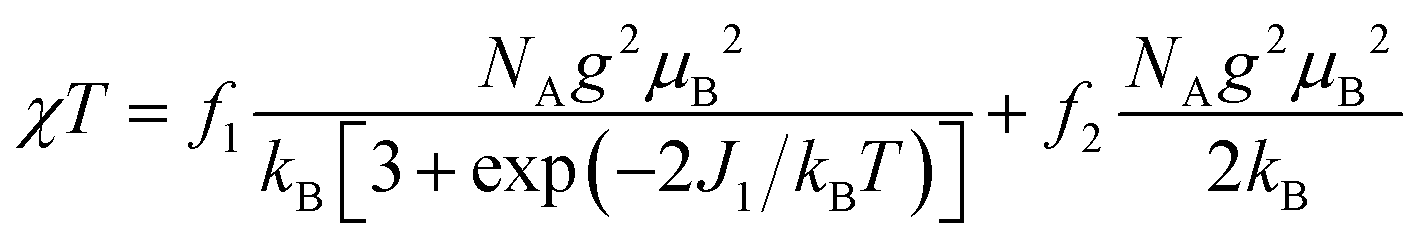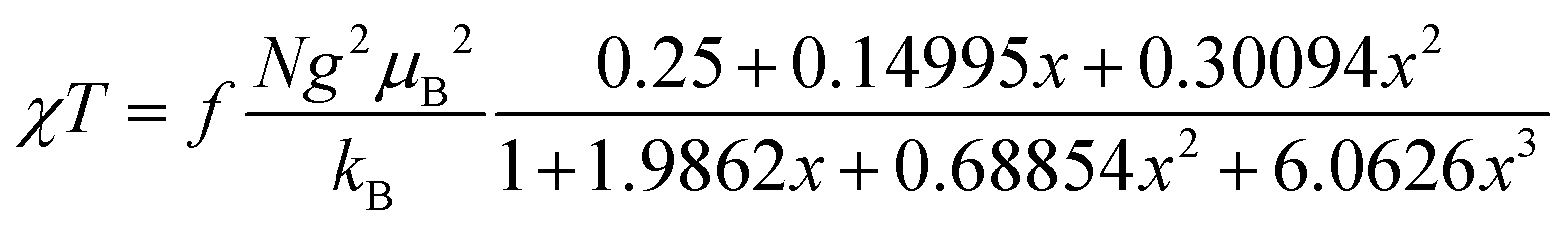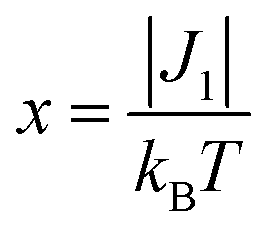DOI:
10.1039/C6SC02721K
(Edge Article)
Chem. Sci., 2017,
8, 189-199
Highly planar diarylamine-fused porphyrins and their remarkably stable radical cations†
Received
21st June 2016
, Accepted 30th July 2016
First published on 1st August 2016
Abstract
Oxidative fusion reactions of meso-phenoxazino Ni(II) porphyrin were found to be temperature dependent, giving rise to either a doubly phenylene-fused product at room temperature or a singly phenoxazine-fused product at 70 °C. The latter was further oxidized to a doubly phenoxazine-fused Ni(II) porphyrin, which was subsequently converted to the corresponding free base porphyrin and Zn(II) porphyrin. Compared to previously reported diphenylamine-fused porphyrins that displayed a molecular twist, doubly phenoxazine-fused porphyrins exhibited distinctly different properties owing to their highly planar structures, such as larger fluorescence quantum yields, formation of an offset face-to-face dimer both in solution and the solid state, and the generation of a mixed-valence π-radical cation dimer upon electrochemical oxidation. One-electron oxidation of the phenoxazine-fused Ni(II) porphyrin with Magic Blue gave the corresponding radical cation, which was certainly stable and could be isolated by separation over a silica gel column but slowly chlorinated at the reactive β-positions in the solid state. This finding led to us to examine β,β′-dichlorinated phenoxazine-fused and diphenylamine-fused Ni(II) porphyrins, which, upon treatment with Magic Blue, provided remarkably stable radical cations to an unprecedented level. It is actually possible to purify these radical cations by silica gel chromatography, and they can be stored for over 6 months without any sign of deterioration. Moreover, they exhibited no degradation even after the CH2Cl2 solution was washed with water. However, subtle structural differences (planar versus partly twisted) led to different crystal packing structures and solid-state magnetic properties.
Introduction
Radical cations of porphyrins have been long studied because they are involved as key intermediates in the crucial steps of photosynthesis and enzymatic oxidation cycles.1 To reveal the key roles and specific molecular attributes that are necessary for relevant functions, fundamental properties of radical cations of porphyrins have been continuously investigated.2–4 As representative examples, Wolberg and Manassen prepared radical cations of Ni(II), Zn(II), Cu(II), Co(III), and Fe(III) tetraphenylporphyrins by electrochemical oxidation, and studied the optical, electrochemical, and magnetic properties.2d,e Furthermore, detailed structures and reactivities of various porphyrin radical cations have also been successively explored.3,4
In recent years, stable organic radicals have attracted increasing attention in light of their potential application in organic electronics, photonics, spintronics, and energy storage devices.5 Considering the rich chemistry of porphyrins, exploration of highly stable radical cations of porphyrins that can be isolated and manipulated as usual organic closed-shell molecules is a promising approach worthy of a challenge. However, such stable radical cations of porphyrins have been quite rare despite the extensive number of investigations mentioned above.
Generally, radical cations of porphyrins are reactive and undergo facile degradation under ambient conditions. For instance, a half-life of a radical cation of Ni(II) tetraphenylporphyrin was found to be only 5 minutes at room temperature in benzonitrile solution containing nBu4NBF4 as a supporting electrolyte.2d Therefore, isolation of such inherently reactive porphyrin radical cations usually required careful manipulations under strictly inert atmosphere, purposefully avoiding conventional work-up procedures and silica-gel column chromatography in open air.3 The instability of radical cations of porphyrins can be ascribed to their high oxidizing power and poor kinetic protection toward a wide range of nucleophiles and reducing reagents. Actually, Tsuchiya reported that stability of a oxo-Fe(IV) porphyrin radical cation was improved by exhaustive introduction of phenyl groups both at the meso- and β-positions.4b However, even in the case of such a sterically protected radical cation, the reactivity remained relatively high, and the physical measurements had to be performed below 8 °C. To provide an extremely stable porphyrin radical cation that can be handled like a usual closed-shell molecule, effective employment of both thermodynamic and kinetic stabilization is of paramount importance as observed in stable neutral radicals of other porphyrins and porphyrinoids.6
As an effective structural motif to realize a stable radical cation, planar triarylamines possessing an embedded nitrogen atom are known (Chart 1).7 While a triphenylaminium radical was reported to be unstable and coupled at the para-positions to give its dimer even under inert atmosphere,8 Hellwinkel and co-workers reported a pioneering work that a methylene-bridged planar triarylamine 1 provided a stable radical cation via one-electron oxidation in concentrated sulfuric acid or trifluoroacetic acid.7a,b Recently, other researchers extended this work to include more stable radical cations of planar triaryamines such as 1 and 2.7c–g However, preparation of these radical cations always required anaerobic and/or anhydrous conditions.
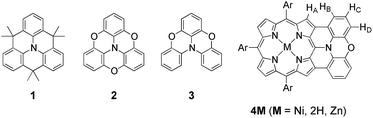 |
| | Chart 1 Structures of internally bridged triphenylamines 1–3 and phenoxazine-fused porphyrin 4M. | |
As an intriguing aspect, Okada et al. found that planarity of radical cations played a crucial role in the packing structures.7c,d Namely, a radical cation of oxygen-linked planar triarylamine 2 packed as a dimer due to the high planarity, exhibiting a strong antiferromagnetic interaction in the solid state, while a radical cation of partially linked twisted congener 3 showed a three-dimensionally connecting weak antiferromagnetic interaction instead of such dimer formation.
With these examples in mind, we envisioned the exploration of very stable radical cations of porphyrins by incorporating a fused diarylamine unit directly at the porphyrin periphery. Here, we report the first synthesis of doubly phenoxazine-fused porphyrins 4M as highly planar nitrogen-embedded porphyrins and the isolation of a radical cation of phenoxazine-fused Ni(II) porphyrin, [4Ni]+, as a certainly stable molecule. Furthermore, we found that β,β′-dichlorination of [4Ni]+ enhanced its chemical stability remarkably, so that it could withstand usual aqueous work up procedures and be stored under ambient conditions without any deterioration over several months.
Results and discussion
Preparation of fully phenoxazine-fused porphyrins
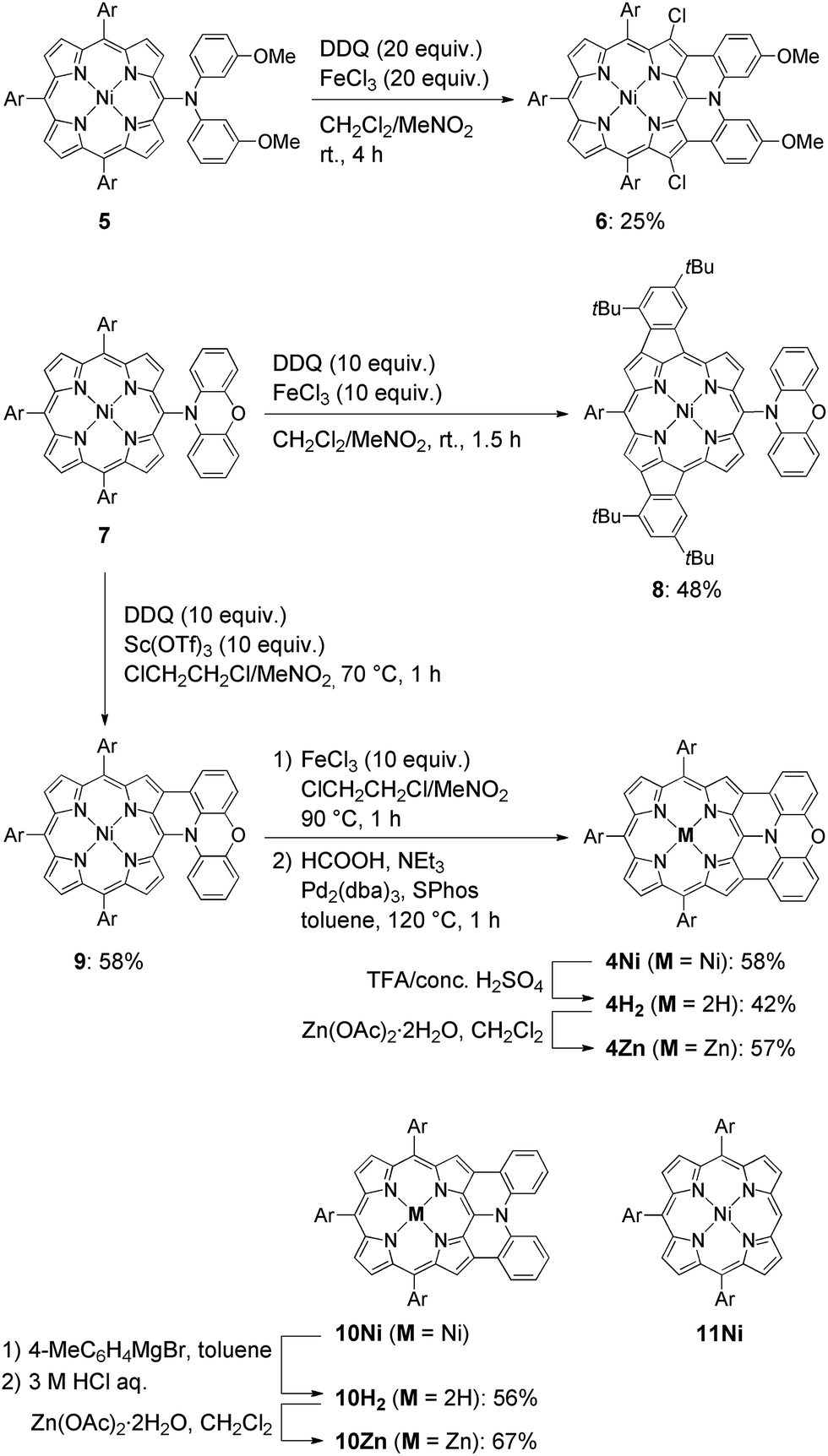
The synthesis of phenoxazine-fused porphyrins 4M is depicted in Scheme 1. Previously, we reported that an oxidative fusion reaction of meso-[bis(3-methoxyphenyl)amino]porphyrin 5 with 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) and FeCl3 at room temperature provided β,β′-dichlorinated diarylamine-fused porphyrin 6.9a,10 In sharp contrast, the oxidation of 5-phenoxazino Ni(II) porphyrin 7 under similar reaction conditions resulted in fusion of the 10,20-aryl groups at the 12- and 18-positions, yielding doubly phenylene-fused porphyrin 8. We speculated that the formation of 8 might be ascribed to a high rotational barrier of the phenoxazinyl substituent after one-electron oxidation of the phenoxazine segment. The oxidized phenoxazine segment would remain rather perpendicular to the porphyrin plane, hence serving as an electron-withdrawing group and causing the fusion of the 10,20-phenyl groups at the 12- and 18-positions. This consideration came from our previous study that electron-withdrawing substituents at the meso-position induced a similar double phenylene-fusion reaction at the opposite side.11 Therefore, we examined the reaction at higher temperature to facilitate the rotation of the meso-phenoxazinyl substituent. To our delight, the oxidation of 7 with FeCl3 at 90 °C led to the formation of phenoxazine-fused porphyrins together with phenylene-fused porphyrin 8. We chose a combination of DDQ and Sc(OTf)3 as a weaker oxidant in order to suppress over-oxidation. After the extensive optimization of reaction conditions, the reaction at 70 °C proceeded nicely to provide singly phenoxazine-fused porphyrin 9 in 58% yield. Interestingly, the formation of 8 was not observed under these conditions. In 9, the phenoxazine segment is held in coplanar fashion with respect to the porphyrin core, a geometrically ideal arrangement for realizing complete fusion. Complete fusion was easily achieved by further oxidation with FeCl3, giving doubly fused products with partial chlorination at the pyrrolic β-position adjacent to the fusion site as observed in the synthesis of 6. Separation of these doubly fused products was tedious and thus their dechlorination9a,12 was attempted with NEt3 and HCOOH under Pd/SPhos13 catalysis, which furnished 4Ni in 58% yield. Removal of the central nickel atom of 4Ni was achieved upon treatment with concentrated sulfuric acid in trifluoroacetic acid (TFA) at 0 °C to provide free base 4H2 in 42% yield, and subsequent zinc-complexation with Zn(OAc)2·2H2O afforded the Zn complex 4Zn in 57% yield. As a reference, doubly diphenylamine-fused free base porphyrin 10H2 was synthesized by denickelation of previously reported diphenylamine-fused porphyrin 10Ni9b with p-tolylmagnesium bromide followed by demagnesiation with 3 M HCl aq. in 56% yield.14 The zinc complex 10Zn was also prepared by zinc-complexation of with Zn(OAc)2·2H2O in 67% yield.
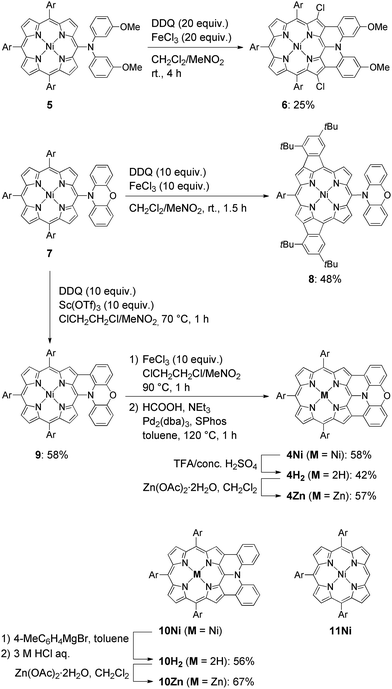 |
| | Scheme 1 Synthesis of β,β′-dichlorinated diarylamine-fused porphyrin 6, phenylene-fused porphyrin 8, doubly phenoxazine-fused porphyrins 4M, and diphenylamine-fused porphyrins 10M. Structure of triaryl Ni(II)-porphyrin 11Ni. Ar = 3,5-di-tert-butylphenyl. | |
Optical properties
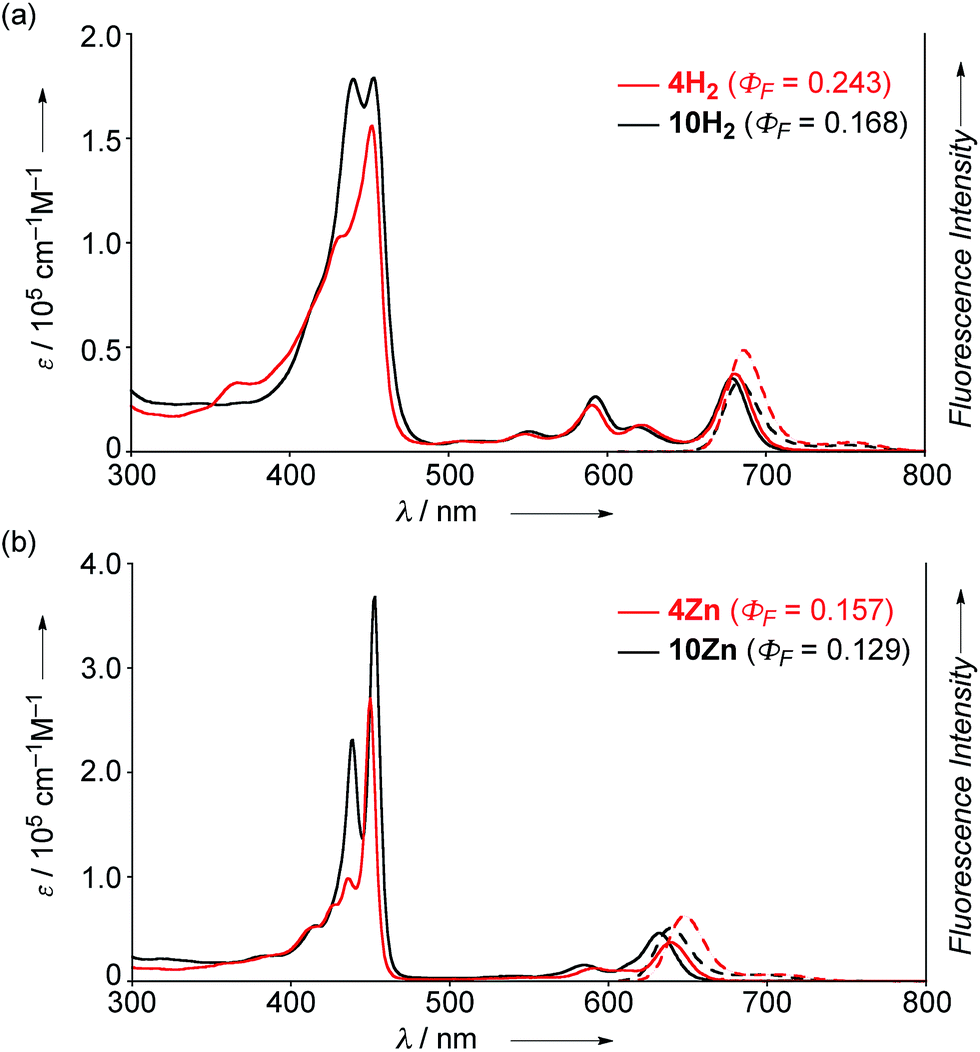
The UV/Vis absorption and emission spectra of 4H2, 10H2, 4Zn and 10Zn in CH2Cl2 are shown in Fig. 1. The absorption spectrum of 10Zn exhibits a split Soret band at 439 and 453 nm, and Q-bands at 585 and 632 nm, while that of 4Zn shows a distinct Soret band at 450 nm and slightly red-shifted Q-bands at 591 and 640 nm. Similarly, 4H2 displays an intense Soret band at 452 nm and red-shifted Q-bands at 549, 591, 620 and 680 nm. Compared to 10H2 and 10Zn, the slightly red-shifted absorption spectral features of 4H2 and 4Zn suggest that the fused phenoxazine group leads to a more planar geometry with efficient extension of π-conjugation over the whole molecule.
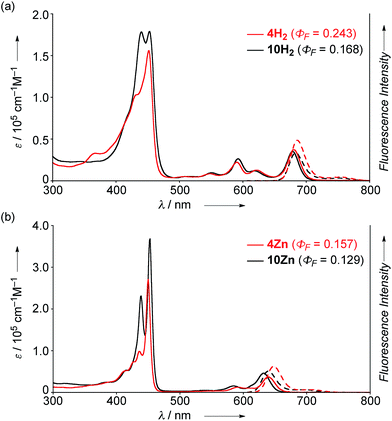 |
| | Fig. 1 UV/Vis absorption and emission spectra of (a) 4H2 and 10H2, and (b) 4Zn and 10Zn in CH2Cl2. ε = extinction coefficient. | |
The fluorescence spectra of 4H2, 4Zn, 10H2 and 10Zn are observed at 686, 648, 683, and 640 nm, respectively. The observed fluorescence quantum yields (ΦF) and fluorescence lifetimes (τ), calculated radiative decay rates (kr), and non-radiative decay rates (knr) are summarized in Table 1. The fluorescence quantum yields of 4H2, 4Zn, 10H2 and 10Zn are distinctly larger than those of tetraphenylporphyrinato zinc(II) (0.033) and tetraphenylporphyrin (0.11).15 As suggested in a previous report,9a effective conjugation of the embedded nitrogen atom with the porphyrin network may increase the radiative decay rate while less conformational flexibility associated with the fused structure is expected to decrease non-radiative decay. Therefore, the larger radiative decay rates of 4H2 and 4Zn compared to those of 10H2 and 10Zn are attributable to effective conjugation of the nitrogen atom due to enhanced planarity, and the smaller non-radiative decay rates can be ascribed to the more rigid structures.
Table 1 Optical properties of 4H2, 4Zn, 10H2, and 10Zn
|
|
Φ
F
|
τ [ns] |
k
r [s−1] |
k
nr [s−1] |
|
4H2
|
0.243 |
6.0 |
4.7 × 107 |
1.3 × 108 |
|
4Zn
|
0.157 |
2.7 |
5.9 × 107 |
3.1 × 108 |
|
10H2
|
0.168 |
4.8 |
3.5 × 107 |
1.7 × 108 |
|
10Zn
|
0.129 |
2.5 |
5.2 × 107 |
3.5 × 108 |
X-ray structures of 4Ni and 10Ni
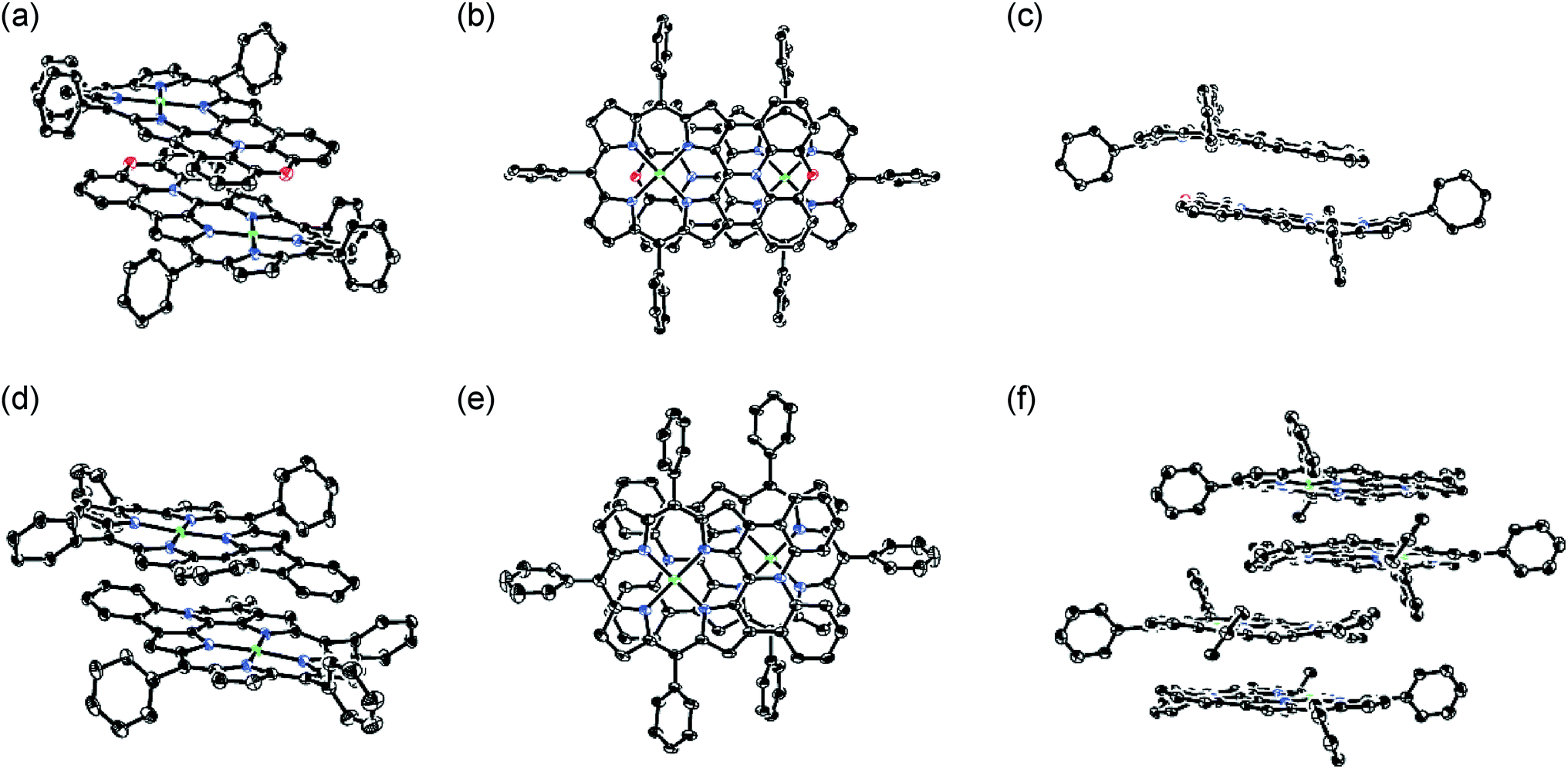
The X-ray crystal structures of 4Ni and 10Ni are shown in Fig. 2.1610Ni exhibits a distorted structure as indicated by a dihedral angle of 32° between the two phenyl groups in the fused-diphenylamine unit. This molecular twist is considered to arise from intramolecular steric congestion between the two ortho-hydrogen atoms of the phenyl groups. In contrast, 4Ni takes on a slightly wavy but more planar structure with a small dihedral angle of 4° between the relevant two phenyl groups. The mean plane deviation (MPD) of 4Ni was calculated to be 0.157 Å, which is apparently smaller than that of 10Ni (0.209 Å).17 In the crystals, both 4Ni and 10Ni form face-to-face dimers with the dipole moment arranged in an offset manner. In the case of 10Ni, such dimer units are packed to form a one-dimensional columnar structure while dimers of 4Ni are isolated from each other. Reflecting the more planar structure, the interporphyrin separation distance in the dimer of 4Ni is distinctly shorter (3.392 Å) than that of 10Ni (3.655–3.734 Å).
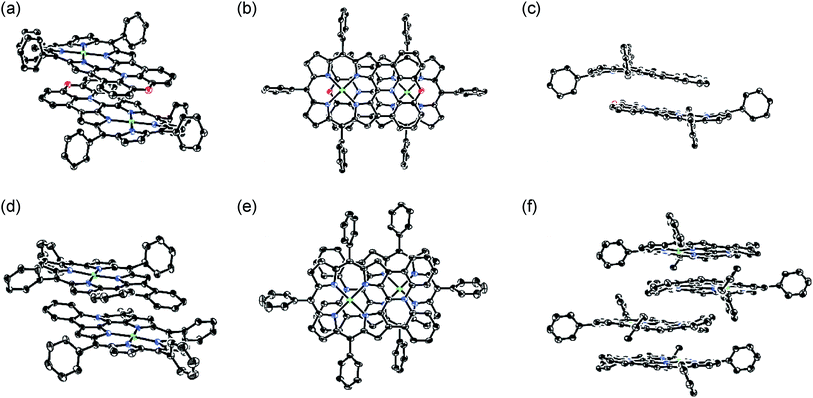 |
| | Fig. 2 X-Ray crystal structures of 4Ni and 10Ni. (a) Over view, (b) top view, and (c) side view of dimeric 4Ni. (d) Over view and (e) top view of dimeric 10Ni. (f) Side view of packing structure of 10Ni. Thermal ellipsoids are drawn at the 50% probability level. Solvent molecules, tert-butyl groups, and all hydrogen atoms are omitted for clarity. | |
Dimerization in CDCl3
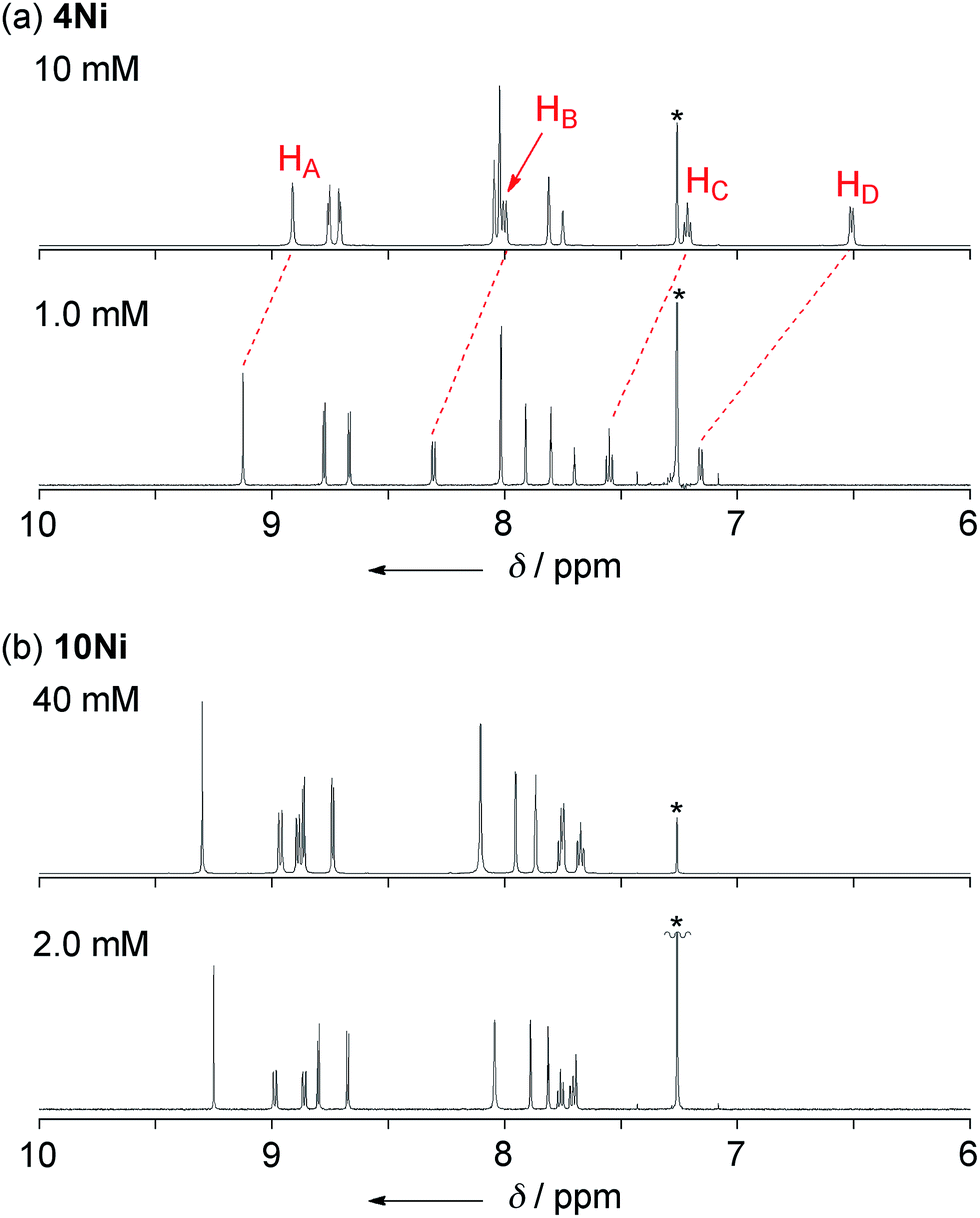
Phenoxazine-fused porphyrin 4Ni was found to exhibit a concentration-dependent 1H NMR spectrum in CDCl3 at 298 K, as observed in other π-stacking dimers (Fig. 3a).18 The 1H NMR spectrum of 4Ni in CDCl3 at 10 mM showed signals of HA, HB, HC, and HD at 8.91, 8.00, 7.22, and 6.51 ppm, respectively. They were downfield shifted at 1.0 mM, being observed at 9.12, 8.31, 7.55, and 7.16 ppm, respectively. The observed down-field shifts at low concentration were larger for signals due to the protons at the fused-phenoxazine moiety; namely in the order of HA (0.23 ppm) < HB (0.31 ppm) < HC (0.33 ppm) < HD (0.65 ppm). In contrast, the down-field shifts were very small or negligible for signals due to the pyrrolic β-protons at the porphyrin moiety. These results suggest that 4Ni formed a slipped face-to-face dimer overlapping at the phenoxazine moiety as observed in the solid state.
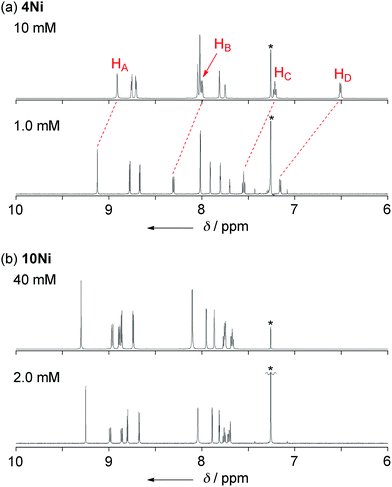 |
| | Fig. 3
1H NMR spectra of (a) 4Ni and (b) 10Ni in CDCl3 under different concentrations at 298 K. * Residual CHCl3. | |
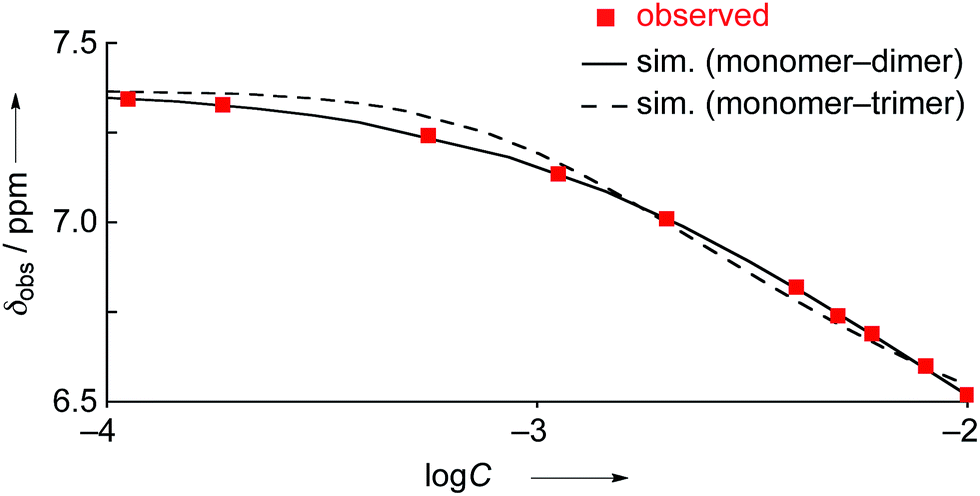
The concentration dependent 1H NMR chemical shifts were analyzed by the Saunders–Hyne method, which led to the conclusion that a monomer–dimer equilibration model was apparently superior to fit the observed spectra over a monomer–trimer equilibration model (Fig. 4).19 Then, the association constant has been estimated to be 7.2 × 10 M−1 for 4Ni by 1H NMR dilution experiment based on the Horman–Doreux model.20 Complexes 4H2 and 4Zn also showed similar concentration-dependent 1H NMR spectra, and the association constants of 4H2 and 4Zn have been estimated to be 2.9 × 10 M−1 and 1.0 × 102 M−1, respectively. In contrast, the 1H NMR spectrum of diphenylamine-fused porphyrin 10Ni showed negligible concentration dependence (Fig. 3b), and the association constant was estimated to be at most below 1 M−1. Similarly, the 1H NMR spectra of 10H2 and 10Zn were also almost independent of the concentration. Therefore, it can be concluded that the structural difference in the fused diarylamine part; a noncyclic diphenylamine in 10Mversus a cyclic phenoxazine in 4M, leads to contrasting assembling behaviors in CDCl3.
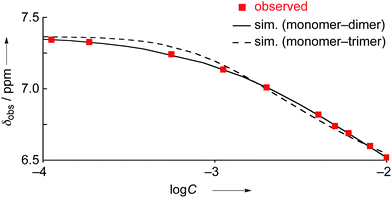 |
| | Fig. 4
1H NMR dilution curves obtained for 4Ni in CDCl3. Red squares: observed values, solid line: simulated curve as the monomer–dimer model, and dashed line: simulated curve as the monomer–trimer model. | |
Electrochemistry
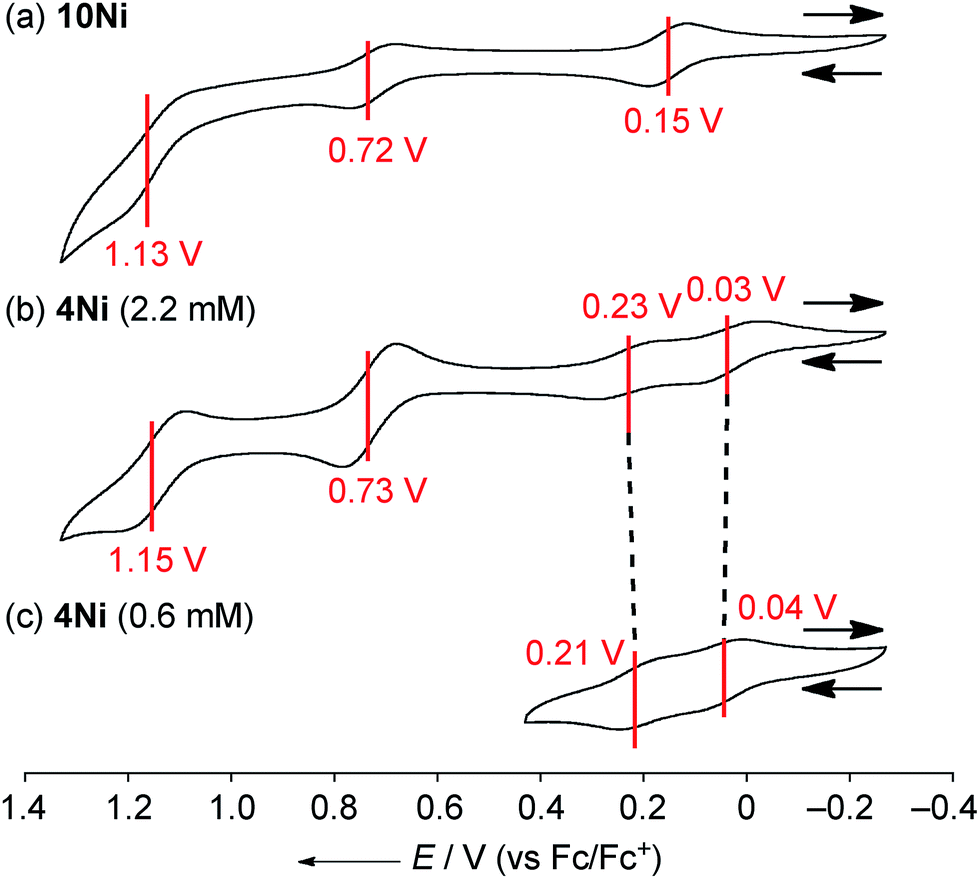
The electrochemical properties of 4Ni and 10Ni were studied by cyclic voltammetry (CV) and differential pulse voltammetry (DPV) in CH2Cl2 (Fig. 5). Diphenylamine-fused porphyrin 10Ni showed three reversible oxidation waves at 0.15, 0.72, and 1.13 V, and one reversible reduction wave at −1.84 V. Phenoxazine-fused porphyrin 4Ni showed four reversible oxidation waves at 0.03, 0.23, 0.73, and 1.15 V, and one reversible reduction wave at −1.83 V. These oxidation potentials of 4Ni showed apparent negative shifts compared to those of meso-phenoxazino Ni(II) porphyrin 7 (0.34 and 0.89 V)9a and 5,10,15-tris(3,5-di-tert-butylphenyl)porphyrinato nickel(II) 11 (0.56 and 0.82 V).21 These results indicate that the embedding of a nitrogen atom directly at the periphery led to an increase of the electron-donating ability of the substituent. On the other hand, the reduction potential of 4Ni remains almost unchanged compared to those of 7 (−1.80 V) and 11 (−1.75 V).
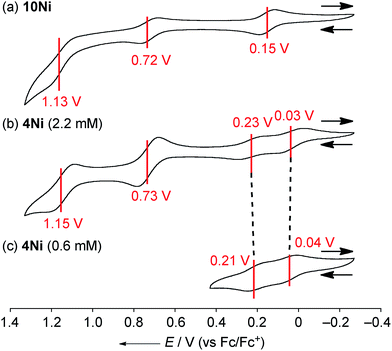 |
| | Fig. 5 Cyclic voltammograms of (a) 10Ni, (b) 4Ni at 2.2 mM, and (c) 4Ni at 0.6 mM. The redox potentials were measured by cyclic voltammetry in anhydrous CH2Cl2 with 0.1 M nBu4NPF6 as a supporting electrolyte, and Ag/AgClO4 as a reference electrode. The ferrocene/ferrocenium ion couple was used as an external reference. Scan rate: 0.05 V s−1. | |
DPV experiments revealed that the electrochemical oxidation of 10Ni proceeded via usual 1e/1e/1e processes. On the other hand, those of 4Ni proceeded through 0.5e/0.5e/1e/1e oxidation process for one porphyrin unit. These waves were similar to electrochemical responses of the other porphyrin complex,22a phthalocyanine,22b Ru(II) polypyridyl complexes,22c–e and tetrathiafulvalene derivatives,22f,g all containing planar aromatic units capable of forming π-dimers. An association constant of 4Ni in CH2Cl2 has been estimated to be 8.2 × 102 M−1 by dilution experiment of the UV/Vis absorption spectra, which is almost 10 times larger than that in CDCl3. In addition, ΔE values (Eox21/2 − Eox11/2) of 4Ni are only weakly concentration-dependent; 0.20 V at 2.2 mM and 0.17 V at 0.6 mM, as observed in other π-systems.22 Thus, the split first oxidation waves of 4Ni can be interpreted in terms of the generation of a mixed-valence π-radical cation dimer comprised of neutral and radical cation species.3d,22,23 Therefore, the ΔE values (0.17–0.20 V), which represent the degree of the electronic interaction in the dimer, are comparable to those of previously reported porphyrin dimers (0.07–0.31 V).24 After the second oxidation, the generated cation radicals are dissociated due to coulombic repulsion and undergo the third and fourth oxidations similarly to 10Ni.
Isolation of radical cations
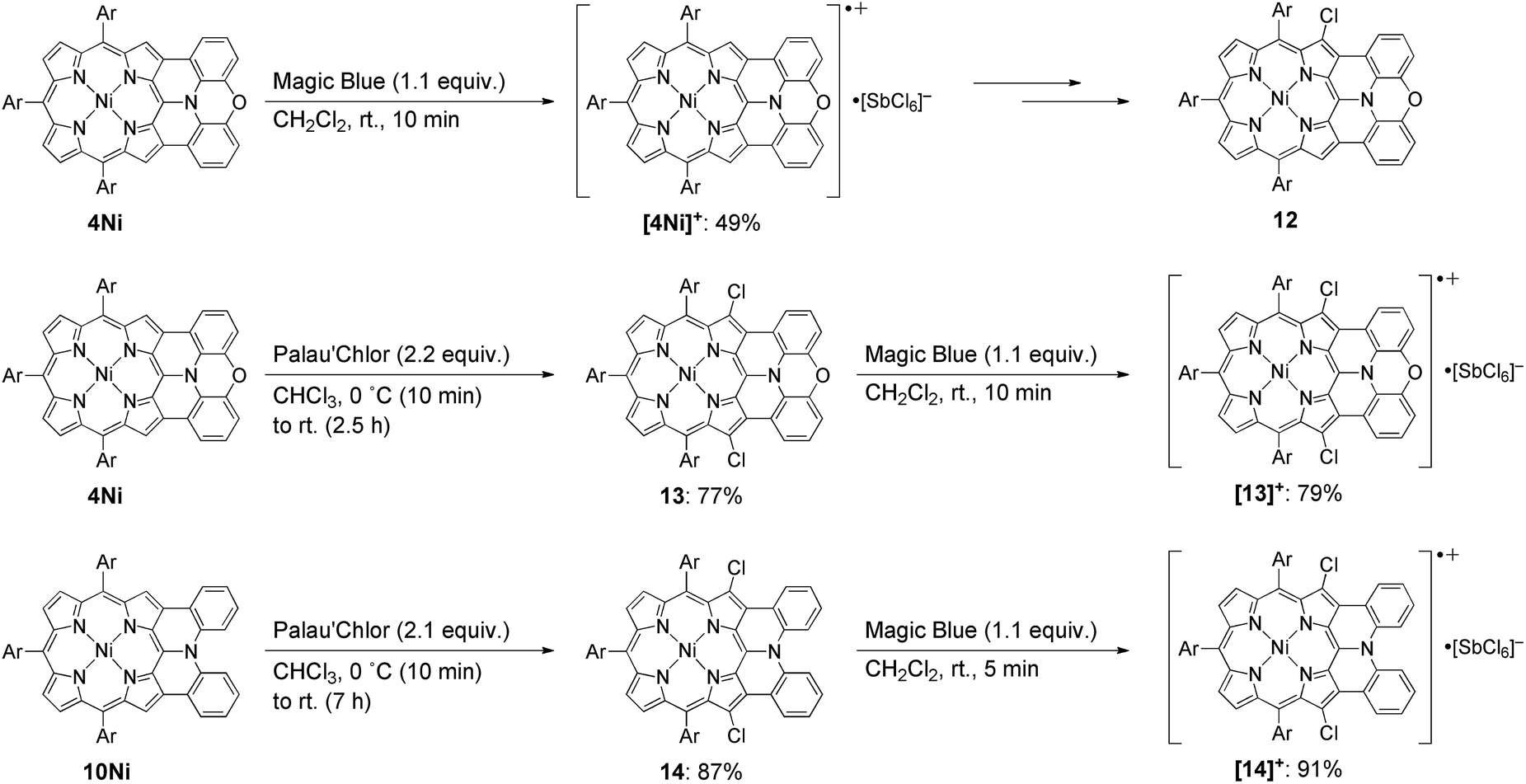
Isolation of the radical cation of 4Ni was attempted (Scheme 2). Porphyrin 4Ni was oxidized with tris(4-bromophenyl)aminium hexachloroantimonate (Magic Blue), and radical cation [4Ni]+ was obtained in 49% yield after separation over a silica gel column in the open air. Radical cation [4Ni]+ was certainly stable but was susceptible to gradual chlorination in the solid state, providing β-chlorinated porphyrin 12. Therefore, we protected the most reactive β-positions of 4Ni by chlorination with 2-chloro-1,3-bis(methoxycarbonyl)guanidine (Palau'Chlor)25 to produce 13 in 77% yield. The dimerization constant of 13 in CDCl3 has been evaluated to be 7.0 × 102 M−1 by 1H NMR titration experiments. This association constant is considerably larger than those of 4M, because of the electron-withdrawing chloro groups that facilitate π–π stacking through reducing the π-electron density of the porphyrin core.26 Then, one-electron oxidation of 13 was attempted with Magic Blue under similar conditions. The oxidation proceeded smoothly to produce radical cation [13]+ in 79% yield after separation over a silica gel column. Surprisingly, radical cation [13]+ is remarkably stable under ambient conditions, which is underlined by the facts that (1) [13]+ can be stored under ambient conditions over 6 months without any detectable decomposition, (2) the absorption spectrum of [13]+ remained unchanged after being kept as a tetrachloroethane solution at 70 °C for 1 h, (3) [13]+ can be purified by silica-gel column chromatography, and (4) [13]+ was inert to moisture, and when a solution of [13]+ in CH2Cl2 was washed with water, no decomposition was observed. Similarly, porphyrin 10Ni was converted to the corresponding β,β′-dichloroporphyrin 14 by chlorination with Palau'Chlor, and subsequent oxidation with Magic Blue gave [14]+ in 79% yield in two steps. Radical cation [14]+ is also extremely stable, being comparable to [13]+.
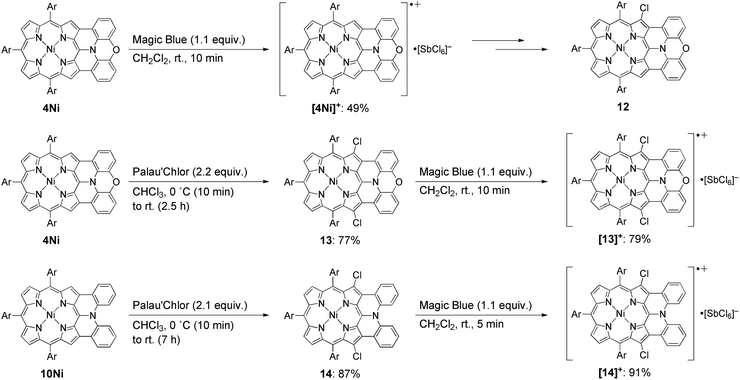 |
| | Scheme 2 Synthesis of radical cations [4Ni]+, [13]+, and [14]+. Ar = 3,5-di-tert-butylphenyl. | |
X-ray crystal structures of radical cations
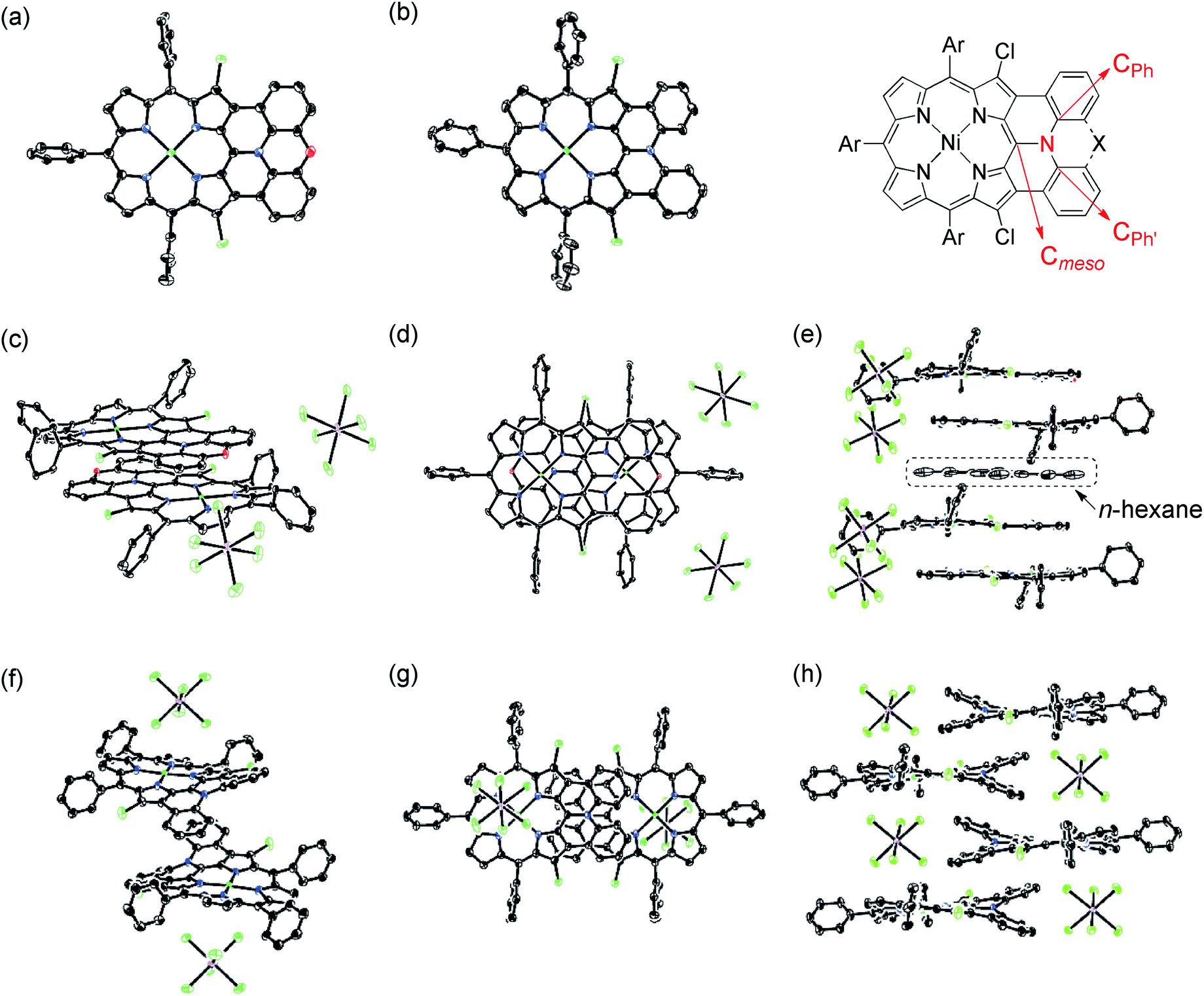
X-Ray diffraction analysis unambiguously revealed the structures of 13, 14, [13]+, and [14]+ (Fig. 6).27 In the structures of [13]+ and [14]+, each porphyrin unit carries one counter anion. Selected structural factors are summarized in Table 2. In each compound, the sum of C–N–C bond angles around the fused-diarylamine unit approaches 360°, suggesting sp2-like conformations of the embedded nitrogen atoms. The bond distances around the embedded nitrogen atoms of 13 remains unchanged even after one-electron oxidation due to the inherently rigid structure. On the other hand, the MPD of [13]+ (0.092 Å) is slightly smaller than that of the neutral species 13 (0.104 Å).
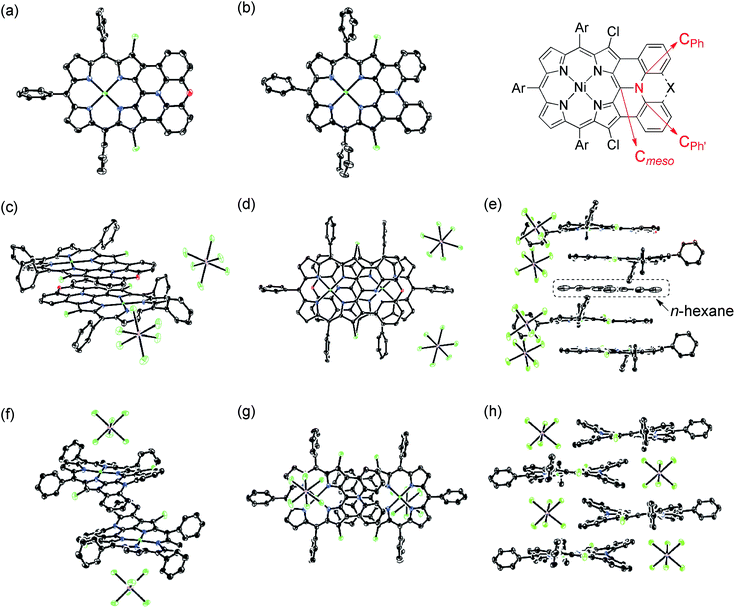 |
| | Fig. 6 X-Ray crystal structures of 13, 14, [13]+, and [14]+. (a) Top view of 13. (b) Top view of 14. (c) Over view and (d) top view of dimeric [13]+. (e) Side view of the packing structure of [13]+. (f) Over view and (g) top view of dimeric [14]+. (h) Side view of the packing structure of [14]+. Thermal ellipsoids are drawn at the 50% probability level for 13, 14 and [13]+, and at the 30% probability level for [14]+. Solvent molecules (except for n-hexane in (e)), tert-butyl groups, and all hydrogen atoms are omitted for clarity. | |
Table 2 Selected bond lengths (Å)a, inner angles (deg)a, MPD (Å), and interporphyrin separation (Å) for 13, [13]+, 14, and [14]+ from crystal structures
|
|
N–Cmeso |
N–CPh |
∠Cmeso–N–CPh |
∠CPh–N–CPh′ |
MPD |
Interporphyrin separation |
|
The values were averaged for C2v symmetry. The estimated standard deviations of mean values were calculated from the experimental values using the following equation: δ(l) = 1/(Σ(1/δi2))1/2.
The values were averaged for two crystallographically independent molecules. The estimated standard deviations of mean values were calculated from the experimental values using the following equation: δ(l) = 1/(Σ(1/δi2))1/2.
|
|
13
|
1.393(4) |
1.413(2) |
121.1(2) |
117.9(3) |
0.104 |
3.511 |
|
[13]+
|
1.396(7) |
1.425(7) |
120.8(5) |
118.4(7) |
0.092 |
3.354 |
|
14
|
1.401(5) |
1.423(4) |
119.7(2) |
120.5(3) |
0.398 |
3.390 |
|
[14]+
|
1.388(10) |
1.423(6) |
120.2(4) |
119.7(6) |
0.409 |
4.212–4.353 |
In the crystal, [13]+ forms an offset face-to-face dimer with an interporphyrin separation of 3.354 Å, which is slightly smaller than a sum of van der Waals radii of two carbon atoms (3.40 Å). Importantly, the interporphyrin separation of [13]+ is distinctly shorter than that of neutral 13 (3.511 Å) despite Coulomb's repulsion between the two cationic π-systems. The observed short interporphyrin separation of [13]+ imply some associative interactions like SOMO–SOMO interaction as observed in planar radical cations.28 On the other hand, [14]+ shows a one-dimensional columnar packing with an interporphyrin separation of 4.212 Å, which is longer than that of 14 (3.390 Å). The different packing structure between [13]+ and [14]+ can be ascribed to the structural difference; almost planar for [13]+ and partially twisted for [14]+. In accordance with this interpretation, overlap of the two π-planes is larger in [13]+ than [14]+.
