
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentifying a cobalt catalyst for highly selective hydrosilylation of allenes†
Zheng
Yang‡
ab,
Dongjie
Peng‡
ab,
Xiaoyong
Du
ab,
Zheng
Huang
*ab and
Shengming
Ma
 *abc
*abc
aState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Lu, Shanghai 200032, P. R. China. E-mail: masm@sioc.ac.cn; huangzh@sioc.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
cDepartment of Chemistry, Fudan University, 220 Handan Lu, Shanghai 200433, P.R. China
First published on 18th July 2017
Abstract
An efficient method of cobalt-catalyzed allene-hydrosilylation is developed. The reaction enjoys an excellent regio- and stereoselectivity and a broad scope affording Z-allylic silanes. Many synthetically useful functional groups can be tolerated. A Co(I)-species involved mechanism is proposed.
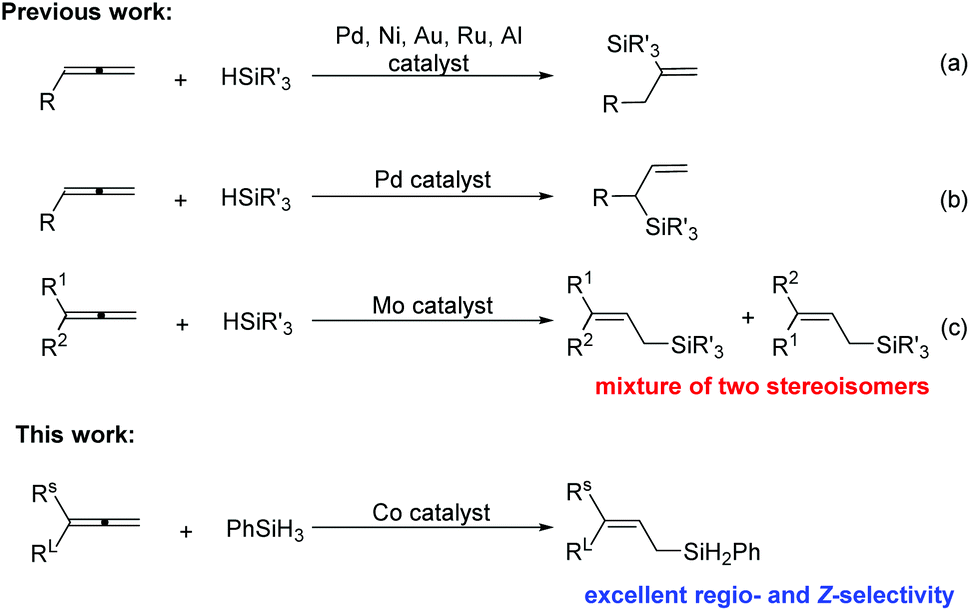
Featuring reasonable stability, low-toxicity, and ease of handling, silane reagents are valuable intermediates for synthetic transformations, such as Hiyama cross-couplings1 and Tamao oxidation reactions to form the carbonyl group.2 The hydrosilylation of unsaturated C–C bonds such as alkenes,3 alkynes,4 and dienes,5 which is considered to be one of the most synthetically efficient approaches to organosilane compounds, has been achieved with various catalysts. However, reports on the hydrosilylation of allenes are relatively rare, partially due to the difficulty in controlling the regio- and stereoselectivity. Most of the reported allene hydrosilylations occurred at the non-terminal C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, affording vinylsilanes using Pd,6 Ni,7 Au,8 Ru9 or Al10 catalysis (Scheme 1a) or branched allylsilanes using Pd6,7 catalysis (Scheme 1b). In 2016, Asako and Takai reported a molybdenum-catalyzed hydrosilylation at the terminal CC bonds of allenes that yielded linear allylic silanes (Scheme 1c).11 However, the stereoselectivity of this reaction is rather poor. To the best of our knowledge, the allene hydrosilylation catalyzed by earth-abundant base metal iron or cobalt has not been established yet. Here we report a cobalt-catalyzed hydrosilylation of allenes to form linear (Z)-allylsilanes, enjoying an excellent regio- and stereoselectivity.
C bond, affording vinylsilanes using Pd,6 Ni,7 Au,8 Ru9 or Al10 catalysis (Scheme 1a) or branched allylsilanes using Pd6,7 catalysis (Scheme 1b). In 2016, Asako and Takai reported a molybdenum-catalyzed hydrosilylation at the terminal CC bonds of allenes that yielded linear allylic silanes (Scheme 1c).11 However, the stereoselectivity of this reaction is rather poor. To the best of our knowledge, the allene hydrosilylation catalyzed by earth-abundant base metal iron or cobalt has not been established yet. Here we report a cobalt-catalyzed hydrosilylation of allenes to form linear (Z)-allylsilanes, enjoying an excellent regio- and stereoselectivity.
 | ||
| Scheme 1 Hydrosilylation of allenes: previous work and our observation. | ||
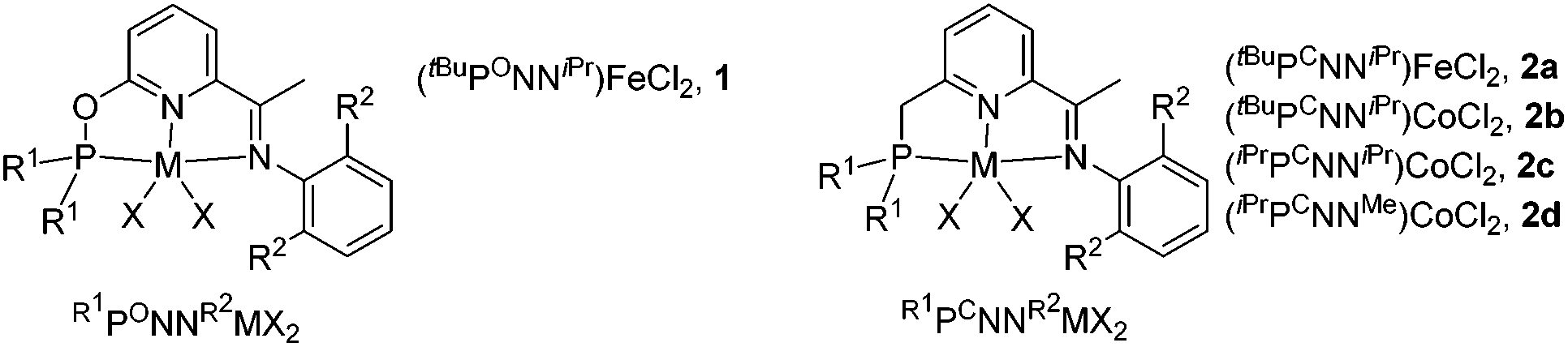
Non-precious metal iron and cobalt catalysts in general offer lower activity than Pd and other precious metal catalysts, thus identifying that a suitable ligand is the key to achieve an efficient and selective hydrosilylation of allenes. We employed iron and cobalt complexes of phosphinite-iminopyridine12 (PONN) and phosphine-iminopyridine13 (PCNN) ligands developed in one of our laboratories (Chart 1), which exhibited an excellent catalytic activity in the hydrosilylation of alkenes.
 | ||
| Chart 1 Fe and Co complexes with PONN and PCNN ligands. | ||
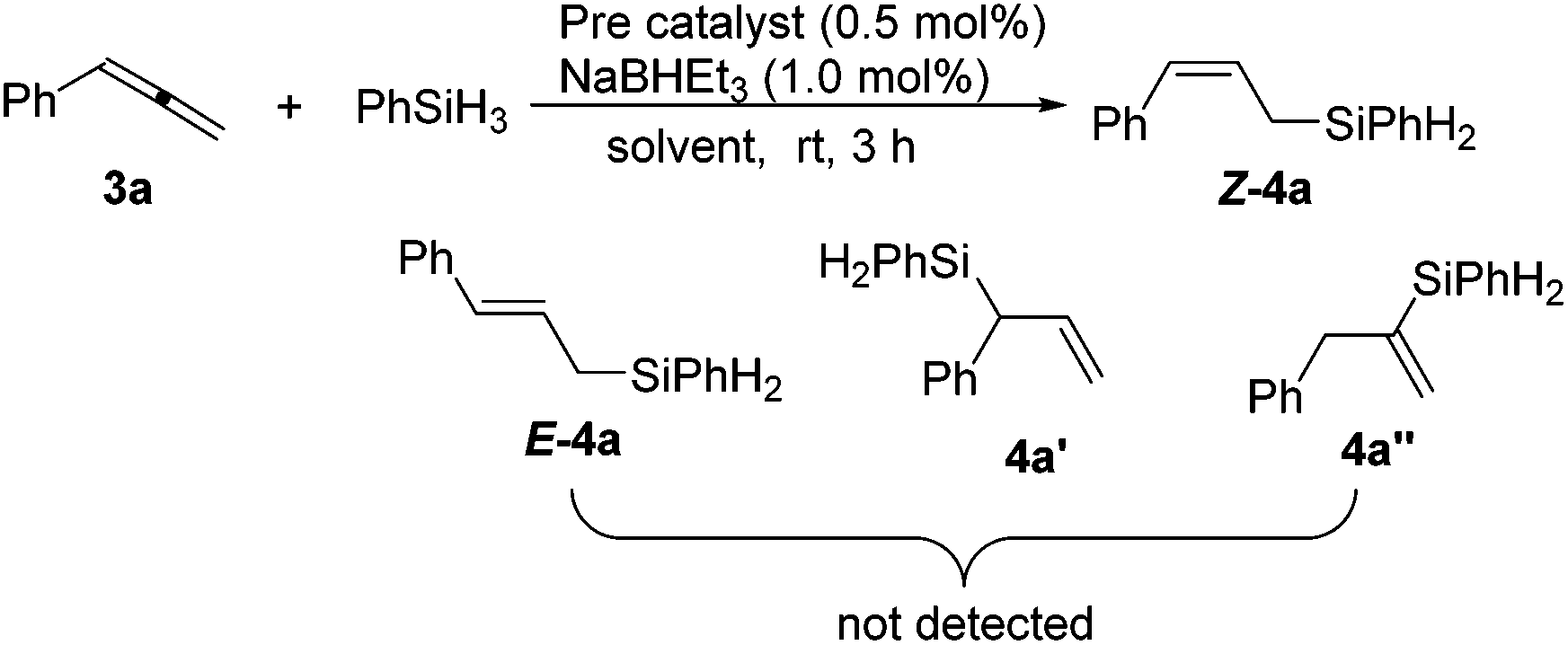
We commenced the study by examining an iron catalyst generated from (tBuPONNiPr)FeCl2 (1) and NaBHEt3 for the hydrosilylation of phenyl allene (3a) with phenylsilane. The initial run in toluene using only 0.5 mol% of the catalyst yielded (Z)-phenyl(3-phenylallyl)silane Z-4a in 93% yield as determined by 1H NMR analysis. Neither the regioisomers 4a′ and 4a′′ nor the stereoisomer E-4a was detected. Solvent variations have a large effect on the cobalt-mediated hydrosilylation. The yield dropped when the reaction was conducted in THF, hexane or acetonitrile (Table 1, entries 2–4), while the reaction did not occur in dichloromethane or DMF (Table 1, entries 5 and 6). Next, we screened a series of complexes of Fe and Co ligated by PCNN ligands (Table 1, entries 7–10) which revealed that (tBuPCNNiPr)CoCl2 (2b) gave the best result (Table 1, entry 8) and the steric effect of the complexes greatly affects the reaction. Lower yields were observed in the runs using the complexes with less bulky substituents on the ligands (Table 1, entries 9 and 10). No conversion was observed when running the reaction in the absence of the metal complex or NaBHEt3, or replacing cobalt complexes with CoCl2, indicating the important effect of the ligand on the hydrosilylation (Table 1, entries 11–13). Notably, a minor impurity was observed in the isolated product when using complex 1 as the precatalyst (entry 1), while the reaction with the precatalyst 2b afforded the product in a very high purity (entry 8). Therefore, the parameters used in entry 8 have been chosen as the optimized reaction conditions for further study.
|
|
||||
|---|---|---|---|---|
| Entry | Precatalyst | Solvent | Yield of (Z)-4ab (%) | Recovery of 3ab (%) |
| a Reaction conditions: 3a (0.5 mmol), PhSiH3 (0.5 mmol), 2.5 μmol of precatalyst, 5.0 μmol of NaBHEt3, in solvent (0.5 mL). b Determined by 1H NMR analysis with CH3NO2 as the internal standard. Values in parentheses are yields of the isolated products. c No NaBHEt3 was added. d Isolated recovery of 3a. | ||||
| 1 | 1 | Toluene | 93 (91) | — |
| 2 | 1 | THF | 70 | — |
| 3 | 1 | Hexane | 81 | — |
| 4 | 1 | MeCN | 9 | 62 |
| 5 | 1 | DCM | 0 | 76 |
| 6 | 1 | DMF | 0 | 64 |
| 7 | 2a | Toluene | 34 | 44 |
| 8 | 2b | Toluene | 94 (92) | — |
| 9 | 2c | Toluene | 54 | 25 |
| 10 | 2d | Toluene | 13 | 39 |
| 11 | — | Toluene | 0 | 94 |
| 12c | 2b | Toluene | 0 | 82 |
| 13 | CoCl2 | Toluene | 0 | 89d |
Next, we investigated the scope of the cobalt-catalyzed hydrosilylation with respect to the allene substrates. All the reactions of 3-aryl or alkyl substituted 1,2-dienes employed 0.5 mol% 2b as the precatalyst, furnishing linear (Z)-allylsilanes in high isolated yields with an excellent (Z)-selectivity (Schemes 2 and 3). Aryl-substituted allenes bearing either electron-donating (4b and 4g) or electron-withdrawing groups (4c) were hydrosilylated with high regio- and stereoselectivity, and substituents in the ortho, meta, and para positions of the phenyl ring are compatible with the reaction conditions (4d–f). The naphthyl substituent is also tolerated, giving a high yield of 4h and no isomeric product was observed (Scheme 2).
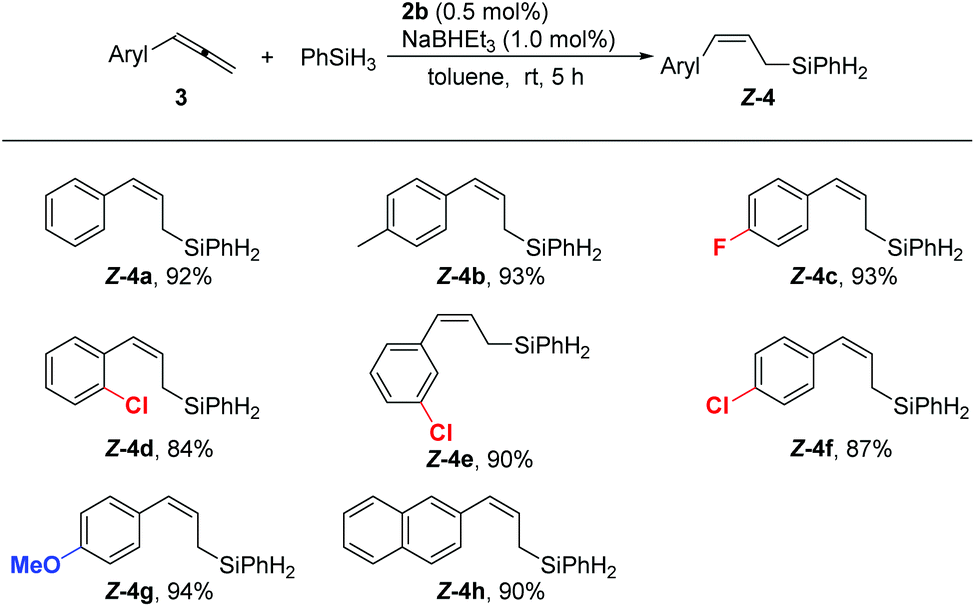 | ||
| Scheme 2 Highly regio- and stereoselective hydrosilylation of aryl substituted allenes. The reaction was carried out with 1.0 mmol of 3, 1.0 mmol of PhSiH3, 5.0 μmol of 2b, 10.0 μmol of NaBHEt3, in 1 mL toluene at room temperature for 5 h. Yields of the isolated products are given. | ||
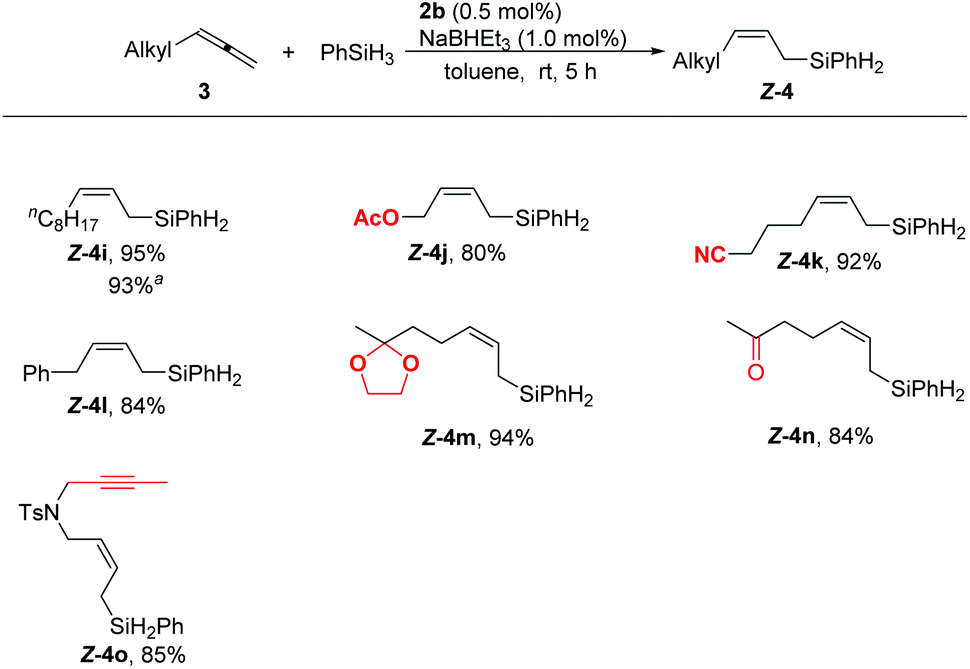 | ||
Scheme 3 Highly regio- and stereoselective hydrosilylation of alkyl substituted allenes. The reaction was carried out with 1.0 mmol of 3, 1.0 mmol of PhSiH3, 5.0 μmol of 2b, 10.0 μmol of NaBHEt3, in 1 mL toluene at room temperature for 5 h. Yields of the isolated products are given. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) The reaction was carried out with 5 mmol of 3i to afford 1.2 g of Z-4i. The reaction was carried out with 5 mmol of 3i to afford 1.2 g of Z-4i. | ||
Alkyl substituted allenes also reacted with phenylsilane smoothly under the standard reaction conditions. Synthetically useful functional groups such as acetoxy (4j), cyano (4k), benzyl (4l), and ketal (4m) could be tolerated. Notably, even the reactive acetyl (4n) could be accommodated in this hydrosilylation reaction. Furthermore, the allene showed a higher reactivity than the internal alkyne as demonstrated by the isolation of 4o in a high yield with an exclusive chemoselectivity towards the allene unit. No side-products resulting from the hydrosilylation of the acetyl and internal alkyne groups were observed in these reactions. Moreover, these reactions could be carried out on a one-gram scale, affording 4i in 93% yield.
The reaction of unsymmetric 1,1-disubstituted allene 3p also proceeded smoothly to afford linear allylsilanes Z-4p in a decent yield (Scheme 4). Significantly, only the Z-isomer was formed, demonstrating the capability of the cobalt catalyst to discriminate between Me and a larger substituent in the 1,1-disubstituted allene substrate.
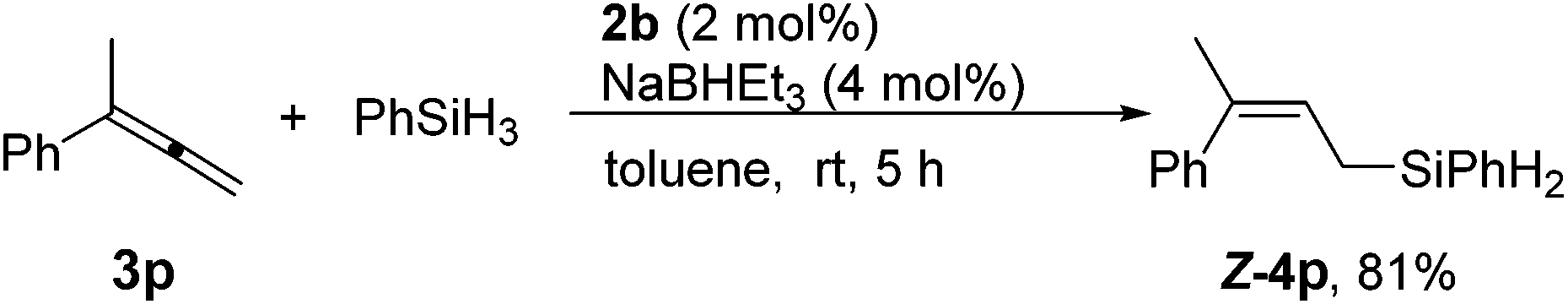 | ||
| Scheme 4 Highly regio- and stereoselective hydrosilylation of 1,1-disubstituted allenes. | ||
Other silanes such as Ph2SiH2, Et2SiH2, and Et3SiH were also tried for this reaction but only complicated mixtures were obtained.
We propose a rationale for the cobalt-catalyzed hydrosilylation of allenes on the basis of the precedents of the relevant (PCNN)Co-catalyzed alkene and alkyne hydrosilylations13,14 (Scheme 5). The hydrosilylation process starts with the activation of 2b by using NaBHEt3, followed by the reaction with PhSiH3 to form a cobalt(I) silyl intermediate Int-1.15 Most likely due to the steric repulsion between the PCNN ligand and the substituent groups of the allenes, the terminal CC double bond of substrate 3 coordinates to the metal center of Int-1 from the less hindered side, resulting in the generation of the allene adduct Int-2. The subsequent insertion of the CC double bond into the Co–Si bond would afford the vinyl cobalt intermediate Int-3 with the Co center located at the cis position relative to the small substituent due to the steric effect. The vinyl complex Int-3 then reacts with PhSiH3, via either sigma-bond metathesis or a silane oxidative addition/reductive elimination pathway, to deliver the hydrosilylation product and regenerate Int-1. Alternatively, the catalytic process may involve a cobalt(I) hydride intermediate Int-1′.16 The corresponding allene adduct Int-2′ then undergoes insertion to form the allyl cobalt intermediate Int-3′, which further reacts with PhSiH3 to form the desired product and regenerate the cobalt(I) hydride. Further detailed mechanistic studies are ongoing to establish unambiguously the real mechanistic nature of the reaction.
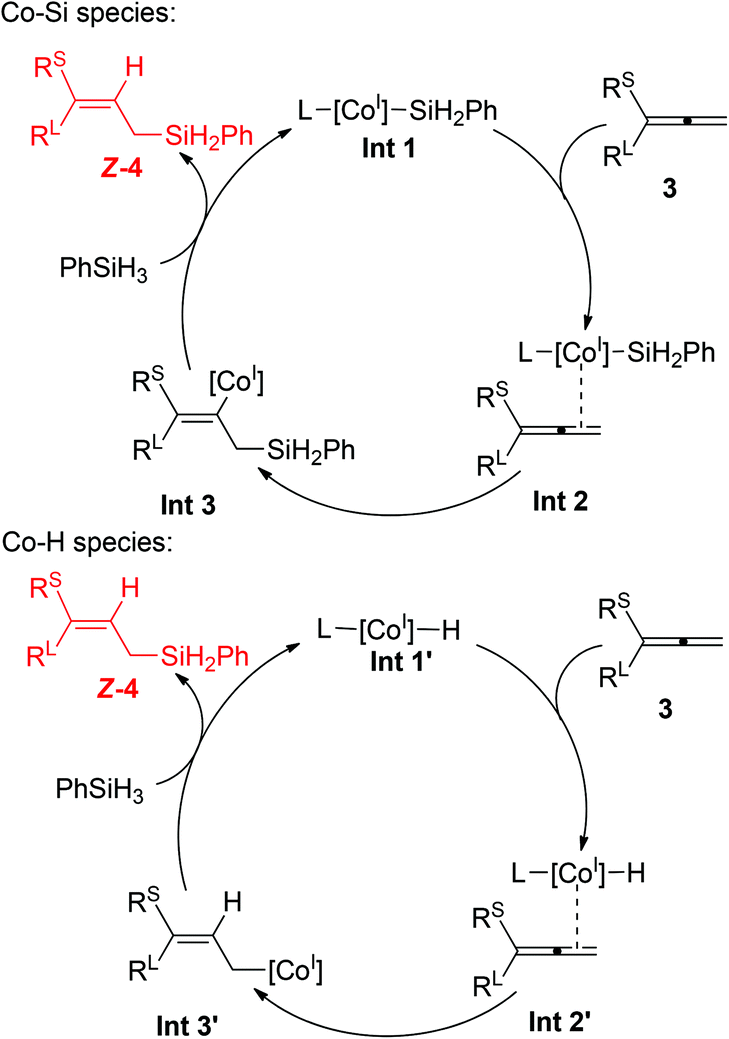 | ||
| Scheme 5 Proposed mechanisms. | ||
In conclusion, we have developed a highly regio- and stereoselective cobalt-catalyzed allene-hydrosilylation method for the synthesis of linear (Z)-allylsilanes. Both mono and 1,1-disubstituted allenes are applicable for this transformation and a variety of synthetically useful functional groups could be tolerated. Further investigations including mechanistic studies and synthetic applications of the allylsilane products have been pursued in this laboratory.
Acknowledgements
Financial support from the National Natural Science Foundation of China (21690063 for S. Ma; 21432011 and 21422209 for Z. Huang) is greatly appreciated. We thank Mr Guangyu Li and Mr Huping Zhu for helpful NMR spectra characterization and analysis. We also thank Yizhan Zhai for reproducing the results of Z-4e, Z-4j, and Z-4k.Notes and references
- (a) Y. Hatanaka and T. Hiyama, J. Org. Chem., 1988, 53, 918 CrossRef CAS; (b) Y. Nakao and T. Hiyama, Chem. Soc. Rev., 2011, 40, 4893 RSC.
- (a) K. Tamao, M. Kumada and K. Maeda, Tetrahedron Lett., 1984, 25, 321 CrossRef CAS; (b) D. Limnios and C. G. Kokotos, ACS Catal., 2013, 3, 2239 CrossRef CAS.
- For selected examples, see: (a) I. Buslov, J. Becouse, S. Mazza, M. Montandon-Clerc and X. Hu, Angew. Chem., Int. Ed., 2015, 54, 14523 CrossRef CAS PubMed; (b) A. M. Tondreau, C. C. H. Atienza, K. J. Weller, S. A. Nye, K. M. Lewis, J. G. P. Delis and P. J. Chirik, Science, 2012, 335, 567 CrossRef CAS PubMed; (c) O. Buisine, G. Berthon-Gelloz, J.-F. Brière, S. Stérin, G. Mignani, P. Branlard, B. Tinant, J.-P. Declercq and I. E. Markó, Chem. Commun., 2005, 3856 RSC; (d) A. J. Chalk and J. F. Harrod, J. Am. Chem. Soc., 1967, 89, 1640 CrossRef CAS; (e) J. L. Speier, J. A. Webster and G. H. Barnes, J. Am. Chem. Soc., 1957, 79, 974 CrossRef CAS.
- For selected examples, see: (a) D. A. Rooke and E. M. Ferreira, Angew. Chem., Int. Ed., 2012, 51, 3225 CrossRef CAS PubMed; (b) G. Berthon-Gelloz, J. Schumers, G. De Bo and I. E. Markó, J. Org. Chem., 2008, 73, 4190 CrossRef CAS PubMed; (c) M. R. Chaulagain, G. M. Mahandru and J. Montgomery, Tetrahedron, 2006, 62, 7560 CrossRef CAS; (d) Z. T. Ball and B. M. Trost, J. Am. Chem. Soc., 2005, 127, 17644 CrossRef PubMed; (e) S. E. Denmark and Z. G. Wang, Org. Lett., 2001, 3, 1073 CrossRef CAS PubMed; (f) B. M. Trost and Z. T. Ball, J. Am. Chem. Soc., 2001, 123, 12726 CrossRef CAS PubMed.
- For selected examples, see: (a) J. Y. Wu, B. N. Stanzl and T. Ritter, J. Am. Chem. Soc., 2010, 132, 13214 CrossRef CAS PubMed; (b) S. Onozawa, T. Sakakura and M. Tanaka, Tetrahedron Lett., 1994, 35, 8177 CrossRef CAS; (c) I. Ojima and M. Kumagai, J. Organomet. Chem., 1978, 157, 359 CrossRef CAS; (d) J. Tsuji, M. Hara and K. Ohno, Tetrahedron, 1974, 30, 2143 CrossRef CAS.
- (a) Z. D. Miller and J. Montgomery, Org. Lett., 2014, 16, 5486 CrossRef CAS PubMed; (b) H. Tafazolian and J. A. R. Schmidt, Chem. Commun., 2015, 51, 5943 RSC.
- Z. D. Miller, W. Li, T. R. Belderrain and J. Montgomery, J. Am. Chem. Soc., 2013, 135, 15282 CrossRef CAS PubMed.
- M. Kidonakis and M. Stratakis, Org. Lett., 2015, 17, 4538 CrossRef CAS PubMed.
- S. Kang, Y. Hong, J. Lee, W. Kim, I. Lee and C. Yu, Org. Lett., 2003, 5, 2813 CrossRef CAS PubMed.
- T. Sudo, N. Asao, V. Gevorgyan and Y. Yamamoto, J. Org. Chem., 1999, 64, 2494 CrossRef CAS.
- S. Asako, S. Ishikawa and K. Takai, ACS Catal., 2016, 6, 3387 CrossRef CAS.
- D. Peng, Y. Zhang, X. Du, L. Zhang, X. Leng, M. D. Walter and Z. Huang, J. Am. Chem. Soc., 2013, 135, 19154 CrossRef CAS PubMed.
- X. Du, Y. Zhang, D. Peng and Z. Huang, Angew. Chem., Int. Ed., 2016, 55, 6671 CrossRef CAS PubMed.
- X. Du, W. Hou, Y. Zhang and Z. Huang, Org. Chem. Front., 2017 10.1039/c7qo00250e.
- Note that Co(I) silyl complexes have been documented. (a) Z. Mo, J. Xiao, Y. Gao and L. Deng, J. Am. Chem. Soc., 2014, 136, 17414 CrossRef CAS PubMed; (b) C. C. H. Atienza, T. Diao, K. J. Weller, S. A. Nye, K. M. Lewis, J. G. P. Delis, J. L. Boyer, A. K. Roy and P. J. Chirik, J. Am. Chem. Soc., 2014, 136, 12108 CrossRef CAS PubMed.
- Related pincer-ligated Co(I) hydride species have been reported. (a) Z. Zhang and Z. Huang, J. Am. Chem. Soc., 2015, 137, 15600 CrossRef PubMed; (b) See also ref. 15b.
Footnotes |
| † Electronic supplementary information (ESI) available: Preparation and characterisation data as well as 1H and 13C NMR spectra of all compounds. See DOI: 10.1039/c7qo00497d |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2017 |