
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultrathin g-C3N4 nanosheets with an extended visible-light-responsive range for significant enhancement of photocatalysis
Yanfang Yang†
a,
Jingjing Chen†b,
Zhiyong Mao*a,
Na Anb,
Dajian Wang*b and
Bradley D. Fahlmanc
aSchool of Materials Science and Engineering, Tianjin University of Technology, Tianjin 300384, P. R. China. E-mail: mzhy984@163.com
bTianjin Key Laboratory for Photoelectric Materials and Devices, Tianjin University of Technology, Tianjin 300384, P. R. China. E-mail: djwang@tjut.edu.cn
cDepartment of Chemistry & Biochemistry and Science of Advanced Materials Program, Central Michigan University, Mount Pleasant, MI, USA 48859
First published on 12th January 2017
Abstract
Ultrathin graphitic carbon nitride (g-C3N4) nanosheets were synthesized via thermal exfoliation of bulk urea-derived g-C3N4 under an argon atmosphere. As a visible-light responsive photocatalyst, this material exhibits a much superior photocatalytic activity in pollution degradation and H2 evolution than bulk g-C3N4, as a result of the extended region of visible light response and the enhanced surface area of ultrathin g-C3N4 nanosheets. These findings may provide a promising and facile approach to the design of high-performance photocatalysts.
Introduction
With constant threats related to the energy crisis and environmental pollution issues, photocatalysis technology has emerged as a feasible alternative to address these important societal challenges.1 The landmark event of photocatalytic water splitting into H2 and O2 using an n-type TiO2 single-crystal electrode under ultraviolet (UV) light was first reported by Honda–Fujishima in 1972.2 Four years later, the photocatalytic degradation of organic pollutants was reported by Carey et al. in the presence of TiO2 under UV-light irradiation in aqueous suspensions.3 To summarize, each photocatalytic reaction basically involves three primary processes: photon absorption, electron–hole-pair generation/separation/transportation, and catalytic surface reactions.4,5 Hence, these three aforementioned processes need to be optimized in order to improve photocatalytic performance. However, traditional TiO2-based catalysts encounter problems in satisfying the requirements of visible-light absorption, which results in an inefficient utilization of solar energy. Therefore, numerous highly efficient, stable and visible-light responsive semiconductor-based photocatalysts have been exploited by researchers, such as WO3,6 CoO,7 and CdS.8,9Polymeric graphitic carbon nitride (g-C3N4), with a graphitic π-conjugated stacked structure consisting of tri-s-triazine repeating units, is recognized as the next generation of photocatalysts with a suitable visible-light response. Wang and co-workers first demonstrated that g-C3N4 can produce hydrogen from water under visible-light irradiation in the presence of triethanolamine-electron donors in 2009.10 Since then, g-C3N4 has been a hot research topic due to its cost effectiveness, facile preparation, metal-free composition, appealing electronic band structure, high physicochemical stability, and environmental benignity. Nevertheless, bulk g-C3N4 suffers from such shortcomings as a high probability of photo-generated electron–hole pair recombination, a small specific surface area, and low visible light utilization efficiency.11–14 So far, researchers have made numerous efforts to optimize its photoactivity by exploring facile and dependable strategies to synthesize modified g-C3N4-based photocatalysts, including new synthetic techniques,15 electronic band structure modulation,16–19 and nanostructure design.20–23
Nanostructural engineering was widely utilized to tune the morphology, porosity, structure, surface area, and size in g-C3N4 photocatalysts. A series of hard templates, such as silica nanospheres, ordered mesoporous silica, SBA-15, and so forth, were employed to develop various nanostructural g-C3N4 materials with a higher photocatalytic activity.24–27 However, the use of extremely hazardous, toxic, and costly fluoride-containing etchants such as NH4HF2 or HF solutions are needed to remove the silica template, which limits the practical applications for scaled-up industrial processes. Apart from the hard-template synthesis of nanostructural g-C3N4, the relatively “greener” soft-template process offers advantages such as altering various morphologies of g-C3N4 via template selection, which simplifies the synthetic route of g-C3N4.17,28,29 However, it encounters issues related to the complete removal of carbon and other heteroatom residues originating from certain soft-template chemicals. Thus, it is urgently and of paramount importance desired to develop a facile, template-free synthetic approach for advanced photochemical reactions. Zhang et al. prepared a porous g-C3N4 nanosheets by the polycondensation of urea at 550 °C for 3 h under air atmosphere.30 Alternatively, g-C3N4 nanosheets can be easily prepared by exfoliating bulk g-C3N4, similar to the development of 2D graphene from bulk graphite. Exfoliation routes such as ultrasonication-assisted liquid exfoliation,31–33 acid exfoliation,34,35 and post-thermal oxidation etching36–38 have been reported to date. Among these, post-thermal oxidation etching of bulk g-C3N4 offers the advantages of cost-effectiveness, easy scale up, and environmental friendliness. It was well recognized that the hydrogen-bond cohered strands of polymeric melon structure in the layers are unstable against oxidation processes. This led to a gradual decrease in the thickness of bulk g-C3N4, achieving nanoscale range by a layer-by-layer thermal etching process. For example, Liang et al. demonstrated the fabrication of holey g-C3N4 nanosheets by thermally treating bulk g-C3N4 under an NH3 atmosphere, thus extending the light absorption to the near-infrared region.39 Kang et al. prepared an amorphous g-C3N4 by thermally annealing dicyandiamide-derived g-C3N4 under an argon atmosphere, decreasing the band gap of g-C3N4 from 2.82 to 1.90 eV.40 Very recently, the same group synthesized dicyandiamide-derived ultrathin g-C3N4 nanosheets with crystalline structure in an argon atmosphere.41 Therefore, the construction of g-C3N4 nanostructure is a fascinating strategy for enhancement of photocatalysis through harnessing additional visible light absorption.
In this paper, ultrathin g-C3N4 nanosheets with extended visible-light-responsive range successfully prepared using a facile post-thermal etching route to exfoliate porous urea-derived g-C3N4 nanosheets in an argon atmosphere. The phase structure, chemical composition, morphology, and photocatalytic performance for both H2 generation and rhodamine B (RhB) degradation were investigated in detail. A comprehensive comparison between g-C3N4 and ultrathin g-C3N4 nanosheets is also provided to discuss the enhancement mechanism of photocatalytic activity for our as-prepared ultrathin g-C3N4 nanosheets.
Experimental
Sample preparation
Urea, triethanolamine (TEOA), isopropanol (IPA), rhodamine B (RhB), and hexachloroplatinum(IV) acid hydrate were purchased from Aladdin Industrial Corporation (Shanghai, China), and were used without purification. Graphitic carbon nitride powder was prepared by the polycondensation of urea. 10 g urea was put into a crucible with a cover semiclosed and calcined in a muffle furnace at 550 °C for 6 h using a heating rate of 2.5 °C min−1 and cooling naturally to room temperature. The obtained graphitic carbon nitride sample was denoted as GCN. To prepare the ultrathin g-C3N4 nanosheets, the obtained light-yellow g-C3N4 powder was post-heated at 600 °C for 2 h using a heating rate of 10 °C min−1 under a flow of argon at 200 mL min−1 and cooling naturally to room temperature. The post-treated graphitic carbon nitride sample was denoted as UGCN.Sample characterization
The crystalline structure of the prepared g-C3N4 samples was analyzed by an X-ray diffractometer (XRD) (Rigaku, RINT Ultima-III, Japan) with Cu-Kα radiation. The morphology and elemental analysis was measured by a scanning electron microscope (SEM) equipped with energy-dispersive spectroscopy (EDS) (Hitachi, S-4800), and a transmission electron microscope (TEM, JEM-6700F, Japan). X-ray photoelectron spectroscopy (XPS, Thermal Fisher Scientific 250XI, America) was used to characterize the chemical speciation of g-C3N4. The chemical bonding status was assessed by Fourier transform infrared spectroscopy (FTIR, Bruker, WQE-410). Ultraviolet-visible (UV-vis) absorption spectra were measured by a UV-visible spectrometer (TU-1901, China) with a 60 mm diameter integration sphere. Photoluminescence spectra (PL) were measured using a fluorescence spectrometer (Hitachi F-4600, Japan). Specific surface area measurements were taken using the BET method (N2 absorption, Kangta AUTOSORB-1, America). The electron spin resonance (ESR) signals of radicals spin-trapped by spin-trap reagent DMPO (5,5′-dimethyl-1-pyrroline-N-oxide) were tested in methanol (for superoxide radical) and water (for hydroxyl radical), respectively. Ultraviolet photoelectron spectroscopy (UPS, Thermo scientific, ESCALAB 250Xi, America) was used to characterize the redox potential of g-C3N4. Electrochemistry impedance spectroscopy (EIS) measurements were performed on an electrochemical workstation (CHI600E, China) based on a conventional three-electrode system with the as-prepared photocatalyst, carbon black and PVDF (mass ratio 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:1) coated on nickel foam as a working electrode, platinum foil as a counter electrode, and saturated calomel electrode (SCE) as a reference electrode, respectively. The electrolyte was immersed in 1 M Na2SO4 aqueous solution, the frequency range was from 0.01 Hz to 100 kHz, and the amplitude of the applied sine wave potential in each case was 5 mV.
:3:1) coated on nickel foam as a working electrode, platinum foil as a counter electrode, and saturated calomel electrode (SCE) as a reference electrode, respectively. The electrolyte was immersed in 1 M Na2SO4 aqueous solution, the frequency range was from 0.01 Hz to 100 kHz, and the amplitude of the applied sine wave potential in each case was 5 mV.
Photocatalytic H2 generation testing
Photocatalytic hydrogen production studies were carried out in a Pyrex top-irradiation reaction vessel connected to a closed glass gas-circulation system (Lab-Solar-III, AG, Perfectlight, Beijing). A 300 W xenon lamp (PLS-SXE 300C, Perfectlight, Beijing) with a 400 nm cut-off filter was chosen as a visible light source, and the light intensity was 100 mW cm−2 (determined by PL-MW2000, Perfectlight, Beijing). 100 mg of photocatalyst was added to an aqueous solution (100 mL) that contained 90 mL water and 10 mL triethanolamine. Then, 3 wt% of Pt nanoparticles were loaded onto the surface of the catalyst by in situ photodeposition using H2PtCl6·6H2O as the precursor. Next, the reactant solution was evacuated several times to remove air prior to the irradiation experiments. The temperature of the reaction solution was maintained at 7 °C by a flow of cooling water during the photocatalytic reaction. The evolved gases were analyzed by gas chromatography (GC 7900, Shanghai) equipped with a thermal conductivity detector (TCD) and a 5 Å molecular sieve column, using argon as the carrier gas.Photocatalytic RhB degradation testing
Rhodamine B (RhB) with high photostability and water solubility was used as a model pollutant to demonstrate the photocatalytic activity of the prepared photocatalysts in pollution degradation. For testing, a photocatalyst (100 mg) was suspended in 100 mL of RhB (aq.) (20 mg L−1) within a Pyrex photocatalytic reactor with a circulating water system maintained at 7 °C by a flow of cooling water during the photocatalytic reaction. Prior to irradiation, the suspensions were magnetically stirred for 30 min in the dark to ensure that RhB could reach the absorption–desorption equilibrium on the photocatalyst surface. Then, suspensions were irradiated by a 300 W xenon lamp with an optical filter (>420 nm). At certain time intervals, 4 mL aliquots were sampled and centrifuged to remove the photocatalyst particles. The filtrates were analyzed by recording variations of the absorption band maximum (554 nm) in the UV-vis spectra of RhB using a UV-vis spectrophotometer (TU-1901, China).Results and discussion
The crystal structure of as-synthesized g-C3N4 samples was studied by X-ray diffraction (XRD). As shown in Fig. 1, the as-prepared g-C3N4 samples give two typical diffraction peaks, 2θ = 27.1° and 13.1° as reported elsewhere. The former is attributed to the long-range interplanar stacking of aromatic systems identified as the (002) peak;42 the latter with a much weaker intensity is related to an in-plane structural packing motif.43 Compared to GCN, the diffraction peak (002) of UGCN shifts from ∼27.1° to 27.4°, corresponding to a decreased distance between layers. This feature is likely due to the formation of ultrathin g-C3N4 nanosheets through exfoliating g-C3N4 at elevated temperature. In addition, a weaker diffraction intensity of the two peaks is observed for UGCN, suggesting that post-treatment with argon gas could decrease the crystallinity of GCN and weaken the long-range order in the atomic arrangements in UGCN. This can been attributed to the breaking of in-plane hydrogen bonds between strands of polymeric melon units, and the outward twisting of melon units due to large outward movement of the NH2 groups from the original g-C3N4 plane after annealing (the inset of Fig. 1), also interpreted as an amorphous carbon nitride with the absence of long-range order in the atomic arrangements by tempering the synthesized dicyandiamide-derived g-C3N4 at 620 °C for 2 h in an argon atmosphere with a flow rate of 50 mL min−1.40 It has been suggested that the temperature limit without depleting the amount of nitrogen in g-C3N4 under an argon atmosphere. | ||
| Fig. 1 XRD patterns for as-prepared GCN and UGCN samples (inset: the structure of g-C3N4). | ||
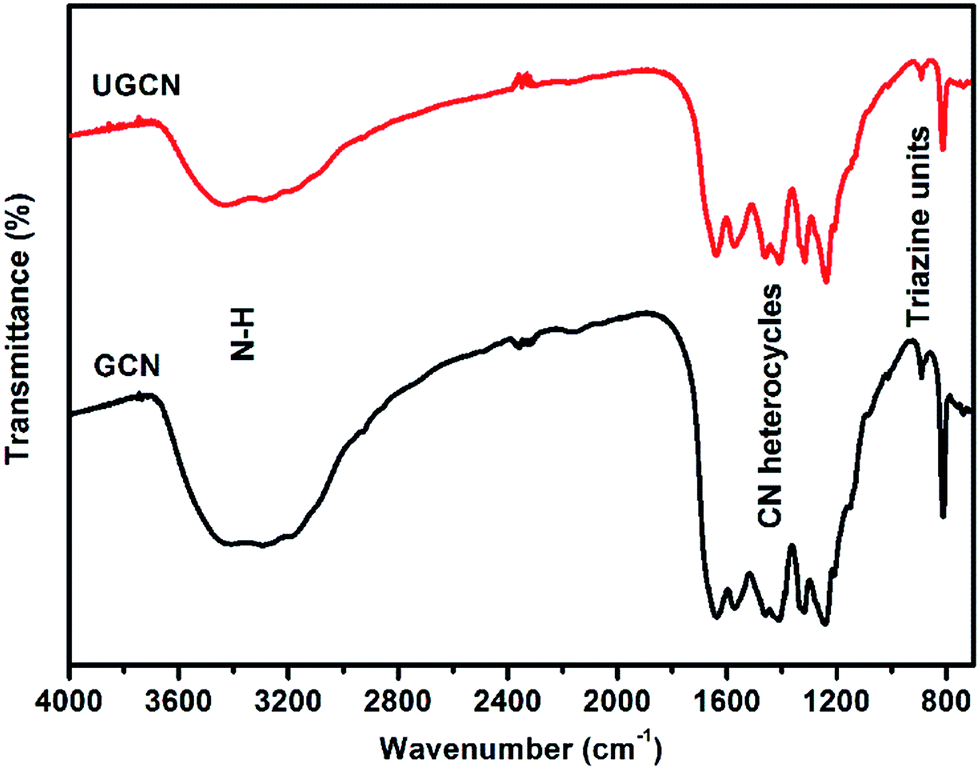
To investigate the chemical state of the as-prepared GCN and UGCN samples, FTIR analysis in the range of 700–4000 cm−1 was performed as shown in Fig. 2. The broad absorption peak at 3000–3600 cm−1 is assigned to the stretching vibration of N–H group. The peaks in the region 1200–1700 cm−1 are typical characteristic stretching modes exhibited by CN heterocycles, and the sharp peak positioned at 810 cm−1 corresponds to the typical breathing mode of the triazine units, revealing their similar chemical structures. It is worth noting that the peak for triazine units in UGCN is reduced relative to GCN, which is caused by the less-ordered packing of triazine motifs in the post-treated sample.
 | ||
| Fig. 2 FTIR spectra for as-prepared GCN and UGCN samples. | ||
To further investigate the chemical composition and speciation of the as-obtained GCN and UGCN, X-ray photoelectron spectroscopy (XPS) was performed. As shown in Fig. 3(a), the XPS spectra of both GCN and UGCN samples contain three sharp peaks at 286, 396, and 529 eV, which are assigned to C 1s, N 1s, and O 1s signals, respectively. From high-resolution XPS analysis, the C 1s spectrum shown in Fig. 3(b) can be fitted by Gaussian curves with dominant components centered at 282.6 and 286 eV, which are respectively attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and N–CN bonds. The N 1s peak (Fig. 3(c)) can similarly be deconvoluted into three peaks centered at 396.2, 397.2 and 398.3 eV, corresponding to CN–C, N–[C]3, and C–NHx bonds, respectively. The binding energies of C 1s and N 1s core electrons remain almost the same, suggesting similar chemical states for carbon and nitrogen. Quantitative analysis from XPS spectra reveals that the C/N atomic ratio of UGCN (1.24) is much larger than that of GCN (1.14). This means the loss of some extra nitrogen atoms from the pristine GCN with amine groups during thermal exfoliation, which is also supported by the decrease in the C/N atomic ratio (0.78 in UGCN vs. 0.67 in GCN) determined by the EDS elemental analysis (Fig. 3(d)). Notably, hybridized sp3 nitrogen not only has three chemical bonds to carbon, but because of hybridization, can also be bonded to hydrogen perpendicular to the direction of the graphitic layer.44 Thus, bulk g-C3N4 can be exfoliated into ultrathin g-C3N4 nanosheets.
C and N–CN bonds. The N 1s peak (Fig. 3(c)) can similarly be deconvoluted into three peaks centered at 396.2, 397.2 and 398.3 eV, corresponding to CN–C, N–[C]3, and C–NHx bonds, respectively. The binding energies of C 1s and N 1s core electrons remain almost the same, suggesting similar chemical states for carbon and nitrogen. Quantitative analysis from XPS spectra reveals that the C/N atomic ratio of UGCN (1.24) is much larger than that of GCN (1.14). This means the loss of some extra nitrogen atoms from the pristine GCN with amine groups during thermal exfoliation, which is also supported by the decrease in the C/N atomic ratio (0.78 in UGCN vs. 0.67 in GCN) determined by the EDS elemental analysis (Fig. 3(d)). Notably, hybridized sp3 nitrogen not only has three chemical bonds to carbon, but because of hybridization, can also be bonded to hydrogen perpendicular to the direction of the graphitic layer.44 Thus, bulk g-C3N4 can be exfoliated into ultrathin g-C3N4 nanosheets.
 | ||
| Fig. 3 (a) XPS survey spectra; (b) high resolution C 1s; (c) high resolution N 1s for GCN and UGCN samples; and (d) EDS elemental analysis of GCN and UGCN. | ||
The morphologies of GCN and UGCN were investigated by SEM and TEM, as shown in Fig. 4. The low-magnification SEM image (Fig. 4(a)) shows that the obtained GCN sample is composed of irregular thick sheets with thickness of 20–50 nm. The high-magnification SEM image (inset in Fig. 4(a)) clearly shows that the GCN sheets have a large number of pores with nanoscale diameters, which are produced by gases released at elevated temperatures. As compared to GCN, UGCN samples feature ultrathin nanosheets with a thickness of a few nanometers (Fig. 4(b)). High-magnification SEM image reveals the presence of in-plane pores within the high curled ultrathin nanosheets (inset in Fig. 4(b)). This phenomenon can be attributed to the weakened interaction between adjacent layers and interruption of hydrogen bonding at elevated temperatures. Transmission electron microscope (TEM) images (Fig. 4(c) and (d)) also confirm the ultrathin nanosheet morphology for UGCN, and clearly shows more curled edges than GCN samples.
 | ||
| Fig. 4 SEM images of (a) GCN and (b) UGCN samples; and TEM images of (c) GCN and (d) UGCN samples. | ||
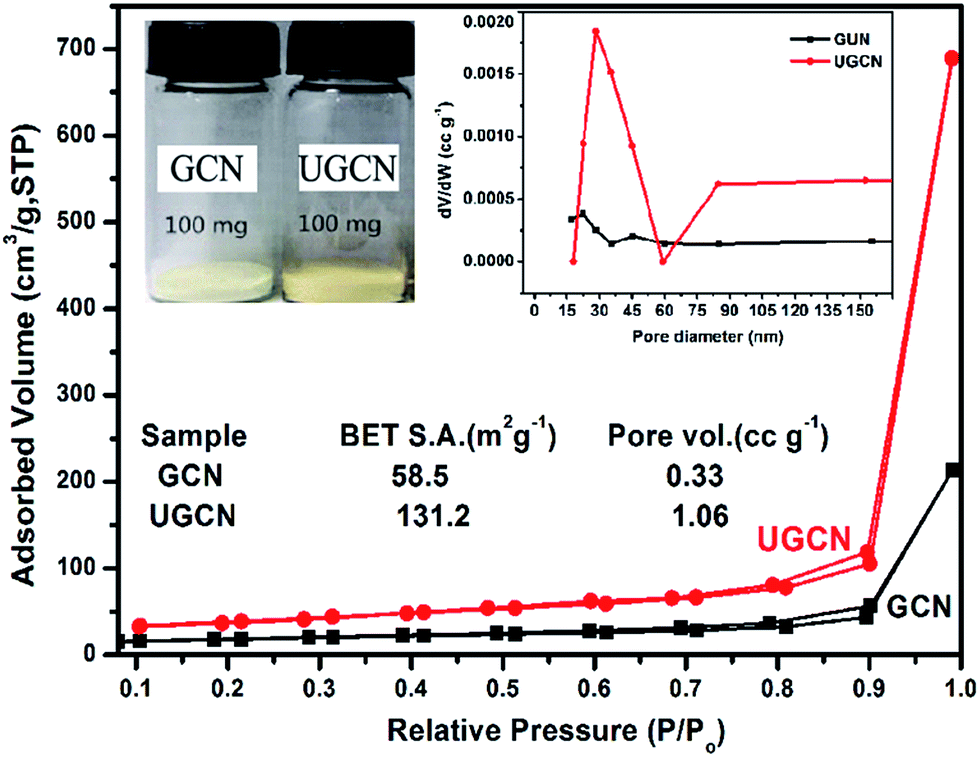
The pore structure and surface area of GCN and UGCN samples are obtained from N2 adsorption–desorption measurements. As shown in Fig. 5, UGCN sample exhibits a type-IV isotherm with an extremely high adsorption capacity at relatively high pressures (P/P0: 0.9–1), suggesting the presence of abundant pores. It is clearly seen that the BET of UGCN (131.2 m2 g−1) is 2.24 times higher than that of GCN (58.5 m2 g−1). The pore size distribution (PSD) curve of UGCN has a sharp peak at 30 nm (inset of Fig. 5 (right)), which is larger than the pore size of GCN (∼18 nm). At a result, the UGCN display the 3.21 times larger pore volume than the GCN at P/P0 = 0.99, indicating the porous have been constructed on the g-C3N4. The higher BET and larger pore volume results in a much larger volume of UGCN with the same weight as GCN (inset of Fig. 5 (left)). It was well known that the larger specific surface area enable more active species and reactants can be absorbed on its surface, and it may effectively promote the kinetics of photocatalytic reaction by facilitating mass transfer. It was assumed the UGCN sample may display more efficiency photocatalytic activity than GCN sample.
 | ||
| Fig. 5 Nitrogen adsorption–desorption isotherms (the inset: the photograph of 100 mg of both GCN and UGCN powders (left) and the corresponding pore size distribution curves (right)). | ||
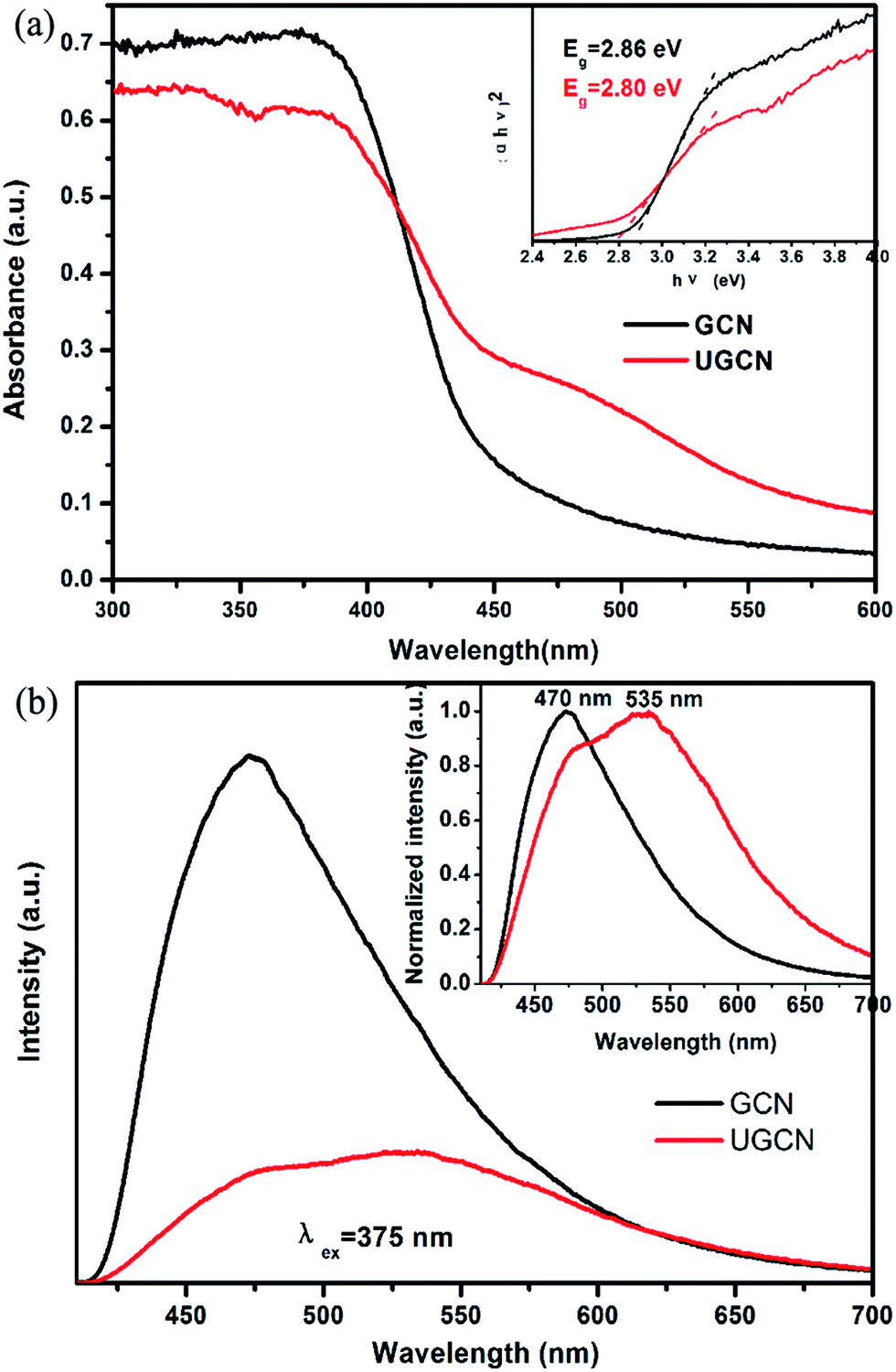
To identify the photoelectric properties of GCN and UGCN samples, UV-vis diffuse reflectance spectroscopy (DRS) and photoluminescence (PL) were measured, as shown in Fig. 6(a) and (b), respectively. Compared to GCN, the absorption threshold of UGCN is significantly shifted towards the low energy region (inset of Fig. 6(a)). The intrinsic absorption band edge (band gap) for GCN was evaluated as 2.80 eV, in agreement with urea-derived g-C3N4 reported by Martin et al. in elsewhere.15 As compared with GCN, the intrinsic absorption edge for UGCN shows no obvious change (Eg = 2.86 eV), but a new absorption band in the region of 450 nm to 550 nm was clearly observed (Fig. 6(a)). The photograph for UGCN sample shown in the inset of Fig. 5 depicts the color change of the photocatalysts from light yellow to brown yellow, corresponding to an expanded absorption in the blue-green region (450–550 nm). Namely, the light absorbance in the visible light region for UGCN has been remarkably enhanced, which is useful for photocatalytic performance.
 | ||
| Fig. 6 (a) UV-vis diffuse reflectance spectroscopy (DRS); and (b) photoluminescence (PL) spectra for as-synthesized GCN and UGCN samples (inset of (a) is Tauc plots, inset of (b) is the normalized PL spectrum). | ||
The photoluminescence (PL) spectra for GCN and UGCN samples under an excitation wavelength of 375 nm are shown in Fig. 6(b). GCN shows a strong intrinsic emission band with a peak at 470 nm, which is attributed to direct electron–hole recombination of band transition. Compared with GCN, UGCN shows a much weaker emission profile, implying the recombination of charge carriers may be effectively inhibited by the post-thermal method. The normalized PL spectrum (inset of Fig. 6(b)) of UGCN sample depicts a new emission band positioned at 535 nm, apart from the intrinsic emission band at 470 nm. This new emission band at 535 nm may be caused by the multiple reflection of incident light in pores and holes in the UGCN structure.
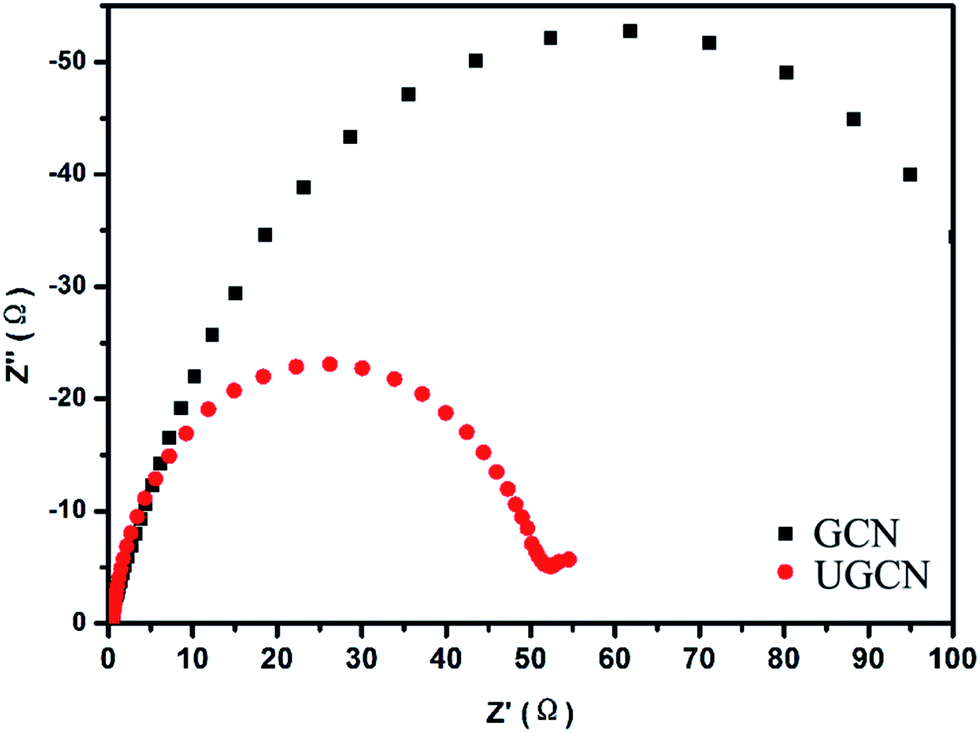
To further study the photogenerated charge separation and transfer performance, electrochemical impedance spectroscopy (EIS) was conducted. The experimental Nyquist impedance plots for GCN and UGCN are shown in Fig. 7. The semicircle at high frequencies in the Nyquist diagrams is in accordance with the electron-transfer-limited process and the semicircle diameter is equivalent to electron-transfer resistance.45 Obviously, UGCN has a smaller arc radius than GCN. This also means that UGCN has a more effective separation efficiency of photogenerated charge carriers and a faster interfacial charge transfer. The EIS analysis is consistent with the results of PL analysis, indicating UGCN will exhibit better photocatalytic performance.
 | ||
| Fig. 7 EIS of GCN and UGCN samples in a 1 M Na2SO4 aqueous solution. | ||
Apart from an appropriate band gap, the proper matching of conduction band and valence band levels of a photocatalyst with the redox potentials of the photocatalytic reactions is also important. To research the redox potentials of UGCN during the photocatalytic process, the ultraviolet photoelectron spectroscopy (UPS) was measured, as shown in Fig. 8. The valence band energy (Ev) of UGCN was calculated to be 7.02 eV by subtracting the width of the HeIUPS spectra from the excitation energy (21.22 eV). The conduction band energy Ec is thus estimated at 4.22 eV from Ev − Eg. The Ev and Ec values of UGCN in electron volts could be converted to electrochemical energy potential in volts at −0.22 V and 2.58 V respectively, according to the reference standard for which 0 V versus RHE (reversible hydrogen electrode) equals −4.44 eV versus evac (vacuum level).46 This means that the reduction level for H2 is positioned below the conduction band of UGCN, and the oxidation level for H2O to O2 is above the valence band. These bands are properly positioned to permit transfer of electrons and holes for water splitting in UGCN.
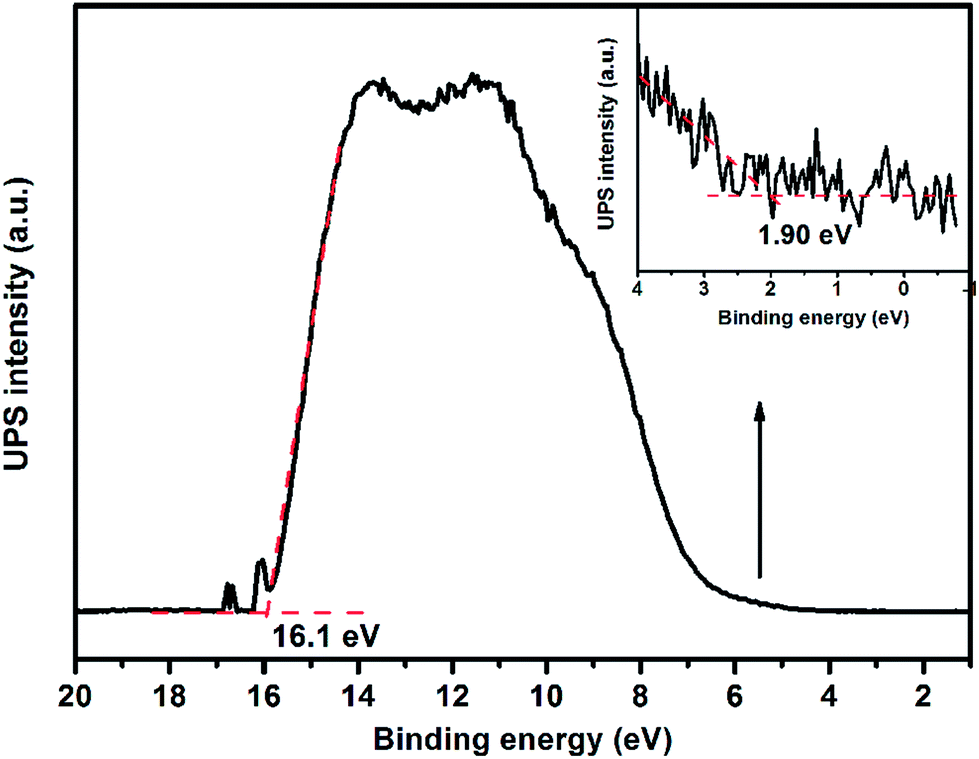 | ||
| Fig. 8 UPS spectra of UGCN. | ||
The photocatalytic performance of the UGCN was evaluated with RhB degradation and H2 production from water under visible light irradiation. Fig. 9(a) shows the photocatalytic activity of the as-synthesized samples for the degradation of RhB under visible light (≥420 nm). One can see that UGCN has a higher photocatalytic performance, compared to GCN. It should be noted that RhB self-degradation is almost negligible in the absence of photocatalyst. After irradiation of 40 min, the degradation rate of RhB is 36% and 99% for GCN and UGCN, respectively, implying the excellent photocatalytic activity of UGCN.
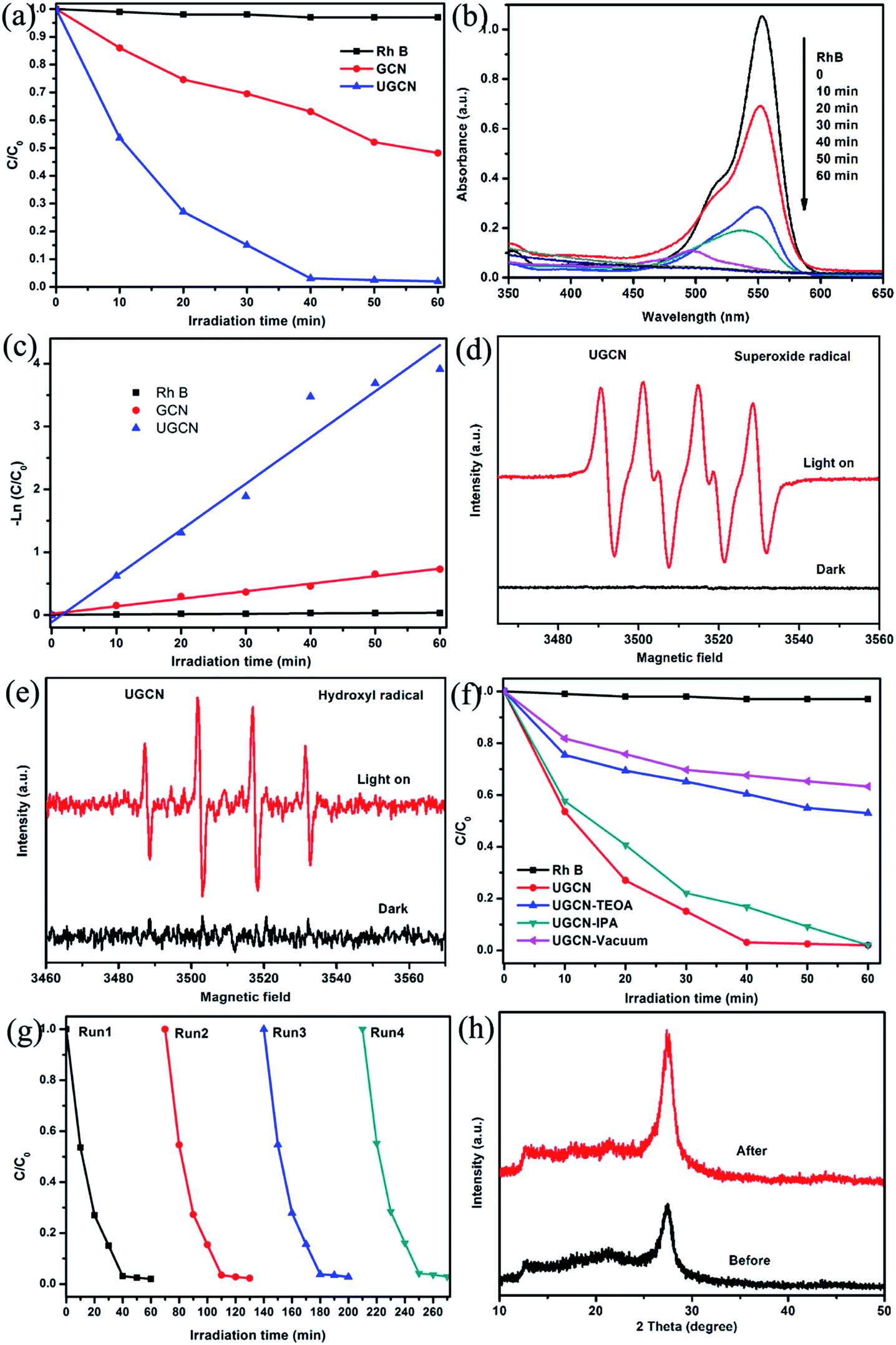 | ||
| Fig. 9 (a) The photocatalytic degradation of RhB dye by 100 mg GCN and UGCN photocatalysts (≥420 nm); (b) changes in UV-visible absorption spectra of RhB solution with variable photodegradation reaction time for UGCN; (c) kinetic fit for the degradation of RhB with GCN and UGCN; ESR spectra of DMPO–˙O2− (d) and DMPO–˙OH (e) adducts in UGCN aqueous dispersion systems before and after visible light irradiation; (f) the effects of different scavengers on the degradation of RhB in the presence of UGCN under visible-light irradiation; (g) recyclability of UGCN photocatalyst for the degradation of RhB under visible-light irradiation; (h) XRD diffraction patterns for UGCN sample before and after photodegradation reaction. | ||
To further demonstrate the mineralization degree of RhB during photodegradation process, the absorbance spectra were given (Fig. 9(b)). It was found that the characteristic absorption peak of RhB solution at 554 nm shifted to shorter wavelength and the intensity decreased under visible light irradiation. These changes indicate the remove of ethyl group from RhB. And one can see the absorption peak disappeared at last, implying the destroy of benzene ring of RhB.47
At the same time, the photocatalytic degradation kinetic was studied (Fig. 9(c)) and the degradation of RhB can be attributed to pseudo-first-order reaction with a simplified Langmuir–Hinshelwood model when C0 was very small: −ln(C/C0) = kt. The UGCN has higher rate constant of 0.07346 min−1 and displayed 6.1 times higher than GCN for the degradation of RhB. Notably, this is due to the larger specific surface area and extended visible light absorption profile of UGCN compared to that of GCN.
During the photocatalytic process, the electrons and holes can be produced in CB and VB when the photocatalyst is irradiated by light. And the photodegradation process almost directly utilize the produced holes with sufficient oxidative ability, or use the formed other active species such as superoxide radical (˙O2−) and hydroxyl radical (˙OH), which originated from reaction of electrons with surface adsorbed molecular oxygen and holes with surface adsorbed water or hydroxyl group, respectively.48 ESR spin-trap technique was employed to determine the active species in UGCN. As shown in Fig. 9(d) and (e), the characteristic peaks of DMPO–˙O2− and DMPO–˙OH can be detected obviously for UGCN under visible light, compared with UGCN for 4 min in dark. These results revealed that the ˙O2− and ˙OH active species could be generated, which might play an important role in the photocatalytic degradation process.
To further confirm the role of reactive species including h+, ˙OH and ˙O2− in the degradation of RhB, triethanolamine (TEOA), isopropanol (IPA), and vacuum conditions were used as h+, ˙OH and ˙O2− scavengers, respectively. As shown in Fig. 9(f), the photocatalytic activity of UGCN was greatly suppressed by the addition of TEOA as a h+ scavenger, and vacuum conditions as ˙O2− scavengers, which indicates that h+ and ˙O2− are the main active species in the photocatalytic reaction. A unpronounced decrease in the photocatalytic activity was also observed by the addition of IPA as ˙OH scavenger. In summary, the main reactive species involved in the photocatalytic degradation of RhB over UGCN are ˙O2− > h+ > ˙OH. From Fig. 9(g), one can see there is no obvious decrease in the performance of photocatalytic degradation of RhB after four cycles, confirming the high stability of UGCN. The XRD diffraction patterns (Fig. 9(h)) for the UGCN photocatalyst before and after photodegradation reaction maintained the crystalline structure of g-C3N4, further affirming the stability of UGCN.
Fig. 10(a) shows UGCN has a better photocatalytic activity with an average H2 evolution rate of 459.94 μmol h−1 g−1, compared to that of GCN (224.09 μmol h−1 g−1), indicating that the average H2 evolution rate of UGCN is 2.03 times higher than that of GCN. Fig. 10(b) gives the dependence of the wavelength of the incident light on the hydrogen generation rate of UGCN together with its UV-vis absorption spectrum. The fact that the hydrogen generation is wavelength dependent is consistent with the optical absorption spectrum and suggests that the observed hydrogen generation of UGCN comes from a photocatalysis process. The maximum wavelength of visible light to excite UGCN for photocatalytic hydrogen generation is determined to be in the range of 450–600 nm, indicating enhanced photocatalytic performance may be attribute to the enlarged visible light response.
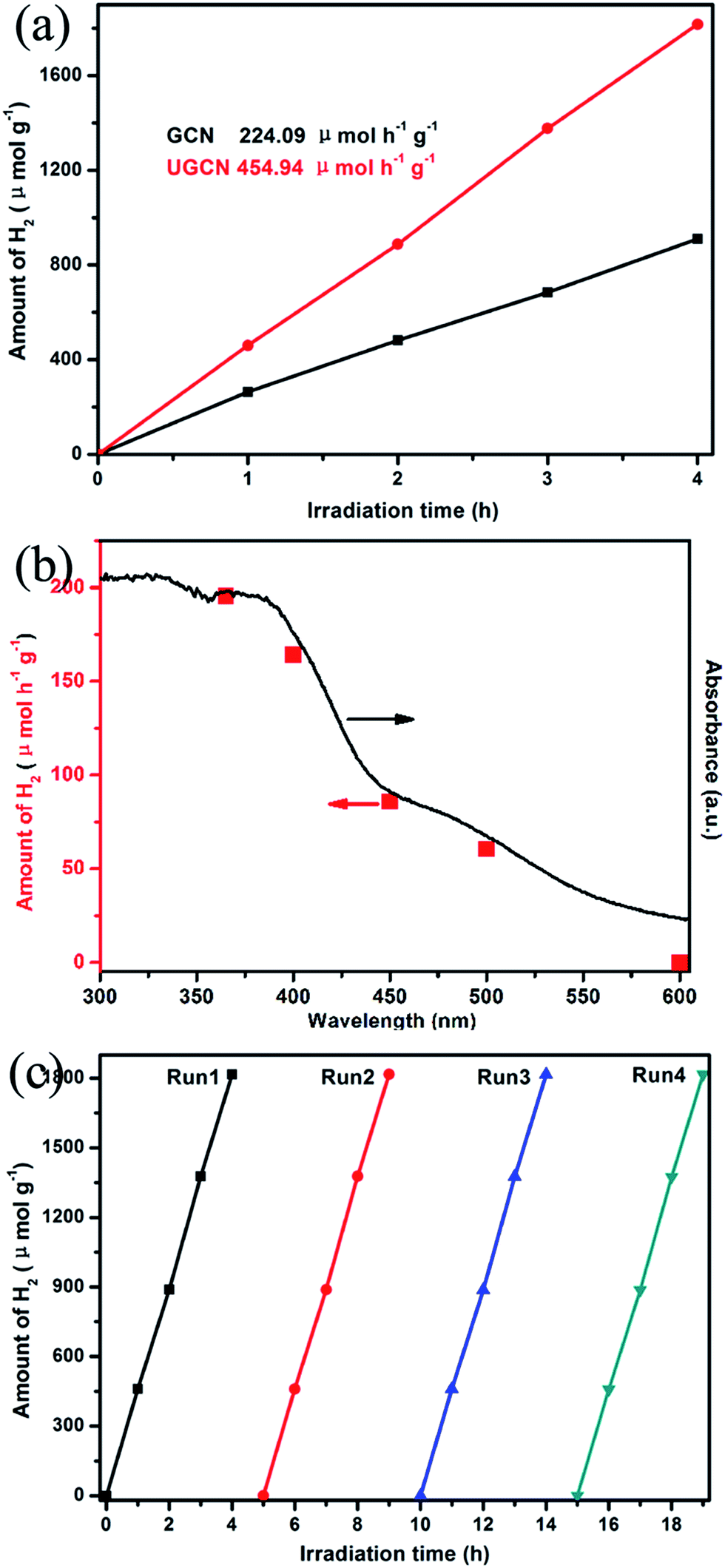 | ||
| Fig. 10 (a) photocatalytic H2 evolution with 10 vol% TEOA, 3 wt% Pt, and 100 mg photocatalysts under visible light (≥400 nm); (b) wavelength dependent hydrogen generation from water for UGCN photocatalyst; (c) recyclability of UGCN photocatalyst for the photocatalytic H2 evolution under visible-light irradiation. | ||
From Fig. 10(c), one can see there is no obvious decrease in the performance of photocatalytic H2 evolution from water after four cycles, confirming the high stability of UGCN. The above results clearly demonstrate the photocatalytic activity of UGCN has been significantly improved compared to those of GCN, which may be attributed to the enlarged surface area to support more active sites, the enhanced visible-light harvesting to extended the utilization of solar energy.
Conclusions
Ultrathin graphitic carbon nitride (g-C3N4) nanosheets were constructed by a post-thermal treatment of urea-derived g-C3N4 under an argon atmosphere. This process exfoliates g-C3N4 into ultrathin nanosheets with a high specific surface area of 131.2 m2 g−1 and a large pore volume of 1.06 cm3 g−1 in UGCN. The band-gap decreased from 2.86 eV to 2.80 eV, increasing the light-harvesting ability in the visible light region. Furthermore, the photogenerated charge carriers could be separated effectively. Compared to GCN, this post-treatment results in UGCN with a higher rate constant of 0.07346 min−1 and displayed 6.1 times higher than GCN for the degradation of RhB, and a two-fold increase in H2 evolution. The significant enhancement of photocatalytic properties are attributed to the higher accessible specific surface area, improved light harvesting and increased separation efficiency of photoexcited carriers. The outstanding photocatalytic performance described herein indicates that these materials show promising prospects as a high-performance photocatalyst for alleviating global environmental and energy issues.Acknowledgements
We gratefully acknowledge the financial support by the National Natural Science Foundation of China (no. 50872091 and 51102265) and Program of Discipline Leader of Colleges and Universities (Tianjin, China) and “Foreign Experts” Thousand Talents Program (Tianjin, China).Notes and references
- S. W. Zhang, Q. H. Fan, H. H. Gao, Y. S. Huang, X. Liu, J. X. Li, X. J. Xu and X. K. Wang, J. Mater. Chem. A, 2016, 4, 1414–1422 CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- J. H. Carey, J. Lawrence and H. M. Tosine, Bull. Environ. Contam. Toxicol., 1976, 16, 697–701 CrossRef CAS PubMed.
- L. Vayssieres, Y. Tachibana and J. R. Durrant, Nat. Photonics, 2012, 6, 511–518 CrossRef.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed.
- D. W. Hwang, J. Kim, T. J. Park and J. S. Lee, Catal. Lett., 2002, 80, 53–57 CrossRef CAS.
- L. Liao, Q. Zhang, Z. Su, Z. Zhao, Y. Wang, Y. Li, X. Lu, D. Wei, G. Feng and Q. Yu, Nat. Nanotechnol., 2013, 9, 69–73 CrossRef PubMed.
- P. V. Kamat, J. Phys. Chem. C, 2007, 111, 2834–2860 CAS.
- Q. Xiang, B. Cheng and J. Yu, Appl. Catal., B, 2013, 138–139, 299–303 CrossRef CAS.
- X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- X. Ding, Y. Li, J. Zhao, Y. Zhu, Y. Li, W. Deng and C. Wang, APL Mater., 2015, 3, 104410 CrossRef.
- X. Wang, S. Blechert and M. Antonietti, ACS Catal., 2012, 2, 1596–1606 CrossRef CAS.
- X. Wang, K. Maeda, X. Chen, K. Takanabe, K. Domen, Y. Hou, X. Fu and M. Antonietti, J. Am. Chem. Soc., 2009, 131, 1680–1681 CrossRef CAS PubMed.
- Z. Zhao, Y. Sun and F. Dong, Nanoscale, 2015, 7, 15–37 RSC.
- D. J. Martin, K. P. Qiu, S. A. Shevlin, A. D. Handoko, X. W. Chen, Z. X. Guo and J. W. Tang, Angew. Chem., Int. Ed., 2014, 53, 9240–9245 CrossRef CAS PubMed.
- Y. P. Zhu, T. Z. Ren and Z. Y. Yuan, ACS Appl. Mater. Interfaces, 2015, 7, 16850–16856 CAS.
- W. Yong, J. Zhang, X. Wang, A. Markus and H. Li, Angew. Chem., 2010, 49, 3356–3359 CrossRef PubMed.
- S. Zhang, J. Li, M. Zeng, G. Zhao, J. Xu, W. Hu and X. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 12735–12743 CAS.
- S. W. Zhang, J. X. Li, X. K. Wang, Y. S. Huang, M. Y. Zeng and J. Z. Xu, J. Mater. Chem. A, 2015, 3, 10119–10126 CAS.
- Y. Zhang, J. Liu, G. Wu and W. Chen, Nanoscale, 2012, 4, 5300–5303 RSC.
- X. H. Li, X. Wang and M. Antonietti, ACS Catal., 2012, 2, 2082–2086 CrossRef CAS.
- K. Dai, L. Lu, Q. Liu, G. Zhu, X. Wei, J. Bai, L. Xuan and H. Wang, Dalton Trans., 2014, 43, 6295–6299 RSC.
- X. F. Chen, Y. S. Jun, K. Takanabe, K. Maeda, K. Domen, X. Z. Fu, M. Antonietti and X. C. Wang, Chem. Mater., 2009, 21, 4093–4095 CrossRef CAS.
- F. Goettmann, A. Fischer, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2006, 45, 4467–4471 CrossRef CAS PubMed.
- X. Li, A. F. Masters and T. Maschmeyer, ChemCatChem, 2015, 7, 121–126 CrossRef CAS.
- A. Du, S. Sanvito, Z. Li, D. Wang, Y. Jiao, T. Liao, Q. Sun, Y. H. Ng, Z. Zhu, R. Amal and S. C. Smith, J. Am. Chem. Soc., 2012, 134, 4393–4397 CrossRef CAS PubMed.
- Y. Fukasawa, K. Takanabe, A. Shimojima, M. Antonietti, K. Domen and T. Okubo, Chem.–Asian J., 2011, 6, 103–109 CrossRef CAS PubMed.
- Y. Wang, X. Wang, M. Antonietti and Y. Zhang, ChemSusChem, 2010, 3, 435–439 CrossRef CAS PubMed.
- W. Shen, J. Mater. Chem., 2011, 21, 3890–3894 RSC.
- Y. W. Zhang, J. H. Liu, G. Wu and W. Chen, Nanoscale, 2012, 4, 5300–5303 RSC.
- Q. Lin, L. Li, S. Liang, M. Liu, J. Bi and W. Ling, Appl. Catal., B, 2015, 163, 135–142 CrossRef CAS.
- X. Zhang, X. Xie, H. Wang, J. Zhang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 18–21 CrossRef CAS PubMed.
- X. D. Zhang, H. X. Wang, H. Wang, Q. Zhang, J. F. Xie, Y. P. Tian, J. Wang and Y. Xie, Adv. Mater., 2014, 26, 4438–4443 CrossRef CAS PubMed.
- J. Xu, L. Zhang, R. Shi and Y. Zhu, J. Mater. Chem. A, 2013, 1, 14766–14772 CAS.
- W. Fan, Q. Zhang and Y. Wang, Phys. Chem. Chem. Phys., 2013, 15, 2632–2649 RSC.
- D. Zheng, C. Huang and X. Wang, Nanoscale, 2015, 7, 465–470 RSC.
- P. Niu, L. Zhang, G. Liu and H. M. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS.
- P. Qiu, H. Chen, C. Xu, N. Zhou, F. Jiang, X. Wang and Y. Fu, J. Mater. Chem. A, 2015, 3, 154–162 Search PubMed.
- Q. H. Liang, Z. Li, Z. H. Huang, F. Y. Kang and Q. H. Yang, Adv. Funct. Mater., 2015, 25, 6885–6892 CrossRef CAS.
- Y. Y. Kang, Y. Q. Yang, L. C. Yin, X. D. Kang, G. Liu and H. M. Cheng, Adv. Mater., 2015, 27, 4572–4577 CrossRef CAS PubMed.
- Y. Kang, Y. Yang, L.-C. Yin, X. Kang, L. Wang, G. Liu and H.-M. Cheng, Adv. Mater., 2016, 28, 6471–6477 CrossRef CAS PubMed.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J. O. Muller, R. Schlogl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC.
- B. V. Lotsch, M. Doblinger, J. Sehnert, L. Seyfarth, J. Senker, O. Oeckler and W. Schnick, Chem.–Eur. J., 2007, 13, 4969–4980 CrossRef CAS PubMed.
- F. Dong, Y. J. Sun, L. W. Wu, M. Fu and Z. B. Wu, Catal. Sci. Technol., 2012, 2, 1332–1335 CAS.
- F. Dong, Z. Y. Wang, Y. H. Li, W. K. Ho and S. C. Lee, Environ. Sci. Technol., 2014, 48, 10345–10353 CrossRef CAS PubMed.
- J. Liu, Y. Liu, N. Y. Liu, Y. Z. Han, X. Zhang, H. Huang, Y. Lifshitz, S. T. Lee, J. Zhong and Z. H. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- J. Zhuang, W. Dai, Q. Tian, Z. Li, L. Xie, J. Wang, P. Liu, X. Shi and D. Wang, Langmuir, 2010, 26, 9686–9694 CrossRef CAS PubMed.
- G. Liu, L. Wang, C. Sun, X. Yan, X. Wang, Z. Chen, S. C. Smith, H.-M. Cheng and G. Q. Lu, Chem. Mater., 2009, 21, 1266–1274 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work and should be considered co-first authors. |
| This journal is © The Royal Society of Chemistry 2017 |