reaction was allowed to proceed for 24 h. The resulting Au–Pd colloidosomes were then collected by centrifugation and washed with H2O 5 times.Surface-enhanced Raman scattering (SERS)
The substrates for SERS were prepared by dropping and drying a known quantity of Au colloidosomes (or Au nanospheres) on a clean silicon wafer (8 mm × 8 mm). In order to clean the surface of the nanoparticles, acetic acid (50%) was added onto the above silicon substrate. After 10 min, excess acetic acid was cleaned by rinsing with H2O 5 times. The substrate was then dried at ambient temperature. In a typical SERS analysis, 25 μL of crystal violet (10−6 M) was dropped and dried on the substrate in the absence of light. Raman spectra were recorded using a LabRAM HR800 confocal Raman spectrophotometer equipped with a 633 nm He–Ne laser line at room temperature. For all measurements, laser spots were 0.85 μm under a 100× objective, the power of the laser spot was 0.3 mW and the signal acquisition time was fixed to be 1 s.Electrochemical measurements
Electrochemical measurements were conducted with an Autolab PGSTAT302N electrochemical workstation with a three-electrode system (Pine Research Instrumentation). A glassy carbon rotating disk electrode (RDE; diameter: 5 mm; geometric area: 0.196 cm2) equipped on a Pine rotator, Pt foil, and a Ag/AgCl (3 M KCl) electrode (calibrated before use) were employed as the working, counter and reference electrodes, respectively. All potentials were converted into values with reference to a reversible hydrogen electrode (RHE). The ECSAs of the Pd-based catalysts were estimated by the underpotential deposition (UPD) of Cu in N2-saturated solution containing 0.05 M H2SO4 and 0.05 M CuSO4, with a reference value of 420 μC cm−2.66,67Formic acid oxidation reaction (FAOR)
The as-prepared catalysts cleaned with H2O were dispersed in acetic acid, heated at 60 °C for 12 h and washed with ethanol/H2O 4 times to clean the surface of the nanoparticles.54 The catalysts were then re-dispersed in H2O. Aqueous dispersions of the catalysts, such as commercial Pd/C (Pd, 10 wt%, Sigma-Aldrich), Pd colloidosomes and Au–Pd colloidosomes with different Pd–Au ratios, were ultrasonicated for 30 min to make a homogeneous ink, with the Pd concentration fixed to be 0.2 g L−1 (determined by ICP-MS). Then, 20 μL (Pd, 4 μg) of the ink was dropped onto the RDEs and dried at room temperature in air. After that, 15 μL of Nafion (0.05 wt%, Sigma-Aldrich) was dropped onto the RDEs and dried again. Before electrocatalytic measurements, the working electrode was conditioned by continuous cyclic voltammetric cycles in 0.5 M H2SO4 (100 mV s−1) until a stable CV curve was obtained. The FAOR was conducted by recording CV curves at a sweep rate of 50 mV s−1 in N2-saturated 0.5 M H2SO4 with 0.25 M HCOOH at room temperature. The durability of the catalysts was determined by investigating the CV curves before and after 500 cycles of sweeping at a rate of 50 mV s−1 at room temperature.Characterization
TEM was performed on a Hitachi HT-7700 microscope equipped with a LaB6 filament, operated at 100 kV. High-resolution TEM (HRTEM) was performed on an FEI Tecnai F20 FEG-TEM, operating at 200 kV. The PVP emulsion was visualized by TEM after staining with Lugol’s iodine (5% I2 + 10% KI). Typically, 160 μL of DEA and 40 μL of Lugol’s iodine were added to 16 mL of 10 × diluted PVP/seeds in sequence with stirring. After stirring for 1 h, the resulting emulsion was dropped onto a copper grid and dried for TEM imaging. SEM and EDS were performed on a field-emission JEOL JSM-7800F microscope operating at 15 kV. DLS was performed on a Beckman Coulter Delsa Nano C particle analyzer. XRD patterns were recorded on a Rigaku SmartLab Powder X-ray diffractometer equipped with a Cu Kα X-ray source and a D/teX Ultra detector, scanning at a rate of 5° min−1 over a range of 10–90° (2θ). UV-vis spectroscopy was recorded on an Ocean Optics HR2000 + ES UV-vis-NIR spectrophotometer with a DH-2000-Bal light source. ICP-MS was performed on an Agilent 7500ce.Acknowledgements
C. G. acknowledges the support from the National Natural Science Foundation of China (21671156, 21301138) and the startup fund from Xi’an Jiaotong University. Y. Y. acknowledges the support from the U.S. National Science Foundation (CHE-1308587). The authors thank Prof. Liqing Huang at School of Science, Xi’an Jiaotong University, and Zhongbo Yang at Chongqing Institute of Green and Intelligent Technology, Chinese Academy of Sciences, for help with FDTD simulations.Notes and references
- E. V. Shevchenko, D. V. Talapin, N. A. Kotov, S. O’Brien and C. B. Murray, Nature, 2006, 439, 55–59 CrossRef CAS PubMed.