Optical properties and excited-state decay-dynamics of radical cations
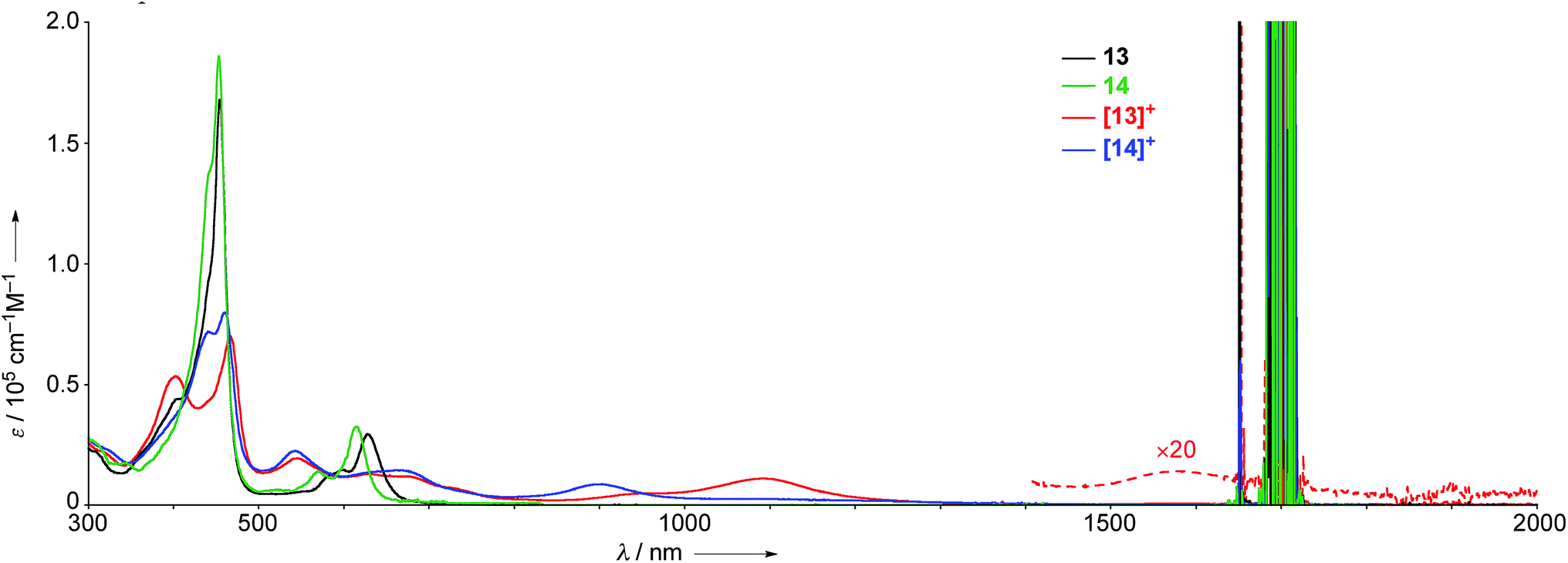
The UV/Vis absorption spectra of 13, 14, [13]+, and [14]+ in CH2Cl2 are shown in Fig. 7. The neutral species 13 exhibits a Soret band at 455 nm with a shoulder at 402 nm, and Q bands at 598 and 628 nm. In contrast, the radical cation [13]+ displays an ill-defined and largely red-shifted absorption spectrum with peaks at 403, 467, 543, 609, 677, 1092, and 1583 nm. The faint broad absorption at around 1583 nm is characteristic of porphyrinoid radicals.6 Similarly, the radical cation of β,β′-dichloro diphenylamine-fused porphyrin [14]+ also shows a broad and largely red-shifted absorption spectrum with peaks at 441, 460, 543, 663, and 903 nm, while the neutral species 14 displays a Soret band at 453 nm and Q bands at 570 and 615 nm. The observed red-shifted absorption bands of [13]+ compared to those of [14]+ can be ascribed to the effective conjugation of the fused phenoxazine moiety.
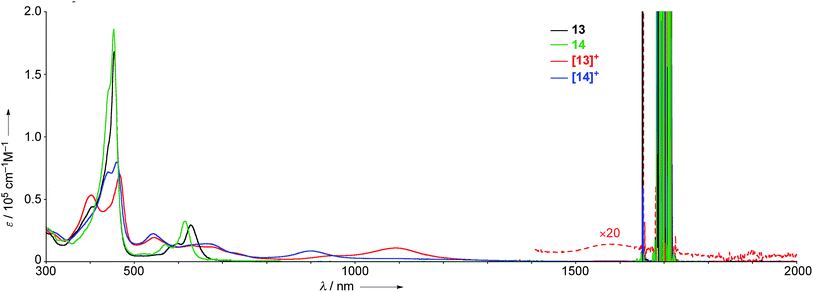 |
| | Fig. 7 UV/Vis absorption spectra of 13, 14, [13]+, and [14]+ in CH2Cl2. ε = extinction coefficient. | |
We have investigated the excited-state decay-dynamics of 13, 14, [13]+ and [14]+ by femtosecond transient absorption (TA) measurements. The TA spectra of 13 exhibited solvent-dependent decay-dynamics with time constants of 0.8 and 31 ps in methylcyclohexane, 0.5 and 18 ps in toluene, and 0.4 and 10 ps in CH2Cl2. Similarly, 14 also displayed solvent-dependent decay-dynamics with time constants of 0.9 and 70 ps in methylcyclohexane, 0.5 and 34 ps in toluene, and 0.4 and 10 ps in CH2Cl2. Then, their decay time constants increase according to a decrease of solvent polarity, suggesting the occurrence of an intramolecular charge transfer (CT) process in 13 and 14. Particularly, faster decay time constants were observed in 13 compared to 14, indicating a faster charge recombination process facilitated by effective π-conjugation in planar conformation of 13. The TA spectra of [13]+ and [14]+ decay rapidly with time constants of 0.7 and 6.5 ps for [13]+ and 0.6 and 8.0 ps for [14]+, respectively. These fast relaxation processes are assignable to internal conversion processes to the lowest excited state and the open-shell ground state, which is well-matched with the ultrafast relaxation dynamics of radical species due to their high density of states.6
To obtain more insights into the π-conjugation and radical nature, we conducted two photon absorption (TPA) measurements by using open-aperture Z-scan methods (see Fig. S68 and 69 in the ESI†). The maximum TPA cross-section values of 13 and 14 were measured as 260 and 140 GM, respectively, at 1200 nm. Compared to a small TPA cross-section value of typical porphyrins (<100 GM),29 larger TPA cross-section values of 13 and 14 are comprehended by the extension of π-conjugation.9a,30 In the same regard, the larger TPA cross-section value of 13 compared with that of 14 may arise from π-system extension due to the fused phenoxazine group. For the radical cations [13]+ and [14]+, the maximum TPA cross-section values were measured as 500 GM at 2200 nm, and 410 GM at 1800 nm, respectively, which are almost twofold larger than their non-radical analogues 13 and 14. These largely enhanced TPA cross-section values of [13]+ and [14]+ can be ascribed to their radical characters.6f
Magnetic properties and DFT calculations
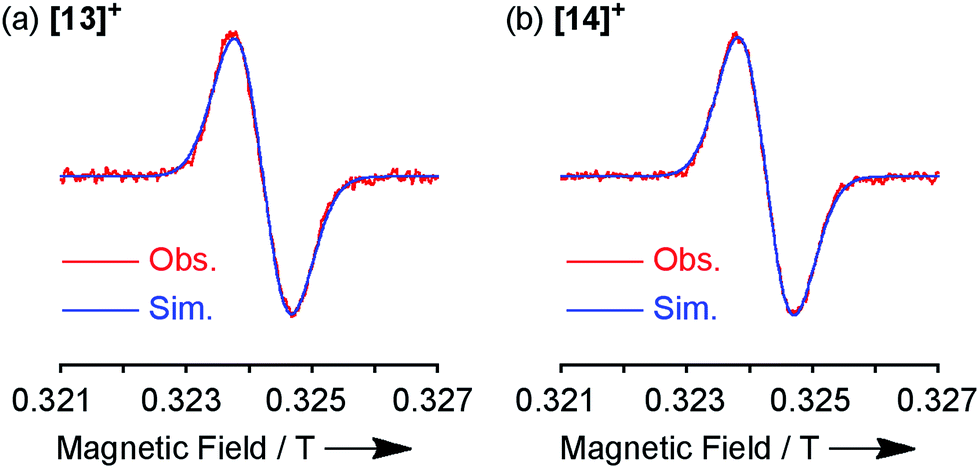
Both the 1H NMR spectra of [13]+ and [14]+ (from −2 to 12 ppm) in CDCl3 exhibited no signal except for two broad singlets due to the tert-butyl groups (ESI, Fig. S32 and S34†). Instead, the electron spin resonance (ESR) spectra of [13]+ and [14]+ in degassed toluene solutions at room temperature showed sharp signals at g = 2.0031 and 2.0029, respectively, suggesting their π-radical characters (Fig. 8). The lack of hyperfine coupling may be attributed to the fast equilibrium between the monomer and dimer and/or small contribution of the H and N atoms to the spin delocalization.| | 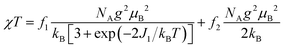 | (1) |
| | 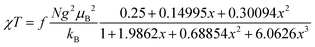 | (2) |
| | 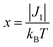 | (3) |
 |
| | Fig. 8 ESR spectra observed at room temperature and the corresponding simulated spectra of (a) [13]+ and (b) [14]+. | |
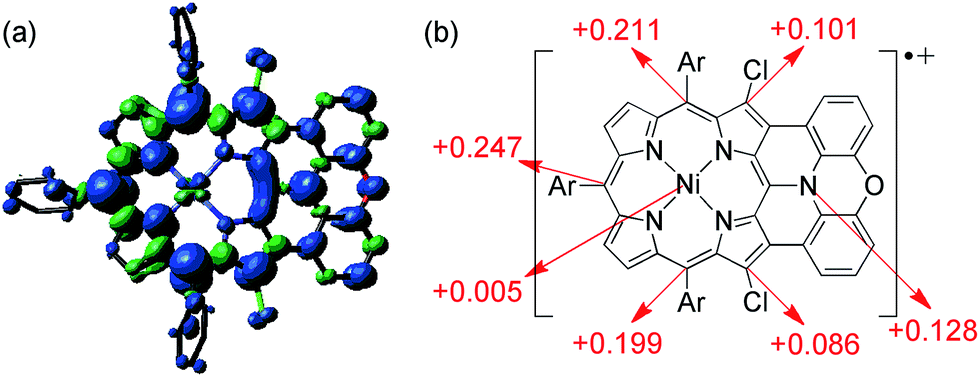
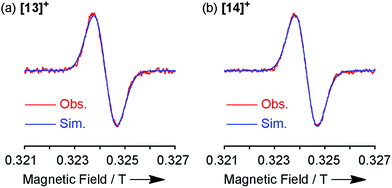
The spin density of [13]+ was calculated at the UB3LYP/6-31*(C,H,N,O,Cl) + LANL2DZ(Ni) level using the Gaussian 09 program package31 excluding the counteranion, which was found to be delocalized over the whole π-system (Fig. 9). The spin density value of the embedded nitrogen atom (+0.132) is significantly smaller than those of planarized triarylamines7c,g (+0.328 and +0.366) due to the effective spin delocalization onto the porphyrin unit. The large spin densities at the three meso-positions apart from the fused nitrogen atom have been observed in other porphyrin π-radicals.6a,j,k In addition, chlorine-substituted β-positions show moderate contribution to the spin delocalization (+0.101 and +0.086). Attempted calculations on [4Ni]+ also exhibit similar spin density at the reactive β-positions (+0.111 and +0.095) (ESI, Fig. S62†), emphasizing the necessity for steric protection.6g In contrast, the central nickel atom displays negligible spin density, suggesting that [13]+ is a π-radical having a two-valent nickel center.
 |
| | Fig. 9 (a) Spin density distribution and (b) selected values of [13]+ calculated at the UB3LYP/6-31G*(C,H,N,O,Cl) + LANL2DZ(Ni) level (isovalue: 0.001; the molecular geometry was optimized based on the X-ray crystal structure). | |
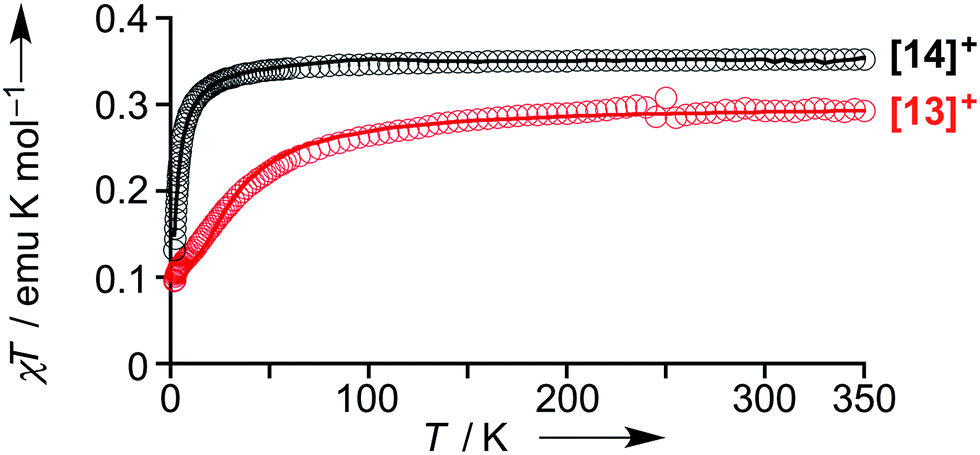
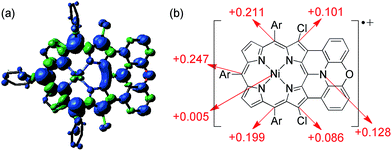
The magnetic properties of [13]+ and [14]+ in the solid (powder) states were examined by temperature-dependent magnetic susceptibility measurements (SQUID) at 0.5 T from 2 to 350 K for [13]+ and from 2 to 300 K for [14]+ (Fig. 10). The observed χT values of [13]+ and [14]+ clearly indicated the different degrees of magnetic interaction. The observed low χT values of [13]+ might be induced by the break down of the crystal structure originating from the defect of the solvent molecules. Although the magnetic analyses were conducted for the powder samples, it may be considered that the molecules should take packing structures similar to their single crystals in a microscopic sense. Namely, [13]+ will adopt an offset face-to-face dimer while [14]+ will take a one-dimensional columnar arrangement. According to these assumptions, the χT values of [13]+ and [14]+ obeyed the Bleaney–Bowers singlet-triplet model32 (eqn (1)) and Bonner–Fisher model33 (eqn (2) and (3)), respectively. The parameters J1/kB of [13]+ and [14]+ are −30.4 and −1.6 K, respectively. The small absolute value of J1/kB in [14]+ indicates the very weak intermolecular antiferromagnetic interaction. In contrast, [13]+ exhibits relatively strong antiferromagnetic interaction, reflecting the large overlap and short separation between the two π-systems in the crystal structure.
 |
| | Fig. 10 Temperature-dependent magnetic susceptibility in the solid states of [13]+ (red) and [14]+ (black). Circles: observed; solid line: simulated. | |
Conclusions
Oxidative fusion reaction of meso-phenoxazino Ni(II) porphyrin 7 was found to be temperature dependent. Doubly phenoxazine-fused porphyrin 4Ni was synthesized by stepwise oxidation of meso-phenoxazino Ni(II) porphyrin 7 at high temperature, and was converted to the corresponding free base porphyrin 4H2 and Zn(II) porphyrin 4Zn. The X-ray structure analysis of 4Ni revealed its fairly planar structure and an offset pairing with a small interporphyrin separation in the crystal. Similar effective π–π stacking dimerization in CDCl3 was also indicated by 1H NMR titration. The cyclic voltammetry of 4Ni exhibited a characteristic split first oxidation wave, indicating the generation of a mixed-valence π-radical cation dimer upon electrochemical oxidation. The fused porphyrins 4H2 and 4Zn show large fluorescence quantum yields as compared to those of the diphenylamine-fused counterpart 10H2 and 10Zn.
One-electron oxidation of 4Ni with Magic Blue gave stable radical cation [4Ni]+, which was isolated via separation over a silica gel column in open air but was slowly chlorinated at the reactive β-positions. β,β-Dichlorinated radical cations [13]+ and [14]+ were much more stable and could be stored for over several months without any degradation under ambident conditions. In the solid states, [13]+ showed distinctly larger antiferromagnetic interaction than that of [14]+ owing to the more tightly packed dimeric unit arising from its planar structure. Further investigations are underway to create other stable porphyrin radical cations in our laboratory.
Acknowledgements
The work at Kyoto was supported by Grants-in-Aid from MEXT (No. 25107002 “Science of Atomic Layers”), from JSPS (No. 25220802 (Scientific Research (S)), 24685007 (Young Scientists (A)), 26620081 (Exploratory Research)), and ACT-C, JST. N. F. acknowledges a JSPS Fellowship for Young Scientists. H. Y. acknowledges Kansai Research Foundation for Technology Promotion and Asahi Glass Foundation for financial support. The work at Yonsei was supported by the Global Research Laboratory (GRL) Program (2013K1A1A2A02050183) of the Ministry of Education, Science and Technology (MEST) of Korea.
Notes and references
- Selected reviews for photosynthesis:
(a) M. R. Wasielewski, Chem. Rev., 1992, 92, 435 CrossRef CAS;
(b) D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2001, 34, 40 CrossRef CAS PubMed;
(c) S. Fukuzumi, K. Ohkubo and T. Suenobu, Acc. Chem. Res., 2014, 47, 1455 CrossRef CAS PubMed. Selected reviews for enzymatic oxidation cycles of cytochrome P-450:
(d) R. E. White and M. J. Coon, Annu. Rev. Biochem., 1980, 49, 315 CrossRef CAS PubMed;
(e) M. Sono, M. P. Roach, E. D. Coulter and J. H. Dawson, Chem. Rev., 1996, 96, 2841 CrossRef CAS PubMed;
(f) P. Hlavica, Eur. J. Biochem., 2004, 271, 4335 CrossRef CAS PubMed.
-
(a)
R. H. Felton, Primary Redox Reactions of Metalloporphyrins: The Porphyrins, ed. D. Dolphin, Academic Press, New York, 1978, vol. 5, pp. 53–125 Search PubMed;
(b) J.-H. Fuhrhop and D. Mauzerall, J. Am. Chem. Soc., 1968, 90, 3875 CrossRef CAS PubMed;
(c) R. H. Felton, D. Dolphin, D. C. Borg and J. Fajer, J. Am. Chem. Soc., 1969, 91, 196 CrossRef CAS;
(d) A. Wolberg and J. Manassen, J. Am. Chem. Soc., 1970, 92, 2982 CrossRef CAS PubMed;
(e) A. Wolberg and J. Manassen, Inorg. Chem., 1970, 9, 2365 CrossRef CAS;
(f) J. Fajer, D. C. Borg, A. Forman, A. D. Adler and V. Varad, J. Am. Chem. Soc., 1974, 96, 1238 CrossRef CAS PubMed.
-
(a) W. F. Scholz and C. A. Reed, J. Am. Chem. Soc., 1982, 104, 6791 CrossRef CAS;
(b) P. Gans, G. Buisson, E. Duée, J.-C. Marchon, B. S. Erler, W. F. Scholz and C. A. Reed, J. Am. Chem. Soc., 1986, 108, 1223 CrossRef CAS;
(c) H. Song, R. D. Orosz, C. A. Reed and W. R. Scheidt, Inorg. Chem., 1990, 29, 4274 CrossRef CAS;
(d) W. R. Scheidt, K. E. Brancato-Buentello, H. Song, K. V. Reddy and B. Cheng, Inorg. Chem., 1996, 35, 7500 CrossRef CAS;
(e) K. M. Barkigia, M. W. Renner and J. Fajer, J. Phys. Chem. B, 1997, 101, 8398 CrossRef CAS;
(f) R.-J. Cheng, C.-H. Ting, T.-C. Chao, T.-H. Tseng and P. P.-Y. Chen, Chem. Commun., 2014, 50, 14265 RSC.
-
(a) J. T. Groves, R. C. Haushalter, M. Nakamura, T. E. Nemo and B. J. Evans, J. Am. Chem. Soc., 1981, 103, 2884 CrossRef CAS;
(b) S. Tsuchiya, J. Chem. Soc., Chem. Commun., 1991, 716 RSC.
-
R. G. Hicks, Stable Radicals: Fundamentals and Applied Aspects of Odd-Electron Compounds, Wiley-Blackwell, New York, 2010 Search PubMed.
-
(a) R. G. Khoury, L. Jaquinod, A. M. Shachter, N. Y. Nelson and K. M. Smith, Chem. Commun., 1997, 215 RSC;
(b) S. Hiroto, K. Furukawa, H. Shinokubo and A. Osuka, J. Am. Chem. Soc., 2006, 128, 12380 CrossRef CAS PubMed;
(c) T. Koide, G. Kashiwazaki, M. Suzuki, K. Furukawa, M.-C. Yoon, S. Cho, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2008, 47, 9661 CrossRef CAS PubMed;
(d) M. Ishida, J.-Y. Shin, J. M. Lim, B. S. Lee, M.-C. Yoon, T. Koide, J. L. Sessler, A. Osuka and D. Kim, J. Am. Chem. Soc., 2011, 133, 15533 CrossRef CAS PubMed;
(e) D. Shimizu, J. Oh, K. Furukawa, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2015, 54, 6613 CrossRef CAS PubMed;
(f) Y. Hisamune, K. Nishimura, K. Isakari, M. Ishida, S. Mori, S. Karasawa, T. Kato, S. Lee, D. Kim and H. Furuta, Angew. Chem., Int. Ed., 2015, 54, 7323 CrossRef CAS PubMed;
(g) P. Schweyen, K. Brandhorst, R. Wicht, B. Wolfram and M. Bröring, Angew. Chem., Int. Ed., 2015, 54, 8213 CrossRef CAS PubMed;
(h) Y. Tanaka, T. Yoneda, K. Furukawa, T. Koide, H. Mori, T. Tanaka, H. Shinokubo and A. Osuka, Angew. Chem., Int. Ed., 2015, 54, 10908 CrossRef CAS PubMed;
(i) T. Yoshida, W. Zhou, T. Furuyama, D. B. Leznoff and N. Kobayashi, J. Am. Chem. Soc., 2015, 137, 9258 CrossRef CAS PubMed;
(j) D. Shimizu, J. Oh, K. Furukawa, D. Kim and A. Osuka, J. Am. Chem. Soc., 2015, 137, 15584 CrossRef CAS PubMed;
(k) L. J. Esdaile, L. Rintoul, M. S. Goh, K. Merahi, N. Parizel, R. M. Wellard, S. Choua and D. P. Arnold, Chem.–Eur. J., 2016, 22, 3430 CrossRef CAS PubMed.
-
(a) D. Hellwinkel and M. Melan, Chem. Ber., 1974, 107, 616 CrossRef CAS;
(b) D. Bamberger, D. Hellwinkel and F. A. Neugebauer, Chem. Ber., 1975, 108, 2416 CrossRef;
(c) M. Kuratsu, M. Kozaki and K. Okada, Angew. Chem., Int. Ed., 2005, 44, 4056 CrossRef CAS PubMed;
(d) M. Kuratsu, S. Suzuki, M. Kozaki, D. Shiomi, K. Sato, T. Takui and K. Okada, Inorg. Chem., 2007, 46, 10153 CrossRef CAS PubMed;
(e) M. Takase, V. Enkelmann, D. Sebastiani, M. Baumgarten and K. Müllen, Angew. Chem., Int. Ed., 2007, 46, 5524 CrossRef CAS PubMed;
(f) M. Takase, T. Narita, W. Fujita, M. S. Asano, T. Nishinaga, H. Benten, K. Yoza and K. Müllen, J. Am. Chem. Soc., 2013, 135, 8031 CrossRef CAS PubMed;
(g) X. Zheng, X. Wang, Y. Qiu, Y. Li, C. Zhou, Y. Sui, Y. Li, J. Ma and X. Wang, J. Am. Chem. Soc., 2013, 135, 14912 CrossRef CAS PubMed.
- E. T. Seo, R. F. Nelson, J. M. Fritsch, L. S. Marcoux, D. W. Leedy and R. N. Adams, J. Am. Chem. Soc., 1966, 98, 3498 CrossRef.
-
(a) N. Fukui, W.-Y. Cha, S. Lee, S. Tokuji, D. Kim, H. Yorimitsu and A. Osuka, Angew. Chem., Int. Ed., 2013, 52, 9728 CrossRef CAS PubMed;
(b) N. Fukui, H. Yorimitsu and A. Osuka, Angew. Chem., Int. Ed., 2015, 54, 6311 CrossRef CAS PubMed.
- Gryko et al. also reported a singly dinaphthylamine-fused porphyrin, independently. A. Nowak-Król and D. T. Gryko, Org. Lett., 2013, 15, 5618 CrossRef PubMed.
- N. Fukui, S.-K. Lee, K. Kato, D. Shimizu, T. Tanaka, S. Lee, H. Yorimitsu, D. Kim and A. Osuka, Chem. Sci., 2016, 7, 4059 RSC.
-
(a) N. A. Cortese and R. F. Heck, J. Org. Chem., 1977, 42, 3491 CrossRef CAS;
(b) A. K. Sahoo, Y. Nakamura, N. Aratani, K. S. Kim, S. B. Noh, H. Shinokubo, D. Kim and A. Osuka, Org. Lett., 2006, 18, 4141 CrossRef PubMed.
- 2-(2′,6′-Dimethoxybiphenyl)dicyclohexylphosphine (SPhos): T. E. Barder, S. D. Walker, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 4685 CrossRef CAS PubMed.
- K. Murakami, Y. Yamamoto, H. Yorimitsu and A. Osuka, Chem.–Eur. J., 2013, 19, 9123 CrossRef CAS PubMed.
- J.-P. Strachan, S. Gentemann, J. Seth, W. A. Kalsbeck, J. S. Lindsey, D. Holten and D. F. Bocian, J. Am. Chem. Soc., 1997, 119, 11191 CrossRef CAS.
- Crystallographic data for 2Ni: C74H77N5Ni·CHCl3·CH3CN, monoclinic, space group C2/c (no. 15), a = 36.653(7) Å, b = 21.504(4) Å, c = 17.115(3) Å, β = 98.833(2)°, V = 13330(4) Å3, T = 93 K, Z = 8, R1 = 0.0768 (I > 2σ(I)), Rw = 0.2444 (all data), GOF = 1.047. CCDC number: 1469154. Crystallographic data for 3Ni: C74H75N5NiO·(C6H5CH3)1.5, monoclinic, space group P21/n (no. 14), a = 21.181(7) Å, b = 10.751(4) Å, c = 29.482(10) Å, β = 100.128(9)°, V = 6609(4) Å3, T = 93 K, Z = 4, R1 = 0.0714 (I > 2σ(I)), Rw = 0.2075 (all data), GOF = 1.029. CCDC number: 1469155.
- The MPD of 10Ni is defined by the 38 core atoms consisting of meso-carbons, four pyrrole units, central nickel atom, and fused-diphenylamine unit. The MPD of 4Ni is defined by the 39 core atoms consisting of four pyrrole units, central nickel atom, and fused-phenoxazine unit.
-
(a) A. S. Shetty, J. Zhang and J. S. Moore, J. Am. Chem. Soc., 1996, 118, 1019 CrossRef CAS;
(b) Y. Tobe, N. Utsumi, K. Kawabata, A. Nagano, K. Adachi, S. Araki, M. Sonoda, K. Hirose and K. Naemura, J. Am. Chem. Soc., 2002, 124, 5350 CrossRef CAS PubMed;
(c) J. Wu, A. Fechtenkötter, J. Gauss, M. D. Watson, M. Kastler, C. Fechtenkötter, M. Wagner and K. Müllen, J. Am. Chem. Soc., 2004, 126, 11311 CrossRef CAS PubMed;
(d) C.-J. Wallentin, T. Wixe, O. F. Wendt, K.-E. Bergquist and K. Wärnmark, Chem.–Eur. J., 2010, 16, 3994 CrossRef CAS PubMed.
- M. Saunders and J. B. Hyne, J. Chem. Phys., 1958, 20, 1319 CrossRef.
- I. Horman and B. Dreux, Helv. Chim. Acta, 1984, 67, 754 CrossRef CAS.
- K. Fujimoto, J. Oh, H. Yorimitsu, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2016, 55, 3196 CrossRef CAS PubMed.
-
(a) K. M. Kadish, D. Sazou, Y. M. Liu, A. Saoiabi, M. Ferhat and R. Guilard, Inorg. Chem., 1988, 27, 686 CrossRef CAS;
(b) A. Giraudeau, A. Louati, M. Gross, J. J. Andre, J. Simon, C. H. Su and K. M. Kadish, J. Am. Chem. Soc., 1983, 105, 2917 CrossRef CAS;
(c) D. Gut, I. Goldberg and M. Kol, Inorg. Chem., 2003, 42, 3483 CrossRef CAS PubMed;
(d) S. D. Bergman, I. Goldberg, A. Barbieri, F. Barigelletti and M. Kol, Inorg. Chem., 2004, 43, 2355 CrossRef CAS PubMed;
(e) N. R. d. Tacconi, R. Chitakunye, F. M. MacDonnell and R. O. Lezna, J. Phys. Chem. A, 2008, 112, 497 CrossRef PubMed;
(f) M. A. Christensen, C. R. Parker, T. J. Sørensen, S. d. Graaf, T. J. Morsing, T. Brock-Nannestad, J. Bendix, M. M. Haley, P. Rapta, A. Danilov, S. Kubatkin, O. Hammerich and M. B. Nielsen, J. Mater. Chem. C, 2014, 2, 10428 RSC;
(g) M. A. Christensen, G. E. Rudebusch, C. R. Parker, C. L. Anderson, A. Kadziola, M. M. Haley, O. Hammerich and M. B. Nielsen, RSC Adv., 2015, 5, 49748 RSC.
- R. Sakamoto, M. Nishikawa, T. Yamamura, S. Kume and H. Nishihara, Chem. Commun., 2010, 46, 2028 RSC.
-
(a) Y. L. Mest, M. L'Her, N. H. Hendricks, K. Kim and J. P. Collman, Inorg. Chem., 1992, 31, 835 CrossRef;
(b) Y. Matano, K. Matsumoto, Y. Terasaka, H. Hotta, Y. Araki, O. Ito, M. Shiro, T. Sasamori, N. Tokitoh and H. Imahori, Chem.–Eur. J., 2007, 13, 891 CrossRef CAS PubMed.
- R. A. Rodriguez, C.-M. Pan, Y. Yabe, Y. Kawamata, M. D. Eastgate and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 6908 CrossRef CAS PubMed.
-
(a) F. Cozzi and J. S. Siegel, Pure Appl. Chem., 1995, 67, 683 CrossRef CAS;
(b) M. J. Rashkin and M. L. Waters, J. Am. Chem. Soc., 2002, 124, 1860 CrossRef CAS PubMed.
- Crystallographic data for 8: (C74H73Cl2N5NiO)2·(CHCl3)1.5, triclinic, space group P
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 18.141(3) Å, b = 20.0791(19) Å, c = 20.299(2) Å, α = 62.0374(10)°, β = 79.030(16)°, γ = 87.782(14)°, V = 6400.6(14) Å3, T = 93 K, Z = 2, R1 = 0.0791 (I > 2σ(I)), Rw = 0.2451 (all data), GOF = 1.045. CCDC number: 1469157. Crystallographic data for [8]+: C74H73Cl2N5NiO·SbCl6·(C6H14)2·CH2Cl2, tetragonal, space group I41/a (no. 88), a = 57.941(6) Å, b = 57.941(6) Å, c = 10.6631(12) Å, V = 35798(8) Å3, T = 93 K, Z = 16, R1 = 0.0992 (I > 2σ(I)), Rw = 0.2692 (all data), GOF = 1.079. CCDC number: 1469158. Crystallographic data for 9: (C74H75Cl2N5Ni)2·(CHCl3)1.5·C3H7OH, monoclinic, space group C2/c (no. 15), a = 72.720(9) Å, b = 13.4081(16) Å, c = 33.152(4) Å, β = 102.869(4)°, V = 31513(7) Å3, T = 93 K, Z = 8, R1 = 0.0996 (I > 2σ(I)), Rw = 0.2800 (all data), GOF = 1.064. CCDC number: 1469159. Crystallographic data for [9]+: C74H75Cl2N5Ni·SbCl6·(CH2Cl2)0.5, monoclinic, space group P21/n (no. 14), a = 27.888(11) Å, b = 9.758(3) Å, c = 29.846(11) Å, β = 113.508(6)°, V = 7448(5) Å3, T = 93 K, Z = 4, R1 = 0.0826 (I > 2σ(I)), Rw = 0.2363 (all data), GOF = 1.004. CCDC number: 1469160.
(no. 2), a = 18.141(3) Å, b = 20.0791(19) Å, c = 20.299(2) Å, α = 62.0374(10)°, β = 79.030(16)°, γ = 87.782(14)°, V = 6400.6(14) Å3, T = 93 K, Z = 2, R1 = 0.0791 (I > 2σ(I)), Rw = 0.2451 (all data), GOF = 1.045. CCDC number: 1469157. Crystallographic data for [8]+: C74H73Cl2N5NiO·SbCl6·(C6H14)2·CH2Cl2, tetragonal, space group I41/a (no. 88), a = 57.941(6) Å, b = 57.941(6) Å, c = 10.6631(12) Å, V = 35798(8) Å3, T = 93 K, Z = 16, R1 = 0.0992 (I > 2σ(I)), Rw = 0.2692 (all data), GOF = 1.079. CCDC number: 1469158. Crystallographic data for 9: (C74H75Cl2N5Ni)2·(CHCl3)1.5·C3H7OH, monoclinic, space group C2/c (no. 15), a = 72.720(9) Å, b = 13.4081(16) Å, c = 33.152(4) Å, β = 102.869(4)°, V = 31513(7) Å3, T = 93 K, Z = 8, R1 = 0.0996 (I > 2σ(I)), Rw = 0.2800 (all data), GOF = 1.064. CCDC number: 1469159. Crystallographic data for [9]+: C74H75Cl2N5Ni·SbCl6·(CH2Cl2)0.5, monoclinic, space group P21/n (no. 14), a = 27.888(11) Å, b = 9.758(3) Å, c = 29.846(11) Å, β = 113.508(6)°, V = 7448(5) Å3, T = 93 K, Z = 4, R1 = 0.0826 (I > 2σ(I)), Rw = 0.2363 (all data), GOF = 1.004. CCDC number: 1469160.
-
(a) T. Nishinaga and K. Komatsu, Org. Biomol. Chem., 2005, 3, 561 RSC;
(b) M. Tateno, M. Takase, M. Iyoda, K. Komatsu and T. Nishinaga, Chem.–Eur. J., 2013, 19, 5457 CrossRef CAS PubMed.
- T. K. Ahn, K. Kim, D. Y. Kim, S. B. Noh, N. Aratani, C. Ikeda, A. Osuka and D. Kim, J. Am. Chem. Soc., 2006, 128, 1700 CrossRef CAS PubMed.
-
(a) P. Kim, S. Ham, J. Oh, H. Uoyama, H. Watanabe, K. Tagawa, H. Uno and D. Kim, Phys. Chem. Chem. Phys., 2013, 15, 10612 RSC;
(b) W. Zeng, S. Lee, M. Son, M. Ishida, K. Furukawa, P. Hu, Z. Sun, D. Kim and J. Wu, Chem. Sci., 2015, 6, 2427 RSC.
-
M. J. Frisch, et al., Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- B. Bleaney and K. D. Bowers, Proc. R. Soc. London, Ser. A, 1952, 214, 451 CrossRef CAS.
-
(a) J. C. Bonner and M. E. Fisher, Phys. Rev., 1964, 135, A640 CrossRef;
(b) W. E. Estes, D. P. Gavel, W. E. Hatfield and D. J. Hodgson, Inorg. Chem., 1978, 17, 1415 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1469154–1469160. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc02721k |
|
| This journal is © The Royal Society of Chemistry 2017 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence