
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and properties of magnetic-optical core–shell nanoparticles
Elyahb Allie Kwizera
,
Elise Chaffin
,
Yongmei Wang
and
Xiaohua Huang
 *
*
Department of Chemistry, The University of Memphis, Memphis, TN 38152, USA. E-mail: xhuang4@memphis.edu; Fax: +1 901 678 3744; Tel: +1 901 678 1728
First published on 20th March 2017
Abstract
Due to their high integrity, facile surface chemistry, excellent stability, and dual properties from the core and shell materials, magnetic–plasmonic core–shell nanoparticles are of great interest across a number of science, engineering and biomedical disciplines. They are promising for applications in a broad range of areas including catalysis, energy conversion, biological separation, medical imaging, disease detection and treatment. The technological applications have driven the need for high quality nanoparticles with well controlled magnetic and optical properties. Tremendous progress has been made during past few decades in synthesizing and characterizing magnetic–plasmonic core–shell nanoparticles, mainly iron oxide–gold core–shell nanoparticles. This review introduces various approaches for the synthesis of spherical and anisotropic magnetic–plasmonic core–shell nanoparticles focusing on iron oxide–gold core–shell nanoparticles. Growth mechanisms are discussed to provide understanding of the key factors controlling shape-controlled synthesis. Magnetic and optical properties are summarized from both computational and experimental studies.
 Elyahb Allie Kwizera | Elyahb A. Kwizera received his bachelor's degree in chemistry and mathematics from the University of Central Arkansas in 2014. He is currently a Ph.D. student under the supervision of Dr Xiaohua Huang. His research is focused on controlled synthesis, properties and applications of magnetic–plasmonic core–shell nanoparticles as well as development of novel nanotechnology-based technologies for detection and analysis of circulating vesicles. |
 Elise Chaffin | Dr Elise Chaffin received her Ph.D. degree in chemistry from the University of Memphis in 2016. She is currently a postdoctoral fellow at the University of Memphis. Under the mentorship of Dr Wang and Huang, her research has been focused on modelling the optical properties of noble metal nanoparticles using discrete dipole approximation and Mie theory in collaboration with the synthesis and characterization by the Huang group. |
 Yongmei Wang | Dr Yongmei Wang is a tenured full professor at Department of Chemistry, The University of Memphis. She received her PhD in Chemistry from University of Notre Dame in 1990 and postdoctoral training at University of Akron from 1990 to 1993 and University of Houston from 1994 to 1996. She began her academic career in 1996 as an Assistant Professor at North Carolina Agricultural and Technical State University and moved to the University of Memphis in 2003. Her research interests are theoretical/computational studies for polymer characterization, polymer based gene delivery and nanomedicines. |
 Xiaohua Huang | Dr Xiaohua Huang is an associate professor of chemistry at The University of Memphis, USA. She received her Ph.D. degree in chemistry from Georgia Institute of Technology in 2006 and postdoctoral training at Georgia Institute of Technology and Emory University from 2006 to 2010. She has published 50 peer-reviewed journal articles including 11 reviews with over 15 |
1. Introduction
Nanoscale materials are a topic of considerable interest across a number of science, engineering and biomedical disciplines. The basic rationale is that nanoscale materials, typically 1–100 nm, exhibit exceptional structural and functional properties that are not available in bulk materials or discrete molecules. Two major classes of functional nanoplatforms have been extensively studied and widely used in a variety of fields: plasmonic nanoparticles (NPs) and magnetic NPs (MNPs). Plasmonic NPs are typically composed of noble metals, generally gold (Au) and silver (Ag). They exhibit unique localized surface plasmon resonance (LSPR), the collective oscillation of the conduction electrons of the NPs in resonance with the electric field of the incident light.1–3 This LSPR leads to strongly enhanced radiative (e.g. absorption and scattering) and nonradiative (e.g. photothermal and energy transfer) properties.4,5 Compared to Ag NPs, Au NPs are more stable under ambient conditions. Additionally, the LSPR of Au NPs can be tuned from the visible to near infrared region (NIR) by adjusting the particle's size, shape and structure.5–9 These intriguing optical properties have made Au NPs highly favorable for sensing, optical imaging, photothermal cancer therapy, catalysis, and many other material and biomedical applications.10–14MNPs, commonly consisting of magnetic elements such as iron (Fe) and cobalt (Co) and their chemical compounds, show alignment of their magnetic moment in the presence of an external magnetic field and concentrate the external magnetic flux density.15 This magnetic response causes the attraction of the MNPs in the direction of applied magnetic gradient. This magnetic property makes the MNPs useful for many applications including data storage, spintronics, molecular and cellular isolation, magnetic resonance imaging (MRI) and hyperthermia treatment of cancer.16–19 The most common MNPs are iron oxide nanoparticles (IO NPs) including magnetite (Fe3O4) or maghemite (γ-Fe2O3). These MNPs have advantages of ease-of-preparation, biodegradability, excellent stability, and the tunability of magnetic properties through changes in size and shape, making them the most attractive MNP platforms. When the sizes of these MNPs are smaller than 30 nm, they are superparamagnetic, otherwise they are ferromagnetic.20 Superparamagnetic NPs avoid the induced aggregation associated with the residual magnetization of ferromagnetic NPs. MNPs can be spherical or anisotropic such as rods, cubes and stars. Thus, they provide versatile structural platforms for generations of different nanostructures.
However, each of these NP types displays distinct limitations. For example, plasmonic NPs lack the ability to separate analytes, which is usually required for the analysis of rare molecules and cells in a complex milieu. MNPs do not exhibit the properties needed for highly sensitive optical imaging. Thus, hybrid NPs with combined magnetic and optical properties are much more powerful and can be used in a broad range of applications such as magnetic resonance imaging,21–27 optical imaging,26–28 biological separation,29–37 molecular/cellular detection,33–40 and cancer treatment.23–25,27,31,41–43 Additionally, these hybrid NPs offer new modalities that neither plasmonic NPs nor MNPs exhibit. For example, Jin et al. have demonstrated the use of IO–Au core–shell NPs for magnetomotive photoacoustic imaging.44 This novel imaging mode shows remarkable contrast enhancement compared with photoacoustic images obtained using solid Au NPs. Magnetic–plasmonic core–shell NPs can consist of any magnetic core such as Fe, Co or their oxides and a plasmonic shell such as Au, Ag or platinum. However, IO–Au core–shell NPs have become the primary platform because of their remarkable advantages. The IO NPs are stable and easy to synthesize, and the Au surface offers facile surface modification, excellent stability, and biosafety. The optical properties of the IO–Au core–shell NPs can be precisely tuned by changing the core size, shell thickness as well as the core and shell shapes. A key to the technological applications is the synthesis of high quality IO–Au core–shell NPs with desirable magnetic and optical properties. During the past decade, intense research has been directed to make IO–Au core–shell NPs with different core sizes and shell thicknesses. Synthesizing anisotropic IO–Au core–shell NPs requires precise control of the thermodynamic and kinetic parameters of the growth solution. In this review, we introduce various approaches for the synthesis of monodisperse spherical and anisotropic IO–Au core–shell NPs. Growth mechanisms are discussed to provide understanding of the key factors controlling the uniform formation of the Au shell and shape-controlled synthesis. Magnetic and optical properties of these core–shell NPs are summarized in both computational and experimental results to better understand the structural–functional relationship of IO–Au core–shell NPs. This review focusses on IO–Au core–shell NPs, but the information can be applied to other types of core–shell NPs as well.
2. Synthesis
2.1 Synthesis of spherical IO–Au core–shell NPs
Methods to make spherical IO–Au core–shell NPs can be divided into two major categories. In the first Au atoms are directly deposited onto IO NPs via the reduction of Au precursor in a growth solution containing IO NPs. In this direct deposition method, the surface of the IO NPs may be chemically modified to adsorb Au ions to facilitate Au shell formation. In the second method, Au atoms are deposited onto Au-seeded IO NPs in which Au seeds serve as nucleation sites to facilitate the growth of Au shell. This Au-seeded growth method requires more steps than the direct deposition method, but it provides flexibility in tuning the morphology of Au shell by fine control the kinetic and thermodynamic parameters of the growth solution.| (hkl) index | Fe3O4 (Å) | γ-Fe2O3 (Å) | Au (Å) |
|---|---|---|---|
| 111 | 4.85 | 4.82 | 2.35 |
| 220 | 2.97 | 2.95 | 1.44 |
| 311 | 2.53 | 2.52 | 1.23 |
| 400 | 2.10 | 2.09 | 1.02 |
| 422 | 1.71 | 1.70 | 0.83 |
To synthesize IO–Au core–shell NPs with uniform Au surface, one approach is to iteratively deposit Au atoms onto IO NPs via repeated reduction of Au3+ by hydroxylamine (NH2OH).27,28,33,39,49,50 NH2OH is known to promote Au3+ surface catalyzed reduction.51 Thus, Au atoms are deposited only on the surfaces of IO NPs rather than forming self-nucleated Au NPs. This method was first reported by Lyon et al. in 2004 (Fig. 1A).49 In their studies, γ-Fe2O3 or oxidized Fe3O4 NPs were immersed in sodium citrate for 10 min to exchange adsorbed hydroxide anions with citrate anions. Au atoms were deposited onto the IO NPs via the reduction of HAuCl4 by NH2OH at room temperature. Five additions of HAuCl4 and NH2OH were performed to achieve continuous Au shells on the IO NP surfaces. Characterization by transmission electron microscopy (TEM) showed that Au atoms were initially deposited onto specific sites of the IO core, which led to a jagged Au surface. Au atoms from the subsequent reduction steps filled the empty sites on the IO surface, producing a continuous Au shell and, thus, uniform IO–Au core–shell NPs. This process also explained why the size of the core–shell NPs did not change between the first and fifth depositions having similar sizes of around 60 nm in diameter following each step. It is interesting to note that they were not able to form an Au shell on the freshly prepared Fe3O4 NPs. This mechanism remains to be investigated. The unit cell parameters for both Fe3O4 and γ-Fe2O3 based cubic settings are almost identical with a = 0.845 nm for γ-Fe2O3 and a = 0.840 for Fe3O4.52,53 Therefore, the success of the Au shell formation on Fe2O3/oxidized Fe3O4 NPs but not on freshly prepared Fe3O4 NPs cannot be ascribed to the lattice dissimilarity between the IO NPs and Au. Recently, Tamer et al. modified the method by treating the oxidized Fe3O4 NPs with ethylenediaminetetraacetic acid (EDTA) before Au coating and reduced HauCl4 with NH2OH in the presence of EDTA-immobilized Fe3O4 NPs and cetyltrimethylammonium bromide (CTAB) in a basic solution was added.54 Uniform, highly stable IO–Au core–shell NPs with a narrow size distribution were produced with one-step reductive Au deposition. In a different study by Zhang et al., Fe3O4 NPs were modified with 3-aminopropyltrimethoxysilane (APTMS) before reductive deposition of Au with NH2OH.55 APTMS modification was used to facilitate Au ion adsorption because of its positive charge.
 | ||
| Fig. 1 Synthesis of IO–Au core–shell NPs by the direct deposition method. (A) Synthesis of IO–Au core–shell NPs by iterative hydroxylamine seeding. Top: Schematic of the preparation procedure. Bottom: TEM images of IO–Au core–shell NPs with zero, one, three, and five times of deposition of Au atoms via reduction of Au3+ with hydroxylamine, respectively. Scale bar: XX nm. Reprinted with permission from ref. 49. Copyright (2009) American Chemical Society. (B) Synthesis of IO–Au core–shell NPs by hydroxylamine reduction of Au precursor on the surface of polymer-coated IO NPs. Left: Schematic of the preparation procedure. Right: TEM image of Fe3O4 NPs and Fe3O4–Au core–shell NPs. Reprinted by permission from Macmillan Publishers Ltd: [Nature Communications] (ref. 44), copyright (2010). | ||
Another technique for forming IO–Au core–shell NPs is to perform the Au deposition in a microemulsion56 or nanoemulsion57 system that confines the Au precursor and IO NPs in close proximity to facilitate Au shell formation. The emulsion consists of an organic solvent, a surfactant such as CTAB or poly(vinylpyrrolidone) (PVP), and an aqueous mixture of FeCl3, FeCl2 and HAuCl4. IO NPs are first formed inside the micelle by coprecipitation through the addition of basic solution such as sodium hydroxide. Then, a reducing agent, typically NaBH4, is added to reduce HAuCl4 on the surface of the IO NPs to form Au shells.