- M. A. Boles, M. Engel and D. V. Talapin, Chem. Rev., 2016, 116, 11220–11289 CrossRef CAS PubMed.
- M. P. Pileni, Acc. Chem. Res., 2007, 40, 685–693 CrossRef CAS PubMed.
- X. Shen, L. Chen, D. Li, L. Zhu, H. Wang, C. Liu, Y. Wang, Q. Xiong and H. Chen, ACS Nano, 2011, 5, 8426–8433 CrossRef CAS PubMed.
- H. Wang, L. Chen, X. Shen, L. Zhu, J. He and H. Chen, Angew. Chem., Int. Ed., 2012, 51, 8021–8025 CrossRef CAS PubMed.
- M. Pang, Q. Wang and H. C. Zeng, Chem.–Eur. J., 2012, 18, 14605–14609 CrossRef CAS PubMed.
- S. Kinge, M. Crego-Calama and D. N. Reinhoudt, ChemPhysChem, 2008, 9, 20–42 CrossRef CAS PubMed.
- H. Ma and J. Hao, Chem. Soc. Rev., 2011, 40, 5457–5471 RSC.
- L. Yao, C. Liu, W. H. Chong, H. Wang, L. Chen and H. Chen, Small, 2015, 11, 232–238 CrossRef CAS PubMed.
- A. Tao, P. Sinsermsuksakul and P. Yang, Nat. Nanotechnol., 2007, 2, 435–440 CrossRef CAS PubMed.
- A. R. Tao, J. Huang and P. Yang, Acc. Chem. Res., 2008, 41, 1662–1673 CrossRef CAS PubMed.
- H.-L. Nie, X. Dou, Z. Tang, H. D. Jang and J. Huang, J. Am. Chem. Soc., 2015, 137, 10683–10688 CrossRef CAS PubMed.
- A. Courty, A. Mermet, P. A. Albouy, E. Duval and M. P. Pileni, Nat. Mater., 2005, 4, 395–398 CrossRef CAS PubMed.
- E. V. Shevchenko, D. V. Talapin, N. A. Kotov, S. O’Brien and C. B. Murray, Nature, 2006, 439, 55–59 CrossRef CAS PubMed.
- W. Cheng, M. J. Campolongo, J. J. Cha, S. J. Tan, C. C. Umbach, D. A. Muller and D. Luo, Nat. Mater., 2009, 8, 519–525 CrossRef CAS PubMed.
- G. D. Moon, T. I. Lee, B. Kim, G. Chae, J. Kim, S. Kim, J.-M. Myoung and U. Jeong, ACS Nano, 2011, 5, 8600–8612 CrossRef CAS PubMed.
- J. T. Zhang, L. Wang, D. N. Lamont, S. S. Velankar and S. A. Asher, Angew. Chem., Int. Ed., 2012, 51, 6117–6120 CrossRef CAS PubMed.
- Y. Lin, H. Skaff, T. Emrick, A. D. Dinsmore and T. P. Russell, Science, 2003, 299, 226–229 CrossRef CAS PubMed.
- A. Böker, J. He, T. Emrick and T. P. Russell, Soft Matter, 2007, 3, 1231–1248 RSC.
- O. D. Velev, K. Furusawa and K. Nagayama, Langmuir, 1996, 12, 2374–2384 CrossRef CAS.
- A. D. Dinsmore, M. F. Hsu, M. G. Nikolaides, M. Marquez, A. R. Bausch and D. A. Weitz, Science, 2002, 298, 1006–1009 CrossRef CAS PubMed.
- T. Bollhorst, K. Rezwan and M. Maas, Chem. Soc. Rev., 2017, 46, 2091–2126 RSC.
- J. Wu and G. H. Ma, Small, 2016, 12, 4633–4648 CrossRef CAS PubMed.
- K. L. Thompson, M. Williams and S. P. Armes, J. Colloid Interface Sci., 2015, 447, 217–228 CrossRef CAS PubMed.
- J. Song, P. Huang, H. Duan and X. Chen, Acc. Chem. Res., 2015, 48, 2506–2515 CrossRef CAS PubMed.
- Y. Liu, Y. Liu, J. J. Yin and Z. Nie, Macromol. Rapid Commun., 2015, 36, 711–725 CrossRef CAS PubMed.
- D. Patra, A. Sanyal and V. M. Rotello, Chem.–Asian J., 2010, 5, 2442–2453 CrossRef CAS PubMed.
- P. F. Noble, O. J. Cayre, R. G. Alargova, O. D. Velev and V. N. Paunov, J. Am. Chem. Soc., 2004, 126, 8092–8093 CrossRef CAS PubMed.
- D. Lee and D. A. Weitz, Adv. Mater., 2008, 20, 3498–3503 CrossRef CAS.
- K. L. Thompson, P. Chambon, R. Verber and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 12450–12453 CrossRef CAS PubMed.
- J. He, Z. Wei, L. Wang, Z. Tomova, T. Babu, C. Wang, X. Han, J. T. Fourkas and Z. Nie, Angew. Chem., Int. Ed., 2013, 52, 2463–2468 CrossRef CAS PubMed.
- X. W. Xu, X. M. Zhang, C. Liu, Y. L. Yang, J. W. Liu, H. P. Cong, C. H. Dong, X. F. Ren and S. H. Yu, J. Am. Chem. Soc., 2013, 135, 12928–12931 CrossRef CAS PubMed.
- M. Cui, T. Emrick and T. P. Russell, Science, 2013, 342, 460–463 CrossRef CAS PubMed.
- G. C. Phan-Quang, H. K. Lee, I. Y. Phang and X. Y. Ling, Angew. Chem., Int. Ed. Engl., 2015, 54, 9691–9695 CrossRef CAS PubMed.
- D. Liu, F. Zhou, C. Li, T. Zhang, H. Zhang, W. Cai and Y. Li, Angew. Chem., Int. Ed., 2015, 54, 9596–9600 CrossRef CAS PubMed.
- B. Y. Guan, L. Yu and X. W. Lou, Adv. Mater., 2016, 28, 9596–9601 CrossRef CAS PubMed.
- X. C. Yang, B. Samanta, S. S. Agasti, Y. Jeong, Z. J. Zhu, S. Rana, O. R. Miranda and V. M. Rotello, Angew. Chem., Int. Ed., 2011, 50, 477–481 CrossRef CAS PubMed.
- P. Huang, J. Lin, W. Li, P. Rong, Z. Wang, S. Wang, X. Wang, X. Sun, M. Aronova, G. Niu, R. D. Leapman, Z. Nie and X. Chen, Angew. Chem., Int. Ed., 2013, 52, 13958–13964 CrossRef CAS PubMed.
- J. Song, L. Cheng, A. Liu, J. Yin, M. Kuang and H. Duan, J. Am. Chem. Soc., 2011, 133, 10760–10763 CrossRef CAS PubMed.
- T. Bollhorst, T. Grieb, A. Rosenauer, G. Fuller, M. Maas and K. Rezwan, Chem. Mater., 2013, 25, 3464–3471 CrossRef CAS.
- T. Bollhorst, S. Shahabi, K. Worz, C. Petters, R. Dringen, M. Maas and K. Rezwan, Angew. Chem., Int. Ed., 2015, 54, 118–123 CrossRef CAS PubMed.
- Z. Nie, D. Fava, E. Kumacheva, S. Zou, G. C. Walker and M. Rubinstein, Nat. Mater., 2007, 6, 609–614 CrossRef CAS PubMed.
- S. Sihler, A. Schrade, Z. Cao and U. Ziener, Langmuir, 2015, 31, 10392–10401 CrossRef CAS PubMed.
- S. Li, B. A. Moosa, J. G. Croissant and N. M. Khashab, Angew. Chem., Int. Ed., 2015, 54, 6804–6808 CrossRef CAS PubMed.
- M. Maas, T. Bollhorst, R. N. Zare and K. Rezwan, Part. Part. Syst. Charact., 2014, 31, 1067–1071 CrossRef CAS.
- M. Karg, Y. Lu, E. Carbó-Argibay, I. Pastoriza-Santos, J. Pérez-Juste, L. M. Liz-Marzán and T. Hellweg, Langmuir, 2009, 25, 3163–3167 CrossRef CAS PubMed.
- A. Schrade, Z. Cao, K. Landfester and U. Ziener, Langmuir, 2011, 27, 6689–6700 CrossRef CAS PubMed.
- Y. Liu, Y. Li, J. He, K. J. Duelge, Z. Lu and Z. Nie, J. Am. Chem. Soc., 2014, 136, 2602–2610 CrossRef CAS PubMed.
- J. He, Y. Liu, T. Babu, Z. Wei and Z. Nie, J. Am. Chem. Soc., 2012, 134, 11342–11345 CrossRef CAS PubMed.
- J. Wei, K. Niikura, T. Higuchi, T. Kimura, H. Mitomo, H. Jinnai, Y. Joti, Y. Bessho, Y. Nishino, Y. Matsuo and K. Ijiro, J. Am. Chem. Soc., 2016, 138, 3274–3277 CrossRef CAS PubMed.
- M. R. Rasch, E. Rossinyol, J. L. Hueso, B. W. Goodfellow, J. Arbiol and B. A. Korgel, Nano Lett., 2010, 10, 3733–3739 CrossRef CAS PubMed.
- S. U. Pickering, J. Chem. Soc., Trans., 1907, 91, 2001–2021 RSC.
- X. Liu, Y. Yin and C. Gao, Langmuir, 2013, 29, 10559–10565 CrossRef CAS PubMed.
- L. Zhang, L. T. Roling, X. Wang, M. Vara, M. Chi, J. Liu, S.-I. Choi, J. Park, J. A. Herron, Z. Xie, M. Mavrikakis and Y. Xia, Science, 2015, 349, 412–416 CrossRef CAS PubMed.
- E. Roduner, Chem. Soc. Rev., 2006, 35, 583–592 RSC.
- W. Zhou, J. Wu and H. Yang, Nano Lett., 2013, 13, 2870–2874 CrossRef CAS PubMed.
- D. S. He, D. He, J. Wang, Y. Lin, P. Yin, X. Hong, Y. Wu and Y. Li, J. Am. Chem. Soc., 2016, 138, 1494–1497 CrossRef CAS PubMed.
- X. Sun, K. Jiang, N. Zhang, S. Guo and X. Huang, ACS Nano, 2015, 9, 7634–7640 CrossRef CAS PubMed.
- X. Wang, S.-I. Choi, L. T. Roling, M. Luo, C. Ma, L. Zhang, M. Chi, J. Liu, Z. Xie, J. A. Herron, M. Mavrikakis and Y. Xia, Nat. Commun., 2015, 6, 7594 CrossRef PubMed.
- X. Wang, M. Vara, M. Luo, H. Huang, A. Ruditskiy, J. Park, S. Bao, J. Liu, J. Howe, M. Chi, Z. Xie and Y. Xia, J. Am. Chem. Soc., 2015, 137, 15036–15042 CrossRef CAS PubMed.
- X. Wang, L. Figueroa-Cosme, X. Yang, M. Luo, J. Liu, Z. Xie and Y. Xia, Nano Lett., 2016, 16, 1467–1471 CrossRef CAS PubMed.
- V. Stamenkovic, B. S. Mun, K. J. Mayrhofer, P. N. Ross, N. M. Markovic, J. Rossmeisl, J. Greeley and J. K. Norskov, Angew. Chem., Int. Ed., 2006, 45, 2897–2901 CrossRef CAS PubMed.
- D. Mott, J. Luo, P. N. Njoki, Y. Lin, L. Wang and C.-J. Zhong, Catal. Today, 2007, 122, 378–385 CrossRef CAS.
- K. Tedsree, T. Li, S. Jones, C. W. Chan, K. M. Yu, P. A. Bagot, E. A. Marquis, G. D. Smith and S. C. Tsang, Nat. Nanotechnol., 2011, 6, 302–307 CrossRef CAS PubMed.
- S. Zhang, X. Zhang, G. Jiang, H. Zhu, S. Guo, D. Su, G. Lu and S. Sun, J. Am. Chem. Soc., 2014, 136, 7734–7739 CrossRef CAS PubMed.
- M. Shao, J. H. Odell, S.-I. Choi and Y. Xia, Electrochem. Commun., 2013, 31, 46–48 CrossRef CAS.
- L.-l. Fang, Q. Tao, M.-f. Li, L.-w. Liao, D. Chen and Y.-x. Chen, Chin. J. Chem. Phys., 2010, 23, 543–548 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details, TEM images, FTIR spectra, size distributions, FDTD simulations, SERS analysis, electrochemical measurements, extension of the self-assembly approach to other metals and comparison of the FAOR results with those from the literature. See DOI: 10.1039/c7sc01841j |
| This journal is © The Royal Society of Chemistry 2017 |