
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The value of pyrans as anticancer scaffolds in medicinal chemistry†
Dinesh Kumar
*a,
Pooja Sharma
ae,
Harmanpreet Singh
a,
Kunal Nepali
a,
Girish Kumar Gupta
*b,
Subheet Kumar Jain
a and
Fidele Ntie-Kang
 *cd
*cd
aDepartment of Pharmaceutical Sciences, Guru Nanak Dev University, Amritsar, Punjab-143005, India. E-mail: dinesh_arora_81@yahoo.co.in; Tel: +91-9988902489
bDepartment of Pharmaceutical Chemistry, M. M. College of Pharmacy, Maharishi Markandeshwer University, Mullana, 133203, Haryana, India. E-mail: girish_pharmacist92@rediffmail.com; Tel: +91-80599-30169 Tel: +91-72069-30164
cDepartment of Chemistry, Faculty of Science, University of Buea, P.O. Box 63, Buea, Cameroon. E-mail: fidele.ntie-kang@ubuea.cm; Tel: +237 677915473
dInstitute for Pharmacy, Martin-Luther-Universität Halle-Wittenberg, Wolfgang-Langenbeck-Str. 4, 06120 Halle (Saale), Germany. E-mail: ntiekfidele@gmail.com; fidele.ntie-kang@pharmazie-uni-halle.de; Tel: +49 3455525043
eSri Sai College of Pharmacy Manawala, Amritsar, 143115, Punjab, India
First published on 25th July 2017
Abstract
Pyran is an oxygen-containing heterocyclic moiety, which exhibits an array of pharmacological properties. Pyran is also one of the important structural subunits found widely in natural products, e.g. coumarins, benzopyrans, sugars, flavonoids, xanthones, etc. The diverse anticancer capabilities of pyrans have been additionally evidenced by the fact that this heterocycle has recently been a focal point for researchers worldwide. This review provides a summary of pyran-based anticancer compounds, with emphasis on the past 10 years. It focuses on advancements in the field of naturally occurring pyrans as anticancer agents. The discussion also includes structure–activity relationships, along with the structures of the most promising molecules, their biological activities against several human cancer cell lines, as well as mechanistic insights discovered through the pharmacological evaluation and molecular modeling of pyran-based molecules. The promising activities revealed by these pyran-based scaffolds undoubtedly place them at the forefront for the discovery of prospective drug candidates. Thus, they could therefore be of great interest to researchers working on the synthesis of antitumour drug candidates.
 Dinesh Kumar | Dinesh Kumar graduated in pharmacy in 2005 from the Punjab Technical University, occupying the 4th position in the entire university. In 2008 he obtained his Master's degree in pharmaceutical chemistry from the Guru Nanak Dev University, Amritsar, Punjab, India. He is currently a Ph.D. candidate in pharmaceutical sciences from the Guru Nanak Dev University, Amritsar, where he works on the synthesis and evaluation of chalconoid hybrids as potent anti-proliferative agents. In 2014 he became Assistant Professor of Pharmaceutical Sciences, Guru Nanak Dev University, Amritsar. His current research is focused on the synthesis and evaluation novel heterocycles, along with their application in medicinal chemistry. |
 Pooja Sharma | Pooja Sharma is a graduate in pharmacy from the Punjab Technical University (2005), later obtaining her Master's degree in Pharmacognosy (Phytochemistry) in 2008 from the Department of Pharmaceutical Sciences, Guru Nanak Dev University, Amritsar. During her graduate studies, she emerged 2nd position in the university. She also worked in the Council of Scientific & Industrial Research (CSIR) laboratory of India at Jammu, focusing on the phytochemical investigation and standardization of extracts from Boerhaavia diffusa. She later became Assistant Professor in Sri Sai College of Pharmacy, Manawala, Amritsar, Punjab, India. Currently she is Head of Department at Sri Sai College of Pharmacy, Manawala, Amritsar, Punjab, India, where she focuses on the extraction and isolation of phytochemical constituents of pharmaceutical interest. |
 Harmanpreet Singh | Harmanpreet Singh studied pharmacy at the Punjab Technical University between 2006 and 2010. His post graduate work was within the field of pharmaceutics (ISF college of Pharmacy), where he worked on the transmucosal delivery of docetaxel using nanofiber as a carrier system for the treatment of buccal cancer. He started his Ph.D. in 2014 at the Guru Nanak Dev University, Amritsar, India, where he is currently working on the development of different carrier system for increasing the bioavailability, shelf life and effective delivery of the docosahexanoic acid (DHA). |
 Kunal Nepali | Kunal Nepali obtained his Master's degree in pharmaceutical chemistry from ISF college of Pharmacy Moga, Punjab in 2008. He later obtained his Ph.D. in pharmaceutical chemistry from the Punjab Technical University. His research is focused on designing bioactive products based on SAR studies, developing synthetic methodologies for multi-component synthesis and molecular modeling studies of xanthine oxidase and tubulin inhibitors. In 2011 he became Assistant Professor at the Department of Pharmaceutical Sciences, Guru Nanak Dev University, Amritsar. He has attended many national and international conferences and has published his research in journals of international repute. |
 Girish Kumar Gupta | Girish Kumar Gupta graduated in pharmacy in 2006 from Guru Jambheswar University of Science and Technology, Hisar, India. In 2009 he obtained his Master's degree in pharmaceutical chemistry from the University Institute of Pharmaceutical Sciences, Kurukshetra University, Kurukshetra, Haryana, India. Currently working as Assistant Professor in M. M. College of Pharmacy, Maharishi Markandeshwar University, Mullana, Ambala, India, he has more than 8 years of research experience in pharmaceutical field including academic and industrial research and teaching. He has over 60 publications in the field of heterocyclic chemistry in peer reviewed high impact journals. He is also a member of many pharmaceutical associations and societies. He is also an editorial board member, editor, guest editor and reviewer of many reputed international and national journals. His area of research encompasses drug design, molecular modelling, green synthesis related to nitrogen containing heterocycles. |
 Subheet Kumar Jain | Subheet Kumar Jain obtained his M. Pharmacy in pharmaceutics from Dr H. S. Gour University, Sagar in 2000. He later obtained his Ph.D. in pharmaceutical sciences from the same university in 2004. Currently, he is working as Professor & Head in the Department of Pharmaceutical Sciences, Guru Nanak Dev University, Amritsar. His areas of interest include novel drug delivery systems, anti-cancer drug delivery using nano carrier approaches, dermal drug delivery using carrier approaches, and gastro-retentive drug delivery. He has published his research in many national and international journals. |
 Fidele Ntie-Kang | Fidele Ntie-Kang studied Chemistry at the University of Douala in Cameroon between 1999 and 2004, leading to BSc and MSc degrees. His Ph.D. work at the Centre for Atomic Molecular Physics and Quantum Optics (CEPAMOQ) was based on the computer-aided design of anti-tubercular agents. He has an experience in molecular modeling and has been involved in the design and management of databases of natural products from African flora for virtual screening. Fidele has formerly worked as a Scientific Manager/Senior Instructor at the Chemical and Bioactivity Information Centre (CBIC), hosted at the Chemistry Department of the University of Buea, Cameroon. He is currently a Senior Scientist in the group of Prof. Wolfgang Sippl, sponsored by the Alexander von Humboldt Foundation, Germany, under a Georg Forster fellowship. |
1 Introduction
The expression “tumour” induces fear, particularly when one considers recent statistics of cancer cases worldwide.1 Another cause for concern is the mammoth task that physicians must carry out in order to attempt to save patients' lives. A tumour is depicted by the uncontrolled development and spread of abnormal cells. While normal body cells grow, divide and die in an orderly fashion, cancer cells do not follow this norm. They rather continue to grow and divide in a disorderly fashion. The weapons used for this fight generally include specialised surgical operations, radiation therapy and chemotherapy. Despite continued research efforts towards the development of anticancer (chemotherapeutic) drugs, cancer remains a primary cause of death. It is estimated that the number of cancer cases may reach up to 15 million at the end of 2020.2–6According to the World Health Organization (WHO), more than 80% of the world's population relies on traditional medicines for their essential health care needs.7,8 Plants have a long history of their utilization in the treatment of tumors and it is estimated that more than 60% of presently utilised anticancer agents are obtained from nature.8 Heterocyclics represent the most abundant compound classes present among known drugs. Typically, the former need to be decorated with suitable substituents in order to obtain their appropriate biological effects.9
Tremendous progress has been made in the war against cancer, with the development of many novel chemotherapeutic agents. However, due to toxicity and drug-resistance problems encountered with many currently available treatments, it remains a great challenge to discover and develop more effective drugs to treat cancer. We present the structure–activity relationships and their mechanistic insights established during the pharmacological evaluation of selected potent pyrans. The structures of the designed and synthesised molecules discussed in this compilation clearly highlight the interesting and promising anticancer profiles of the compounds. An overview of selected molecular modeling studies has also been incorporated, with the aim of providing insight into the possible binding sites. This classification of the pyrans discussed in this study is based on one of the core functionalities of their chemical architecture. The classification is as follows:
• Benzopyrans and fused pyran-based anticancer scaffolds.
• Flavones and fused flavone-based anticancer scaffolds.
• Coumarins and fused coumarin-based anticancer scaffolds.
• Xanthones and xanthene-based anticancer scaffolds.
• Other scaffolds.
A summary of the most potent compounds have been presented in the ESI (Table S1†).
2 Benzopyrans and fused pyran-based anticancer scaffolds
The pyran ring is the core unit of benzopyran, chromone, flavanoids, coumarin, xanthones, and naphthoquinones, which exhibit diverse pharmacological activities. Pyran heterocycles are both prevalent across compounds classified as of ‘natural origin’ and ‘man-made’. Numerous naturally occurring compounds containing pyrans and benzopyrans, show fascinating therapeutic activities. These have spurred considerable awareness of the synthetic arena based on their structure, reactivity, synthesis and biological properties. The classification of pyran heterocyclic compounds depends on the presence of the 2H or 4H pyran scaffold (Fig. 1). Thus, the benzo derivative of 2H-pyran is named 2H-1-benzopyran (commonly 2H-chromene) and the benzo analogue of 4H-pyran is called 4H-1-benzopyran (commonly 4H-chromene).10 Interrelated naphthyl derivatives are exemplified by 2H-naphtho[1,2,b]pyran and xanthenes. Ketones obtained from pyrans are called pyranones (likewise regularly pyrones), the parent molecules being pyran-2-one and pyran-4-one.11–13 Paltry names are utilised for the related benzo analogues; coumarin, dihydrocoumarin, chromone, xanthone, and chromanone or chroman-4-one. | ||
| Fig. 1 Pyran-based heterocycles. | ||
It is well established that small heterocyclic molecules are predominant building blocks for biologically active compounds,14,15 while an increasing number of structural frameworks have been described as privileged structures.16 Pyran skeletons are important structural units found widely in natural products, e.g. sugars, coumarins,17 flavonoids,18 anthraquinones,19 etc. Examples include flavonoid-based pyran derivatives (Fig. 2), including epicalyxins F and G along with calyxins F, G, L and I (Fig. 2), isolated from the seeds of Alpinia blepharocalyx. Epicalyxin F is the most potent member of this class, as an anticancer agent against human HT-1080 fibrosarcoma and murine 26-L5 carcinoma.20
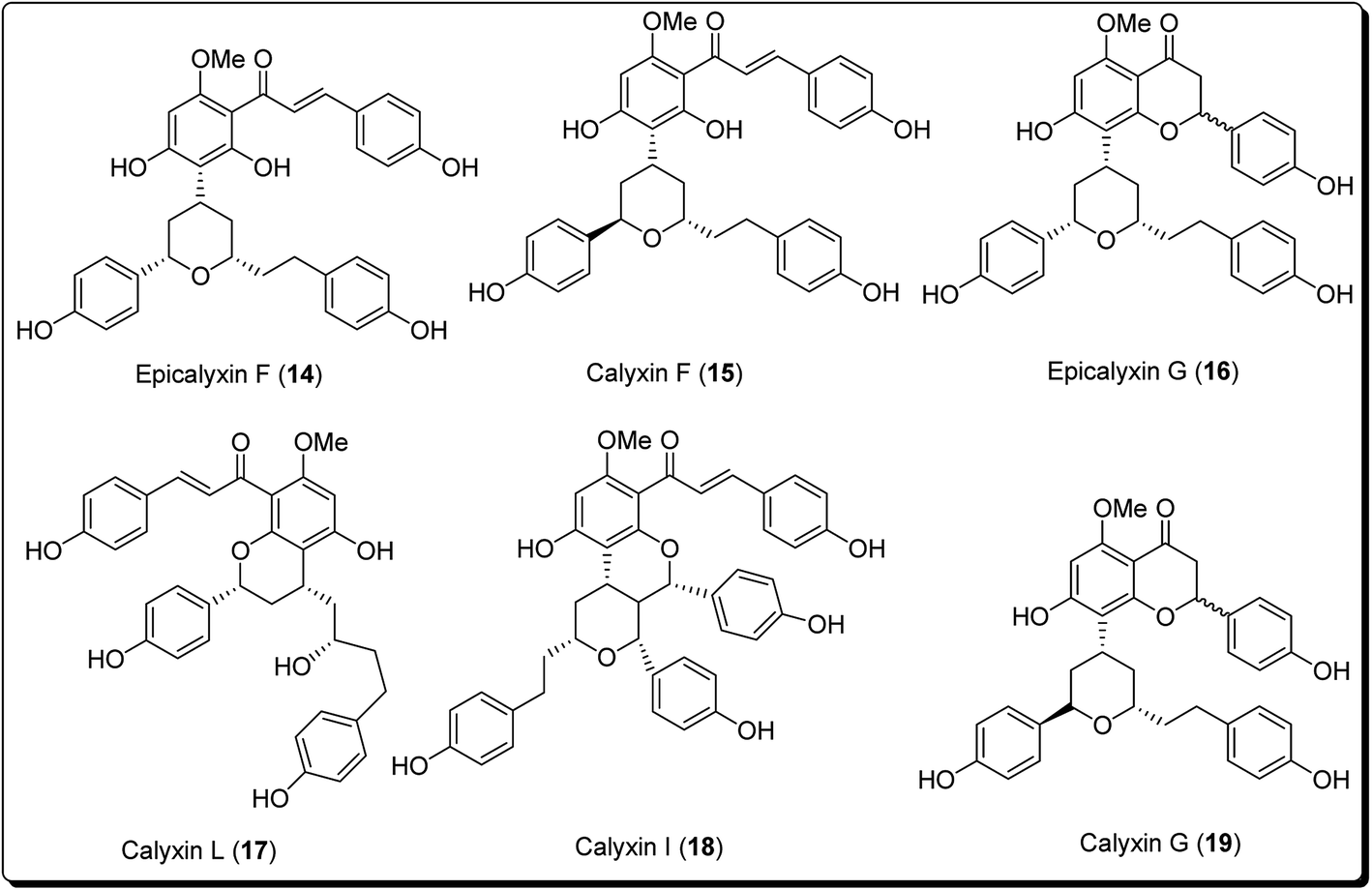 | ||
| Fig. 2 Pyran-based derivatives obtained from natural origin with cell damage potential. | ||
The bioactive metabolite, β-lapachone (20, Fig. 3), is a typical example of a pyran derivative, which generally shows diverse biological activities (e.g. anticancer, antibacterial and anti-inflammatory activities), making it important for drug development. Zanamivir (22, Fig. 3), for example, was approved for prevention of influenza A and B. Moreover, zanamivir was the first commercially developed neuraminidase inhibitor. This drug is currently marketed by GlaxoSmithKline under the trade name of “Relenza”. Laninamivir octanoate is a prodrug of laninamivir (23, Fig. 3), which is structurally similar to zanamivir and is administered orally.21,22 Pyran-based drugs, which are commercially available and/or are in preclinical/clinical trials have been shown in Fig. 3. A literature survey has shown the abundance of commercially available therapeutic agents containing the pyran unit. Benzopyrans and fused pyran-based are an important class of structural motif for many natural and synthetic compounds, possessing high activity profiles, due to their wide range of biological activities, including anticancer properties.23–25
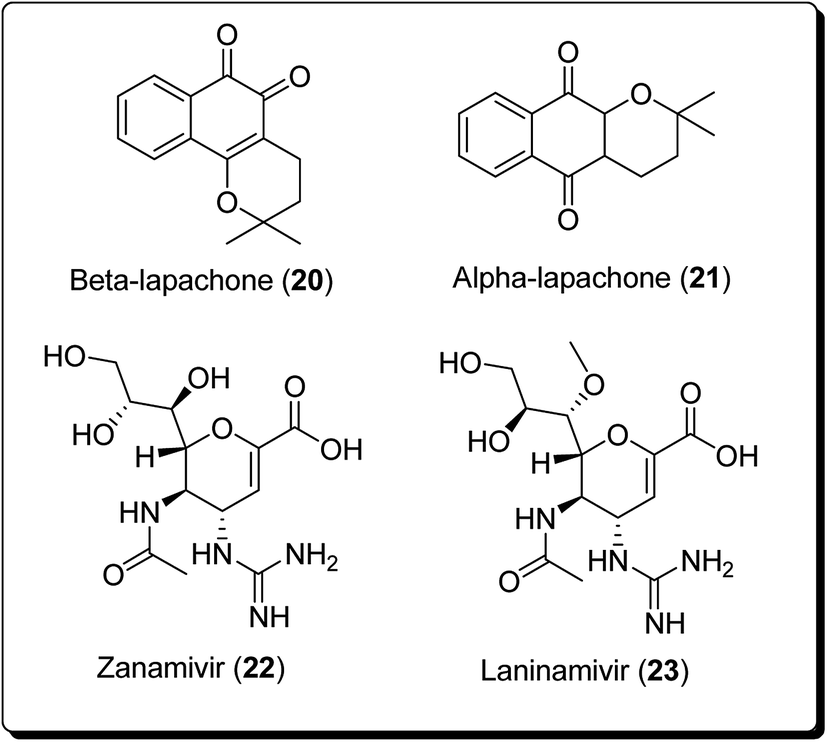 | ||
| Fig. 3 Pyran-based natural and synthetic marketed drugs in preclinical/clinical trials. | ||
Madda et al. synthesised new chromeno-annulated cis-fused pyrano[3,4-c]benzopyran and naphtho pyran analogues, and tested these compounds against different human cancer cell lines. It was shown that compounds 27 and 28 (Fig. 4) had exceptionally high cytotoxicity towards human cervical malignant cells (HeLa). Compound 27, for example, exhibited pronounced inhibitory action against both breast cancer cell lines (MDA-MB-231 and MCF-7). Furthermore, compound 29 displayed high cytotoxicity against only MDA-MB-231, while compound 28 demonstrated promising effects against human lung cancer cell line, A549 with an IC50 value of 2.53 μM.26 Additionally, Morales et al. discovered 5-morpholino-7H-thieno[3,2-b]pyran-7-ones as potential prospective PI3K inhibitors. Substitution of the thiophene for the phenyl core in compound 30 resulted in compound 31, which showed a comparative or better PI3K and mTOR enzymatic inhibition profile than compound 30 (Fig. 5). The former also showed a marginally better aqueous solubility, cell porosity, and better activity when tested in a PC3 cell expansion, while down-regulating the PI3K pathway as shown by restraining pAKT-S473 levels.27
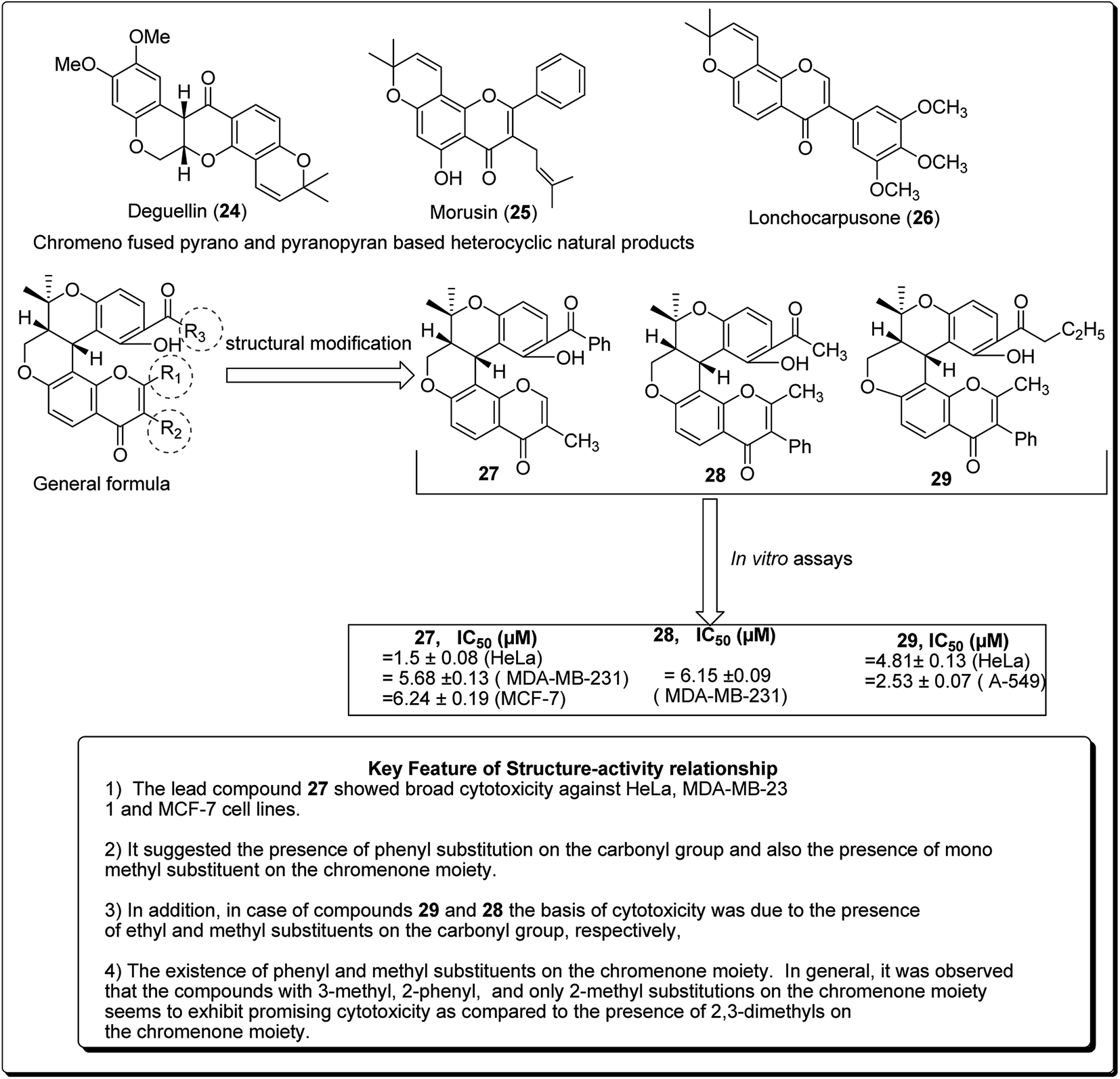 | ||
| Fig. 4 Novel chromeno-annulated cis-fused pyrano[3,4-c]benzopyran and naphtho pyran derivatives along with their SAR. | ||
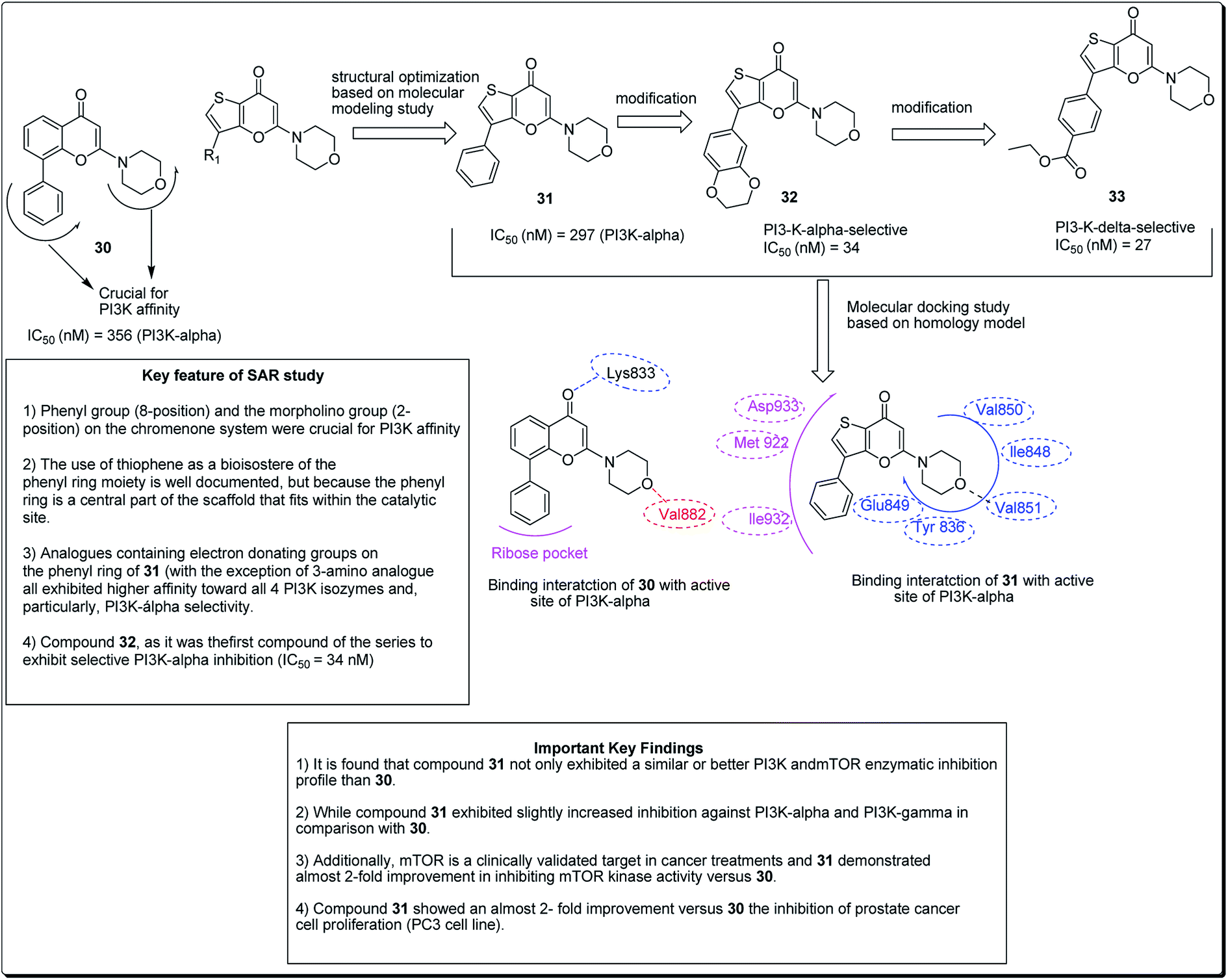 | ||
| Fig. 5 5-Morpholino-7H-thieno[3,2-b]pyran-7-ones designed as next generation PI3K inhibitors along with SARs. | ||
The 4H-pyrano-[2,3-b]naphthoquinone scaffold is known to be a mimetic of an assorted assembly of naturally occurring pyranonaphthoquinones and their engineered analogues, with promising anticancer potentials.28 Natural products within this class include rhinacanthin O (34, Fig. 6) from the Asian medicinal plant Rhinocanthus nasutus, pyranokunthone B (35, Fig. 6) from a marine actinomycete, α- and β-lapachones (21 and 20), isolated from the heartwood of the trees of Bignoniaceae, among others.29 β-Lapachone has been examined for the treatment of tumors connected with hoisted NADH quinone oxidoreductase levels. The compound is currently in stage II clinical trials for the treatment of pancreatic tumour.28–35 Magedov et al. screened a synthesised library of molecules, exhibiting low micromolar antiproliferative activity and initiated apoptosis in human cancerous cells, towards a set of malignant cells.34 Selected analogues exhibited promising activities against cancer cell lines impervious to professional apoptotic stimuli, thus exhibiting their potential in treating tumors with grim anticipations. It was found that compound 36 and 37 showed antiproliferative effects better than those of α-lapachone, even though the latter was optional to the regioisomeric β-lapachone.34
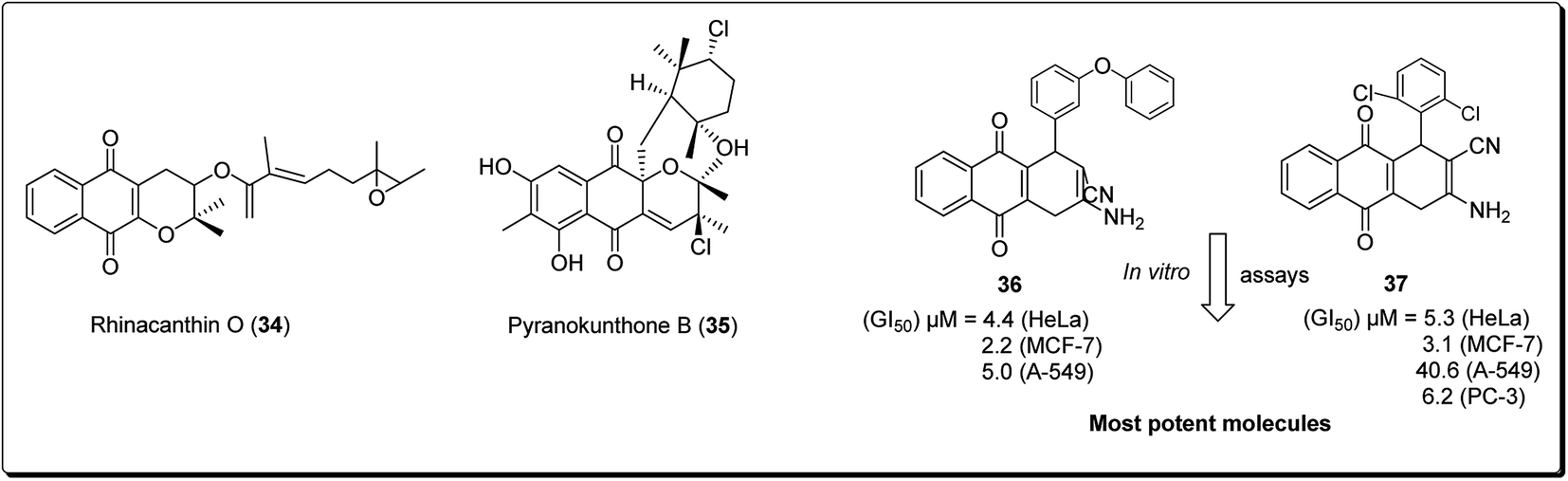 | ||
| Fig. 6 4H-Pyrano-[2,3-b]naphthoquinones with anticancer activity. | ||
Naturally occurring (dihydro) pyranonaphthoquinones can be found in bacteria, fungi, and higher plants, pointing to their biochemical relevance in nature. Many of these pyranonaphthoquinone derivatives have indeed been found to possess diverse and pronounced biological activities, including antimicrobial, antiparasitic, antiviral and anticancer properties.35 Eleutherin (38, Fig. 7) and psychorubrin (39) and pentalongin (40) are typical examples of this class of compounds. Thi et al. carried out the synthesis of new (dihydro) pyranonaphthoquinones (41–44) and their epoxy analogues. The most potent compound (44) showed an IC50 value of 1.5 μM against KB and 3.6 μM in Hep-G2 cell lines.35
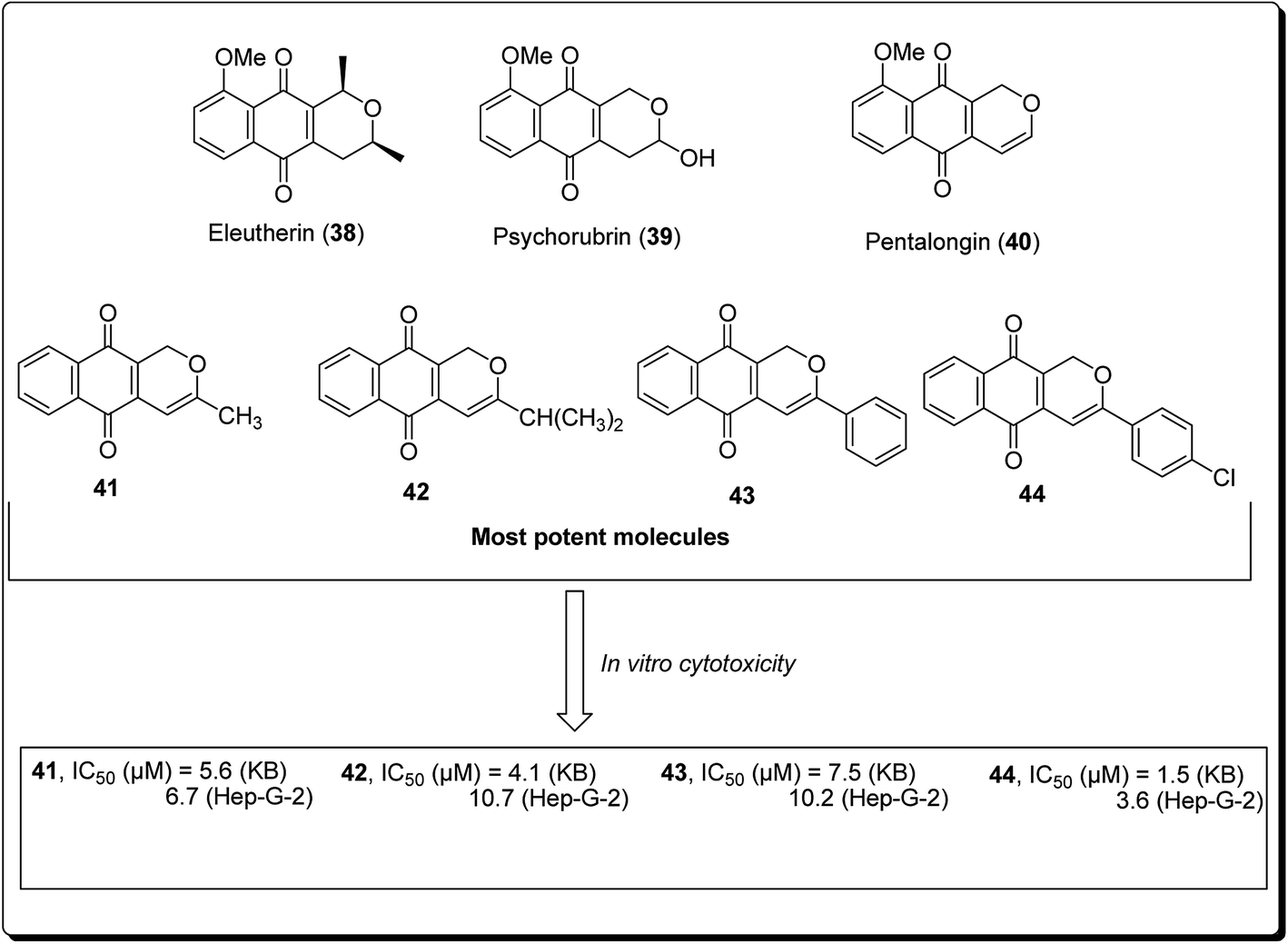 | ||
| Fig. 7 Bioactive (dihydro)pyranonaphthoquinone-derived natural and synthetic anticancer agents. | ||
Natural products bearing the furanone-fused pyranonaphthoquinone skeleton, with the tricyclic pharmacophore (Fig. 8) also play an important role in medicinal chemistry, e.g. kalafungin (45), medermycin (46), griseusin (47) and granaticin (48). Kalafungin, for example, has shown activity against L5178Y mouse leukemic cells, as well as against AKT kinase.36 Meanwhile, medermycin was shown to possess several biological activities, including cytotoxicity against K562 human myeloid leukemia, P-388 murine leukemia and L5178Y murine lymphoblastoma cell lines.37 Griseusin and granaticin also have proven antiprotozoal, antibacterial, and cytotoxic activities.38–40 Based on these evidences, Jiang et al. synthesised several compounds bearing this quinone–pyran–lactone tricyclic pharmacophore and evaluated their anticancer properties against several cell lines, including squamous carcinoma KB cells, vincristine-resistant KB/VCR cells, human lung cancer A549 cells, and human leukemia HL60 cells.41 The most promising compounds were the stereoisomers with the aliphatic amino(piperazinyl) substituent on the tricyclic pharmacophore (49 and 50), with inhibitory potencies in the lower and sub micromolar ranges.41 Meanwhile, fluoro substituted benzo[b]pyran derivated analogues of 6-flurobenzo[b]pyran-4-one (51, Fig. 9) have shown activities against NCI-H460 (lung), MCF7 (breast) and SF-268 (CNS) cancer cell lines.42
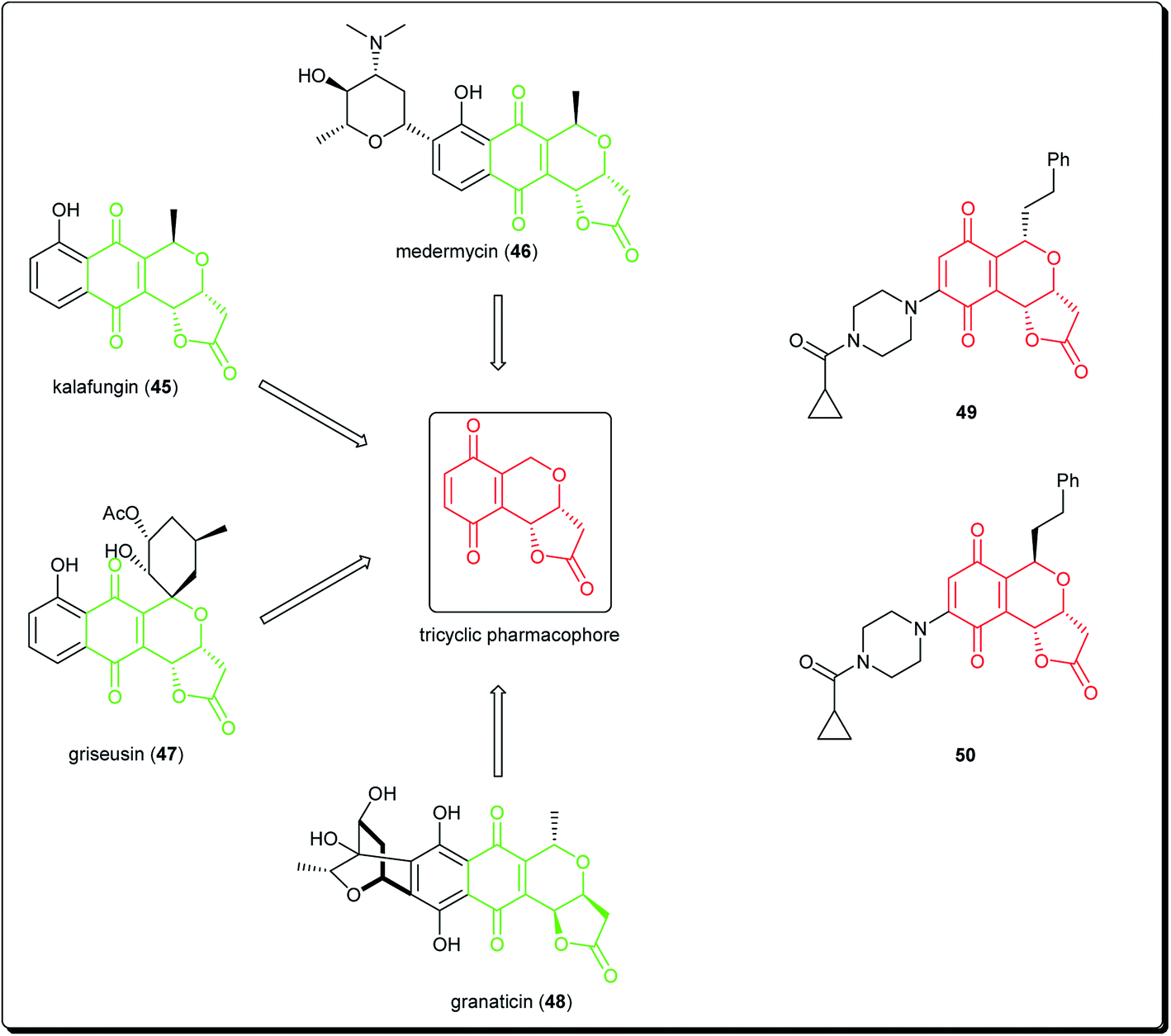 | ||
| Fig. 8 Natural products bearing the furanone-fused pyranonaphthoquinone skeleton with tricyclic pharmacophore, along with synthesized anticancer derivatives. | ||
 | ||
| Fig. 9 Fluorinated pyran-4-one scaffold used for designing potent anticancer agents. | ||
3 Flavone-based scaffolds
The term “flavonoid” refers to a huge class of plant secondary metabolites, which are biosynthesised from common chalcone precursors.43 Flavonoids are members of a much bigger family of more than 5000 naturally occurring polyphenolics, present in several foods of plant origin, which are known to be a rich source of anticancer drugs.44,45 These compounds are often characterised by the presence of a common phenylbenzopyrone linkage (C6–C3–C6) in their structures. Several flavonoid subclasses exist, depending on the saturation level and opening of the central pyran ring, including; flavones, isoflavones, flavonols, flavanonols, flavanols, flavanones and pterocarpans.45,46 Flavonoids exhibit a broad range of biological activities, e.g. anti-mutagenic, antiproliferative and antioxidant activities.47–49 The antioxidants are usually involved in cell signaling, cell cycle regulation, and angiogenesis.50–53Flavanones have been thought to be quite promising in the search for new lead compounds in the field of cancer chemotherapy. About a decade ago, Hsiao et al. established that flavanone and 2′-OH flavanone inhibited cell growth of A549, LLC, AGS, SK-Hepl and HA22T malignant cells, whereas other flavanones (4′-OH flavanone, 6-OH flavanone) showed little or no inhibition.54 Moreover, Choi et al. later reported that 4′,7-dimethoxyflavanone exhibits persuasive anticancer activity by inducing cell cycle arrest and apoptosis in human breast cancer MCF-7 cells.55 The results of another study, published soon afterwards, explored the antiproliferative effects of synthetic flavanone derivatives on human breast cancer cells by way of p53-mediated apoptosis and the induction of cell cycle arrest at the G1 phase.56 Usman et al. had previously reported the cytotoxic activities of flavanones isolated from the bark of Cryptocarya costata.57 A study of eight flavanones on colorectal carcinoma cells indicated that 2′-OH flavanone showed the most potent cytotoxic effect on these cancer cells, and cell death induced by 2′-OH flavanone, was via the occurrence of DNA ladders, apoptotic bodies, and hypodiploid cells, all characteristics of apoptosis.58 Flavonoids (Fig. 10) are also important ingredients of human diet.56,59–62
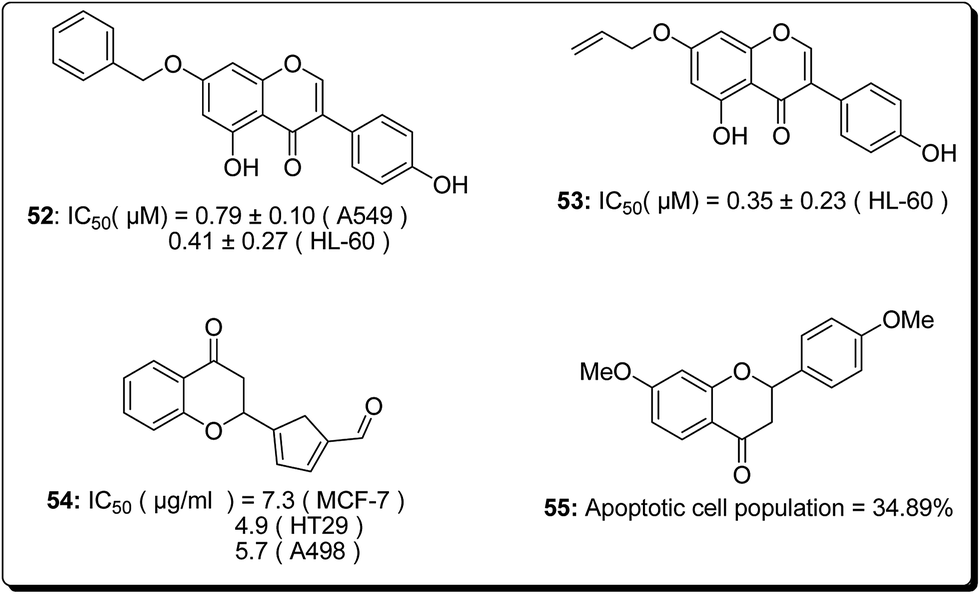 | ||
| Fig. 10 The cell killing potential of some flavonoid-based anticancer agents. | ||
Kumar et al. established the design and synthesis of naphthoflavones (56–59, Fig. 11). All the synthesised compounds were screened towards a panel of human malignant cells. Compound 59 displayed noteworthy cytotoxicity towards MiaPaCa-2 cell lines, with IC50 values of 1.93 μM and 5.63 μM against MCF-7 cell lines. Compound 59 was found to prompt apoptosis, confirmed through phase contrast microscopy, DAPI staining and mitochondrial membrane potential loss (MMP). The cell phase division study demonstrates an increase from 11.26% (control test) to 55.19% (treatment with compound 59 at 20 μM) in the apoptotic population.63
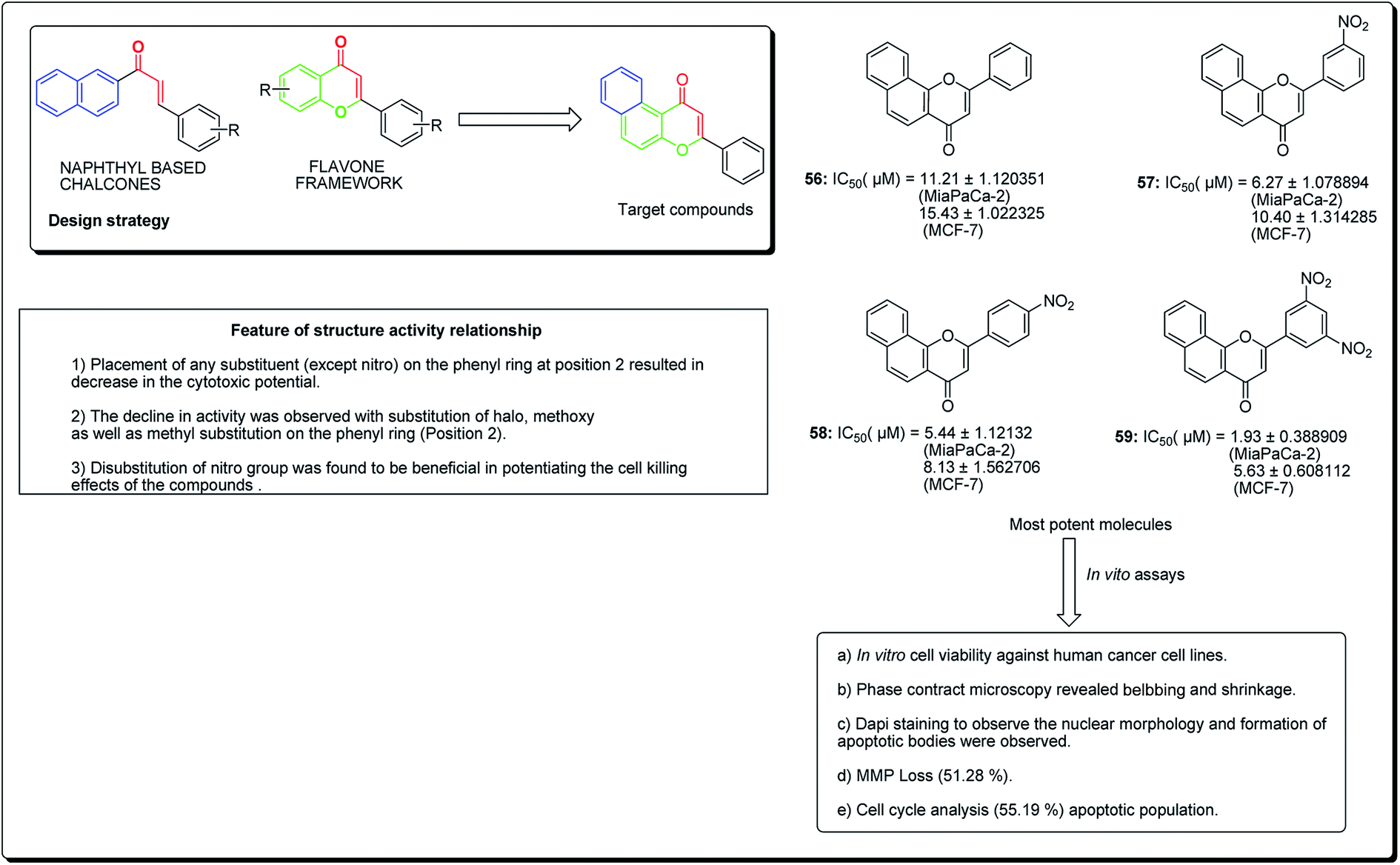 | ||
| Fig. 11 Anticancer potential of most potent naphthoflavone along with their SARs. | ||
Myricetin (60, Fig. 12), one of the flavonoids, is available in a wide assortment of natural sources. Strikingly, those myricetin subordinates are thought to indicate anticancer action, which could diminish pancreatic malignancy development by means of acceptance of cell apoptosis.64,65 On the basis of the previous findings, Xue et al. established a sequence of new myricetin analogues.83 It was experimentally demonstrated that compound 62 affects the growth of human breast cells MDA-MB-231. Results from the telomerase inhibition assay also demonstrated that compound 62 acts against human bosom cells MDA-MB-231, with an IC50 value of 0.91 μM. The docking simulation of compound 75, towards the target site, was performed to get the likely binding mode. The docking pose showed that the heterocyclic ring was profoundly embedded into the dynamic site, forming hydrophobic associations with build-ups of Phe568, Pro627, with four methoxy groups having hydrophobic collaborations with residues Phe568, Pro627, Lys902, Val904 and Pro929 (Fig. 12).66
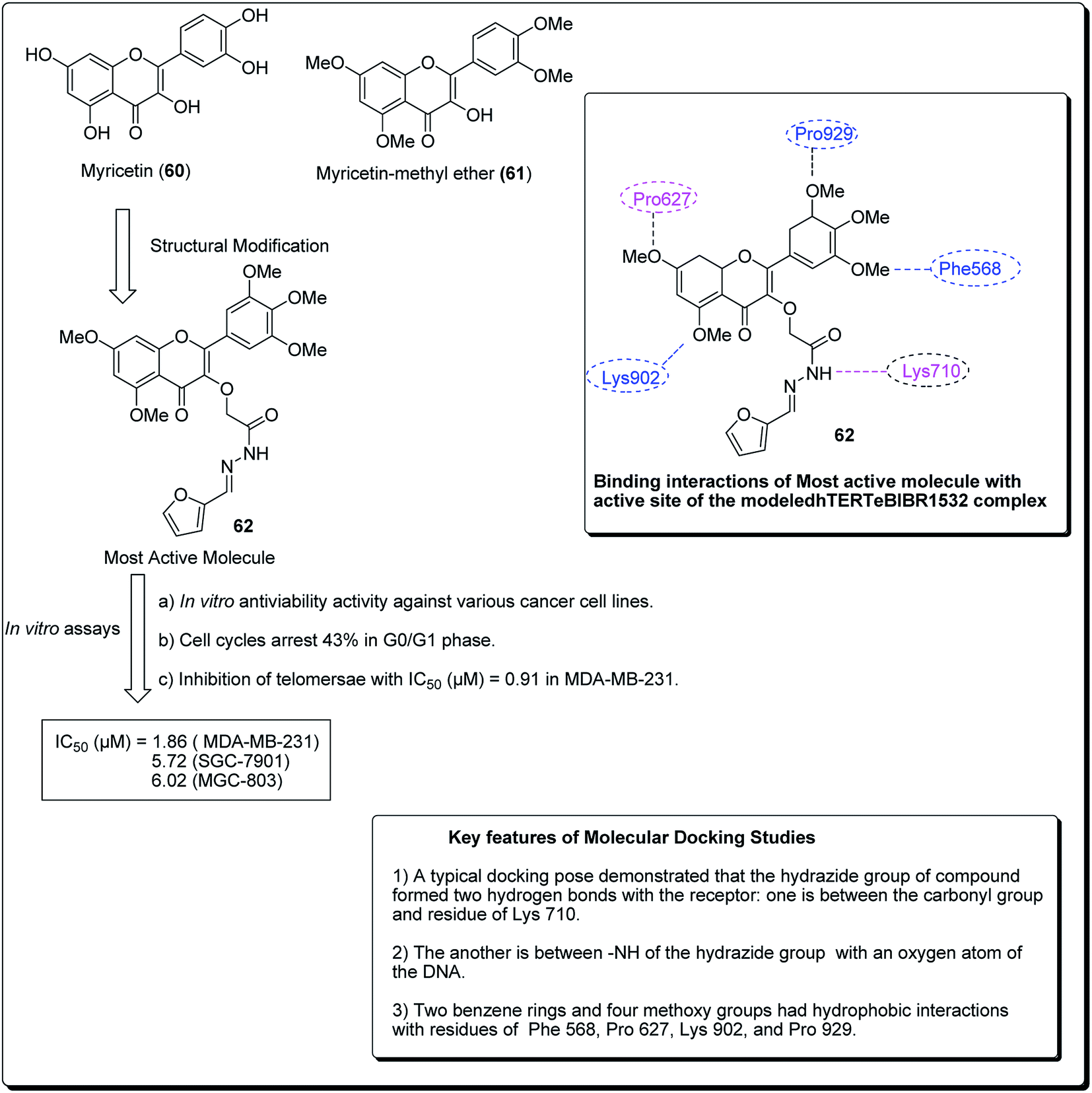 | ||
| Fig. 12 Structure of most active novel myricetin anticancer analogs along with their molecular docking features. | ||
Safavi et al. further carried out the synthesis and testing of the cytotoxicity of halogenated flavanones against a panel of human cancer cell lines.67 Among the synthesised compounds, 3′,7-dichloroflavanone (65) showed the highest activity against MCF-7, LNCaP, PC3, Hep-G2, KB and SK-NMC cells. However, 3′,6-dichloroflavanone (66, Fig. 13), with an IC50 value of 2.9 μM, was the most potent compound against MDA-MB-231 cells, being approximately 12 times more potent, when compared with the reference drug (etoposide). It has been demonstrated that the modulation of the flavanone structure could increase antitumor activity. Thus, chlorine substitution on the chromanone ring and on the C-2 attached phenyl ring was used for structural modification and modulation of the basic pharmacophore of flavanones. Among the synthesised compounds (Fig. 13), 3′,7-dichloroflavanone (65) showed the better profile of cytotoxicity. However, 3′,6-dichloroflavanone (66) with IC50 value of 2.9 μM, was the most potent compound against MDA-MB-231 cells, as previously mentioned. According to the flow-cytometric analysis, compound 66 could be shown to induce apoptosis by 66.19 and 21.37% in PC3 and MDA-MB-231 cells, respectively. The results of acridine orange/ethidium bromide staining and TUNEL assays suggested that the cytotoxic activity of this compound in PC3 and MDA-MB-231 cells occurs via apoptosis.67 Topoisomerases are known to play essential roles in maintaining DNA topology during the processes of DNA replication, transcription and recombination. Thus, topoisomerase inhibitors are cytotoxic agents that bind to free topoisomerase and prevent the formation of a covalent enzyme–DNA complex, and are thereby referred to as topoisomerase antagonists or “topoisomerase poisons”. A number of flavonoids and other polyphenolic compounds are known to inhibit and poison mammalian topoisomerase I and II. These include quercetin, acacetin, apigenin, kaempferol, morin and luteolin.45 The structural features of flavonoids essential for the inhibition of topoisomerase have been described (Fig. 14).45
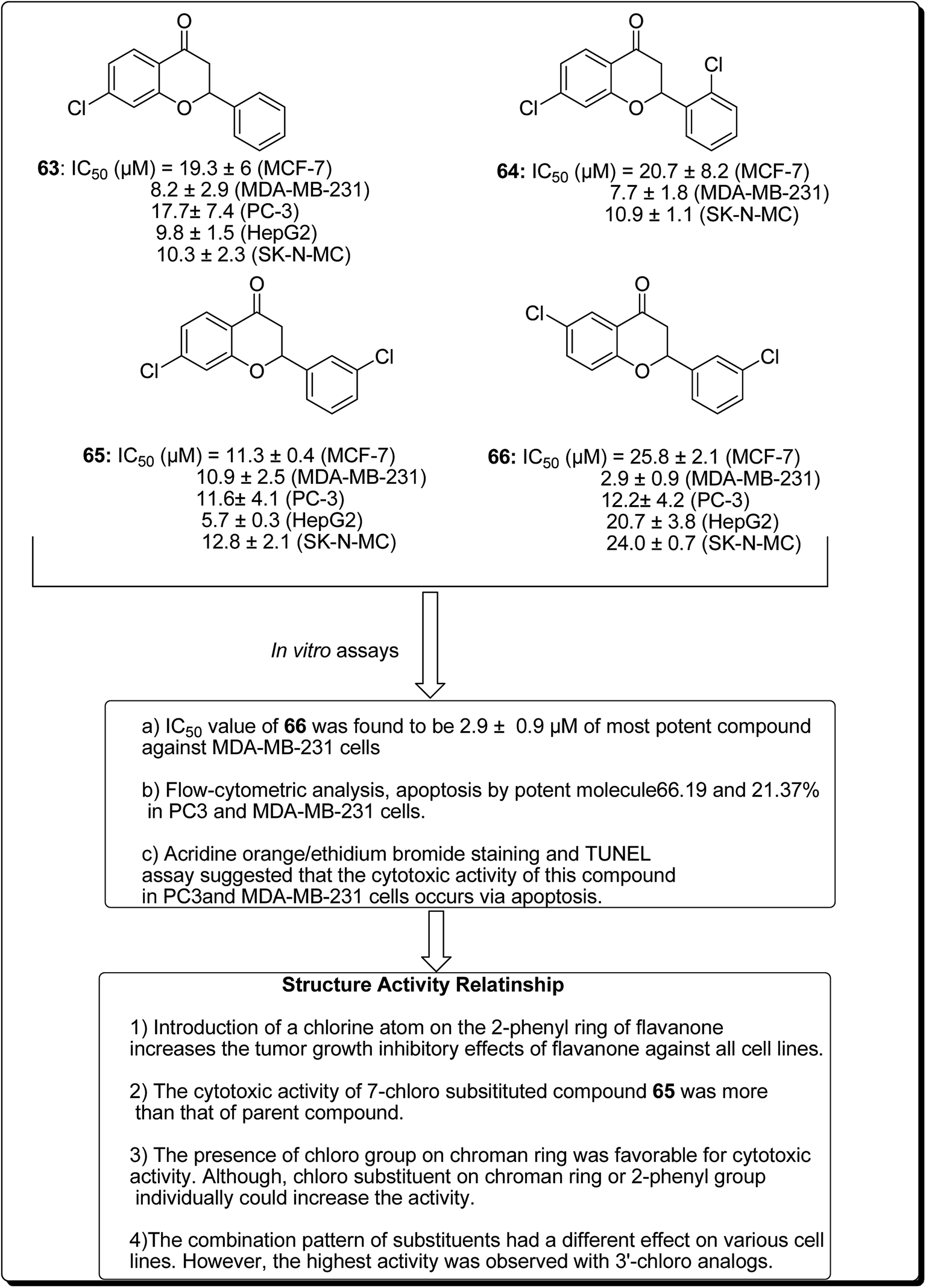 | ||
| Fig. 13 Structure of halogenated flavanones as potential apoptosis-inducing agents along with their SAR studies. | ||
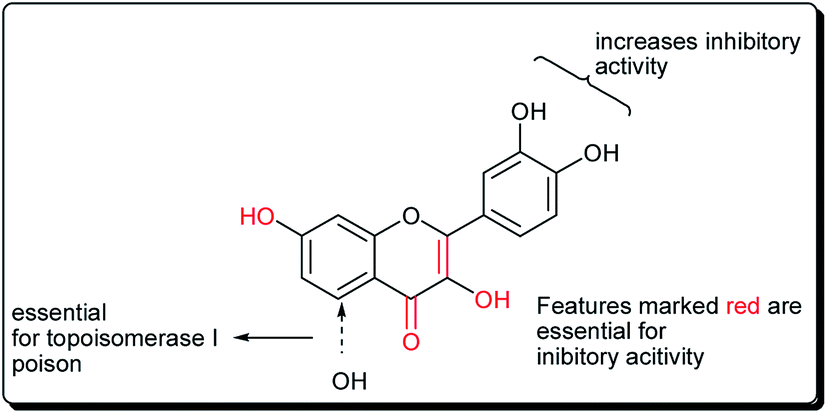 | ||
| Fig. 14 Key structural features essential for inhibitory topoisomerase activity. | ||
4 Coumarin-based scaffolds
Coumarins and pyrans form an exceptional class of oxygen-containing heterocyclic compounds, which play a key role in medicinal chemistry, due to their structural diversity and pharmaceutical properties.68 Coumarins play a special role in nature.69,70 Coumarins scaffolds are present in natural phytoconstituents, exhibiting diverse biological activities, including anticancer properties through diverse mechanisms,71–73 thus making it a privileged structure. These abilities have been explored in detail.74 Coumarin scaffolds have also been explored through the formation of diverse hybrids, with promising biological activities (Fig. 15).75–79 Among the coumarin hybrids from natural sources are pyranocoumarin derivatives, having several structural arrangements between the coumarin and the pyran rings. The few important pyranocoumarins include xanthyletin (74) (predominantly isolated from Zanthoxylum americanum), khellactone (73) (isolated from Ligusticum elatum), arisugacins (75), and pyripyropenes (76) (Fig. 16).80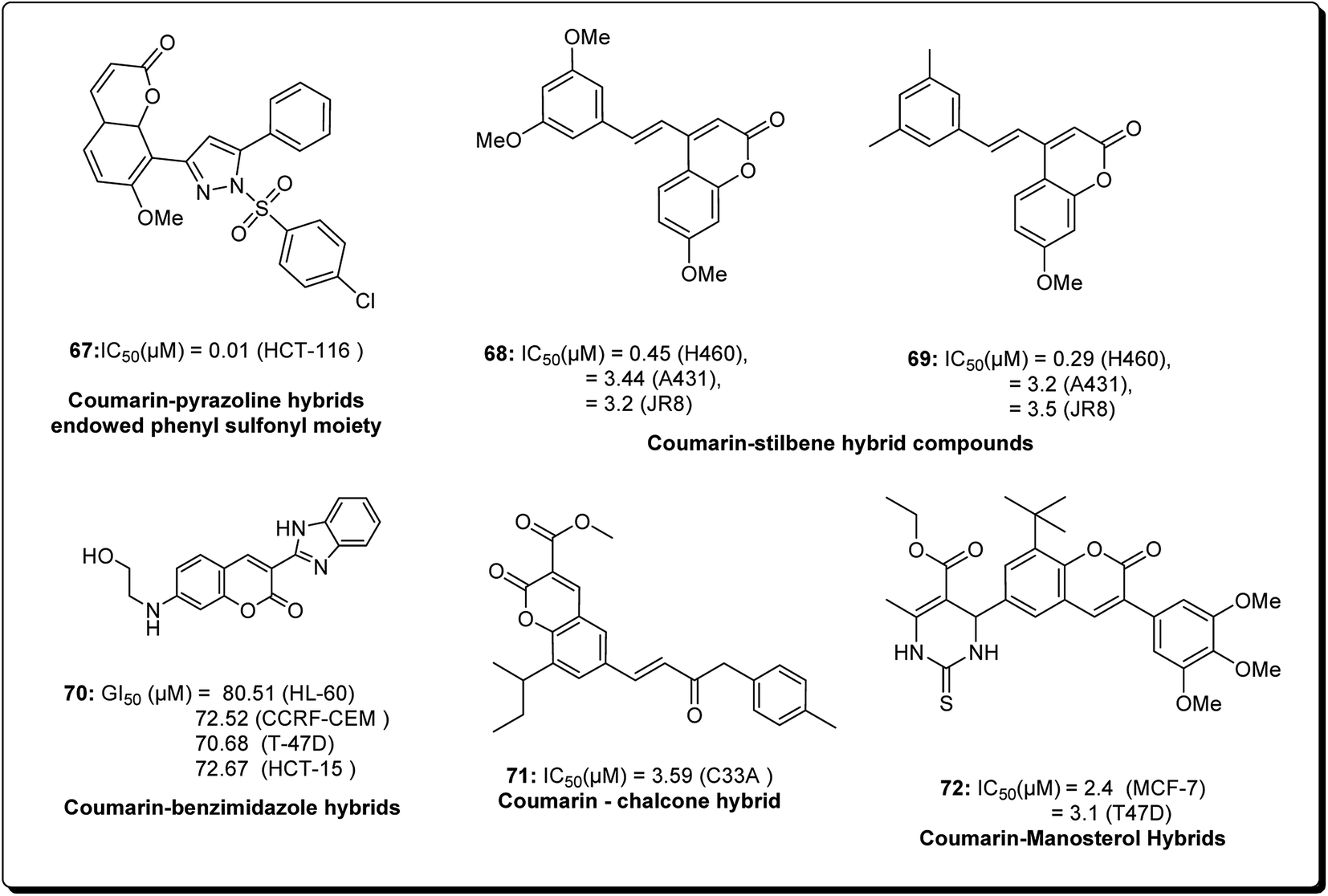 | ||
| Fig. 15 Structure of coumarin hybrids along with their IC50 values against various cancer cell lines. | ||
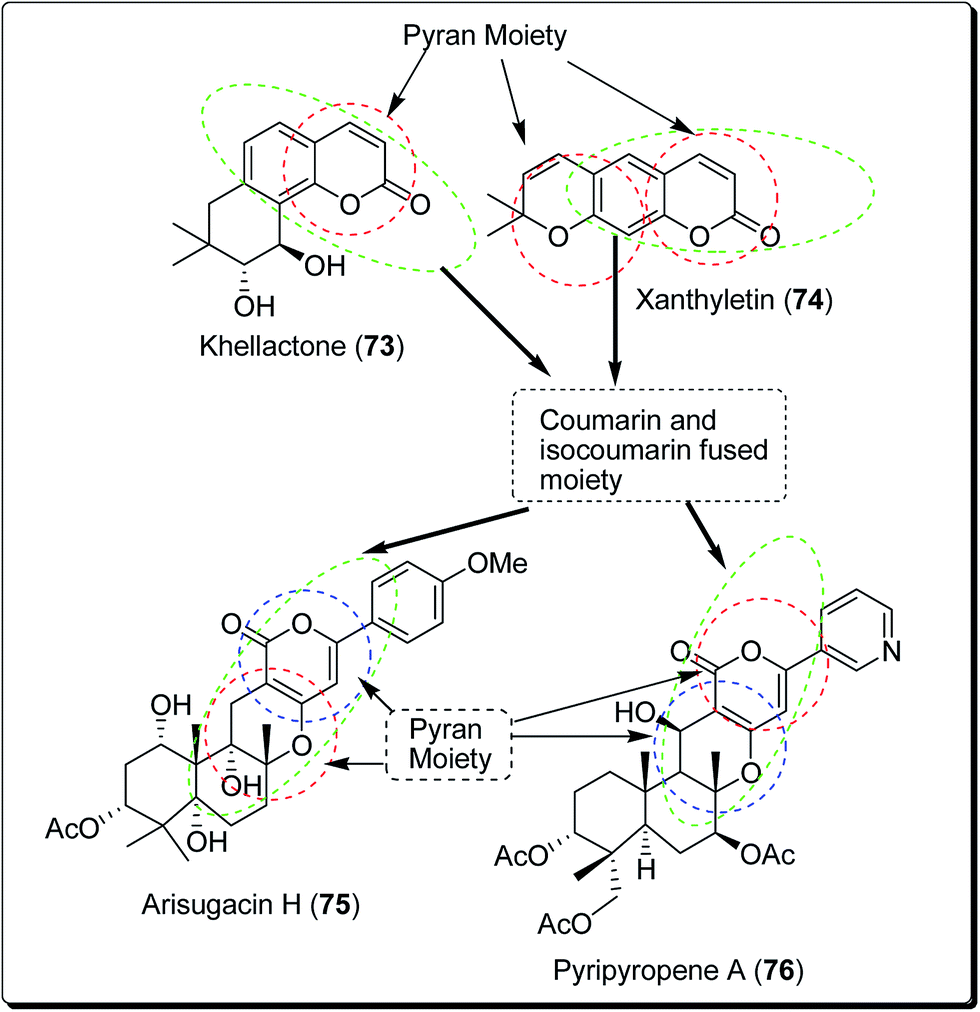 | ||
| Fig. 16 Natural agents containing coumarin and pyran moieties. | ||
Kumar et al. designed and synthesised 2,4-diarylpyrano[3,2-c]chromen-5(4H)-ones.81 The design strategy involved the fusion of coumarin and chalcone, employing pyran as a linker. Among the obtained derivatives, compound 77 (Fig. 17) revealed momentous effects in HCT 116 cell lines, with IC50 values of 1.4 and 4.3 μM towards “MiaPaCa-2” cell lines. This compound was shown to initiate apoptosis as revealed by Hoechst 33258 staining, phase contrast microscopy, and mitochondrial membrane potential (MMP) loss. The cell phase division study indicated that the apoptotic population amplified from 10.22% in the control to 57.19% in a sample treated with compound 77 at a concentration of 20 μM.81
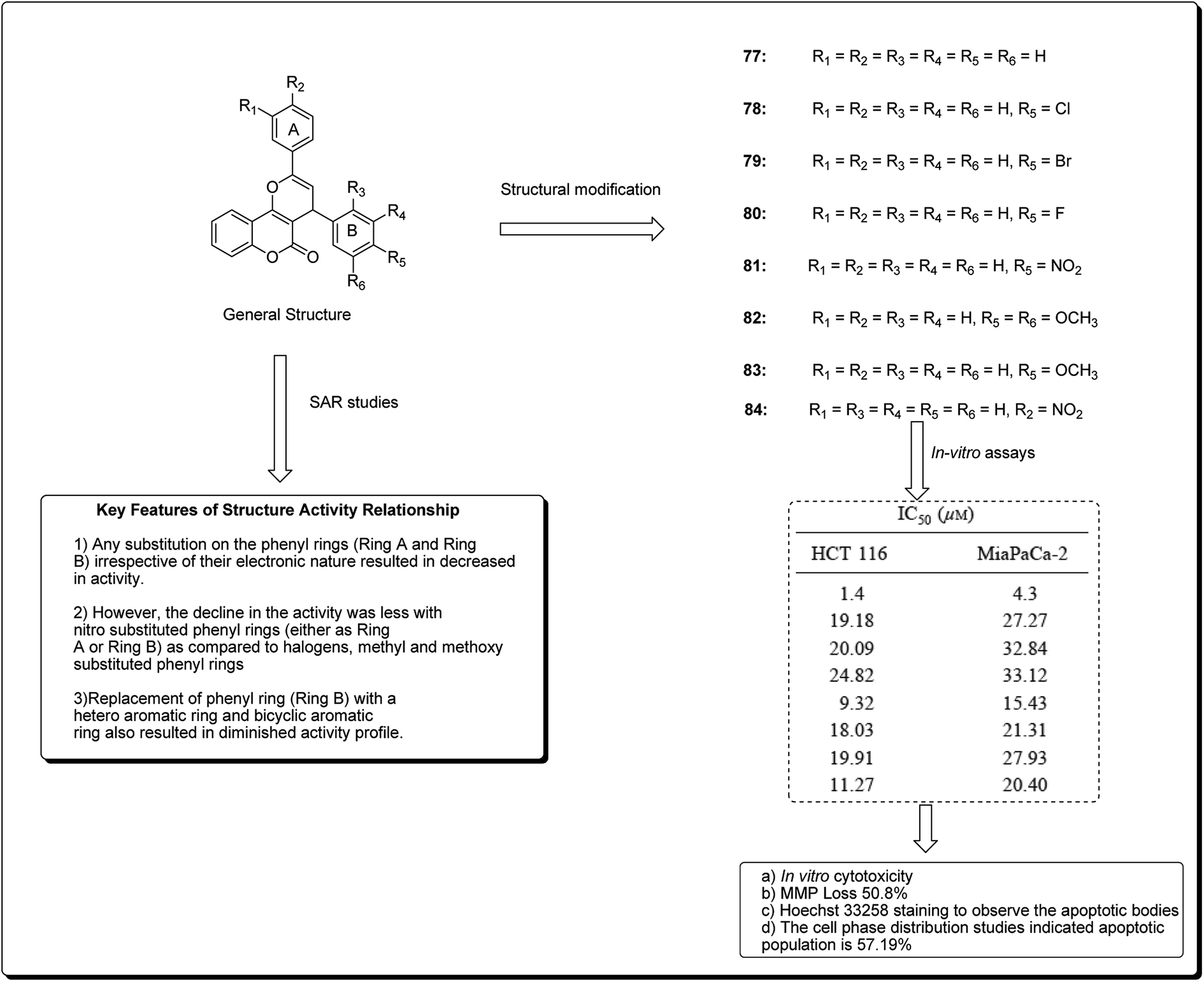 | ||
| Fig. 17 Most potent coumarins scaffolds along with their IC50 values and SARs. | ||
Hussain et al. further conducted a novel synthesis of coumarin derivatives as potent anti-breast cancer agents against ER +ve and ER −ve cell lines.82 Compound 85 was found to be ER-α selective and most dynamic from all synthesised molecules, exhibiting prospective antiproliferative activity. The docking simulation showed that compound 85 could favorably fit well in the receptor cavity of ER-α, following the binding pattern similar to the standard drug. The coumarin nucleus and the p-methoxyphenyl group at the third position formed a hydrophobic interaction with the residues Glu353, Arg394, Phe404 and Leu349. The aroyl substituent at the fourth position, having the amino alkoxy chain, anchored the piperidine ring by forming hydrophobic contacts with Trp383, Asp351, Leu354, Leu536 and Thr347. The methoxy group of coumarin at the seventh position interacted with Glu353 and Arg394 (Fig. 18). Compound 85 had a similar (but non-standard) binding pattern for ER-β binding, the coumarin pharmacophore forming hydrophobic interactions different from that observed in ER-α, i.e. interacting with Glu305, Arg346, Leu301, Leu339, and Leu343. At the third position, the 4-methoxyphenyl group forms a hydrophobic interaction with the amino residues Met421, Gly472, His575, Leu298, Phe356, Met340, and Ile373, which are essential features for ER-β binding.82
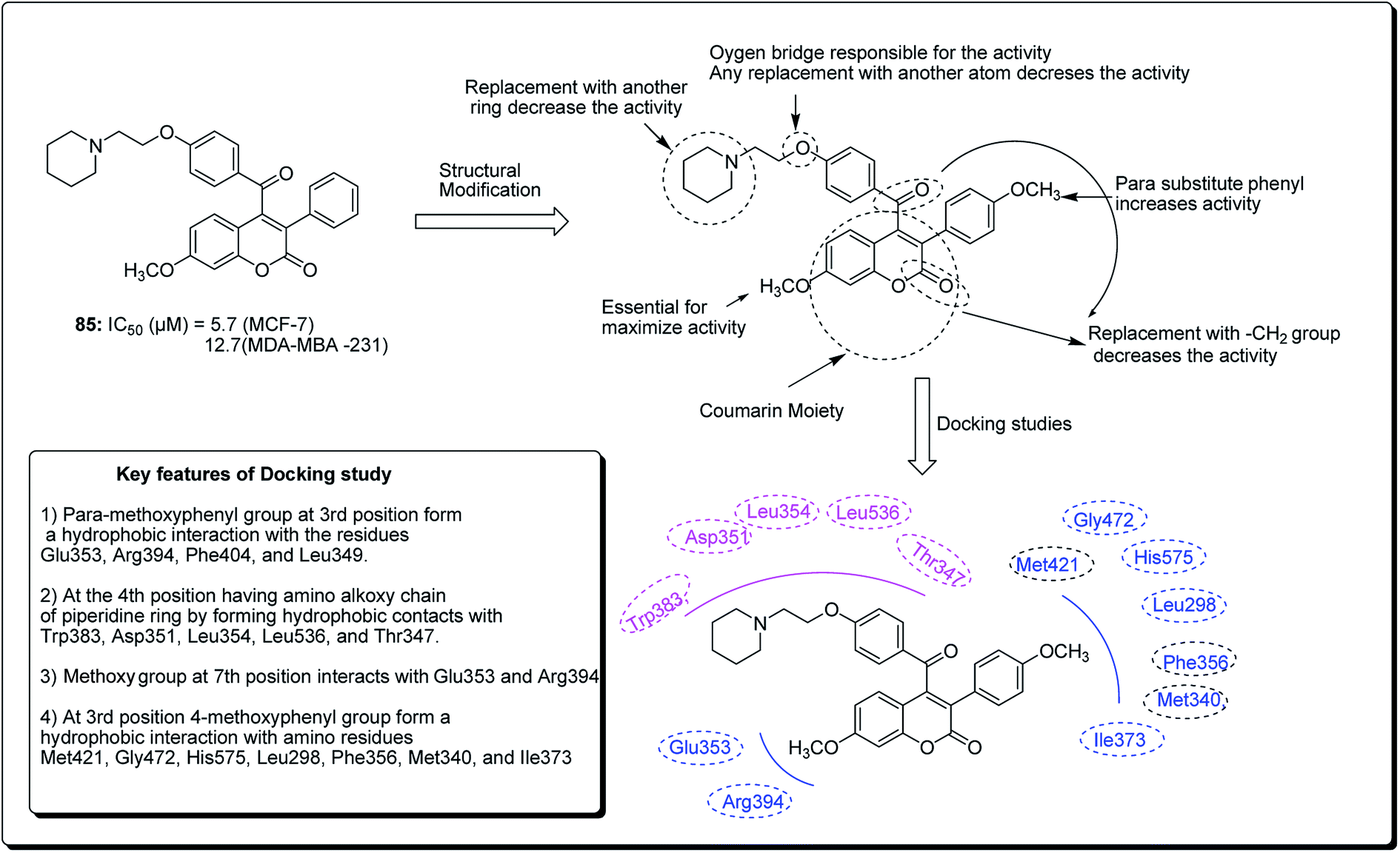 | ||
| Fig. 18 Structure of most potent ERα/ERβ selective coumarin derivative along with their docking study. | ||
Coumarin is a modification of the benzopyran-2-one by directed introduction of a heterocyclic substituent. In most cases, a heteroaryl substituent is introduced at position 3 or 4 of the coumarin ring. Thus, 3- and 4-heteroarylcoumarins are reported to exhibit significant biological activities, including inhibiting the growth of several cancer types.83 Prompted by this, Yana et al. reported the synthesis and anticancer evaluation of a series of novel 6-pyrazolinylcoumarins via NCI60-cell line assay. The outcome of the study revealed that compound 87 showed the highest level of anti-mitotic activity with a mean GI50 value of 10.20 μM and a sensitivity profile toward the Leukemia cell lines CCRF-CEM and MOLT-4 (GI50 values 1.88 μM and 1.92 μM), respectively, as represented in Fig. 19. The SAR study indicated that the antitumor activity of the synthesised compounds depends on substituent at third and fourth positions of the coumarin core. Moreover, it was found that compounds bearing the 3-methoxy-4 hydroxyphenyl and the 4-hydroxyphenyl substituents at position 5 of the pyrazoline fragment were more active than the other analogues.84
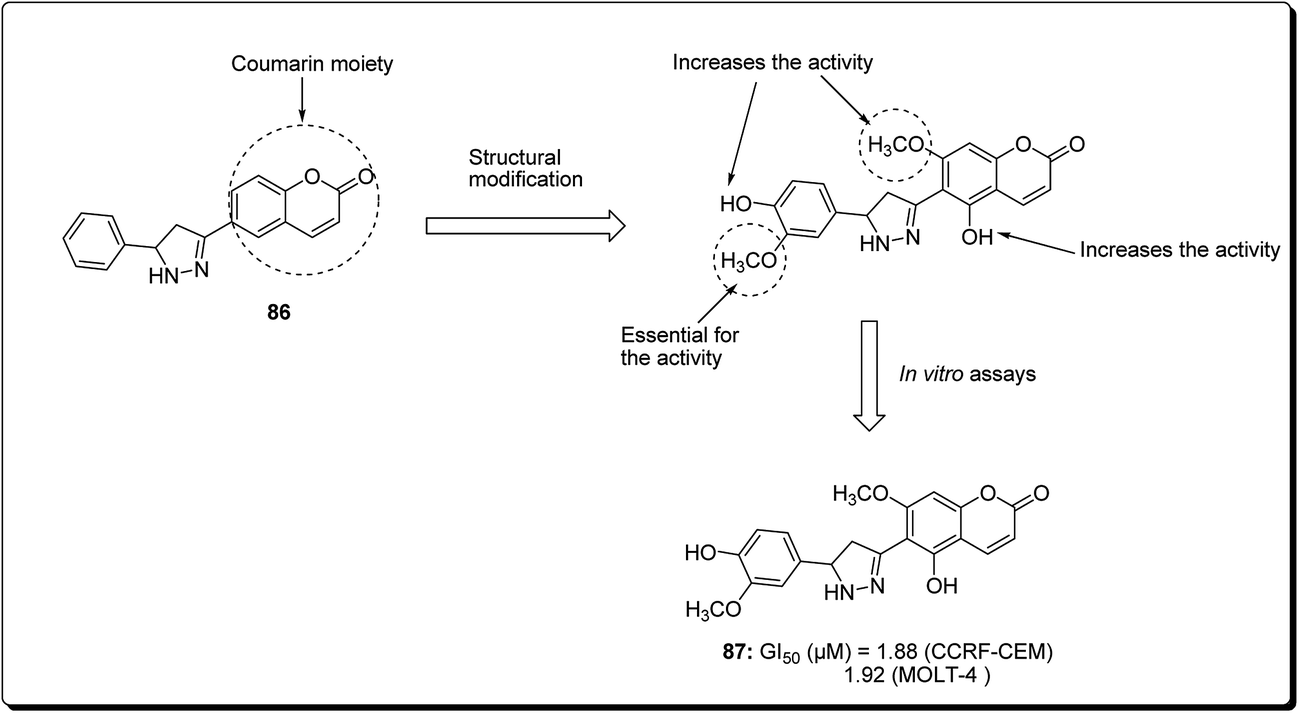 | ||
| Fig. 19 Structure of most active 6-pyrazolinylcoumarin analogue as anticancer agent. | ||
Another study was carried out on the design and synthesis of coumarin derivatives with improved anticancer activity. Among them, oligomerization (di/tri) of coumarin is one of the effective ways.85,86 The derived dimeric natural product was shown to be more effective than the monomeric species (with IC50 ∼70 μmol L−1).87,88 With this inspiration, the concept of molecular oligomerization led to the discovery of two novel series of dimeric derivatives of triphenylethylene–coumarin hybrids.89,90 The dimeric compounds had potent anti-tumor activities, possibly by acting on DNA via the intercalative mode, and higher than their corresponding monomeric compounds,91,92 respectively. The positive results inspired interests to explore the trimeric variants of the triphenylethylene–coumarin hybrid in an effort to produce more efficient antitumour agents. Zhang et al. further discovered new trimers of triphenylethylene–coumarin hybrids, containing two amino side chains. The trimeric compound 88 (Fig. 20) exhibited significant antiproliferative activity against three cancer cells at IC50 of nearly 10 μmol L−1. The outcome of the DNA photocleavage studies revealed that compound 88 had significant interaction with Ct-DNA by the intercalative mode. Overall, the presence of extended linker and piperidinyl substitutions on the side chains were found to be favourable for DNA binding and the antitumour activity.93
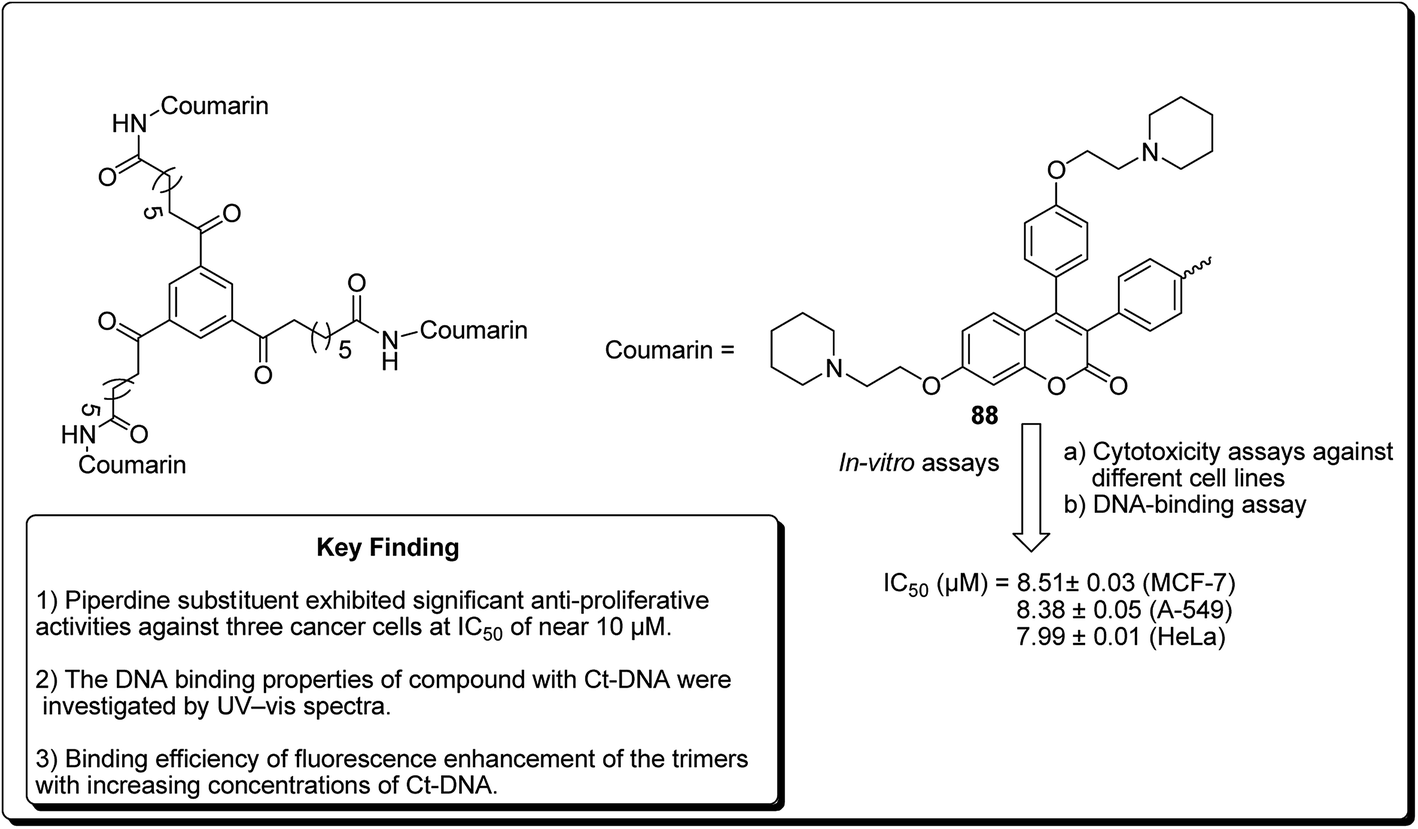 | ||
| Fig. 20 Structure of most potent trimers of triphenylethylene–coumarin hybrids. | ||
5 Xanthones and xanthene-based scaffolds
Xanthones are an outstanding class of oxygenated tricyclic compounds, which display different fascinating pharmacological properties, relying on the nature and types of substitutions.94–96 Recently, xanthones have been valued as having an effective pharmacophore in the field of medicinal chemistry world.97 Prior to this, xanthones were shown to be present in bug sprays, larvicides and ovicides.98 Shortly afterward, several experimental studies established that xanthone analogues could stop the growth of tumor cells and could also possess antioxidant and anti-inflammatory properties.99 Xanthones are mainly found in plants of the Bonnetiaceae and Clusiaceae, in addition to the Podostemaceae, Guttiferae and Gentianaceae.100Many naturally-occurring and man-made xanthene derivatives have been reported to exhibit antitumor activities,101–104 among others. In the recent past, there has been a renewed interest in the synthesis of this class of compounds as the number of its applications have increased, both in the field of medicinal chemistry and material science. Particularly, xanthones (9H-xanthen-9-ones) are well explored heterocyclic derivatives with the dibenzo-γ-pyrone skeleton.105–108 Some xanthone-containing plant extracts are directly used in traditional medicines. Analogous thioxanthone derivatives are also present in anticancer drugs.109–113 Moreover, there are some marketed formulations having xanthone derivatives (89–97, Fig. 21) as one of their active ingredients.
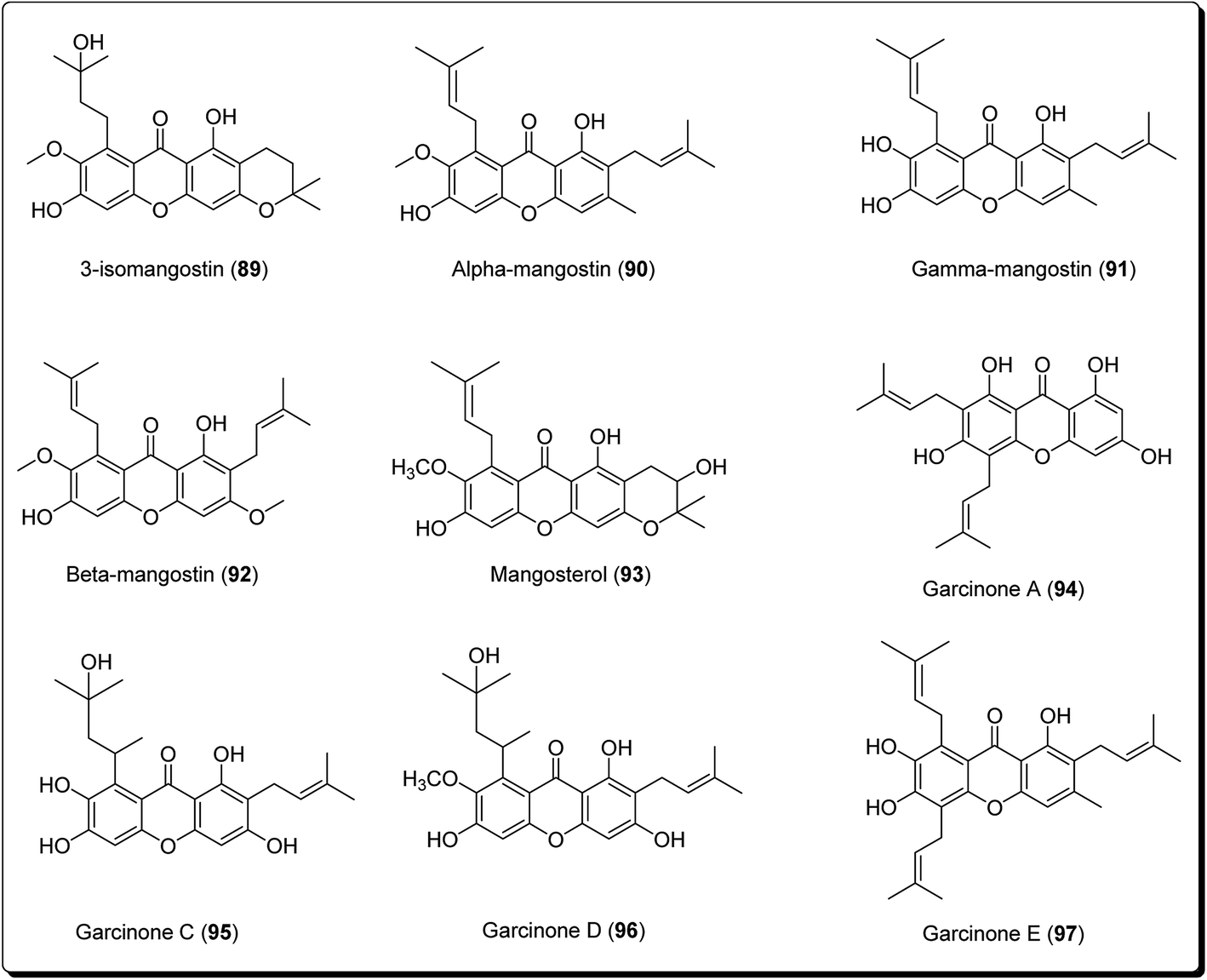 | ||
| Fig. 21 Structure of natural xanthones in marketed formulations. | ||
Lee et al. isolated three coumarin derivatives, theraphins (98–101) are recognised xanthones such as 2-hydroxyxanthone, 1,7-dihydroxyxanthone and 5-hydroxy-1-methoxyxanthone (Fig. 22), from the bark of Kayea assamica (Clusiaceae).114 These were analysed for their cytotoxic activities, based on a panel of human cancer cell lines. Among these compounds, 99–101 displayed cytotoxic action against Col-205, KB, and LNCaP cell lines with IC50 values ranging from 3.5 to 13.1 μM. Meanwhile, the coumarin subsidiaries demonstrated modest effects, with IC50 values in the range 9.7–11.1 μM against the D6 clone, and IC50 values in the range 5.1–10.4 μM against the W2 clone. The result of the study demonstrated that the 7-hydroxycoumarins had an inhibitory impact on human malignant cell lines.110
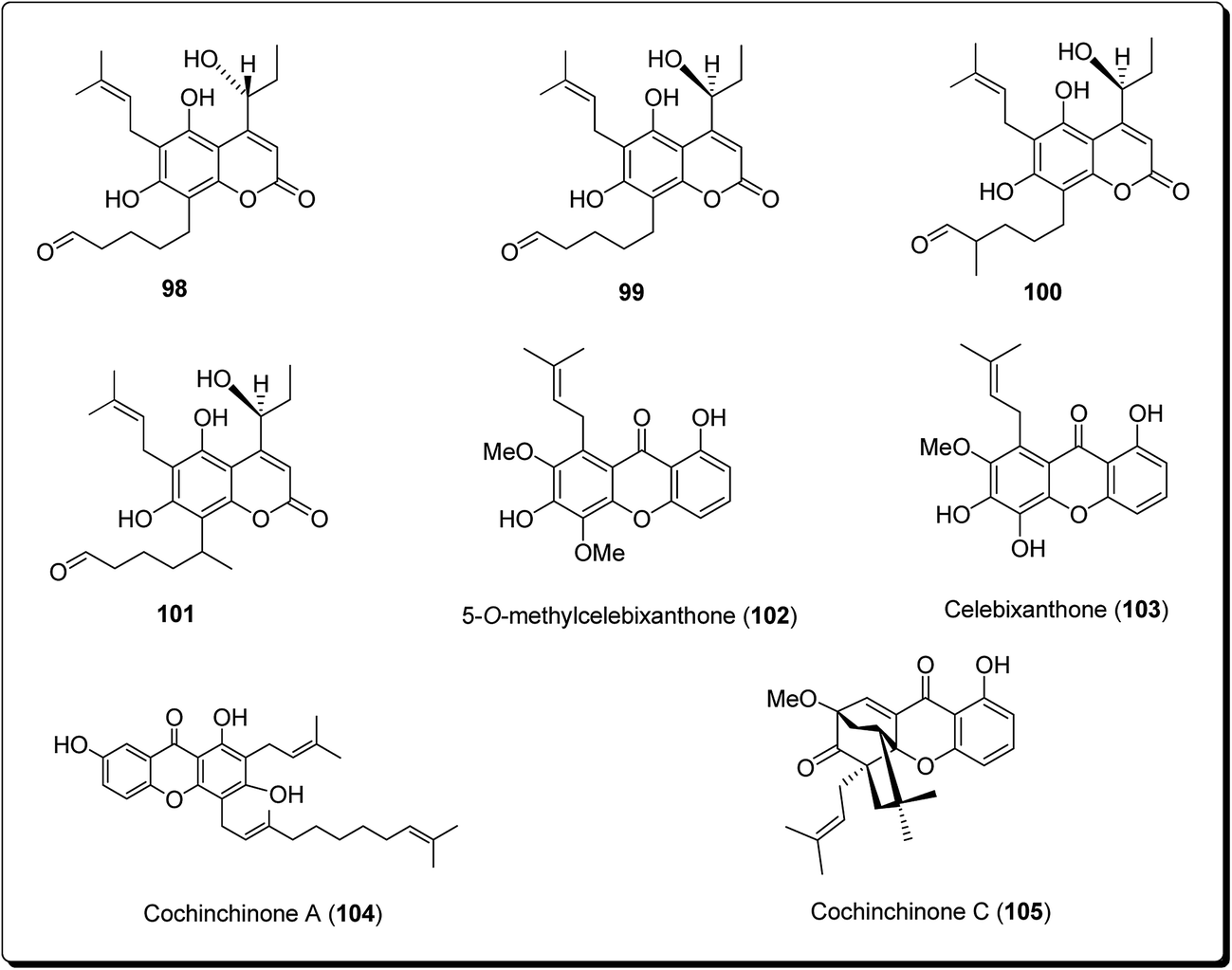 | ||
| Fig. 22 Some potent cytotoxic coumarins and xanthones (98 to 105). | ||
Laphookhieo et al. isolated 5-O-methylcelebixanthone (102), along with six compounds; celebixanthone (103), 1,3,7-trihydroxy-2,4-di(3-methylbut-2-enyl)xanthone, cochinchinone A (104), α-mangostin (90), β-mangostin (92) and cochinchinone C (105) from roots of Cratoxylum cochinchinense.115 These analogues were screened for their cytotoxic effects in NCI-H187 (human lung cancer) cell line. Among these, compounds 90, 103 and 104 showed cytotoxic activities with IC50 values ranging from 0.65 to 5.2 μg mL−1.111 Chantarasriwong et al. also established the series of caged Garcinia xanthones and evaluated them for their anticancer activity using cell proliferation and apoptosis assays against human colon and leukemic HCT-116 and HL-60 cell lines respectively. Compound 106 proved to be the most active compound against colon cancer cells, with an IC50 value of 0.2 μM against HCT-116, while compound 107 was the most active against HL-60 (Leukemia), having an IC50 value 0.4 μM, Fig. 23.116
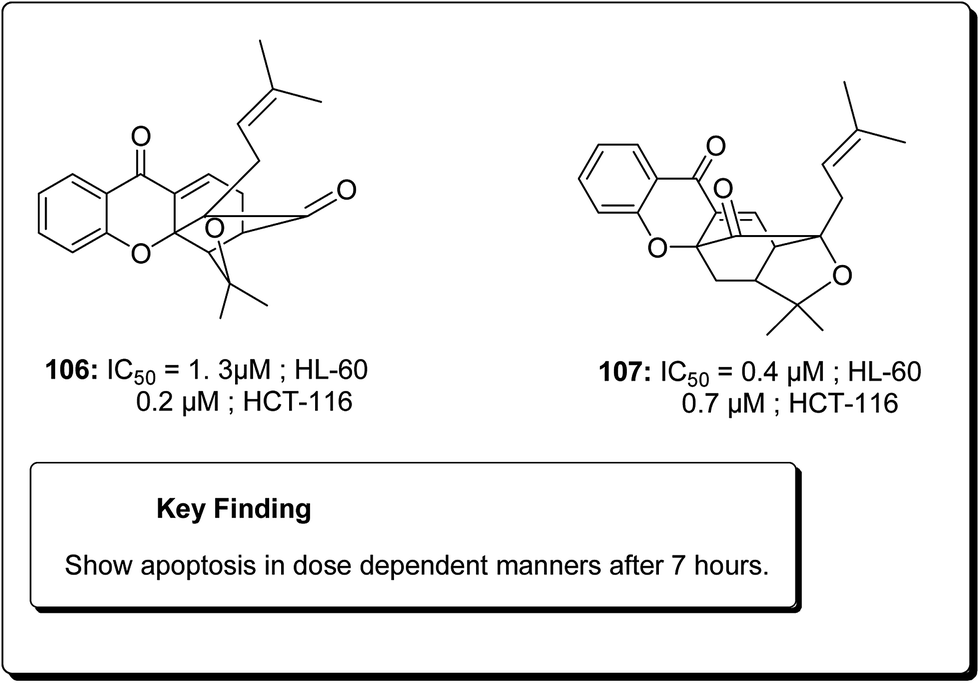 | ||
| Fig. 23 Some potent xanthones (106 and 107). | ||
In a similar study, Matsumoto et al. confirmed that all xanthones obtained from Garcinia mangostana (Fig. 24) demonstrated a noteworthy anticancer activity.117 However, α, β and γ-mangostins (90–92) were particularly active at 10 μM. The most active compound at this concentration was α-mangostin. The anticancer effect of α-mangostin was also shown in other leukemia cell lines: K562, NB4 and U937. Cell development of all these leukemia cell lines was hindered by α-mangostin at 5–10 μM.117 Chiang et al. reported that the heated water concentrate of mangostin-organic product pericarp showed an intense antileukemic activity, with IC50 values of 61 and 159 μg mL−1 against K562 and Raji cells, respectively.118 Balunas et al. have also screened α, β and γ-mangostins by using a non-cell, chemical based the microsomal aromatase hindrance assay with an IC50 value 4.97 μM against SK-BR-3 breast cancer cell lines.119 Recently, Jung et al. determined the antitumor properties of these compounds in pre-neoplastic injuries induced with 7,12-dimethylbenz[a]anthracene (DMBA) in a mouse mammary organ development. It was observed that α-mangostin restrained DMBA-induced preneoplastic sores with an IC50 of 2.44 μM.120 Suksamrarn et al. separated distinctive xanthones from mangosteen fruit pericarp and tested them for antineoplastic activity against three diverse human malignant cells, e.g. mouth carcinoma (KB), breast cancer (BC-1) and small cell lung cancer (NCI-H187), with IC50 values of 2.8, 3.53 and 3.72 μg mL−1, respectively.121 Nonetheless, α-mangostin (90) showed the most pronounced effect on BC-1 cells, with an IC50 value of 0.92 μg mL−1. It was found that an action of α-mangostin was further noteworthy than the standard medication ellipticine (IC50 = 1.46 μg mL−1).121
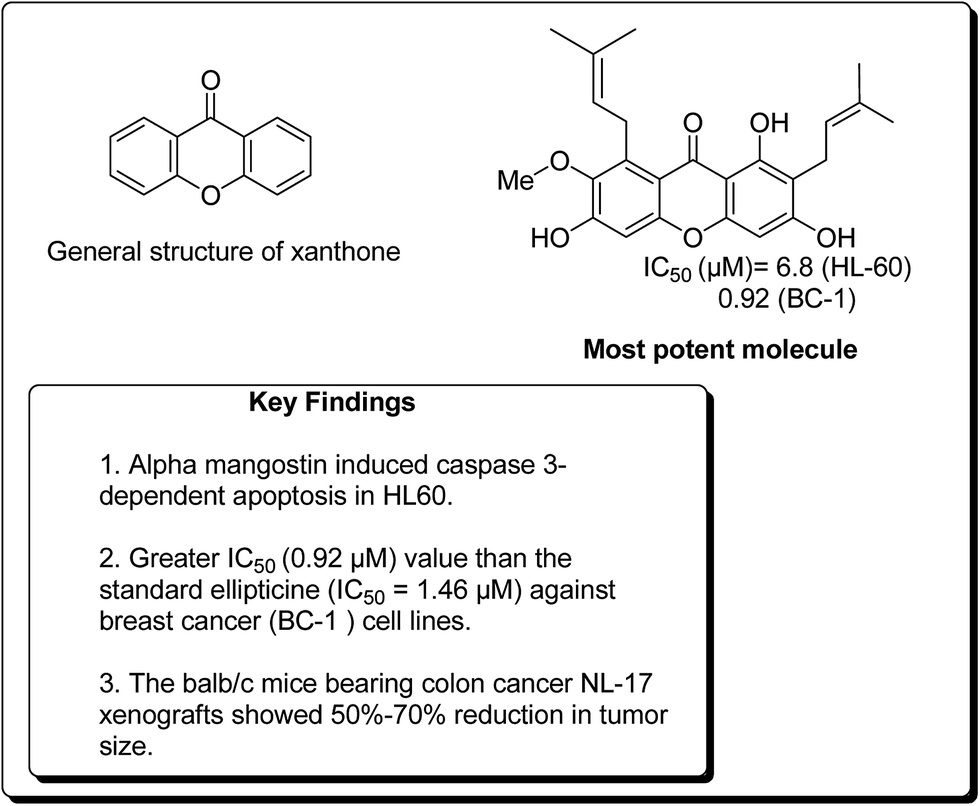 | ||
| Fig. 24 Xanthone nucleus and structure of a potent xanthone. | ||
Chen et al. verified that α- and γ-mangostins appreciably subdued lipopolysaccharide-stimulated NO˙ production and cytotoxic effects when applied to RAW 264 cells.122 The quantity of NO˙ fabrication at 3 to 25 μM was continuously calculated, and the IC50 values were found to be 12.4 and 10.1 μM for α- and γ-mangostins, respectively.122 Watanapokasin et al. examined the antiproliferative effects of mangostin xanthones, focusing on colon malignancy.123 Nutritional administration of α-mangostin altogether hindered the acceptance and improvement of unusual grave foci in an artificially instigated rodent model of colon carcinogenesis. The development of COLO 205 xenografts was totally stifled when mice were infused intraperitoneally with 3 mg of a mangostin extract containing α- and γ-mangostin. In addition, minor doses of the extract were decreased the tumor volume. Atomic component kappa-B (NF-κB) action was also diminished by 30%. The balb/c mice bearing colon tumor NL-17 xenografts indicated 50–70% lessening in tumor size when intraperitoneal treated with a concentrate from mangosteen pericarp containing 25% α-mangostin.123–126
Cao et al. isolated two new cytotoxic xanthones; termicalcicolanone A (108) and termicalcicolanone B (109, Fig. 25) from the ethanolic extract of the Madagascan plant Terminalia calcicola.127 These compounds were evaluated for their antiproliferative activity in the A2780 human ovarian cancer cell line assay and had IC50 values of 40.6 and 8.1 μM, respectively.127 Han et al. isolated three new prenylated xanthones, along with ten known compounds, from the stem bark of Garcinia lancilimba.128 These analogues were tested for their apoptotic effects against HeLa-C3 cells, which produce a biosensor proficient in detecting caspase-3 activation and it had been found out that 7,9,12-trihydroxy-2,2-dimethylpyrano[3,2-b]xanthen-6(2H)-one (110, Fig. 25), also arresting cell mitosis by interfering with microtubule formation and then induce apoptotic cell death.128 Tao et al. isolated new xanthones, a pair of new natural products and known related compounds (Fig. 25) from the resin of Garcinia hanburyi.129 These compounds were evaluated for their cytotoxicity against HeLa cervical carcinoma cells, with adriamycin as the positive control and all except compound 111 (IC50 = 111 μM) was found to display most potent cytotoxicity (Fig. 25).129
 | ||
| Fig. 25 Some potent antiproliferative compounds (108 to 111). | ||
Garcinia hanburyi, resin (named gamboge) is originally used as pigment and folk medicine. In recent years, a special group of xanthones, caged Garcinia xanthones, which have been identified as bioactive compounds with potent biological properties, e.g. antitumor, anti-HIV-1, antibacterial, and anti-inflammatory activities. The compounds occur naturally in the resin, fruit, and other parts of the plant. Han et al. reported 40 different xanthones from G. hanburyi. Furthermore, multiple mechanisms of cytotoxic activity have been reported, such as cell cycle arrest, apoptosis induction, telomerase inhibition, and anti-angiogenesis.130 Mu et al. established an oxidative analogue of gambogic acid as a potential antitumor compound by inducing apoptosis in HepG2 cells. Caged xanthones isolated from G. hanburyi were screened cytotoxic activities against many cell lines, such as human lung carcinoma cells (A 549), henrietta lacks cervical carcinoma tumor cells (HeLa), human hepatoma (SMMC-7221), human leukemia K 562 (K 562/S), doxorubicin-resistant K 562 (K 562/R), human colon carcinoma cells (HCT 116), human breast carcinoma cells (SK-BR-3), human hepatocellular carcinoma cells (HepG2), human liver cancer cells (Hep3B), human liver cancer cells (Huh7), and human neuroblastoma cells (SH-SY5Y).131,132 The modified xanthones were found to exhibit the most potent antitumor activities by inducing apoptosis in HepG2 cell lines in a dose-dependent manner.133 It was found that the efficiency of cell growth inhibition increased dramatically when the concentration of modified xanthones was increased.131,132 Jang et al. reported that modified xanthones (Fig. 26) were selective agonist for TrkA receptor, showing a strong neurotrophic activity by selectively binding to TrkA, inducing its tyrosine phosphorylation, provoking outgrowth in PC12 cells, eliciting PI3-kinase/Akt and MAPK activation, thus preventing neuronal cell death.133
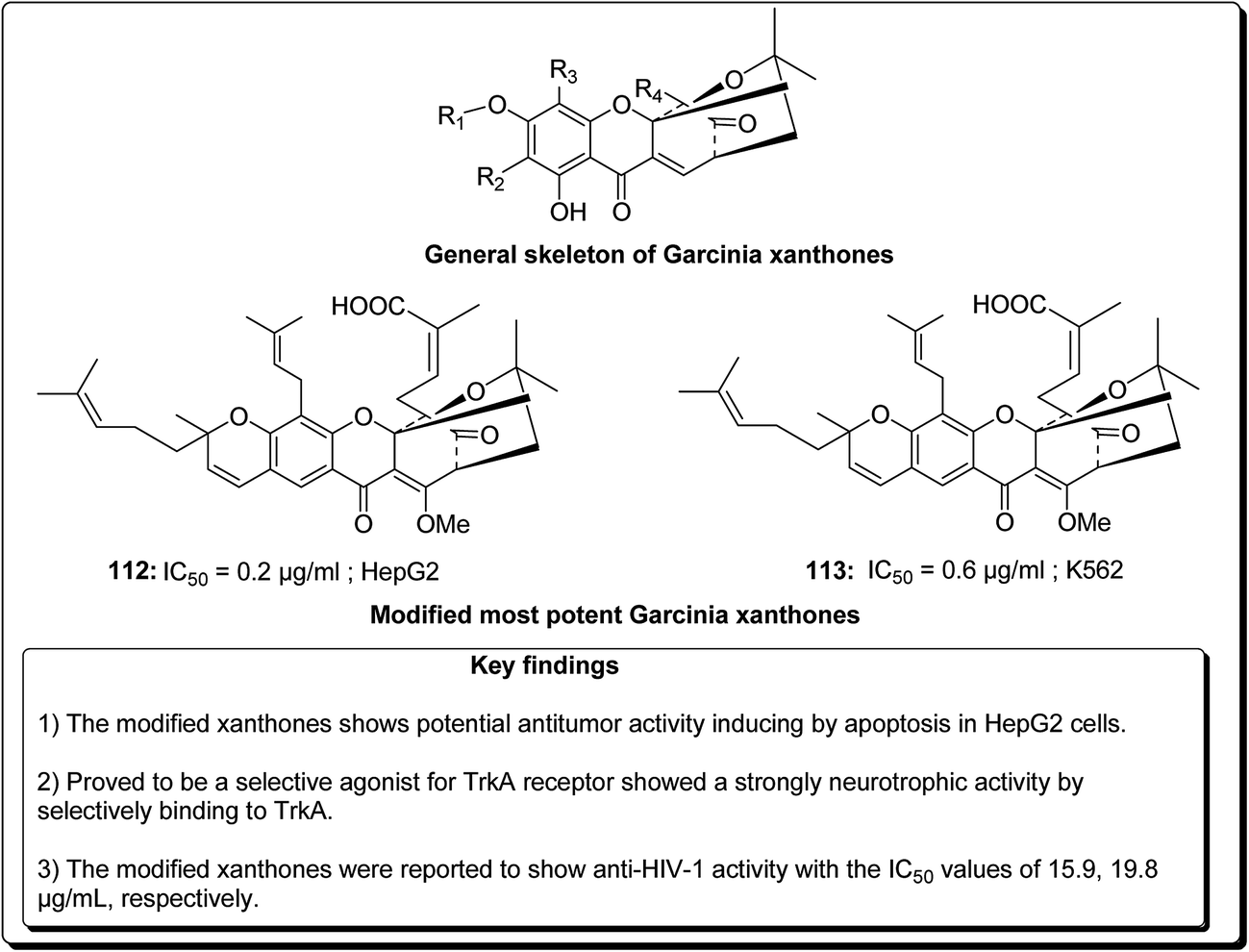 | ||
| Fig. 26 Some reported potent cytotoxic compounds (112 and 113). | ||
Zelefack et al. isolated butyraxanthones A–D, along with four known xanthones (114–117) and a triterpenoid (lupeol) from the shoot bark of Pentadesma butyracea.134 These compounds were evaluated for their in vitro antiplasmodial action towards Plasmodium falciparum chloroquine-resistant strain and for the cytotoxic effect in human breast tumor cell line (MCF-7). It was found out that among all tested compounds, only butyraxanthone D (114) was inactive (IC50 > 10 μg mL−1) but another isolated compound 115 showed the best potency.134
Mosoophon et al. also extracted ruguloxanthones A–C, 14-methoxytajixanthone and tajixanthone ethanoate, a new bicyclo[3.3.1]nona-2,6-diene analogue, rugulosone and seven known compounds, shamixanthone, tajixanthone, 14-methoxytajixanthone-25-acetate, tajixanthone hydrate, tajixanthone methanoate, isoemericellin, and ergosterol, from the fungus Emericella rugulosa. Compound 118 (rugulosone, Fig. 27) also exhibited cytotoxicity against the BC1, KB, and NCI-H187 cancer cell lines, with IC50 values of 1.3, 2.6 and 1.3 μg mL−1, respectively.135
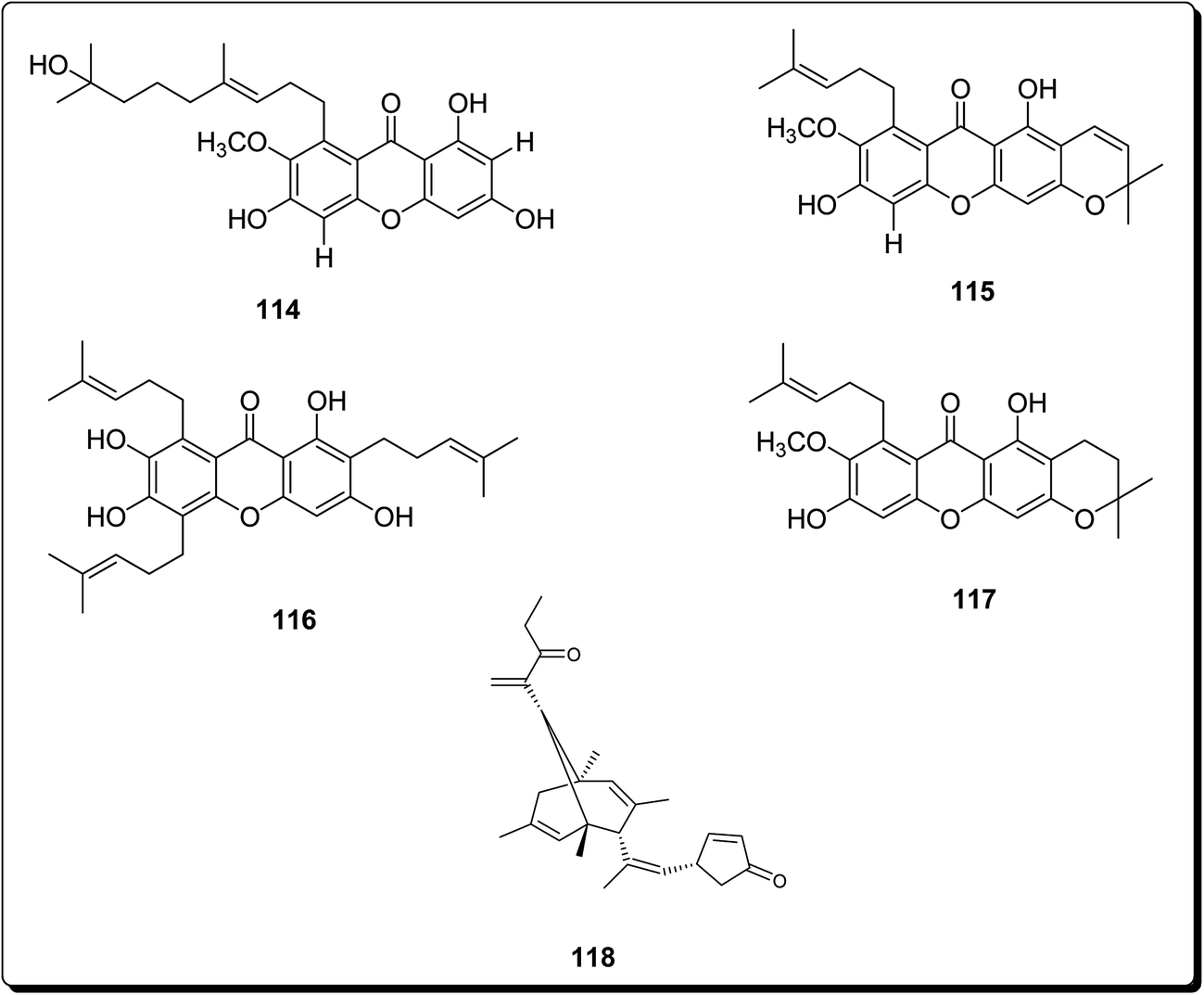 | ||
| Fig. 27 More potent cytotoxic analogues (114 to 118). | ||
Bhattacharya et al. synthesised xanthenes from the one-pot condensation of β-naphthol with aryl aldehydes catalysed by TaCl5 under solvent-free conventional heating.136 The synthesised xanthenes (Fig. 28) were evaluated against a group of six human tumor lines such as SW-620, 502713 and Colo-205 (colon), SKNSH (CNS), A-549 (lung) and PC-3 (prostate), using sulforhodamine B. Compound 119 showed IC50 of 37.9 and 41.3 μM against Colo-205 and 502713 respectively, whereas compound 120 showed an IC50 of 41.9 μM against Colo-205 cell line.136 Niu et al. isolated 1,4,5,6-tetrahydroxanthenes and bracteaxanthenes, together with 26 known compounds from the ethanol extract of stem bark of Garcinia bracteata. These compounds were evaluated for their cell growth inhibiting effect against human leukaemic HL-60 cell lines. The prenylated xanthones (Fig. 28) showed more potent effects. Compounds 121–123 were found most effective via the inhibition of HL-60 cell growth with GI50 values of 2.8, 3.4 and 3.1 μM, respectively.137 Caxanthones A–E, with anticancer properties, which were identified from Codonopsis ovata.139 While coxanthone B showed significant inhibitory activity against SF-295 and MDAMB-435 (IC50 values of 7.0 and 15.0 μM, respectively), coxanthone A showed cytotoxicity against the A549 cell line (IC50 value of 22.5 μM). Meanwhile, the cytotoxic activity of 1-hydroxy-3,5-dimethoxyxanthone, swertiperenine and 1,7,8-trihydroxy-3-methoxyxanthone were shown to be with IC50 values of 3.0, 5.0 and 21.0 μM against A549, MDAMB-435, and A549 cell lines, respectively.138 Among synthesised xanthones with promising anticancer properties, Mulakayala et al. showed that 9-(2-hydroxyphenyl)-3,3,6,6-tetramethyl-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (124) and its analogues could be good starting points for anticancer drug discovery programs, as this compound and its analogues showed good anti-proliferative properties in vitro against three cancer cell lines (with IC50 values between 23 and 38 μM).139
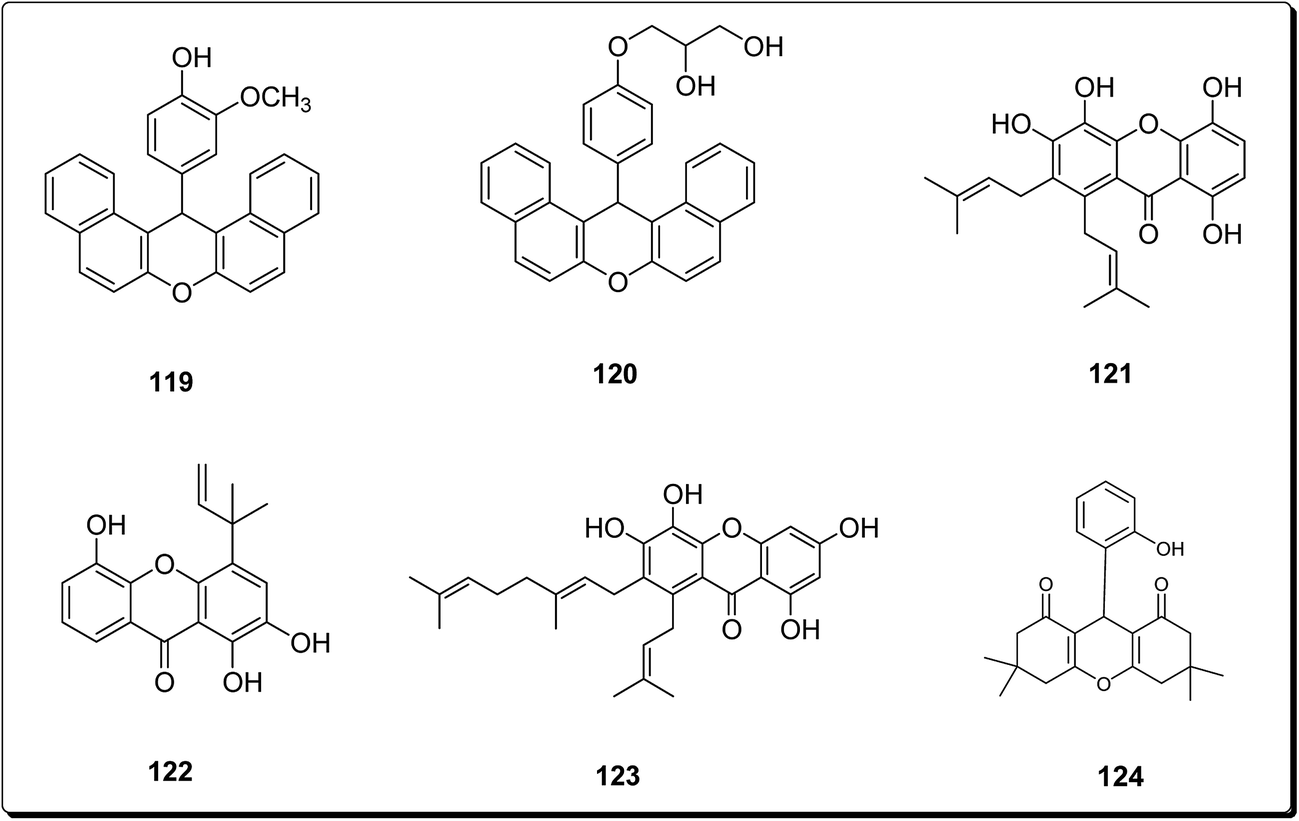 | ||
| Fig. 28 Selected potent anticancer synthesized compounds (119 to 124). | ||
6 Other scaffolds
Mohareb and Schatz described 1,3,4-oxadiazine pyran derivatives with highly potent activities against breast adenocarcinoma (MCF-7), non-small cell lung cancer (NCI-H460) and CNS cancer (SF-268), some of which showed better inhibitory effect towards three cell lines than the standard drug doxorubicin, among which compounds 125 to 128 (Fig. 29) showed submicromolar activities.140 Kumar et al. also designed and synthesised a number of chromeno[4,3-b]quinoxaline derivatives with activities against human metastatic breast cancer cells (MDA-MB 231) and human chronic myeloid leukemia cells (K562).141 The most potent compounds (129 to 133, Fig. 30) inhibited the growth of the cancer cells up to about 50% at 1 μM.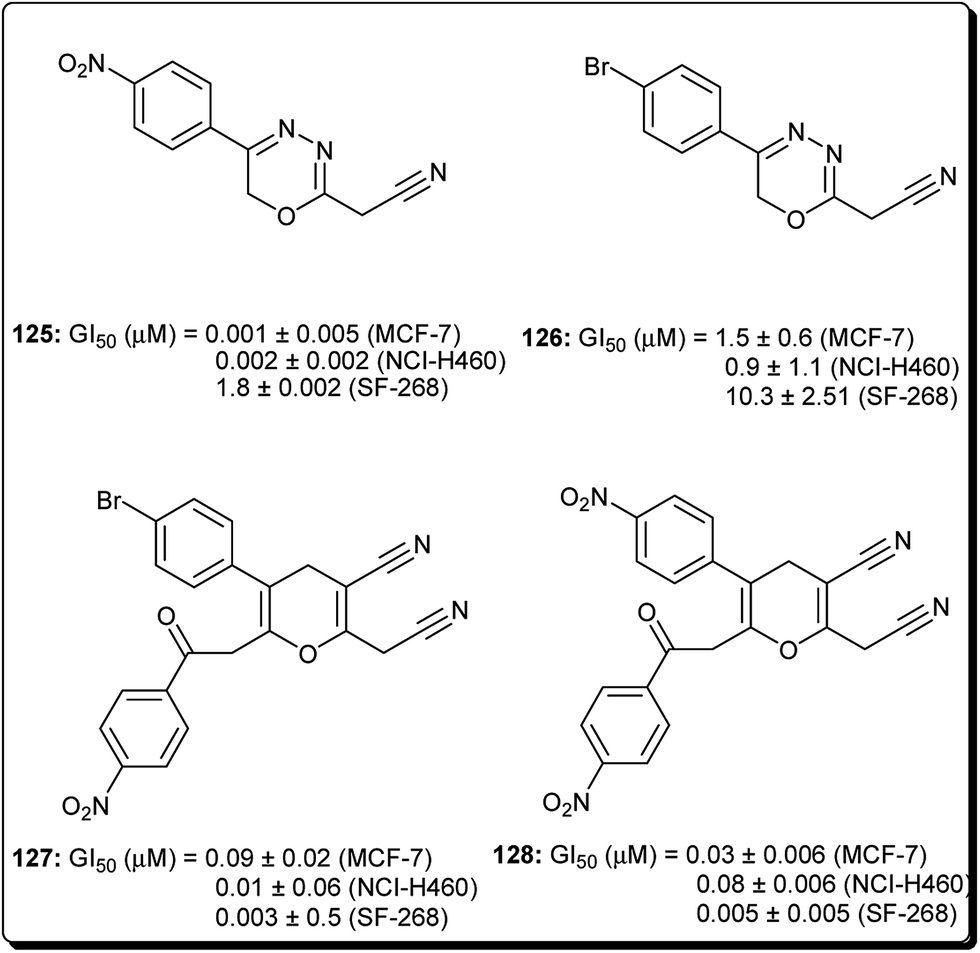 | ||
| Fig. 29 Selected potent 1,3,4-oxadiazine pyran derivatives (125 to 128). | ||
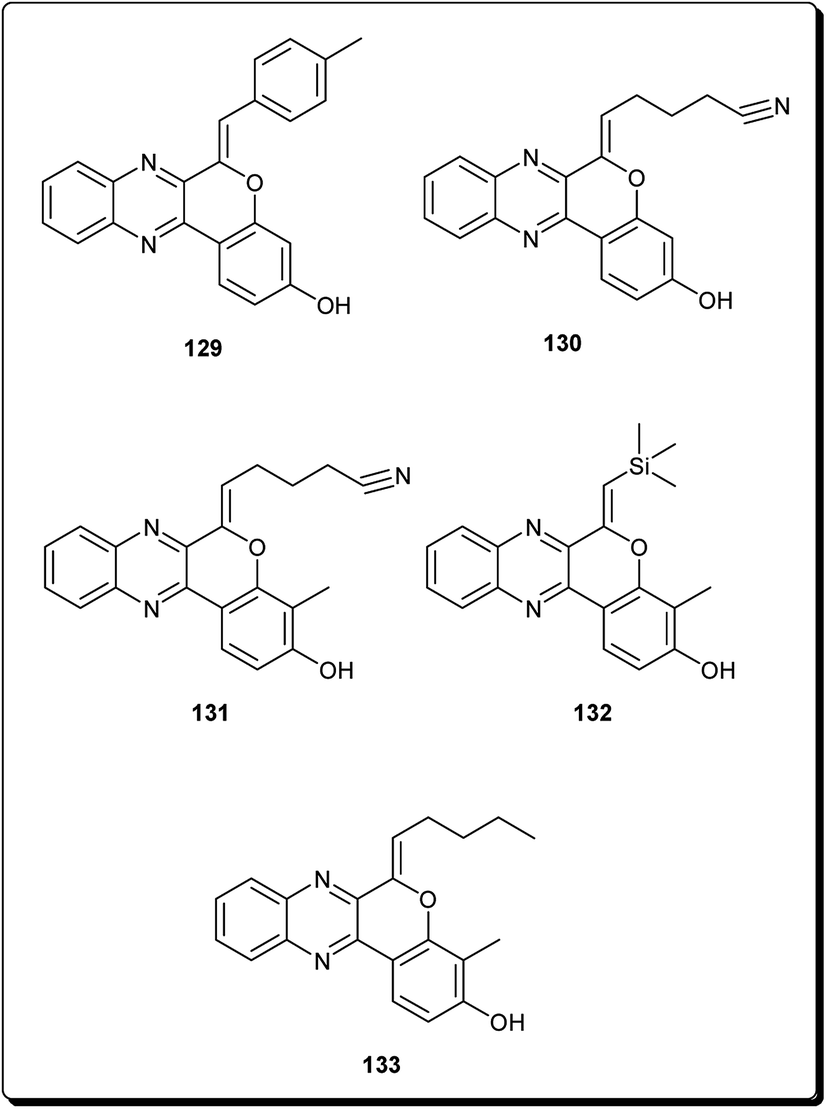 | ||
| Fig. 30 Selected potent chromeno[4,3-b]quinoxaline derivatives (129 to 133). | ||
7 Conclusions
The pyran scaffold has received much attention of researchers both from the pharmaceutical industries and academic organizations in the recent past. As evident from numerous cited papers, the pyran scaffold is the building block of various coumarins, xanthones and flavonoids present in various natural plants. Numerous compounds containing pyran nucleus have displayed inhibitory activities with IC50 values in the micromolar range. The overall conclusion is that pyran being one of the privileged heterocycles has shown a wide array of biological activities, particularly against cancer. There is abundant evidence that the utilization of diversely substituted pyran analogues has provided the platform for identification of new chemical entities which could be drug candidates with diverse biological properties. The in vitro, in vivo, and in silico experiments have shown pyrans to be molecules with potentially exploitable structures for the development of new cytotoxic and anticancer agents. Moreover, structures of designed and synthesised molecules discussed in this compilation clearly highlight the interesting and promising anticancer profiles along with their structure–activity relationships. A discussion of the key interactions with the amino acid in selected binding sites, as demonstrated by molecular modeling studies, has also been provided.Acknowledgements
Authors are thankful to Prof. Ajaib Singh Brar, Vice-chancellor of Guru Nank Dev University, Amritsar for their encouragements. One of the authors Mr Dinesh Kumar is thankful to UGC, New Delhi for providing research fellowship under UPE (Focus Area-Health Care, Drug Development, and Sports Medicines) Scheme (Sanction No. 25994/Estt./A-11). FNK is currently a Georg Forster fellow of the Alexander von Humboldt Foundation, Germany.Notes and references
- J. Ferlay, I. Soerjomataram, M. Ervi, R. Dikshit, S. Eser, C. Mathers, M. Rebelo, D. M. Parkin, D. Forman and F. Bray, GLOBOCAN, 2012, v1.0, Cancer Incidence and Mortality Worldwide: IARC Cancer Base No. 11, International Agency for Research on Cancer, Lyon, France, 2013, http://globocan.iarc.fr, accessed on 29 January 2017 Search PubMed.
- A. Nakhi, R. Adepu, D. Rambabu, R. Kishore, G. R. Vanaja, A. M. Kalle and M. Pal, Bioorg. Med. Chem. Lett., 2012, 22, 4418–4427 CrossRef CAS PubMed.
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2016, 66, 7–30 CrossRef PubMed.
- J. M. Winter, M. F. Brennan, L. H. Tang, M. I. D'Angelica, R. P. DeMatteo, Y. Fong, D. S. Klimstra, W. R. Jarnagin and P. J. Allen, Ann. Surg. Oncol., 2012, 19, 169–175 CrossRef PubMed.
- E. Deutsch, L. Maggiorella, P. Eschwege, J. Bourhis, J. C. Soria and B. Abdulkarim, Lancet Oncol., 2004, 5, 303–313 CrossRef CAS PubMed.
- M. J. Alvarez-Cubero, M. Saiz, L. J. Martinez-Gonzalez, J. C. Alvarez, J. A. Lorente and J. M. Cozar, Urol. Oncol., 2013, 31, 1419–1429 CrossRef CAS PubMed.
- W. C. Willett, Science, 1994, 264, 532–537 CAS.
- V. Duraipandiyan, M. Ayyanar and S. Ignacimuthu, BMC Complementary Altern. Med., 2006, 6, 35 CrossRef PubMed.
- D. Kumar and S. K. Jain, Curr. Med. Chem., 2016, 23, 4338–4394 CrossRef CAS PubMed.
- S. R. Jaggavarapu, A. S. Kamalakaran, J. Nanubolu, V. P. Jalli, S. K. Gangisetty and G. Gaddamanugu, Tetrahedron Lett., 2014, 55, 3670–3673 CrossRef CAS.
- R. Pal, V. Kumar, A. K. Gupta, V. Beniwal and G. K. Gupta, Med. Chem. Res., 2014, 23, 4060–4069 CrossRef CAS.
- G. K. Gupta, V. Kumar and A. Mittal, Lett. Org. Chem., 2014, 11, 273–286 CrossRef CAS.
- A. Kumar, P. Lohan, G. K. Gupta, D. Kaushik and O. Prakash, Eur. J. Med. Chem., 2012, 50, 81–89 CrossRef CAS PubMed.
- L. F. Tietze, Chem. Rev., 1996, 96, 115–136 CrossRef CAS PubMed.
- D. Enders, C. Grondal and M. R. M. Huttl, Angew. Chem., Int. Ed., 2007, 46, 1570–1581 CrossRef CAS PubMed.
- B. Kazemi, S. Javanshir, A. Maleki, M. Safari and H. R. Khavasi, Tetrahedron Lett., 2012, 53, 6977–6981 CrossRef CAS.
- K. C. Nicolaou, J. A. Pfefferkorn and G. Q. Cao, Angew. Chem., Int. Ed., 2000, 39, 734–739 CrossRef CAS PubMed.
- Y. Moumou, J. Vasseur, F. Trotin and J. Dubois, Phytochemistry, 1992, 31, 1239–1241 CrossRef CAS.
- I. E. Soria-Mercado, A. Prieto-Davo, P. R. Jensen and W. J. Fenical, J. Nat. Prod., 2005, 68, 904–910 CrossRef CAS PubMed.
- M. B. Gewali, Y. Tezuka, A. H. Banskota, M. S. Ali, I. Saiki, H. Dong and S. Kadota, Org. Lett., 1999, 1, 1733–1736 CrossRef CAS PubMed.
- K. Koyama, M. Takahashi, M. Oitate, N. Nakai, H. Takakusa, S. Miura and O. Okazaki, Antimicrob. Agents Chemother., 2009, 53, 4845–4851 CrossRef CAS PubMed.
- M. Kiso, S. Kubo, M. Ozawa, Q. M. Le, C. A. Nidom, M. Yamashita and Y. Kawaoka, PLoS Pathog., 2010, 6, e1000786 Search PubMed.
- D. R. da Rocha, A. C. de Souza, J. A. Resende, W. C. Santos, E. A. dos Santos, C. Pessoa, M. O. de Moraes, L. V. Costa-Lotufo, R. C. Montenegro and V. F. Ferreira, Org. Biomol. Chem., 2011, 9, 4315–4322 CAS.
- Y. Dong, Q. Shi, K. Nakagawa-Goto, P. C. Wu, S. L. Morris-Natschke, A. Brossi, K. F. Bastow, J. Y. Lang, M. C. Hung and K. H. Lee, Bioorg. Med. Chem., 2010, 18, 803–808 CrossRef CAS PubMed.
- J. M. Doshi, D. Tian and C. Xing, J. Med. Chem., 2006, 49, 7731–7739 CrossRef CAS PubMed.
- J. Madda, A. Venkatesham, B. N. Kumar, K. Nagaiah, P. Sujitha, C. G. Kumar, T. P. Rao and N. J. Babu, Bioorg. Med. Chem. Lett., 2014, 24, 4428–4434 CrossRef CAS PubMed.
- G. A. Morales, J. R. Garlich, J. Su, X. Peng, J. Newblom, K. Weber and D. L. Durden, J. Med. Chem., 2013, 56, 1922–1939 CrossRef CAS PubMed.
- I. E. Soria-Mercado, A. Prieto-Davo, P. R. Jensen and W. Fenical, J. Nat. Prod., 2005, 46, 904–910 CrossRef PubMed.
- P. Siripong, K. Kanokmedakul, S. Piyaviriyagul, J. Yahuafai, R. Chanpai, S. Ruchirawat and N. Oku, J. Tradit. Med., 2006, 23, 166–172 CAS.
- H. Hussain, K. Krohn, V. U. Ahmad, G. A. Miana and I. R. Greend, Lapachol: an overview, ARKIVOC, 2007, 2, 145–171 Search PubMed.
- A. J. da Silva, C. D. Netto, W. Pacienza-Lima, E. C. Torres-Santos, B. Rossi-Bergmann, S. Maurel, A. Valentin and P. R. Costa, J. Braz. Chem. Soc., 2009, 20, 176–182 CrossRef CAS.
- S. Jiinnez-Alonso, H. C. Orellana, A. Estevez-Braun, A. G. Ravelo, E. Perez-Sacau and F. Machin, J. Med. Chem., 2008, 51, 6761–6772 CrossRef PubMed.
- M. S. Bentle, E. A. Bey, Y. Dong, K. E. Reinicke and D. E. Boothman, J. Mol. Histol., 2006, 37, 203–218 CrossRef CAS PubMed.
- I. V. Magedov, A. S. Kireev, A. R. Jenkins, N. M. Evdokimov, D. T. Lima, P. Tongwa, J. Altig, W. F. Steelant, S. van Slambrouck, M. Y. Antipin and A. Kornienko, Bioorg. Med. Chem. Lett., 2012, 22, 5195–5198 CrossRef CAS PubMed.
- T. A. D. Thi, T. H. V. Thi, H. T. Phuong, T. H. Nguyen, C. P. The, C. V. Duc, Y. Depetter, T. V. Nguyen and M. D'hooghe, Bioorg. Med. Chem. Lett., 2015, 25, 3355–3358 CrossRef PubMed.
- E. J. Salaski, G. Krishnamurthy, W.-D. Ding, K. Yu, S. S. Insaf, C. Eid, J. Shim, J. I. Levin, K. Tabei, T.-B. Lourdes, W.-G. Zhang, L. A. McDonald, E. Honores, C. Hanna, A. Yamashita, B. Johnson, Z. Li, L. Laakso, D. Powell and T. S. Mansour, J. Med. Chem., 2009, 52, 2181 CrossRef CAS PubMed.
- N. Tanaka, T. Okabe, F. Isono, M. Kashiwagi, K. Nomoto, M. Takahashi, A. Shimazu and T. Nishimura, J. Antibiot., 1985, 38, 1327 CrossRef CAS PubMed.
- N. Tsuji, M. Kobayashi, Y. Wakisaka, Y. kawamura, M. Mayama and K. Matsumoto, J. Antibiot., 1976, 29, 7 CrossRef CAS PubMed.
- H. G. Floss, P. Soong and C.-T. Chang, J. Antibiot., 1975, 28, 156 CrossRef.
- R. Corbaz, L. Ettlinger, E. Gaumann, J. Kalvoda, W.-K. Schierlein, F. Kradolfer, B.-K. Manukian, L. Neipp, V. Prelog, P. Reusser and H. Zahner, Helv. Chim. Acta, 1957, 40, 1262 CrossRef CAS.
- X. Jiang, M. Wang, S. Song, Y. Xu, Z. Miao and A. Zhang, RSC Adv., 2015, 5, 27502–27508 RSC.
- A. E. G. Hammam, O. I. A. El-Salam, A. M. Mohamed and N. A. Hafez, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2005, 44, 1887–1893 Search PubMed.
- K. Kowalski, A. K. Chyła, L. Szczupak, P. Hikisz, J. Bernasińska, A. Rajnisz, J. Solecka and B. Therrien, J. Organomet. Chem., 2013, 741, 153–161 CrossRef.
- M. K. Chahar, N. Sharma, M. P. Dobhal and Y. C. Joshi, Pharmacogn. Rev., 2011, 5, 1–12 CrossRef CAS PubMed.
- D. Ravishankar, A. K. Rajora, F. Greco and H. M. I. Osborn, Int. J. Biochem. Cell Biol., 2013, 45, 2821–2831 CrossRef CAS PubMed.
- W. Y. Huang and Y. Z. Cai, Nutr. Cancer, 2010, 62, 1–20 CAS.
- H. De Groot and U. Rauen, Fundam. Clin. Pharmacol., 1998, 12, 249–255 CrossRef CAS PubMed.
- J. E. Middleton, Adv. Exp. Med. Biol., 1998, 439, 175–182 CrossRef PubMed.
- J. S. Yoon, M. K. Lee, S. H. Sung and Y. C. Kim, J. Nat. Prod., 2006, 69, 290–291 CrossRef CAS PubMed.
- M. L. Neuhouser, Nutr. Cancer, 2004, 50, 1–7 CrossRef CAS PubMed.
- L. W. Min, Cancer Treat. Rev., 2009, 35, 57–68 CrossRef PubMed.
- Y. Akao, T. Itoh, K. Ohguchi, M. Iinuma and Y. Nozawa, Bioorg. Med. Chem., 2008, 16, 2803–2810 CrossRef CAS PubMed.
- J. R. Mays, S. A. Hill, J. T. Moyers and B. S. Blagg, Bioorg. Med. Chem., 2010, 18, 249–266 CrossRef CAS PubMed.
- Y. C. Hsiao, Y. S. Hsieh, W. H. Kuo, H. L. Chiou, S. F. Yang, W. L. Chiang and S. C. Chu, J. Biomed. Sci., 2007, 14, 107–119 CrossRef CAS PubMed.
- E. J. Choi, J. I. Lee and G. H. Kim, Int. J. Mol. Med., 2010, 25, 293–298 CAS.
- E. J. Choi, J. I. Lee and G. H. Kim, Arch. Pharmacal Res., 2011, 34, 2125–2130 CrossRef CAS PubMed.
- H. Usman, E. H. Hakim, T. Harlim, M. N. Jalaluddin, Y. M. Syah, S. A. Achmad and H. Takayama, Z. Naturforsch., C: J. Biosci., 2006, 61, 184–188 CAS.
- S. C. Shen, C. H. Ko, S. W. Tseng, S. H. Tsai and Y. C. Chen, Toxicol. Appl. Pharmacol., 2004, 197, 84–95 CrossRef CAS PubMed.
- S. Y. Liao, J. C. Chen, L. Qian, Y. Shen and K. C. Zheng, Eur. J. Med. Chem., 2008, 43, 2159–2170 CrossRef CAS PubMed.
- M. Hirunuma, Y. Shoyama, K. Sasaki, S. Sakamoto, F. Taura, Y. Shoyama, H. Tanaka and S. Morimoto, Phytochemistry, 2011, 72, 752–760 CrossRef CAS PubMed.
- M. Switalska, G. Grynkiewicz, L. Strzadala and J. Wietrzyk, Nutr. Cancer, 2013, 65, 874–884 CrossRef CAS PubMed.
- Y. Murti and P. Mishra, Indian J. Pharm. Sci., 2014, 76, 163–166 CAS.
- D. Kumar, O. Singh, K. Nepali, P. M. S. Bedi, A. Qayum, S. Singh and S. K. Jain, Anti Canc. Agents Med. Chem., 2016, 16, 881–890 CrossRef CAS.
- A. Aghdassi, P. Phillips, V. Dudeja, D. Dhaulakhandi, R. Sharif, R. Dawra, M. M. Lerch and S. Saluja, Cancer Res., 2007, 67, 616–625 CrossRef CAS PubMed.
- M. Mouria, A. S. Gukovskaya, Y. Jung, P. Buechler, O. J. Hines, H. A. Reber and S. J. Pandol, Int. J. Cancer, 2002, 98, 761–769 CrossRef CAS PubMed.
- W. Xue, B. A. Song, H. J. Zhao, X. B. Qi, Y. J. Huang and X. H. Liu, Eur. J. Med. Chem., 2015, 97, 155–163 CrossRef CAS PubMed.
- M. Safavi, N. Esmati, S. K. Ardestani, S. Emami, S. Ajdari, J. Davoodi, A. Shafiee and A. Foroumadi, Eur. J. Med. Chem., 2012, 58, 573–580 CrossRef CAS PubMed.
- A. Al-Kawkabani, B. Boutemeur-Kheddis, M. Makhloufi-Chebli, M. Hamdi, O. Talhi and A. M. Silva, Tetrahedron Lett., 2013, 54, 5111–5114 CrossRef CAS.
- Y. Shi and C. Zhou, Bioorg. Med. Chem. Lett., 2011, 21, 956–960 CrossRef CAS PubMed.
- S. Y. Hur, T. E. Kim, Y. G. Park, J. R. Kim and J. W. Kim, Exp. Mol. Med., 2005, 37, 436–446 CrossRef PubMed.
- T. Devji, C. Reddy, C. Woo, S. Awale, S. Kadota and D. Carrico-Moniz, Bioorg. Med. Chem. Lett., 2011, 21, 5770–5773 CrossRef CAS PubMed.
- N. S. Reddy, M. R. Mallireddigari, S. Cosenza, K. Gumireddy, S. C. Bell, E. P. Reddy and M. V. Reddy, Bioorg. Med. Chem. Lett., 2004, 14, 4093–4097 CrossRef CAS PubMed.
- J. Klenkar and M. Molnar, J. Chem. Pharm. Res., 2015, 7, 1223–1238 CAS.
- K. Nepali, S. Sharma, D. Kumar, A. Budhiraja and K. L. Dhar, Recent Pat. Anti-Cancer Drug Discovery, 2014, 9, 303–339 CrossRef CAS PubMed.
- K. M. Amin, A. A. Eissa, S. M. Abou-Seri, F. M. Awadallah and G. S. Hassan, Eur. J. Med. Chem., 2013, 60, 187–198 CrossRef CAS PubMed.
- F. Belluti, G. Fontana, L. Dal Bo, N. Carenini, C. Giommarelli and F. Zunino, Bioorg. Med. Chem., 2010, 18, 3543–3550 CrossRef CAS PubMed.
- K. Paul, S. Bindal and V. Luxami, Bioorg. Med. Chem., 2013, 23, 3667–3672 CrossRef CAS PubMed.
- K. V. Sashidhara, A. Kumar, M. Kumar, J. Sarkar and S. Sinha, Bioorg. Med. Chem., 2010, 20, 7205–7211 CrossRef CAS PubMed.
- K. V. Sashidhara, S. R. Avula, K. Sharma, G. R. Palnati and S. R. Bathula, Eur. J. Med. Chem., 2013, 60, 120–127 CrossRef CAS PubMed.
- A. K. Bagdi, A. Majee and A. Hajra, Tetrahedron Lett., 2013, 54, 3892–3895 CrossRef CAS.
- D. Kumar, F. Malik, P. M. S. Bedi and S. Jain, Chem. Pharm. Bull., 2016, 64, 399–409 CrossRef CAS PubMed.
- M. K. Hussain, M. I. Ansari, N. Yadav, P. K. Gupta, A. K. Gupta, R. Saxena, I. Fatima, M. Manohar, P. Kushwaha and V. Khedgikar, RSC Adv., 2014, 4, 8828–8845 RSC.
- O. G. Ganina, E. Daras, V. Bourgarel-Rey, V. Peyrot, A. N. Andresyuk, J. P. Finet, A. Y. Fedorov, I. P. Beletskaya and S. Combes, Bioorg. Med. Chem., 2008, 16, 8806–8812 CrossRef CAS PubMed.
- Y. Garazd, M. Garazd and R. Lesyk, Saudi Pharm. J., 2017, 25, 214–223 CrossRef PubMed.
- H. P. Zhao, A. C. Donnelly and B. R. Kusuma, J. Med. Chem., 2011, 54, 3839–3853 CrossRef CAS PubMed.
- Z. N. Siddiqui, T. N. M. Musthafa, A. Ahmad and A. U. Khan, Arch. Pharm., 2011, 344, 394–401 CrossRef CAS PubMed.
- B. R. Kusuma, L. B. Peterson, H. Zhao, G. Vielhauer, J. Holzbeierlein and B. S. Blagg, J. Med. Chem., 2011, 54, 6234–6253 CrossRef CAS PubMed.
- J. A. Burlison and B. S. J. Blagg, Org. Lett., 2006, 8, 4855–4858 CrossRef CAS PubMed.
- G. H. Tan, Y. C. Yao, Y. J. Gu, S. Li, M. Lv, K. Wang, H. Chen and X. Li, Bioorg. Med. Chem. Lett., 2014, 24, 2825–2830 CrossRef CAS PubMed.
- M. Zhu, L. K. Zhou, Y. C. Yao, S. Li, M. Lv, K. Wang, X. Li and H. Chen, Med. Chem. Res., 2015, 24, 2314–2324 CrossRef CAS.
- L. Zhao, Y. C. Yao, S. M. Lv, H. Chen and X. Li, Bioorg. Med. Chem. Lett., 2014, 24, 900–904 CrossRef CAS PubMed.
- H. Chen, S. Li, Y. C. Yao, L. Zhou, J. Zhao, Y. Gu, K. Wang and X. Li, Bioorg. Med. Chem. Lett., 2013, 23, 4785–4789 CrossRef CAS PubMed.
- L. Zhang, Y. C. Yao, M. Y. G. R. X. Rong, K. R. Wang, L. Xiao-Liu and H. Chen, Chin. Chem. Lett., 2016, 27, 1708–1716 CrossRef CAS.
- S. M. Kupchan, D. R. Streelman and A. T. Sneden, J. Nat. Prod., 1980, 43, 296–301 CrossRef CAS.
- D. K. Winter, D. L. Sloman and J. A. Porco Jr, Nat. Prod. Rep., 2013, 30, 382–391 RSC.
- C. N. Lin, S. J. Liou, T. H. Lee, Y. C. Chuang and S. J. Won, J. Pharm. Pharmacol., 1996, 48, 539–544 CrossRef CAS PubMed.
- C. A. Chen, R. H. Yeh and D. S. Lawrence, J. Am. Chem. Soc., 2002, 124, 3840–3841 CrossRef CAS PubMed.
- L. F. Steiner and S. A. Summerland, J. Econ. Entomol., 1943, 36, 435–439 CrossRef CAS.
- J. H. Cheng, A. M. Huang, T. C. Hour, S. C. Yang, Y. S. Pu and C. N. Lin, Eur. J. Med. Chem., 2011, 46, 1222–1231 CrossRef CAS PubMed.
- M. Chase, Angiosperm phylogeny group, Bot. J. Linn. Soc., 2003, 141, 399–436 CrossRef.
- J. K. Burmester, Xanthene compounds for cancer chemotherapy, US Pat. WO, 2006102102 A2, 2006.
- Y. Xiao and R. Ottenbrite, Tumor necrosis factor inhibitors, US Pat. WO, 2006083869, 2006.
- M. I. Greene, R. Murali, X. Cheng, R. Ottenbrite and Y. Xiao, US Pat. WO, 2006083970, 2006.
- M. R. Campos-Esparza, M. V. Sanchez-Gomez and C. Matute, Cell Calcium, 2009, 45, 358–368 CrossRef CAS PubMed.
- A. Martinez, A. Galano and R. Vargas, J. Phys. Chem. B, 2011, 115, 12591–12598 CrossRef CAS PubMed.
- J. Pedraza-Chaverri, L. M. Reyes-Fermin, E. G. Nolasco-Amaya, M. Orozco-Ibarra, O. N. Medina-Campos, O. Gonzalez-Cuahutencos, I. Rivero-Cruz and R. Mata, Exp. Toxicol. Pathol., 2009, 61, 491–501 CrossRef CAS PubMed.
- M. Buelna-Chontal, F. Correa, S. Hernandez-Resendiz, C. Zazueta and J. Pedraza-Chaverri, J. Med. Food, 2011, 14, 1370–1374 CrossRef CAS PubMed.
- L. M. Reyes-Fermín, S. González-Reyes, N. G. Tarco-Álvarez, M. Hernández-Nava, M. Orozco-Ibarra and J. Pedraza-Chaverri, Nutr. Neurosci., 2012, 15, 34–41 CrossRef PubMed.
- M. K. Schwaebe, T. J. Moran and J. P. Whitten, Tetrahedron Lett., 2005, 46, 827–829 CrossRef CAS.
- K. Matsumoto, Y. Akao, H. Yi, K. Ohguchi, T. Ito, T. Tanaka, E. Kobayashi, M. Iinuma and Y. Nozawa, Bioorg. Med. Chem., 2004, 12, 5799–5806 CrossRef CAS PubMed.
- M. Pedro, F. Cerqueira, M. E. Sousa, M. S. J. Nascimento and M. Pinto, Bioorg. Med. Chem., 2002, 10, 3725–3730 CrossRef CAS PubMed.
- C. Gnerre, U. Thull, P. Gailland, P. A. Carrupt, B. Testa, E. Fernandes, F. Silva, M. Pinto, J. L. Wolfender, K. Hostettmann and G. Cruciani, Helv. Chim. Acta, 2001, 84, 552–570 CrossRef CAS.
- S. Poondru, S. Zhou, J. Rake, G. Shackleton, T. H. Corbett, R. E. Parchment and B. R. Jasti, J. Chromatogr. B: Biomed. Sci. Appl., 2001, 759, 175–178 CrossRef CAS.
- K. H. Lee, H. B. Chai, P. A. Tamez, J. M. Pezzuto, G. A. Cordell, K. K. Win and M. Tin-Wa, Phytochemistry, 2003, 64, 535–539 CrossRef CAS PubMed.
- S. Laphookhieo, J. K. Syers, R. Kiattansakul and K. Chantrapromma, Chem. Pharm. Bull., 2006, 54, 745–747 CrossRef CAS PubMed.
- O. Chantarasriwong, W. C. Cho, A. Batova, W. Chavasiri, C. Moore, A. L. Rheingold and E. A. Theodorakis, Org. Biomol. Chem., 2009, 7, 4886–4894 CAS.
- K. Matsumoto, Y. Akao, E. Kobayashi, K. Ohguchi, T. Ito, T. Tanaka, M. Iinuma and Y. Nozawa, J. Nat. Prod., 2003, 66, 1124–1127 CrossRef CAS PubMed.
- L. C. Chiang, H. Y. Cheng, M. C. Liu, W. Chiang and C. C. Lin, LWT--Food Sci. Technol., 2004, 37, 539–544 CrossRef CAS.
- M. J. Balunas, B. Su, R. W. Brueggemeier and A. D. Kinghorn, J. Nat. Prod., 2008, 17, 1161–1166 CrossRef PubMed.
- H. A. Jung, B. N. Su, W. J. Keller, R. G. Mehta and A. D. Kinghorn, J. Agric. Food Chem., 2006, 54, 2077–2082 CrossRef CAS PubMed.
- S. Suksamrarn, O. Komutiban, P. Ratananukul, N. Chimnoi, N. Lartpornmatulee and A. Suksamrarn, Chem. Pharm. Bull., 2006, 54, 301–305 CrossRef CAS PubMed.
- L. G. Chen, L. L. Yang and C. C. Wang, Food Chem. Toxicol., 2008, 46, 688–693 CrossRef CAS PubMed.
- R. Watanapokasin, F. Jarinthanan, A. Jerusalmi, S. Suksamrarn, Y. Nakamura, S. Sukseree, W. Uthaisang-Tanethpongtamb, P. Ratananukul and T. Sano, Appl. Biochem. Biotechnol., 2010, 162, 1080–1094 CrossRef CAS PubMed.
- A. F. Aisha, K. M. Abu-Salah, Z. Ismail and A. M. S. A. Majid, BMC Complementary Altern. Med., 2012, 12, 104 CrossRef.
- N. Kosem, K. Ichikawa, H. Utsumi and P. Moongkarndi, J. Nat. Med., 2013, 67, 255–263 CrossRef CAS PubMed.
- S. J. Kim, E. H. Hong, B. R. Lee, M. H. Park, J. W. Kim, A. R. Pyun, Y. J. Kim, S. Y. Chang, Y. W. Chin and H. J. Ko, Immune Netw., 2012, 12, 253–260 CrossRef PubMed.
- S. Cao, P. J. Brodie, J. S. Miller, R. Randrianaivo, F. Ratovoson, C. Birkinshaw, R. Andriantsiferana, V. E. Rasamison and D. G. Kingston, J. Nat. Prod., 2007, 70, 679–681 CrossRef CAS PubMed.
- Q. B. Han, H. L. Tian, N. Y. Yang, C. F. Qiao, J. Z. Song, D. C. Chang, K. Q. Luo and H. X. Xu, Chem. Biodiversity, 2008, 5, 2710–2717 CAS.
- S. J. Tao, S. H. Guan, W. Wang, Z. Q. Lu, G. T. Chen, N. Sha, Q. X. Yue, X. Liu and D. A. Guo, J. Nat. Prod., 2009, 72, 117–124 CrossRef CAS PubMed.
- Q. B. Han and H. X. Xu, Curr. Med. Chem., 2009, 16, 3775–3796 CrossRef CAS PubMed.
- R. Mu, N. Lu, J. Wang, Y. Yin, Y. Ding, X. Zhang, H. Gui, Q. Sun, H. Duan, L. Zhang and Y. Zhang, Eur. J. Cancer Prev., 2010, 19, 61–67 CrossRef CAS PubMed.
- W. Zhou, B. Cai, J. Shan, S. Wang and L. Di, AAPS PharmSciTech, 2015, 16, 28812–28840 CAS.
- S. W. Jang, M. Okada, I. Sayeed, G. Xiao, D. Stein, P. Jin and K. Ye, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16329–16334 CrossRef CAS PubMed.
- F. Zelefack, D. Guilet, N. Fabre, C. Bayet, S. Chevalley, S. Ngouela, B. N. Lenta, A. Valentin, E. Tsamo and M. G. Dijoux-Franca, J. Nat. Prod., 2009, 72, 954–957 CrossRef CAS PubMed.
- P. Moosophon, S. Kanokmedhakul, K. Kanokmedhakul and K. Soytong, J. Nat. Prod., 2009, 72, 1442–1446 CrossRef CAS PubMed.
- A. K. Bhattacharya, K. C. Rana, M. Mujahid, I. Sehar and A. K. Saxena, Bioorg. Med. Chem. Lett., 2009, 19, 5590–5593 CrossRef CAS PubMed.
- S. L. Niu, Z. L. Li, F. Ji, G. Y. Liu, N. Zhao, X. Q. Liu, Y. K. Jing and Y. M. Hua, Phytochemistry, 2012, 77, 280–286 CrossRef CAS PubMed.
- A. A. Dar, N. A. Dangroo, A. Raina, A. Qayum, S. Singh, A. Kumar and P. L. Sangwan, Phytochemistry, 2016, 132, 102–108 CrossRef CAS PubMed.
- N. Mulakayala, P. V. N. S. Murthy, D. Rambabu, M. Aeluri, R. Adepu, G. R. Krishna, C. M. Reddy, K. R. S. Prasad, M. Chaitanya, C. S. Kumar, M. V. B. Rao and M. Pal, Bioorg. Med. Chem. Lett., 2012, 22, 2186–2191 CrossRef CAS PubMed.
- R. M. Mohareb and J. Schatz, Bioorg. Med. Chem., 2011, 19, 2707–2713 CrossRef CAS PubMed.
- K. S. Kumar, D. Rambabu, B. Prasad, M. Mujahid, R. Krishna, M. V. B. Rao, C. M. Reddy, G. R. Vanaja, A. M. Kallee and M. Pal, Org. Biomol. Chem., 2012, 10, 4774–4781 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05441f |
| This journal is © The Royal Society of Chemistry 2017 |