Coprecipitation is the simplest way to make IO NPs without the need for capping materials. However, making monodisperse IO NPs via coprecipitation is difficult. Monodisperse IO NPs are usually produced by the thermal decomposition of Fe precursors at high temperatures.58,59 These NPs are capped with oleic acid (OA) and/or oleylamine (OAM) and are only dispersible in organic phase. Thus, the direct deposition of Au atoms onto the IO NPs must be performed in organic phase.22,29,60–65 For example, Wang et al. synthesized IO–Au core–shell NPs by heating a mixture of OA-capped Fe3O4 NPs, Au acetate (Au(OOCCH3)3) and 1,2-hexadecanediol in phenyl ether to 180–190 °C for 1.5 h.60 Xu et al. coated Au onto OA- and OAM-capped IO NPs at room temperature by gently reducing HAuCl4 in a chloroform solution containing OAM.62 OAM served as a mild reducing agent as well as a surfactant. To make water-soluble core–shell NPs, the as-prepared IO–Au NPs were dried and then dissolved in an aqueous solution containing CTAB and sodium citrate. The water soluble IO–Au NPs can then be used as seeds for the growth of an Au shell. This subsequent Au shell growth was readily achieved by the reduction of HAuCl4 by ascorbic acid in the presence of CTAB. By adjusting the amount of HAuCl4, the thickness of Au shell can be finely controlled.
Spherical IO–Au core–shell NPs generally exhibit LSPR in the UV-Vis region. In 2010, Jin et al. developed a method to make spherical IO–Au core–shell NPs with NIR absorption by separating IO and Au by an organic gap (Fig. 1B).44 They first modified OA-capped Fe3O4 NPs with amphiphilic phospholipid–polyethylene glycol terminated with carboxylic acid. This allowed the particles to be dissolved into aqueous phase. Then, the particles were coated with poly-L-histidine that chelates Au3+. The poly-L-histidine is capable of immobilizing Au3+ on IO NPs with high packing density. The multilayer organic molecules prevent Au3+ from direct growth on the IO core. Subsequent reduction of chelated Au3+ led to the formation of thin layer of Au shell (1–5 nm) within 1 h, with a clear separation from the IO core. The total size of the IO–Au core–shell NPs was only around 35 nm with LSPR wavelength over 600 nm. This method enables the preparation of compact and monodisperse IO–Au NPs with NIR absorption.
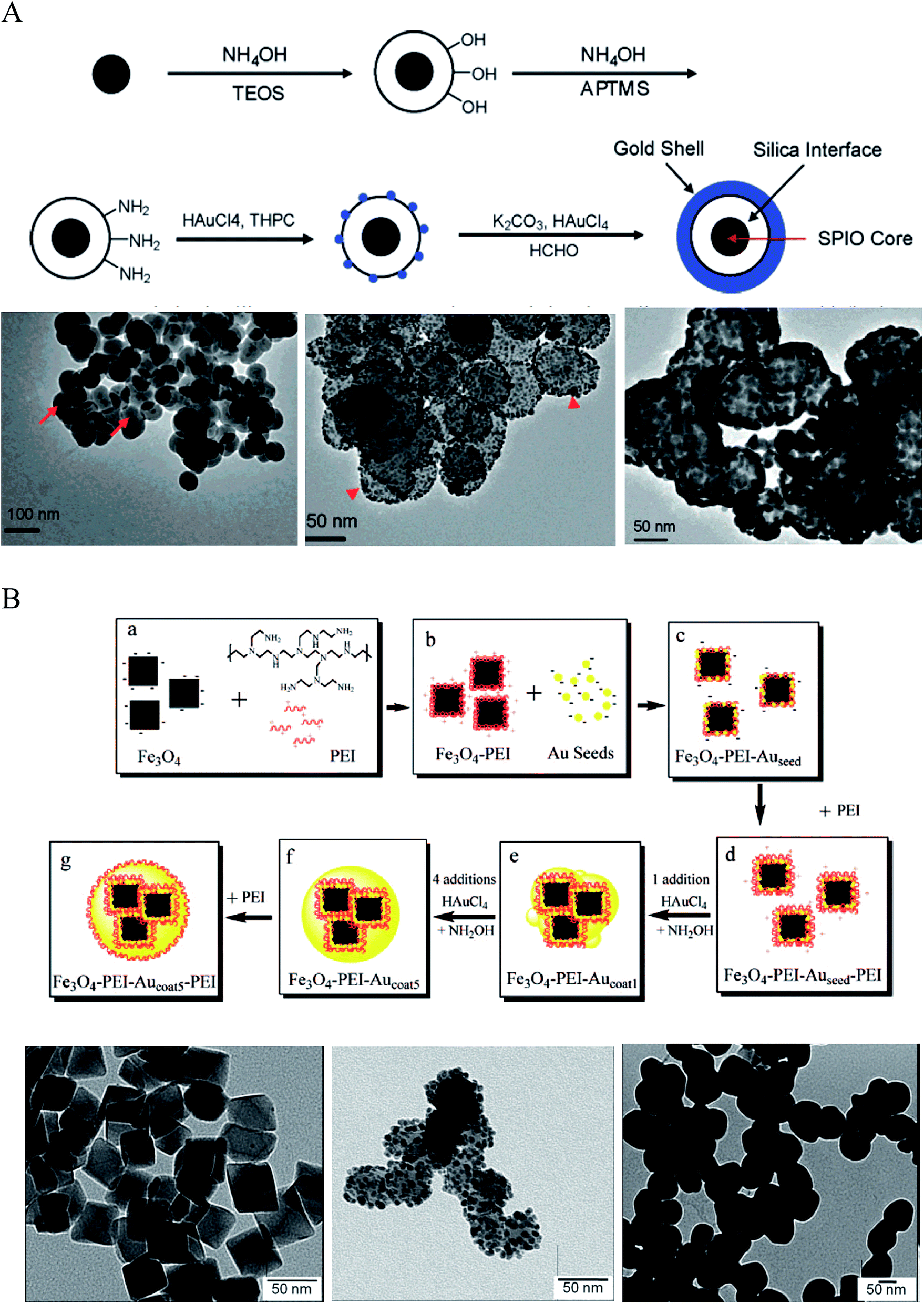 | ||
| Fig. 2 Synthesis of IO–Au core–shell NPs via Au-seeded growth methods. (A) APTMS and (B) PEI are used as the anchor agent for the adsorption of Au seeds on IO NPs. (A) is reprinted with permission from ref. 23. Copyright (2007) American Chemical Society. (B) is reprinted with permission from ref. 71. Copyright (2009) American Chemical Society. | ||
A more expeditious Au-seeded growth method is to use positively charged poly(allylamine hydrochloride) (PAH) to adsorb the Au seeds, an approach developed in 2008 by Wang et al.70 In this approach, Fe3O4 NPs of ∼260 nm in diameter were prepared through a solvothermal method using FeCl3 as the Fe precursor and sodium acetate as the alkali. A thin layer of polymer with carboxyl terminal groups was formed in situ by the copolymerization of acrylamide and methacrylic. The copolymer surface was coated with PAH via electrostatic interaction to introduce amine groups to attract the citrate-capped Au seeds. The gold nanoparticles used as seeds were prepared by reducing HAuCl4 with NaBH4 in the presence of sodium citrate. After adsorption of the citrate-capped Au seeds, the Au shell was formed by reducing HAuCl4 with NH2OH in the presence of the Au-seeded Fe3O4 NPs. Sequential reductive deposition steps led to uniform Au shell formation. These reduction reactions were performed under ultrasonication in an ice bath to prevent aggregation of the ferromagnetic Fe3O4 NPs.
Polyethyleneimine (PEI) is another positively charged polymer that is often used to attract negatively charged Au seeds.36,71,72 In 2009, Goon et al. developed an approach to directly make PEI-coated IO NPs and grew Au shells via repeated hydroxylamine reduction deposition on the PEI-coated IO NPs (Fig. 2B).71 IO NPs were prepared by precipitation of iron sulfate in a basic solution followed by oxidization with potassium nitrate at 90 °C in the presence of branched PEI (MW ∼ 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) in an oxygen-free environment. This method led to 50 nm cubic Fe3O4 NPs capped with PEI, allowing for the direct attachment of citrate-capped Au seeds in aqueous phase. 2 nm citrate-capped Au NPs were attached to the PEI-capped Fe3O4 NPs by mixing via stirring for 2 h. The surface coverage of PEI was critical to the loaded density of Au seeds, with a saturated amount of PEI at 0.88 μg PEI per cm2 on the particle surface leading to the highest Au-seed density (47.7 wt%). The Au-seeded Fe3O4 NPs were further coated with another layer of PEI by mixing the particles in PEI solution for 1 h at 60 °C. Thus, the Au seeds were sandwiched on the surface of IO NPs with two layers of PEI, ensuring a high density of Au seeds. The Au shells were then grown by iterative reduction of HAuCl4 onto the PEI-Au seeds-PEI-IO NPs using NH2OH as the reducing agent. A total of five iterations were performed to achieve a continuous Au shell. It is worthy to mention that the whole process was conducted in aqueous solution. In addition, the Au shell is free of capping agent, allowing for facile surface modification.
000) in an oxygen-free environment. This method led to 50 nm cubic Fe3O4 NPs capped with PEI, allowing for the direct attachment of citrate-capped Au seeds in aqueous phase. 2 nm citrate-capped Au NPs were attached to the PEI-capped Fe3O4 NPs by mixing via stirring for 2 h. The surface coverage of PEI was critical to the loaded density of Au seeds, with a saturated amount of PEI at 0.88 μg PEI per cm2 on the particle surface leading to the highest Au-seed density (47.7 wt%). The Au-seeded Fe3O4 NPs were further coated with another layer of PEI by mixing the particles in PEI solution for 1 h at 60 °C. Thus, the Au seeds were sandwiched on the surface of IO NPs with two layers of PEI, ensuring a high density of Au seeds. The Au shells were then grown by iterative reduction of HAuCl4 onto the PEI-Au seeds-PEI-IO NPs using NH2OH as the reducing agent. A total of five iterations were performed to achieve a continuous Au shell. It is worthy to mention that the whole process was conducted in aqueous solution. In addition, the Au shell is free of capping agent, allowing for facile surface modification.
Other surface chemistry has also been used to adsorb Au seeds onto IO NPs to grow Au shell.30,73–75 For example, Chin et al. modified Fe3O4 NPs with dopamine to introduce positively charged amine groups to attract citrate-capped Au NP seeds.73 Park et al. used 2 nm decanethiol-capped Au NPs prepared by the Brust and Schiffrin method as the Au seeding NPs.30 To attach the nonaqueous Au seeds to the IO NPs, they used a thermal processing treatment in which a mixture of DT-capped Au NPs, OA-capped Fe2O3 NPs and tetraoctylammonium bromide in toluene was heated at 149 °C for 1 h. During this process, the Au seeds first adsorbed onto the Fe3O4 NPs via hydrophobic interactions and then coalesced to form Au shells. To make the particles water soluble, the as-prepared Fe2O3–Au core–shell NPs were subjected to ligand exchange with mercaptoundecanoic acid.
2.2 Synthesis of anisotropic IO–Au core–shell NPs
Anisotropic NPs offer many advantages over spherical NPs due to well-known geometry-dependent LSPR properties. For example, the local field enhancement on anisotropic NPs can be orders of magnitude higher than spherical ones. For example, the E-field enhancement of Au tripod nanocrystals is 20 times higher than that of spherical NPs.76 A direct outcome of this field enhancement is the strong augmentation of the Raman signals of adsorbed molecules as the intensity of Raman signals is proportional to the fourth power of the local field of the metal particle.77 An enhancement factor on the order of 104 to 105 was observed for the Raman signals of adsorbed molecules on Au nanorods (NRs), while no such enhancement has been observed for the adsorbed molecules on Au nanospheres under similar condition.78 In addition, the anisotropic NPs can extend the optical properties from the visible to the NIR region without changing particle size.76 The NIR window is extremely important for biomedical applications as light in this region is tissue penetrative.79 Thus, synthesis of anisotropic NPs with IO cores is extremely attractive.A direct method to make anisotropic IO–Au core–shell NPs is to use anisotropic IO NPs as the core materials; the resulting core–shell NPs preserve the shape of the IO NPs. In 2006, Halas and co-workers reported the synthesis of IO–Au core–shell nanorice using an Au-seeded method (Fig. 3).80 In their studies, monodisperse hematite nanorice with aspect ratio of 6.3 (length = 340 nm. Diameter = 54 nm) were fabricated by heating FeCl3 and potassium dihydrogen phosphate at 100 °C for 72 h. The nanorices were functionalized with APTMS to generate the amine groups to adsorb THPC-capped Au seeds. The growth of the Au shell was performed by the reduction of HAuCl4 by formaldehyde in an aqueous solution containing the Au-seeded nanorice at room temperature. The growth of complete Au shells took only 5–10 min. Au shells from 10 to 30 nm were formed by adjusting the ratio of the hematite nanorice and HAuCl4. The hematite nanorice have the combined plasmonic properties of nanorods and nanoshells in the NIR region, making them promising for biomedical applications.
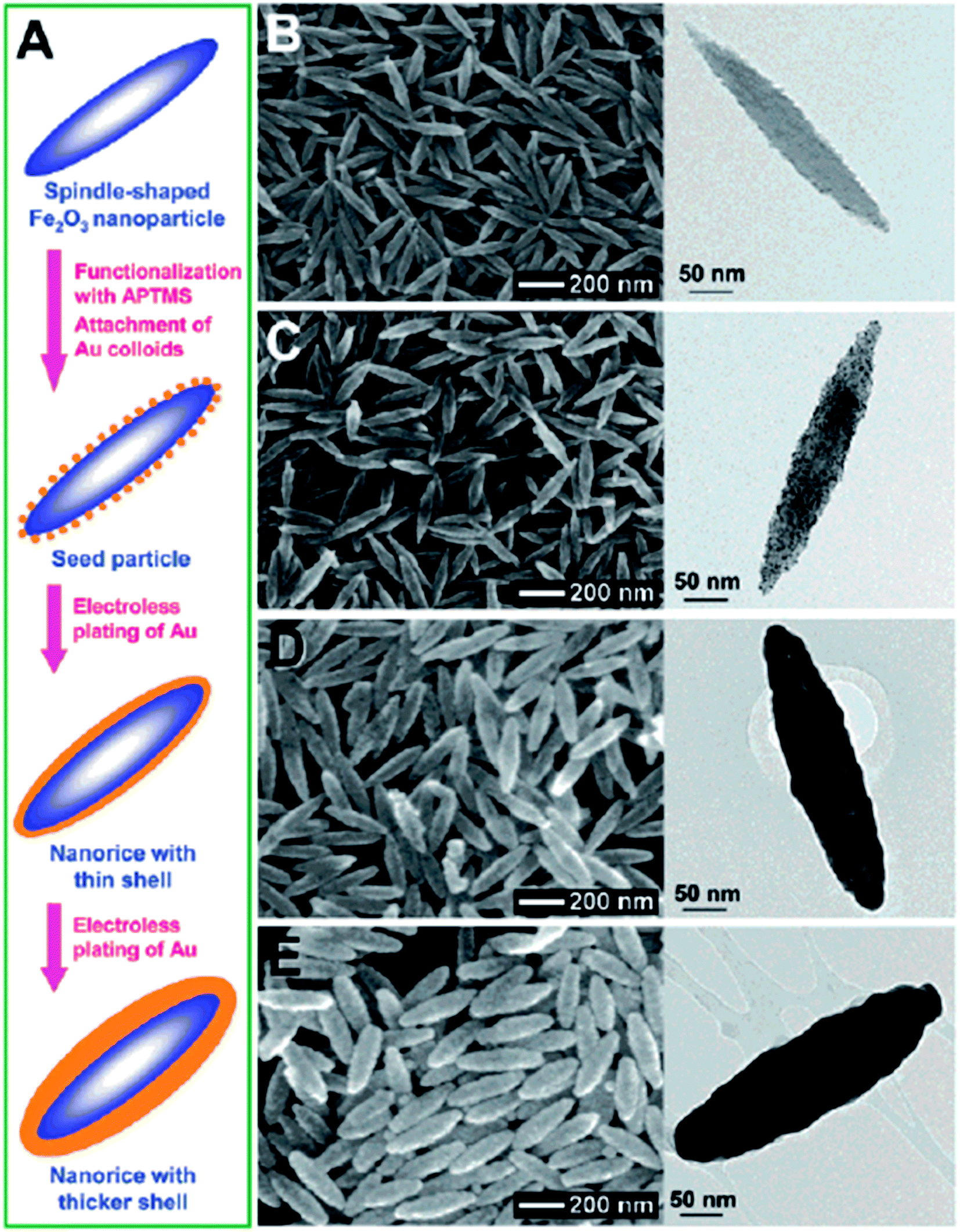 | ||
| Fig. 3 Synthesis of IO–Au core–shell nanorices via an Au-seeded growth method. (A) Schematic of the preparation of IO–Au core–shell nanorices. (B–E) SEM (left) and TEM (right) images of IO–Au core–shell nanorices at different preparation steps. (B) The IO nanorices. (C) Au-seeded IO nanorices. (D) IO–Au core–shell nanorices with thin shells (∼13 nm). (E) IO–Au core–shell nanorices with thick shells (∼28 nm). Reprinted with permission from ref. 80. Copyright (2006) American Chemical Society. | ||
Making anisotropic IO–Au core–shell NPs with anisotropic IO NPs is simple and can preserve the shape of the cores. However, when the size of the IO NPs is small, it is difficult to grow the Au shell while preserving the shape of the core. For example, the use of 60 nm tetracubic IO NPs as the cores resulted in spherical IO–Au core–shell NPs.69 An approach for preparing anisotropic IO–Au nanostars (NSTs) was developed by Wei and co-workers using spherical Fe3O4 NPs as the cores (Fig. 4A).81,82 Spherical Fe3O4–Au core–shell NPs of 8.4–13 nm were first prepared in organic phase at 190 °C by the reduction of gold chloride (AuCl3) by 1,2-hexadecanediol in octyl ether containing 8.4 nm OA- and OAM-caped IO NPs (8.4 nm). This shell was only ∼1.3 nm in thickness. The Fe3O4–Au core–shell NPs with thin Au-shells were introduced into a growth solution containing HAuCl4, CTAB, silver nitrate (AgNO3), and ascorbic acid (AA). This growth solution has been widely used to make anisotropic metallic NPs such as Au NRs due to the shape-directing CTAB surfactant and AgNO3 additive.83 AA is a surface-catalysed reducing agent, which prevents formation of solid Au NPs. Thus, anisotropic IO–Au NSTs were obtained. This is the first report of the synthesis of monodisperse IO–Au core–shell NSTs.
 | ||
| Fig. 4 Synthesis of IO–Au core–shell NSTs. (A) Synthesis of IO–Au core–shell NSTs from small IO–Au core–shell nanospheres. (Top) Schematic of the preparation procedure. (Bottom) TEM image of IO NPs (left), HRTEM image of a tip (middle), and TEM image of IO–Au NSTs. Reprinted with permission from ref. 82. Copyright (2010) American Chemical Society. (B) Synthesis of IO–Au core–shell NSTs by the Au-seeded growth method. (Top) Schematic of the preparation procedure. (Bottom) TEM images of IO NPs (left), Au-seeded IO NPs (middle) and IO–Au NSTs. Reprinted with permission from ref. 72. Copyright (2016) American Chemical Society. | ||
We recently developed a facile method for the synthesis of uniform IO–Au core–shell NSTs (Fig. 4B)72 with all steps performed in aqueous solutions. In our studies, octahedral Fe3O4 NPs capped with PEI (edge length ∼ 35 nm) were synthesized by precipitating ferrous sulfate in a base solution followed by oxidation with KNO3 at 90 °C in the presence of branched PEI (MW ∼ 25000). Citrate-capped Au NPs (<10 nm) were attached to the IO NPs by electrostatic adsorption and the Au-seeded Fe3O4 NPs were further stabilized with PEI. The growth of Au shells was initiated by injecting the Au-seeded IO NPs into a growth solution containing HAuCl2, CTAB, AgNO3 and AA. By adjusting the amount of Au-seeded Fe3O4 NPs, IO–Au core–shell NSTs with base sizes from 70 to 150 nm were obtained. In addition to forming NSTs, this method can be easily adapted to make nanospheres and nanopopcorns by changing the chemical composition of the growth solution. When AgNO3 was removed from the growth solution, IO–Au core–shell nanospheres were formed. When the concentration of AA was tripled, nanopopcorns were obtained. The sizes of the nanospheres and nanopopcorns can also be tuned without affecting particle quality by simply varying the amount of Au-seeded IO NPs in the growth solution. A major advantage of this method is the capability to tune both the size and the shape of the core–shell particles. Later, Shi and co-workers reported another approach to make IO–Au NSTs using Ag-seeded IO NPs.25 In their study, small Ag NPs were synthesized for the seeds via the reduction of AgNO3 by NaBH4 in a partially thiolated PEI aqueous solution. The PEI-stabilized Ag NPs were added into a mixture of FeCl2 and ammonium hydroxide and autoclaved in a sealed pressure vessel at 134 °C for 3 h to obtain Ag-seeded Fe3O4 NPs. This hydrothermal process led to the homogenous coating of Au NPs on the Fe3O4 NPs. Addition of the Ag-seeded Fe3O4 NPs into a growth solution containing HAuCl4, CTAB, AgNO3 and AA resulted in the formation of Fe3O4–Au core–shell NSTs within 1 h.
Using small Ag NPs (2–5 nm) as the seeds and Fe3O4 NPs coated with poly (maleic anhydride-alt-1-octadecene) as the core, we have produced IO–Au core–shell NPs in oval and pin shapes.37,84 The polymer interacts with OA on IO NPs with the hydrophobic alkyl chain while exposing the hydrophilic carboxylate ions to anchor positively charged metal ions. Ag seeds were used as the nucleation sites because the Ag-seeded IO NPs can be formed without aggregation by reduction of the purified Ag(NH3)2+ adsorbed IO NPs. In this way, the formation of un-adsorbed Ag NPs that would lead to solid Au NPs is avoided. Ag has a nearly identical crystal structure to Au, with almost 100% lattice matching. Galvanic replacement between Au ions and Ag seeds during subsequent growth process is prevented by externally introducing a stronger agent, such as ascorbic acid (AA), that is stronger than Ag (EodehydroAA/AA = 0.06 V versus 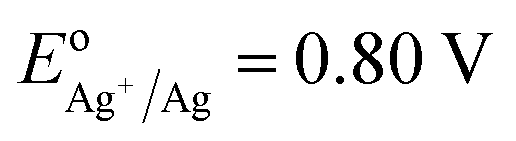 ). Thus, Au can be uniformly deposited onto the Ag nucleation sites to form Au shell.
). Thus, Au can be uniformly deposited onto the Ag nucleation sites to form Au shell.
2.3 Growth mechanism
A common model to understand the growth mechanism of anisotropic solid Au NPs is that the Au seeds are faceted NPs. The formation of various shapes is the outcome of the interplay between the facet binding tendency of the stabilizing agents and the growth kinetics.85 As for the core–shell NPs, Halas and co-workers have studied the growth mechanism of spherical Au nanoshell on silica core by monitoring the progression of Au nanoshell formation with TEM imaging.66 Their studies showed that at the early stages the adsorbed Au seeds gradually grew with time on the silica core, then coalesced on the core surface until finally forming a continuous metallic shell. In the studies of IO–Au NSTs prepared from spherical IO–Au NPs, Wei and co-workers used high resolution TEM (HRTEM) and found multiple twinning defects and overall growth along the (111) directions. As the mismatching of crystal lattices of Au and IO are within 3%, it is believed that the formation of the shell is due to favorable epitaxial growth.We recently investigated the growth mechanisms for shape-controlled synthesis by studying the growth processes of IO–Au nanospheres, popcorn and stars using both experimental and computational and methods (Fig. 5).72 By following the growth process with TEM, it was found that the IO–Au core–shell nanospheres grew at a faster rate than the popcorns and stars. For spheres, the Au seeds coalesced within 5 min and formed continuous Au shells within 20 min. For the popcorns and stars, no visible changes on the Au seeds were observed after 5 min. The Au seeds coalesced at 20 min, with continuous Au shell formation occurring after 30 min. Differences between popcorns and stars were found at 30 min, with the popcorns exhibiting short and wide protrusions and the stars thin spikes.
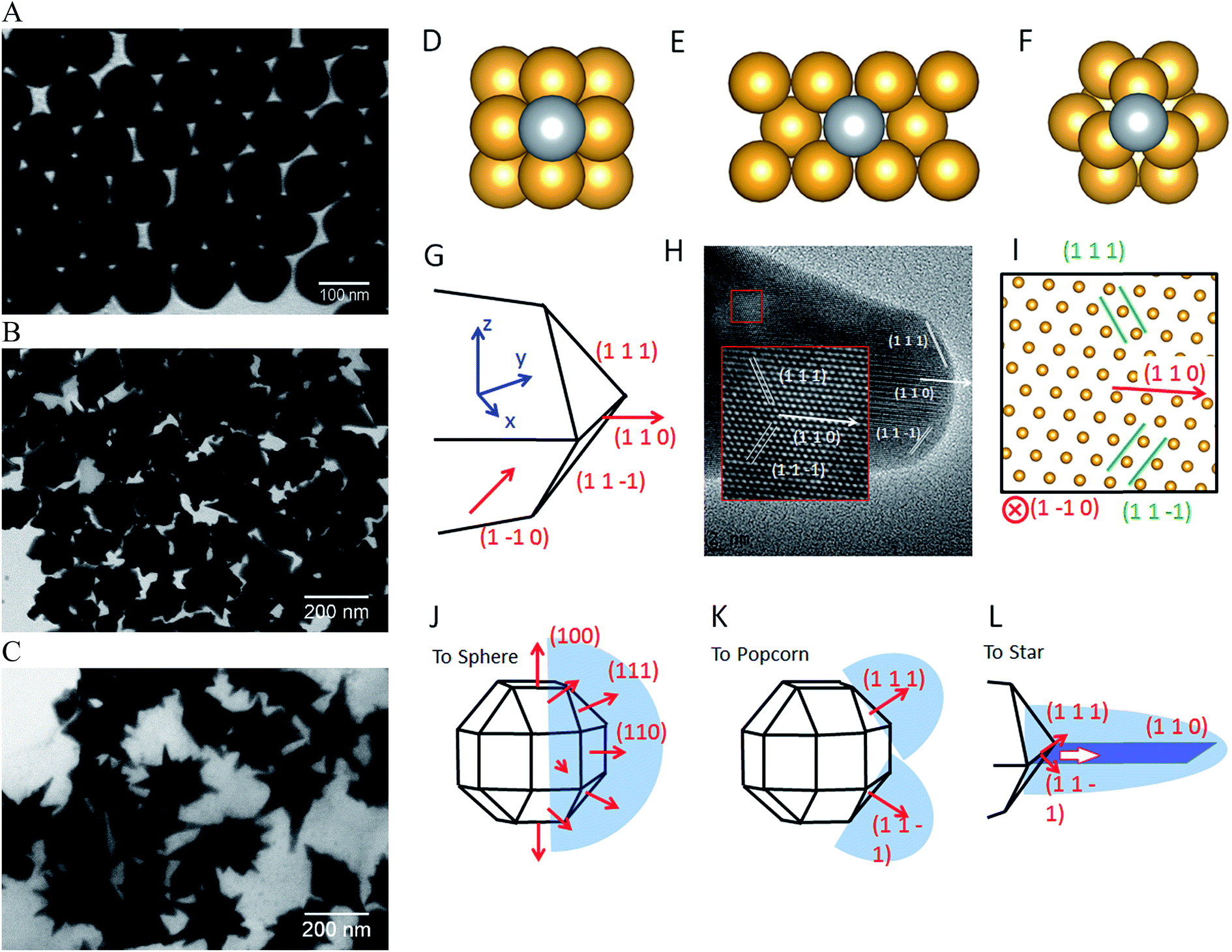 | ||
| Fig. 5 Computational studies on growth mechanisms of IO–Au core–shell nanospheres, nanopopcorns, and nanostars. (A–C) TEM images of IO–Au core–shell nanospheres, nanopopcorns, and nanostars. (D–F) The adsorption of Ag on the Au (100) surface (D), Au (110) surface (E), and Au (111) surface (F). (G) Schematic of the proposed geometry of the tip of the nanostar. (H) HRTEM image of a tip of the nanostar. The inset shows an enlarged view of the lattice, where the position of each atom is visible. (I) The positions of atoms according to the proposed model in (G), which is viewed from the [1–10] direction. (J–L) The growth mechanism of Au nanospheres (J), nanopopcorn (K), and nanostars (L). Reprinted with permission from ref. 72. Copyright (2016) American Chemical Society. | ||
The growth of the Au-seeded IO NPs into anisotropic was due to the effects by AgNO3. AgNO3 has been widely used to assist the preparation of Au NRs and other anisotropic metal NPs.85–87 It has also been used to form spiky Au nanoshells from an Ag-seeded polymer template.88,89 A previous model to explain the role of Ag+ is silver underpotential deposition, the reduction of Ag+ to Ag0 on a metal substrate with a surface potential less than the standard reduction potential.90 Based on this model, Ag+ is reduced and deposited onto the surfaces of Au seeds in the presence of a reducing agent. This may explain the slower growth rate for Au NPCs and NSTs compared with the nanospheres. Using the first principles density functional theory method, we have determined that the adsorption energies (the energy gain from adsorption) of Ag on (110), (100), and (111) are 3.0, 2.7, and 2.2 eV per atom, respectively. Thus, Ag deposition is correspondingly faster on the (110) facet, followed by the (100) and (111) surfaces. With sufficient Ag+ in the solution, Ag covers the (110) and (100) surfaces of Au seeds, blocking the adsorption of Au on these faces. This blocking effect leaves only the (111) surface for Au growth, which leads to the anisotropic growth of the Au.
The branched growth of the IO–Au core–shell NPs into popcorns or stars is a result of kinetic control on the reductive deposition of Au by adjusting the concentration of AA. When the reduction of Au+ to Au0 is fast (higher AA concentration), Au atoms are deposited onto the entire (111) facet, leading to the growth of the Au island along the (111) direction. This mechanism is confirmed by the HRTEM image of the tip of the popcorn, which shows that the tip of popcorn grows along the {111} direction. When the reduction of Au+ to Au0 is slow, Au atoms are mainly deposited at the ridge formed by two {111} planes. This is because the Au atoms at the ridge are more under coordinated than that at the facet center, which makes new Au atoms bind the adsorbed Au atoms more strongly. Also, the electrical field at the ridge is stronger than that at the center of the facet, making the reduction of Au+ faster at the ridge. This cohesive growth of Au atoms from two planes leads to the formation of a twin boundary along the (110) direction. This model was supported by the HRTEM image of the star tip, which shows the tip is made of two of {111} planes growing towards the (110) direction.
3. Properties
3.1 Magnetic properties of IO–Au core–shell NPs
Due to the unpaired electrons in the 3d shell of Fe2+ and Fe3+, nanocrystals formed from Fe2+ and Fe3+ can be in ferromagnetic, ferrimagnetic and antiferromagnetic states. In ferromagnetic materials, the magnetic moments of two sublattices align parallel to each other even without an external field. In ferrimagnetic materials, the magnetic moments align antiparallel, but do not cancel each other out. This is different from the antiferromagnetic materials where the magnetic moments of two sublattices are equal and align antiparallel. Thus, there is no net magnetic moment in zero magnetic field for antiferromagnetic materials. The magnetic moments lose ordering beyond a specific temperature called the Curie temperature TC for ferromagnets and ferrimagnets and the Neel temperature TN for antiferromagnets. Magnetite Fe3O4 is a well-known ferrimagnetic material with a TC of 858 K.91 Maghemite γ-Fe2O3 is ferrimagnetic at room temperature. They are unstable at high temperatures and thus TC is hard to determine. Both Fe3O4 and γ-Fe2O3 NPs are superparamagnetic at room temperature when their sizes are sufficiently small (less than 30 nm).20,92–94 For the superparamagnetic NPs, the magnetization randomly flips directions due to thermal fluctuations and shows zero net magnetization at zero magnetic field. The particles behave like paramagnetic material under an external field but their magnetic susceptibility is much larger than that of paramagnets.The magnetic properties of IO NPs depend on the surface. When the size of the NPs decreases, the magnetic properties decrease due to increased surface effects. Passivation with organic and inorganic layer helps decrease the surface effect, but it could also adversely affect the surface magnetic moment of IO NPs. The surface magnetic moments of IO NPs can be disordered via the interaction with Au electrons.95 The IO–Au core–shell NPs could have larger surface effect than IO NPs by structural distortions that cause spin canting.63,96 This leads to decreased magnetic properties of the IO–Au core–shell NPs compared to the IO NPs. Magnetic properties are commonly measured by a superconducting quantum interference device by scaling to the total mass of the materials. Au has 4 times higher density than IO. However, Au is diamagnetic in bulk states. It is ferromagnetic when the size very small (4–5 nm).97 Therefore, IO–Au core–shell NPs generally give significantly lower saturation magnetization (Ms) than IO NPs due to the mass contribution from the diamagnetic Au shell. For example, coating the tetracubic IO NPs (∼60 nm) with a 10.9 nm Au shell decreased the Ms from 20 to 0.6 emu g−1 at both 100 K and 250 K (Fig. 6A and B).69 Coating 258 nm Fe3O4 NPs with a 7.5 nm Au shell decreased the Ms from 75.6 to 61.0 emu g−1.70
 | ||
| Fig. 6 Magnetic properties of IO–Au core–shell NPs. (A and B) Magnetization as a function of applied field at 100 K and 250 K for uncoated and Au coated tetracubic IO NPs. Reprinted with permission from ref. 69. Copyright (2009) American Chemical Society. (C) Magnetization as a function of applied field at 10 K for IO–Au core–shell nanopopcorns of three different sizes. Reprinted with permission from ref. 72. Copyright (2016) American Chemical Society. (D) Magnetization as a function of applied field at 10 K and 300 K for bare IO NPs (sample A), IO–Au core–shell NPs with low Au (sample B) and high Au (sample C). Reprinted with permission from ref. 46. Copyright (2011) American Chemical Society. | ||
The IO–Au core–shell NPs typically exhibit the same magnetic behaviour as the IO core. Increasing Au shell would lead to a decrease in the magnetic properties of the core–shell NPs due to the mass contribution of the diamagnetic Au. However, in our recent studies, we found that increasing the Au shell thickness led to an increase of the magnetic properties of IO–Au core–shell nanopopcorns at 10 K (Fig. 6C).72 The particles are capped with CTAB that binds to Au via Br− and is counterbalanced by CTA+ in a bilayer structure.98–100 Thus, charge transfer between Br− and Au may occur, which would enhance the electron mobility and, thus, the surface induced magnetism of Au. Similarly, studies by Crespo et al. showed that the charge transfer between a thiol ligand and an Au surface was a major reason for the observed ferromagnetic properties of thiolated Au NPs.97 It has also been recognized that the magnetic material could spin polarize the Au conduction electrons at the IO–Au interface and thus result in magnetization of the surrounding nonmagnetic Au.101 In addition, a large orbital magnetic moment could be induced at the Fe3O4–Au interface. This interface effect leads to the magnetization enhancement of Fe3O4 NPs by a factor of six (Fig. 6D).46
Research has shown that there is no significant difference in the coercivity between coated and uncoated IO NPs.69 However, enhanced coercivity has been reported by Pal et al. on small Fe3O4 NPs (6 nm) with ultrathin shell (1 nm thickness).63 The Au-coated Fe3O4 NPs gave a coercivity Hc of 200 Oe while the uncoated Fe3O4 NPs had Hc of 160 Oe at 5 K. The coercivity enhancement is possibly due to the role of spin disorder at the Fe3O4–Au interface and weak exchange coupling between surface and core spins.
Separation of IO–Au core–shell NPs from suspension usually takes much longer than uncoated IO NPs because of the massive amount of Au added to the IO NPs by the Au shell. This can be theoretically understood by calculating the particle terminal velocity under external magnetic field. The particle terminal velocity V is equal to 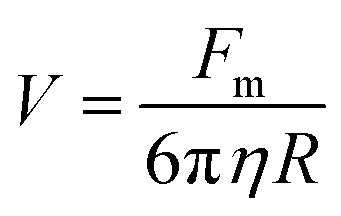 where Fm is the magnetic force exerted onto the particle by the external magnetic field, η is the viscosity of the solution, and R is the hydrodynamic radius of the particle. The Fm of the IO–Au core–shell particle is the same as the IO particle if Au coating does not affect the magnetic property of the IO core. Thus, the particle terminal velocity V is inversely proportional to the hydrodynamic radius R of the particle. The particle also takes time to reach the terminal velocity. The time, called relaxation time τ is equal to
where Fm is the magnetic force exerted onto the particle by the external magnetic field, η is the viscosity of the solution, and R is the hydrodynamic radius of the particle. The Fm of the IO–Au core–shell particle is the same as the IO particle if Au coating does not affect the magnetic property of the IO core. Thus, the particle terminal velocity V is inversely proportional to the hydrodynamic radius R of the particle. The particle also takes time to reach the terminal velocity. The time, called relaxation time τ is equal to 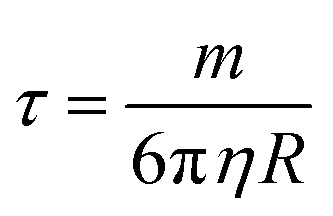 where m is the mass the particle. This means that τ is linearly proportional to the mass of the particle and is inversely proportional to the hydrodynamic radius of the particle. Taking into account of both V and τ from the above two equations, the time taken for a particle to be separated by an external magnetic field is therefore proportional to the mass of the NP. The mass of IO and IO–Au core–shell NP can be calculated based on their sizes measured by TEM and the densities of IO (5.24 g cm−3) and Au (19.32 g cm−3). If an IO NP is 20 nm in diameter and the Au shell is 10 nm, the mass of IO–Au core–shell NPs is 27 times larger than the IO NP. This means that the separation time for a core–shell particle is roughly 27 times longer than that of the IO NP traveling the same distance. Thus, if an IO NP takes 5 min to be separated from the solution, the separation of an IO–Au core–shell NP requires more than 2 h due to the mass added by the diamagnetic Au shell.
where m is the mass the particle. This means that τ is linearly proportional to the mass of the particle and is inversely proportional to the hydrodynamic radius of the particle. Taking into account of both V and τ from the above two equations, the time taken for a particle to be separated by an external magnetic field is therefore proportional to the mass of the NP. The mass of IO and IO–Au core–shell NP can be calculated based on their sizes measured by TEM and the densities of IO (5.24 g cm−3) and Au (19.32 g cm−3). If an IO NP is 20 nm in diameter and the Au shell is 10 nm, the mass of IO–Au core–shell NPs is 27 times larger than the IO NP. This means that the separation time for a core–shell particle is roughly 27 times longer than that of the IO NP traveling the same distance. Thus, if an IO NP takes 5 min to be separated from the solution, the separation of an IO–Au core–shell NP requires more than 2 h due to the mass added by the diamagnetic Au shell.
3.2 Optical properties of IO–Au core–shell NPs
The optical properties of core–shell plasmonic NPs have been a topic of great interest in materials science for many years. Their properties are dependent on the dielectric functions of the core and shell materials, the core size and shape, as well as the shell thickness and geometry. An intensely studied core–shell NP is the Au nanoshell with a silica core pioneered by Halas and co-workers.102 The Si–Au core–shell NPs exhibit strong NIR properties, with increasing thickness of the Au shell leading to an LSPR blue shift.66 They proposed a hybridization model to understand the plasmons of concentric nanoshells, where the hybridization of the plasmons of inner and outer nanoshells determines the LSPR of the Si–Au core–shell NPs.103Using Mie theory, we recently calculated the optical properties of spherical Fe3O4–Au core–shell NPs, and compared them with Si–Au, Co–Au, hollow Au, and solid Au NPs (Fig. 7A).104 Compared with solid Au NPs of the same size, the LSPR peak of IO–Au core–shell NPs is further red shifted than those of silica and hollow NPs. The LSPR peak intensity is comparable to that of solid Au NPs, but much lower than those of the hollow Au and Si–Au core–shell NPs. In contrast, the Co–Au core–shell NPs have very weak plasmon peaks, although cobalt NPs have stronger magnetic properties than magnetite NPs. These optical differences can be traced to the differences in the dielectric properties of the core material, which is related to the refractive index of the material. IO NPs are nontransparent and have complex refractive indices. Silica, on the other hand, is a non-absorbing material and has only a real refractive index. While the LSPR peak wavelength is determined by real component of the refractive index of the material, the peak intensity is determined by the imaginary component of the refractive index. Increasing the real refractive index leads to a red shift of the LSPR peak whereas increasing the imaginary refractive index leads to a reduction of the peak intensity (Fig. 7B). Similar to Si–Au core–shell NPs, the LSPR wavelength blue shifts with the increase of Au shell thickness when the total diameter of the NPs is fixed (Fig. 7C and D). It follows the same universal scaling as the Si–Au core–shell NPs that has been reported by Jain and El-Sayed.105 The shift in LSPR wavelength is determined by the ratio of the thickness of the shell to the radius of the core (t/R). Compared to the Si–Au system, the Fe3O4–Au NPs show a larger decay constant in the plasmon shift versus the ratio of shell thickness. This is consistent with LSPR peaks that are more red-shifted than those of Si–Au NPs having identical shell thicknesses and core radii. In typical IO–Au NP synthesis, the IO-core diameter remains constant; as the Au shell thickness increases, the total particle size increases as well. In this case, the LSPR peak is seen to blue shift first with increasing the thickness of the Au shell (Fig. 7E and F). However, as the thickness of the Au shell continues to grow, a red shift of the LSPR peak occurs. In this case, the peak shift seems do not adhere to the universal scaling equation. In fact, the universal scaling still applies. When the Au NPs with the same diameter of each core–shell NP are used as the references, the data follows the universal scaling nicely.
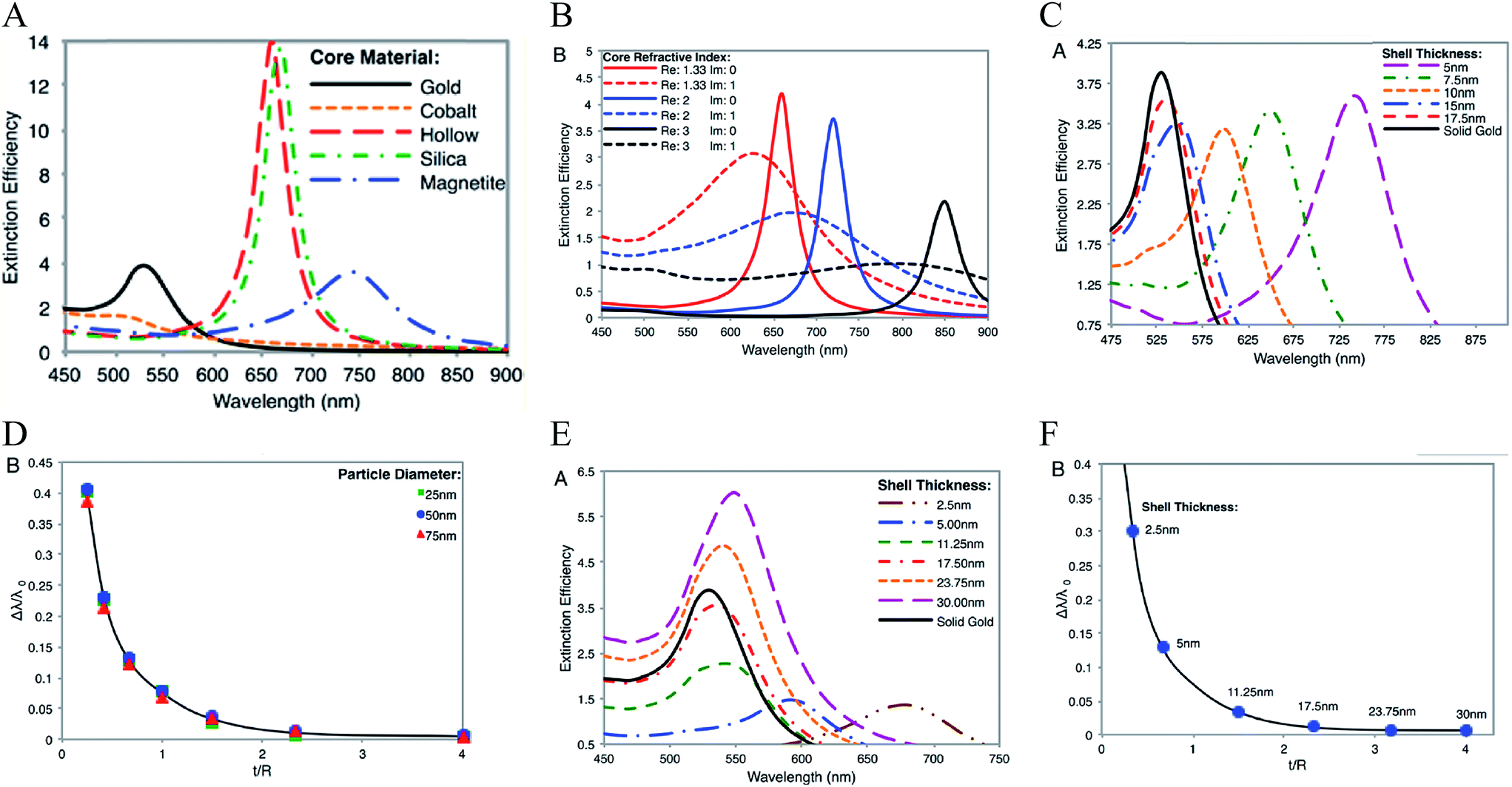 | ||
| Fig. 7 Calculated optical properties of Fe3O4–Au core–shell NPs. (A) Extinction spectrum of Fe3O4–Au core–shell NPs in comparison with other core–shell NPs and solid Au NPs with the same diameter Dtotal = 50 nm of NPs and a shell thickness of 5 nm. (B) Extinction spectra of core–shell NPs with varied core refractive indices. (C) Extinction spectra of Fe3O4–Au core–shell NPs with different shell thickness with fixed total particle size. Dtotal = 50 nm. (D) Fractional shifts (Δλ/λ0) of the LSPR peak maximums of the IO–Au NPs from (C). (E) Extinction spectra of Fe3O4–Au core–shell NPs with different shell thickness with fixed IO core size. Dcore = 15 nm. (F) Fractional shifts (Δλ/λ0) of the LSPR peak maximums of the IO–Au NPs from (E). Reprinted with permission from ref. 104. Copyright (2014) American Chemical Society. | ||
Theoretically, a spherical IO–Au core–shell NP with a 20 nm core and a 15 nm shell has a LSPR peak at 540 nm.104 When the core is increased to 35 nm and Au shell thickness is decreased to 7.5 nm, the LSPR is red shifted to 650 nm. Most existing IO NPs are 30 nm and less. Thus, they have LSPR in the UV-Vis region after Au coating. To make particles with LSPR over 800 nm, the core needs to be larger than 100 nm assuming shell thickness t is equal to or larger than 5 nm.105 NIR-absorbing IO–Au NPs less than 50 nm have been reported by Gao and co-workers (Fig. 8A).44 These particles have a core and shell separated by a few nanometer polymer gap. Although the core is only 25 nm in diameter, the particles absorb over 700 nm when the Au shell is 2–3 nm. When the shell is decreased to 1–2 nm, the particles show a broad absorption band with the plasmon resonance approximately 850 nm. This is consistent with our computational studies that demonstrated a polymer gap induce plasmon red shift.
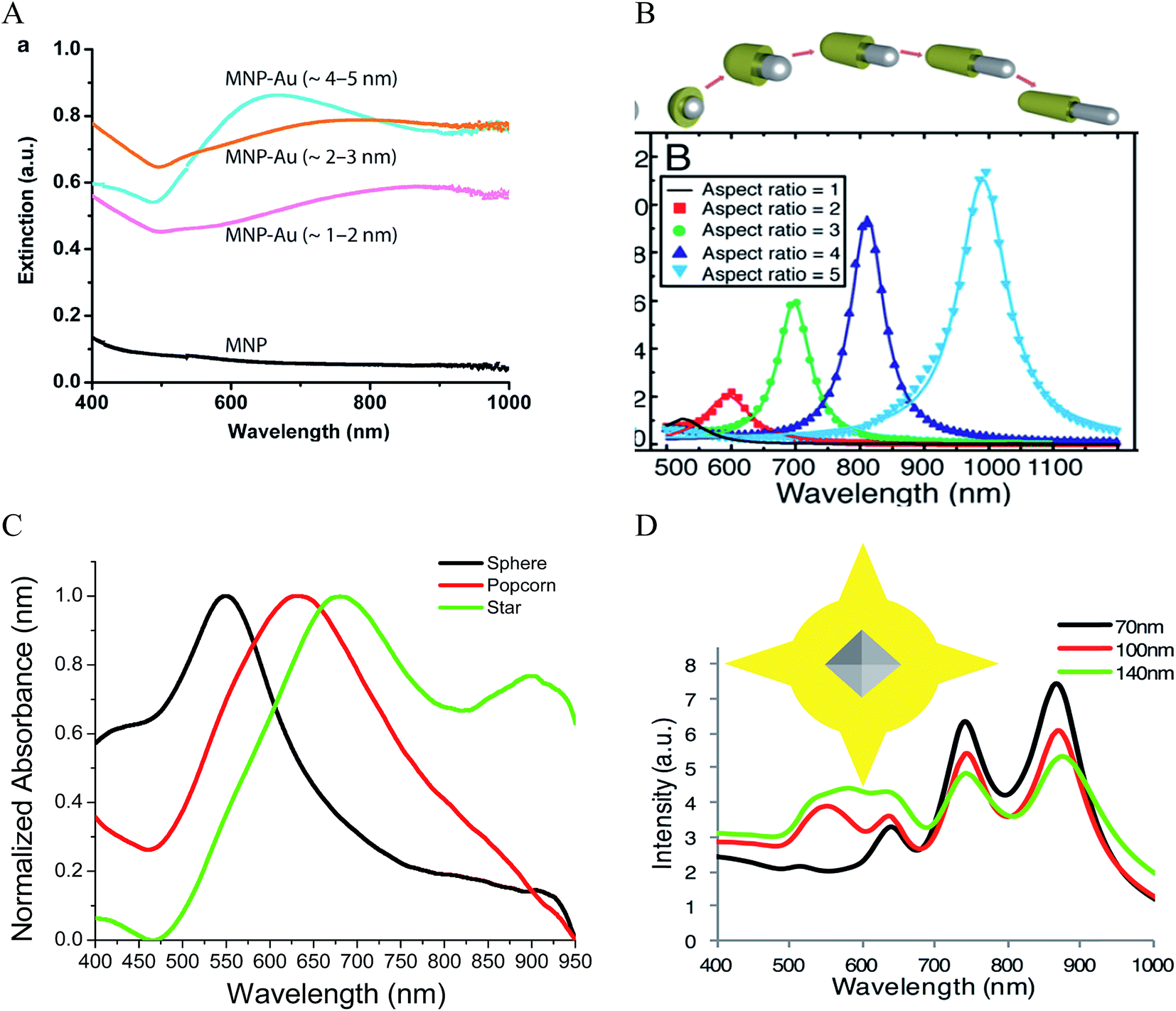 | ||
| Fig. 8 NIR absorbing IO–Au core–shell NPs. (A) Absorption spectra of Fe3O4–Au core–shell NPs with different shell thickness. Dcore = 25 nm. Reprinted by permission from Macmillan Publishers Ltd: [Nature Communications] (ref. 44), copyright (2010). (B) Calculated extinction spectra of IO–Au core–shell NRs with different aspect ratios. Reprinted with permission from ref. 106. Copyright (2011) Elsevier. (C) Absorption spectra of Fe3O4–Au core–shell NPs with different shapes. The Fe3O4 core is octahedral with an edge length of 35 nm. Reprinted with permission from ref. 72. Copyright (2016) American Chemical Society. (D) Calculated extinction spectra of IO–Au core–shell NSTs with different base size. The Fe3O4 core is octahedral with an edge length of 35 nm. Reprinted with permission from ref. 72. Copyright (2016) American Chemical Society. | ||
A common way to tune the LSPR of plasmonic NPs is to change the shape. For example, when an Au NP is changed from sphere to rod, the LSPR is split into two peaks.14 One is around 520 nm (transverse band) resulting from the electron oscillations along the short axis of the rod. The other one (longitudinal band) is at a longer wavelength with much stronger intensity due to the electron oscillations along the long axis. The LSPR of this longitudinal band is very sensitive the particle's aspect ratio (length/width). Increasing the aspect ratio causes large shift of the LSPR wavelength into NIR region. IO–Au core–shell nanorods have not been experimentally produced. Theoretically, Brullot et al. calculated the optical properties of IO–Au core–shell NPs with different aspect ratios using discrete dipole approximation (DDA) (Fig. 8B).106 For an IO–Au NR with fixed size (Reffective = 10 nm) and core to total volume (Vcore/Vtotal = 0.2) the longitudinal LSPR shifts from 520 to 1000 nm when the aspect ratio increases from 1 to 5, accompanied by over 20-fold increase in the extinction efficiency. Core–shell nanorice have been reported by Halas and co-workers.80 The nanorice (core length = 340 nm. Core diameter = 54 nm) have a weak transverse resonance at a wavelength lower than 800 nm and a strong longitudinal resonance over 1000 nm. These plasmon resonances are the result of the hybridization of the parent spheroid and cavity plasmon resonances corresponding to the aspect ratio of the particle. When the thickness of Au shell is increased from 9.8 to 27.5 nm, the longitudinal resonance blueshifts from 1300 nm to 1100 nm.
We recently investigated the optical properties of IO–Au core–shell nanopopcorns and nanostars with both experimental and computation studies.72 The nanopopcorns have LSPR from 600 to 700 nm depending on the size. When the base size (inner spherical IO–Au core) is increased from 65 to 89 and 145 nm, the LSPR redshifts from 600 nm to 633 and 665 nm, respectively. Compared with the IO–Au core–shell nanospheres, the LSPR peaks of the nanopopcorns shift to longer wavelengths and the bands become broader, owing to the anisotropic Au shell of the nanopopcorns (Fig. 8C). The NSTs show multispectral feature, with distinct peaks from 500 nm to 1000 nm depending on the size. They exhibit most red-shifted plasmon resonance compared to the spheres and popcorns due to the elongated tip structure of the stars. DDA calculation using a six-tip star shows that the NSTs are characterized by four distinct peaks, two peaks with LSPR lower than 700 nm and two peaks over 700 nm (Fig. 8D). The core–shell NSTs show similar features to the solid Au NSTs investigated by Nordlander and co-workers.107 These multiple plasmon resonances result from the hybridization of the core and tip plasmons. The most blueshifted peak less than 600 nm could be mainly due to the plasmon resonance of the IO–Au core–shell nanosphere core. The peaks corresponding to additional LSPR modes at longer wavelengths are likely the result of plasmonic coupling of the core and tips. Increasing the base sphere size of the nanostars leads to an increase in the intensities of the first two peaks within 700 nm, but a reduction in the intensities of the two peaks at longer wavelength. No significant plasmon resonance shifts are observed. It should be noted that the number and length of the Au tips are also major players in the optical properties of the nanostars. Further computational studies are needed to better understand the relationship between the optical properties of the core–shell nanostars and the core size and shape, tip length and width, as well as the number of the tips.
4. Concluding remarks and future perspectives
Due to the combined optical and magnetic properties, IO–Au core–shell NPs are of considerable interest in many fields, ranging from materials science to biology and medicine. Synthesis of uniform IO–Au core–shell NPs with desirable magnetic and optical properties are of extreme importance for their technological applications. During the past decade, the majority of studies have been focused on the preparation of spherical core–shell NPs. Two classes of approaches have been used: direct reductive deposition of Au onto IO cores and Au-seeded growth methods. The Au shell is formed either by epitaxial growth or coalescence of Au seeds on the surface of Au NPs. Anisotropic IO–Au core–shell NPs in several shapes have been achieved either using anisotropic IO cores or by controlling the kinetic and dynamic parameters in the growth solutions. IO–Au core–shell NPs generally exhibit the same magnetic behavior as the cores with reduced saturation magnetization due to the mass contribution of the diamagnetic Au. They show optical properties from the visible to the NIR region depending on the core size, shell thickness, and shape. IO–Au core–shell nanostars exhibit multiple plasmon resonances due to the coupling of the core and tip plasmons.Despite the great progress in synthesis methods, the preparation of high quality anisotropic IO–Au core–shell NPs remains a major challenge. This is due to the difficulties in controlling the Au shell geometry. For example, IO–Au core–shell nanostars have been reported by several groups. But the samples are mixtures of stars with different numbers and dimensions of tips. Compared with solid Au NPs, the shape control for IO–Au core–shell NPs is also limited. New shapes with fine tuning of the optical properties will be sought-after in the near future. In addition, new nanostructures should have ultrathin Au shells in order to preserve the advantageous magnetic properties of the core. The effect of the Au shell on the magnetic properties of the IO core at the interface is also much less understood. IO–Au core–shell NPs show similar optical trends to the prototype Si–Au core–shell NPs with their structural variations. But more studies, especially computational studies need to be performed to understand the shape-dependent optical properties. The computational studies can have precise structural controlling on the size and shape of the nanoparticles. Calculated results will greatly help us understand the structure–function relationship as well as the origins of the plasmon peaks.
Acronyms
| AA | Ascorbic acid |
| Ag | Silver |
| AgNO3 | Silver nitrate |
| APTMS | 3-Aminopropyltrimethoxysilane |
| Au | Gold |
| AuCl3 | Gold chloride |
| Co | Cobalt |
| CTAB | Cetyltrimethylammonium bromide |
| DDA | Discrete dipole approximation |
| EDTA | Ethylenediaminetetraacetic acid |
| Fe | Iron |
| Fe3O4 | Magnetite |
| FeCl2 | Ferrous chloride |
| FeCl3 | Ferric chloride |
| γ-Fe2O3 | Maghemite |
| HAuCl4 | Chloroauric acid |
| HRTEM | High resolution transmission electron microscopy |
| IO | Iron oxide |
| IO–Au | Iron oxide–gold |
| LSPR | Localized surface plasmon resonance |
| MNPs | Magnetic nanoparticles |
| MRI | Magnetic resonance imaging |
| Ms | Saturation magnetization |
| NaBH4 | Sodium borohydride |
| NPs | Nanoparticles |
| NH2OH | Hydroxylamine |
| NIR | Near infrared region |
| NR | Nanorod |
| NST | Nanostar |
| OA | Oleic acid |
| OAM | Oleylamine |
| PAH | Poly(allylamine hydrochloride) |
| PEI | Polyethyleneimine |
| PVP | Poly(vinylpyrrolidone) |
| Si | Silica |
| TEM | Transmission electron microscopy |
| THPC | Tetrakis(hydroxymethyl) phosphonium chloride |
Acknowledgements
We gratefully acknowledge the support from the National Institutes of Health (Grant No. 1R15 CA 195509-01).References
- M. Kerker, The scattering of light and other electromagnetic radiation, Academic Press, New York, 1969 Search PubMed.
- G. C. Papavassiliou, Prog. Solid State Chem., 1980, 12, 185 CrossRef.
- C. F. Bohren and D. R. Huffman, Absorption and scattering of light by small particles, Wiley, New York, 1983 Search PubMed.
- S. Link and M. A. El-Sayed, Annu. Rev. Phys. Chem., 2003, 54, 331 CrossRef CAS PubMed.
- S. Link and M. A. El-Sayed, Int. Rev. Phys. Chem., 2000, 19, 409 CrossRef CAS.
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668 CrossRef CAS.
- Y. Xia and N. J. Halas, MRS Bull., 2005, 30, 338 CrossRef CAS.
- K. S. Lee and M. A. El-Sayed, J. Phys. Chem. B, 2005, 109, 20331 CrossRef CAS PubMed.
- S. Eustis and M. A. El-Sayed, Chem. Soc. Rev., 2006, 35, 209 RSC.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293 CrossRef CAS PubMed.
- M. Hu, J. Chen, Z. Y. Li, L. Au, G. V. Hartland, X. Li, M. Marqueze and Y. Xia, Chem. Soc. Rev., 2006, 35, 1084 RSC.
- E. B. Dickerson, E. C. Dreaden, X. Huang, I. H. El-Sayed, H. Chu, S. Pushpanketh, J. F. McDonald and M. A. El-Sayed, Cancer Lett., 2008, 269, 57 CrossRef CAS PubMed.
- S. Lal, S. E. Clare and N. J. Halas, Acc. Chem. Res., 2008, 41, 1842 CrossRef CAS PubMed.
- X. Huang, S. Neretina and M. A. El-Sayed, Adv. Mater., 2009, 21, 4880 CrossRef CAS PubMed.
- R. H. Kodama, J. Magn. Magn. Mater., 1999, 200, 359 CrossRef CAS.
- Q. A. Pankhurst, J. Connolly, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2003, 36, R167 CrossRef CAS.
- V. I. Shubayev, T. R. Pisanic II and S. Jin, Adv. Drug Delivery, 2009, 61, 467 CrossRef CAS PubMed.
- N. A. Frey, S. Peng, K. Cheng and S. Sun, Chem. Soc. Rev., 2009, 38, 2532 RSC.
- S. Singamaneni, V. N. Bliznyuk, C. Binekc and E. Y. Tsymbal, J. Mater. Chem., 2011, 21, 16819 RSC.
- K. J. M. Bishop, C. E. Wilmer, S. Soh and B. A. Grzybowsk, Small, 2009, 5, 1600 CrossRef CAS PubMed.
- S. J. Cho, B. R. Jarrett, A. Y. Louie and S. M. Kauzlarich, Nanotechnology, 2006, 17, 640 CrossRef CAS.
- M. Kumagai, T. K. Sarma, H. Cabral, S. Kaida, M. Sekino, N. Herlambang, K. Osada, M. R. Kano, N. Nishiyama and K. Kataoka, Macromolecules, 2010, 31, 1521 CAS.
- X. Ji, R. Shao, A. M. Elliott, R. J. Stafford, E. Esparza-Coss, J. A. Bankson, G. Liang, Z. P. Luo, K. Park, J. T. Markert and C. Li, J. Phys. Chem. C, 2007, 111, 6245 CAS.
- M. P. Melancon, A. Elliott, X. Ji, A. Shetty, Z. Yang, M. Tian, B. Taylor, R. J. Stafford and C. Li, Invest. Radiol., 2011, 46, 132 CrossRef PubMed.
- J. Li, Y. Hu, J. Yang, P. Wei, W. Sun, M. Shen, G. Zhang and X. Shi, Biomaterials, 2015, 38, 10 CrossRef CAS PubMed.
- H. Cai, K. Li, M. Shen, S. Wen, Y. Luo, C. Peng, G. Zhang and X. Shi, J. Mater. Chem., 2012, 22, 15110 RSC.
- T. A. Larson, J. Bankson, J. Aaron and K. Sokolov, Nanotechnology, 2007, 18, 325101 CrossRef.
- J. S. Aaron, J. Oh, T. A. Larson, S. Kumar, T. E. Milner and K. V. Sokolv, Opt. Express, 2006, 14, 12930 CAS.
- C. H. Wu, Y. Y. Huang, P. Chen, K. Hoshino, H. Liu, E. P. Frenkel, J. X. J. Zhang and K. V. Sokolov, ACS Nano, 2013, 7, 8816 CrossRef CAS PubMed.
- H. Y. Park, M. J. Schadt, L. Wang, I. S. Lim, P. N. Njoki, S. H. Kim, M. Y. Jang, J. Luo and C. J. Zhong, Langmuir, 2007, 23, 9050 CrossRef CAS PubMed.
- Z. Fan, M. Shelton, A. K. Singh, D. Senapati, S. A. Khan and P. C. Ray, ACS Nano, 2012, 6, 1065 CrossRef CAS PubMed.
- T. T. H. Pham, C. Cao and S. J. Sim, J. Magn. Magn. Mater., 2008, 320, 2049 CrossRef.
- F. Bao, J. L. Yao and R. A. Gu, Langmuir, 2009, 25, 10782 CrossRef CAS PubMed.
- U. Tamer, Y. Gündoğdu, I. H. Boyacl and K. Pekmez, J. Nanopart. Res., 2010, 12, 1187 CrossRef CAS.
- L. Lin, E. Crew, H. Yan, S. Shan, Z. Skeete, D. Mott, T. Krentsel, J. Yin, N. A. Chernova, J. Luo, M. H. Engelhard, C. Wang, Q. Li and C. J. Zhong, J. Mater. Chem. B, 2013, 1, 4320 RSC.
- L. Zhang, J. Xua, L. Mi, H. Gong, S. Jiang and Q. Yu, Bioelectronics, 2012, 31, 130 CrossRef CAS PubMed.
- S. Bhana, E. Chaffin, Y. Wang, S. R. Mishra and X. Huang, Nanomedicine, 2014, 9, 593 CrossRef CAS PubMed.
- X. Zhou, W. Xu, Y. Wang, Q. Kuang, Y. Shi, L. Zhong and Q. Zhang, J. Phys. Chem. C, 2010, 114, 19607 CAS.
- C. H. Liang, C. C. Wang, Y. C. Lin, C. H. Chen, C. H. Wong and C. Y. Wu, Anal. Chem., 2009, 81, 7750 CrossRef CAS PubMed.
- H. Chen, F. Qi, H. Zhou, S. Jia, Y. Gao, K. Koh and Y. Yin, Sens. Actuators, B, 2015, 212, 505 CrossRef CAS.
- S. Kayal and R. V. Ramanujan, J. Nanosci. Nanotechnol., 2010, 10, 1 CrossRef.
- R. J. Chung and H. T. Shih, Materials, 2014, 7, 653 CrossRef.
- Y. Guo, Z. Zhang, D. H. Kim, W. Li, J. Nicolai, D. Procissi, Y. Huan, G. Han, R. A. Omary and A. C. Larson, Int. J. Nanomed., 2013, 8, 3437 CrossRef PubMed.
- Y. Jin, C. Jia, S. W. Huang, M. O'Donnel and X. Gao, Nat. Commun., 2010, 1, 1 CrossRef PubMed.
- Q. H. Lu, K. L. Yao, D. Xi, Z. L. Liu, X. P. Luo and Q. Ning, J. Magn. Magn. Mater., 2006, 301, 44 CrossRef CAS.
- S. Banerjee, S. O. Raja, M. Sardar, N. Gayathri, B. Ghosh and A. Dasgupta, J. Appl. Phys., 2011, 109, 123902 CrossRef.
- U. Tamer, Y. Gündoğdu, I. H. Boyac and K. Pekmez, J. Nanopart. Res., 2010, 12, 1187 CrossRef CAS.
- M. Mandal, S. Kundu, S. K. Ghosh, S. Panigrahi, T. K. Sau, S. M. Yusuf and T. Pal, J. Colloid Interface Sci., 2005, 286, 187 CrossRef CAS PubMed.
- J. L. Lyon, et al., Nano Lett., 2004, 4, 719 CrossRef CAS.
- G. V. P. Kumar, N. Rangarajan, B. Sonia, P. Deepika, N. Rohman and C. Narayana, Bull. Mater. Sci., 2011, 34, 207 CrossRef CAS.
- K. R. Brown and M. J. Natan, Langmuir, 1998, 14, 726 CrossRef CAS.
- T. Chen, et al., Earth Planet. Sci. Lett., 2005, 240, 790 CrossRef CAS.
- V. Kuncser, et al., J. Phys.: Condens. Matter, 2007, 19, 016205 CrossRef.
- U. Tamer, et al., Int. J. Mol. Sci., 2013, 14, 6223 CrossRef CAS PubMed.
- D. A. Wheeler, S. A. Adams, T. Lopez-Luke, A. Torres-Castro and J. Z. Zhang, Ann. Phys., 2012, 524, 670–679 CrossRef CAS.
- M. Mikhaylova, D. K. Kim, N. Bobrysheva, M. Osmolowsky, V. Semenov, T. Tsakalakos and M. Muhammed, Langmuir, 2004, 20, 2472 CrossRef CAS PubMed.
- H. Liu, P. Hou, W. X. Zhang and J. H. Wu, Colloids Surf., A, 2010, 356, 21 CrossRef CAS.
- W. W. Yu, J. C. Falkner, C. T. Yavuz and V. L. Colvin, Chem. Commun., 2004, 2306 RSC.
- J. Park, K. An, Y. Hwang, J. G. Park, H. J. Noh, J. Y. Kin, J. H. Park, N. M. Hwang and T. Hyeon, Nature, 2004, 3, 891 CrossRef CAS PubMed.
- L. Wang, J. Luo, M. M. Maye, Q. Fan, Q. Rendeng, M. H. Engelhard, C. Wang, Y. Lin and C. J. Zhong, J. Mater. Chem., 2005, 15, 1821 RSC.
- L. Y. Wang, J. Luo, Q. Fan, M. Suzuki, I. S. Suzuki, M. H. Engelhard, Y. Lin, N. Kim, J. Q. Wang and C. J. Zhong, J. Phys. Chem. B, 2005, 109, 21593 CrossRef CAS PubMed.
- Z. Xu, Y. Hou and S. Sun, J. Am. Chem. Soc., 2007, 129, 8698 CrossRef CAS PubMed.
- S. Pal, M. Morales, P. Mukherjee and H. Srikanth, J. Appl. Phys., 2009, 105, 07B504 CrossRef.
- I. Robinson, L. D. Tung, S. Maenosono, C. Walti and N. T. K. Thanh, Nanoscale, 2010, 2, 2624 RSC.
- E. Skoropata, R. D. Desautels, C. C. Chi, H. Ouyang, J. W. Freeland and J. van Lierop, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 024410 CrossRef.
- S. J. Oldenburg, R. D. Averitt, S. L. Westcott and N. J. Halas, Chem. Phys. Lett., 1998, 288, 243 CrossRef CAS.
- L. R. Hirsch, R. J. Stafford, J. A. Bankson, S. R. Sershen, R. E. Price, J. D. Hazle, N. J. Halas and J. L. West, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13549 CrossRef CAS PubMed.
- M. R. Rasch, K. V. Sokolov and B. A. Korgel, Langmuir, 2009, 25, 11777 CrossRef CAS PubMed.
- C. S. Levin, C. Hofmann, T. A. Ali, A. T. Kelly, E. Morosan, P. Nordlander, K. H. Whitmire and N. J. Halas, ACS Nano, 2009, 3, 1379 CrossRef CAS PubMed.
- L. Wang, J. Bai, Y. Li and Y. Huang, Angew. Chem., Int. Ed., 2008, 47, 2439 CrossRef CAS PubMed.
- I. Y. Goon, L. M. H. Lai, M. Lim, P. Munroe, J. J. Gooding and R. Amal, Chem. Mater., 2009, 21, 673 CrossRef CAS.
- E. A. Kwizera, E. Chaffin, X. Shen, J. Chen, Z. Gai, S. Bhana, R. O'Connor, L. Wang, H. Adhikari, S. Mishra, Y. Wang and X. Huang, J. Phys. Chem. C, 2016, 120, 10530 CAS.
- S. F. Chin, K. S. Iyer and C. L. Raston, Cryst. Growth Des., 2009, 9, 2685 CAS.
- J. K. Lim, S. A. Majetich and R. D. Tilton, Langmuir, 2009, 25, 13384 CrossRef CAS PubMed.
- Q. Zhang, J. Ge, J. Goebl, Y. Hu, Y. Sun and Y. Yin, Adv. Mater., 2010, 22, 1905 CrossRef CAS PubMed.
- E. Hao, G. Schatz and J. Hupp, J. Fluoresc., 2004, 14, 331 CrossRef CAS PubMed.
- K. Kneipp, H. Kneipp, H. Itzkan, R. R. Dasari and M. S. Feld, J. Phys.: Condens. Matter, 2002, 14, R597 CrossRef CAS.
- B. Nikoobakht, J. Wang and M. A. El-Sayed, Chem. Phys. Lett., 2002, 366, 17 CrossRef CAS.
- R. Weissleder, Nat. Biotechnol., 2001, 19, 316 CrossRef CAS PubMed.
- H. Wang, D. W. Brandl, F. Le, P. Nordlander and N. J. Halas, Nano Lett., 2006, 6, 827 CrossRef CAS PubMed.
- Q. Wei, H. M. Song, A. P. Leonov, J. A. Hale, D. Oh, Q. K. Ong, K. Ritchie and A. Wei, J. Am. Chem. Soc., 2009, 131, 9728 CrossRef CAS PubMed.
- H. M. Song, Q. Wei, Q. K. Ong and A. Wei, ACS Nano, 2010, 4, 5163 CrossRef CAS PubMed.
- C. J. Murphy, T. K. Sau, A. M. Gole, C. J. Orendorff, J. Gao, L. Gou, S. E. Hunyadi and T. Li, J. Phys. Chem. B, 2005, 109, 13857 CrossRef CAS PubMed.
- S. Bhana, B. K. Rai, S. R. Mishra, Y. Wang and X. Huang, Nanoscale, 2012, 4, 4939 RSC.
- T. K. Sau and C. J. Murphy, J. Am. Chem. Soc., 2004, 126, 8648 CrossRef CAS PubMed.
- B. Nikoobakht and M. A. El-Sayed, Chem. Mater., 2003, 15, 1957 CrossRef CAS.
- C. J. Orendorff and C. J. Murphy, J. Phys. Chem. B, 2006, 110, 3990 CrossRef CAS PubMed.
- B. L. Sanchez-Gaytan and S. J. Park, Langmuir, 2010, 26, 19170 CrossRef CAS PubMed.
- B. Sanchez-Gaytan, P. Swanglap, T. J. Lamkin, R. J. Hickey, Z. Fakhraai, S. Link and S. J. Park, J. Phys. Chem. C, 2012, 116, 10318 CAS.
- M. Liu and P. Guyot-Sionnest, J. Phys. Chem. B, 2005, 109, 22192 CrossRef CAS PubMed.
- Magnetism: from fundamentals to nanoscale dynamics, ed. J. Stohr and H. C. Siegmann, Springer, New Yorks, 2006 Search PubMed.
- A. S. Teja and P. Y. Koh, Prog. Cryst. Growth Charact. Mater., 2009, 55, 22 CrossRef CAS.
- R. Ramprasad, P. Zurcher, M. Petras, M. Miller and P. Renaud, J. Appl. Phys., 2004, 96, 519 CrossRef CAS.
- P. Guardia, A. Labarta and X. Batlle, J. Phys. Chem. B, 2001, 115, 390 Search PubMed.
- L. Wang, et al., J. Phys. Chem. B, 2005, 109, 21593 CrossRef CAS PubMed.
- M. P. Morales, S. Veintemillas-Verdaguer, M. I. Montero, C. J. Serna, A. Roig, L. I. Casas, B. Martinez and F. Sandiumenge, Chem. Mater., 1999, 11, 3058 CrossRef CAS.
- P. Crespo, R. Litrán, T. C. Rojas, M. Multigner, J. M. de la Fuente, J. C. Sánchez-López, M. A. García, A. Hernando, S. Penadés and A. Fernández, Phys. Rev. Lett., 2004, 93, 087204 CrossRef CAS PubMed.
- B. Nikoobakht and M. A. El-Sayed, Langmuir, 2001, 17, 6368 CrossRef CAS.
- J. Gao, C. M. Bender and C. J. Murphy, Langmuir, 2003, 19, 9065 CrossRef CAS.
- M. Grzelczak, J. Pérez-Juste, P. Mulvaney and L. M. Liz-Marzán, Chem. Soc. Rev., 2008, 37, 1783 RSC.
- J. J. Hauser, Phys. Rev., 1969, 187, 580 CrossRef CAS.
- C. H. Loo, A. Lin, L. R. Hirsch, M. H. Lee, J. Barton, N. J. Halas, J. West and R. A. Drezek, Technol. Cancer Res. Treat., 2004, 3, 33 CrossRef CAS PubMed.
- E. Prodan, C. Radloff, N. J. Halas and P. Nordlander, Science, 2003, 302, 419 CrossRef CAS PubMed.
- E. Chaffin, S. Bhana, X. Huang and Y. Wang, J. Phys. Chem. B, 2014, 118, 14076 CrossRef CAS PubMed.
- P. K. Jain and M. A. El-Sayed, Nano Lett., 2007, 7, 2854 CrossRef CAS PubMed.
- W. Brullot, V. K. Valev and T. Verbiest, Nanomedicine, 2012, 8(5), 559 CAS.
- F. Hao, C. L. Nehl, J. H. Hafner and P. Nordlander, Nano Lett., 2007, 7, 729 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |