
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ luminescence analysis: a new light on monitoring calcium phosphate phase transitions†‡
H.
Terraschke
 *a,
M.
Rothe
a,
A.-M.
Tsirigoni
a,
P.
Lindenberg
a,
L.
Ruiz Arana
a,
N.
Heidenreich
ab,
F.
Bertram
b and
M.
Etter
b
*a,
M.
Rothe
a,
A.-M.
Tsirigoni
a,
P.
Lindenberg
a,
L.
Ruiz Arana
a,
N.
Heidenreich
ab,
F.
Bertram
b and
M.
Etter
b
aInstitut für Anorganische Chemie, Christian-Albrechts-Universität zu Kiel, Max-Eyth-Str. 2, 24118 Kiel, Germany. E-mail: hterraschke@ac.uni-kiel.de
bDESY Photon Science, Notkestr. 85, 22607 Hamburg, Germany
First published on 5th May 2017
Abstract
In this work, in situ luminescence analysis was applied for the first time for monitoring the phase transitions of calcium phosphate (CaP) and confirmed by synchrotron in situ X-ray diffraction in addition to in situ infrared spectroscopy, with simultaneous measurements of pH and ion conductivity. Applying doped Ce3+ and Eu3+ as local coordination sensors, the high sensitivity of their emission spectra upon the changes in the coordination sphere of the doped cation sites enabled to detect the formation of amorphous calcium phosphate (ACP) and Ca5(PO4)3OH, besides their subsequent transitions to CaHPO4·2H2O and Ca8H2(PO4)6·5H2O under real reaction conditions. Calcium phosphates are widely present in mammals and understanding their phase transitions is important to comprehend the conversion between healthy and diseased tissues. In situ luminescence measurements are advantageous for allowing monitoring these phase transitions in a fast and sensitive fashion also in conventional laboratories, independent of synchrotron radiation.
1. Introduction
Efficiently monitoring the events occurring during the formation of solids in solutions including nucleation, crystal growth and formation of reaction intermediates as well as consequent phase transitions culminating in the crystallization of the final product require the application of in situ characterization techniques under real reaction conditions.1–10 Studying the mechanism of chemical reactions by removing samples from the reactor during the synthesis process and analysing them ex situ is rather disadvantageous for offering only snapshots of the process with very limited time resolution, besides probably influencing the sample while preparing them for the ex situ analysis.1,2 The formation and phase transitions of calcium phosphate (CaP) systems offer a critical example for the importance of applying in situ techniques. Calcium phosphate derivatives are one of the main inorganic components in mammals and are widely used for producing medical implants and prosthesis.11,12 For instance, amorphous calcium phosphate (ACP) is found in pathological tissues like heart valve calcifications,11 CaHPO4·2H2O is proposed as an intermediate in bone mineralization,11 Ca8H2(PO4)6·5H2O is present in human dental and urinary calculi,13 CaHPO4 is used in bone cements,14 while Ca5(PO4)3OH is used for coating orthopaedic and dental implants.15 Therefore, understanding the formation and transition of the CaP phases is important for comprehending the transitions between healthy and diseased tissue as well as for predicting and preventing the degradation of implants and prosthesis in our bodies. In addition, studying the formation of CaP is essential for understanding the formation of amorphous pre-nucleation clusters, which is still a challenge for the currently accepted nucleation theories.16–18 Since the phase transitions of CaP are extremely sensitive to variations in the environmental conditions such as temperature, pH and concentration, they should be ideally studied only in situ under real reaction conditions, in order to not disturb the investigated processes.12 Up to now, different powerful in situ techniques have been reported in the literature for monitoring the mineralization mechanisms of CaP systems, as recently summarized by Pan et al.12 Some of these in situ techniques are in situ pH measurements,19 UV/Vis absorption spectroscopy,20 Raman,21 turbidity,22 quartz crystal microbalance (QCM),23 X-ray diffraction (XRD)24 and X-ray absorption spectroscopy (XAS).25 However, many of these techniques provide information about phenomena occurring in the solution and not in the solid material, offer reduced time resolution or depend on synchrotron radiation, limiting their availability. A promising technique for monitoring chemical reactions consists of the in situ luminescence analysis of coordination sensors (ILACS) approach.26 Within this technique, lanthanide ions as Eu3+, Ce3+ and Tb3+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27–29 are incorporated into the investigated materials during synthesis as local coordination sensors. Due to the sensitivity of their spectroscopic properties assigned to 5d → 4f or 4f → 4f electronic transitions to the coordination environment,30,31 changes in the coordination of the cation sites during nucleation, crystal growth and phase transitions can be detected by measuring in situ luminescence applying fast charge-coupled device (ccd)-based detectors under real reaction conditions.26 The fine structure and intensities of the luminescence bands assigned to the 4f → 4f electronic transitions e.g. the 5D0 → 7F0–6 in Eu3+ are often applied as a spectroscopic probe for the local environment of these ions, especially due to the slight influence of the crystal field splitting on the respective energy levels.32 However, a very high resolution of the luminescence spectra of the 4f → 4f transitions is necessary for analyzing the splitting behavior of their energy levels in detail and acquiring comprehensive information, for example, about the point symmetry and coordination polyhedron,33 requiring often cooling the samples to liquid helium or liquid nitrogen temperatures. The parity allowed and highly intensive 5d → 4f electronic transitions e.g. in Ce3+, on the other hand, are largely influenced by the covalence of the ligands (nephelauxetic effect) and crystal field splitting around the emissive ions.34 Therefore, the luminescence bands assigned to the 5d → 4f transitions can vary from the UV to the red spectral range, depending on the host lattice and are able to reflect slight changes in the coordination environment around the emissive ions,35 being ideally applied as a coordination sensor. Due to the above-mentioned reasons, the ILACS method is able to characterize not only ions in solution and amorphous or crystalline materials with high sensitivity and time resolution, but can be also flexibly applied at conventional laboratories or synchrotron facilities, for complementing other characterization techniques.26
27–29 are incorporated into the investigated materials during synthesis as local coordination sensors. Due to the sensitivity of their spectroscopic properties assigned to 5d → 4f or 4f → 4f electronic transitions to the coordination environment,30,31 changes in the coordination of the cation sites during nucleation, crystal growth and phase transitions can be detected by measuring in situ luminescence applying fast charge-coupled device (ccd)-based detectors under real reaction conditions.26 The fine structure and intensities of the luminescence bands assigned to the 4f → 4f electronic transitions e.g. the 5D0 → 7F0–6 in Eu3+ are often applied as a spectroscopic probe for the local environment of these ions, especially due to the slight influence of the crystal field splitting on the respective energy levels.32 However, a very high resolution of the luminescence spectra of the 4f → 4f transitions is necessary for analyzing the splitting behavior of their energy levels in detail and acquiring comprehensive information, for example, about the point symmetry and coordination polyhedron,33 requiring often cooling the samples to liquid helium or liquid nitrogen temperatures. The parity allowed and highly intensive 5d → 4f electronic transitions e.g. in Ce3+, on the other hand, are largely influenced by the covalence of the ligands (nephelauxetic effect) and crystal field splitting around the emissive ions.34 Therefore, the luminescence bands assigned to the 5d → 4f transitions can vary from the UV to the red spectral range, depending on the host lattice and are able to reflect slight changes in the coordination environment around the emissive ions,35 being ideally applied as a coordination sensor. Due to the above-mentioned reasons, the ILACS method is able to characterize not only ions in solution and amorphous or crystalline materials with high sensitivity and time resolution, but can be also flexibly applied at conventional laboratories or synchrotron facilities, for complementing other characterization techniques.26
In this work, the ILACS approach utilizes the 5d → 4f transitions of Ce3+ and the 4f → 4f transitions of Eu3+ for monitoring the formation of ACP and Ca5(PO4)3OH as well as their conversion to CaHPO4·2H2O and Ca8H2(PO4)6·5H2O. These processes have been confirmed here by ex situ and synchrotron-based in situ XRD, besides in situ measurements of pH value, ion conductivity and infrared (IR) spectroscopy. To the best of our knowledge, the application of in situ luminescence and IR techniques for monitoring CaP phase transitions as well as the optical properties of Ce3+ and Eu3+-doped CaHPO4·2H2O and Ca8H2(PO4)6·5H2O are reported here for the first time.
2. Experimental
For the synthesis of the different calcium phosphate phases, Ca(NO3)2·4H2O (98.5+%, Merck KGaA, Darmstadt, Germany), Eu(NO3)3·6H2O (99.9%, ChemPur Feinchemikalien und Forschungsbedarf GmbH, Karlsruhe, Germany), Ce(NO3)3·6H2O (99.99%, Alfa Aesar GmbH & Co KG, Karlsruhe, Germany) and anhydrous (NH4)2HPO4 (99+%, Merck KGaA, Darmstadt, Germany) have been used without further purification. The synthesis methods reported in this work consist of simplified co-precipitation techniques and the solutions have been freshly prepared for every trial, in which the (Ca,Ln):PO43− ratio was approximately 3:2. The concentrations and temperatures used specifically for each experiment are listed in Table 1. A detailed explanation of the assemblies used on experiments at the University of Kiel and at the Deutsches Elektronen-Synchrotron (DESY, Hamburg, Germany), respectively Setup A and Setup B, is supplied in the ESI.‡
| Experiment number | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| Added Ca(NO3)2·4H2O/mmol | 1.06 | 1.06 | 1.06 | 1.06 | 1.06 | 1.06 | 1.06 | 3.53 | 7.60 | 6.33 |
| Added Ln(NO3)3·6H2O/mmol | 0.00 | 0.03 | 0.03 | 0.05 | 0.08 | 0.05 | 0.05 | 0.19 | 0.40 | 0.48 |
| Volume of Ca/Ln solution/ml | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 10 | 5 | 5 |
| Ce3+ doping concentration | 0% | 3% | 3% | 5% | 7% | — | — | — | — | 7% |
| Eu3+ doping concentration | — | — | — | — | — | 3% | 5% | 5% | 5% | — |
| Added (NH4)2HPO4/mmol | 0.7 | 0.7 | 0.7 | 0.7 | 0.7 | 0.7 | 0.7 | 2.9 | 5.3 | 4.5 |
| Volume of (NH4)2HPO4 solution/ml | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 40 | 40 |
| Temperature/°C | 10–80 | 10–80 | 10–80 | 10–80 | 10–80 | 10–80 | 10–80 | 10–80 | ∼30–90 | ∼30–90 |
| Excitation wavelength/nm | — | 280 | — | — | 280 | 395 | 395 | — | 365 | 365 |
| Emission wavelength/nm | — | — | 365 | — | — | — | — | — | — | — |
| Beamline | — | — | — | — | — | — | — | — | P08 | P02.1 |
| Energy of synchrotron X-ray beam/keV | — | — | — | — | — | — | — | — | 25 | 60 |
2.1. Synthesis procedure with Setup A
In a typical synthesis procedure applying Setup A (experiments 1–8, Table 1), 20 mL of an aqueous solution of diammonium hydrogen phosphate are placed inside the reactor. Subsequently, 5–10 mL of an aqueous solution of calcium and cerium or europium nitrates were added to the reactor during the first 10 minutes of the reaction. During the dosing process, the temperature was kept at 10 °C and increased after few minutes to 80 °C. The reaction was then monitored by in situ measurements of pH value, ion conductivity, photoluminescence and infrared spectroscopy (Fig. S1 and S2‡). Because 80 °C is the temperature limit for the use of the pH and conductivity sensors, it is the highest temperature employed with this setup.2.2. Synthesis procedure with Setup B
Similar to Setup A, 5 mL of a solution of calcium and cerium or europium nitrate in water was added with a rate of 0.5 mL min−1 to the reactor containing 40 mL of an aqueous solution of diammonium hydrogen phosphate (experiments 9 and 10, Table 1). In this context, it is important to mention that since the DESY reactor holder does not comprise a cooling system, the initial temperature of the reactor content was 30–35 °C. This high temperature is most probably caused by additional heating effects of the synchrotron X-ray beams and external excitation source for the luminescence measurements. For the DESY experiments (Fig. S3 and S4‡), it was necessary to increase the volume of the (NH4)2HPO4 solution to 40 mL, in order to reach the filling volume required for the in situ XRD analysis applying the adapted glass reactor (Fig. S3 and S4‡). Within these experiments, the concentration of the solutions was also increased for improving the signal-to-noise ratio of the in situ XRD patterns.3. Results and discussion
3.1. Preliminary ex situ experiments
For obtaining preliminary information on the changes of the calcium phosphate phase transitions during synthesis, samples were removed from the reactor of Setup A at determined reaction times and analysed by ex situ X-ray diffraction. Ex situ XRD patterns show the initial formation of a mixture of amorphous calcium phosphate and Ca5(PO4)3OH phase, which increasingly crystallizes to Ca5(PO4)3OH11 (Scheme 1) with an advanced reaction time and transforms after 7–10 min to CaHPO4·2H2O, changing again to Ca8H2(PO4)6·5H2O for temperatures above 57 °C (Fig. S5 and S6a,‡ experiment 1, Table 1). This trend is maintained, doping the Ca2+ sites with Ce3+ as a coordination sensor for concentrations up to 3% (Fig. S6b,‡ experiment 2, Table 1). On increasing the doping concentration to 5% (Fig. S6c,‡ experiment 5, Table 1), the transition between Ca5(PO4)3OH and CaHPO4·2H2O is shifted to 10–15 min. Interestingly, on further increasing the doping concentration to 7% (Fig. S6d,‡ experiment 5, Table 1), an additional transition from Ca5(PO4)3OH to CaHPO4 at 7–10 min is observed, before the next conversion from CaHPO4 to CaHPO4·2H2O at 10–15 min. For 5% and 7% Ce3+, Ca8H2(PO4)6·5H2O was also formed after 50 min. In this context, it is important to mention that increasing the doping concentration to 7% Ce3+, caused the enhancement of the amorphisation, increasing the reaction period in which the ACP phase is stable. A probable explanation for this fact is the increase of the disorder on the calcium phosphate host lattice due to the introduction of Ce3+ ions within the Ca2+ site, due to their differences in ionic radii and charge.36 Moreover, additional experiments have been carried out, for testing the influence of changing the type of coordination sensors from Ce3+ (experiment 4, Table 1) to Eu3+ (experiment 7, Table 1) on the calcium phosphate phase transitions.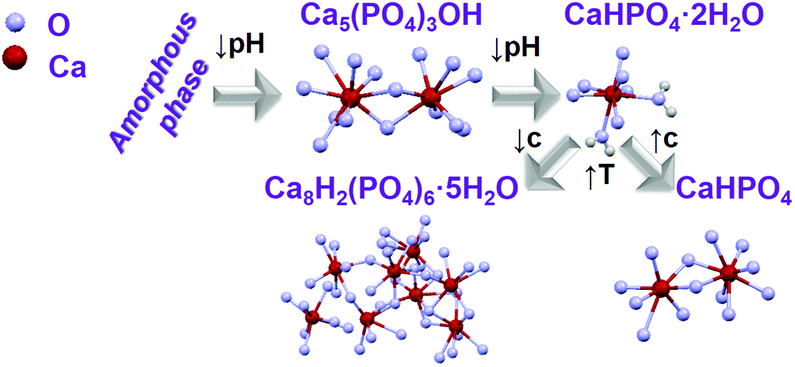 | ||
| Scheme 1 Schematic representation of transition between different calcium phosphate phases by changing synthesis parameters as pH, temperature (T) and concentration (c).11,16,37 | ||
As shown in Fig. S7,‡ the same phases have been obtained for the same reaction time such as a mixture of ACP and Ca5(PO4)3OH for t = 10 min as well as CaHPO4·2H2O for t = 15 min and t = 30 min, for doping concentrations of 5%, showing no significant differences in the powder X-ray diffraction patterns measured for both coordination sensors Ce3+ and Eu3+. In contrast, the concentration of the reactant solution strongly influences the calcium phosphate phase transitions (Fig. S8 and S9,‡ experiment 8, Table 1). On increasing the total concentration of the reactant solutions, reflections assigned to CaHPO4 are observed within the ACP and Ca5(PO4)3OH phases, before the conversion to CaHPO4·2H2O. On increasing the temperature of the CaHPO4·2H2O solution, the product loses two water molecules and is converted to anhydrous CaHPO4 (Scheme 1). Scanning electron microscopy (SEM) images (Fig. S10‡) show the development of elongated needle-formed crystals agglomerated in discrete bundles at reaction time t = 1 min, identified as an ACP and Ca5(PO4)3OH mixture by ex situ XRD (Fig. S8‡). At t = 20 min, isolated single needle-formed crystals with a diameter of ca. 600 nm are observed, together with laminated plates, corresponding to the ex situ identified CaHPO4·2H2O phase. At t = 60 min (CaHPO4), rod-shaped crystals are formed, and the needles are still observed. All the samples doped with Eu3+ show a homogeneous red luminescence (Fig. S11‡), indicating that the coordination sensor was incorporated for all reaction times and ex situ luminescence measurements of the 5D0 → 7F0 Eu3+ transition at ca. 580 nm show the double of the width of this peak for Ca5(PO4)3OH (two Ca2+ sites) in comparison with this peak measured for CaHPO4·2H2O (one Ca2+ site), most probably due to the different numbers on crystallographic doping sites for Eu3+ (Fig. S12‡).31
3.2. In situ luminescence experiments
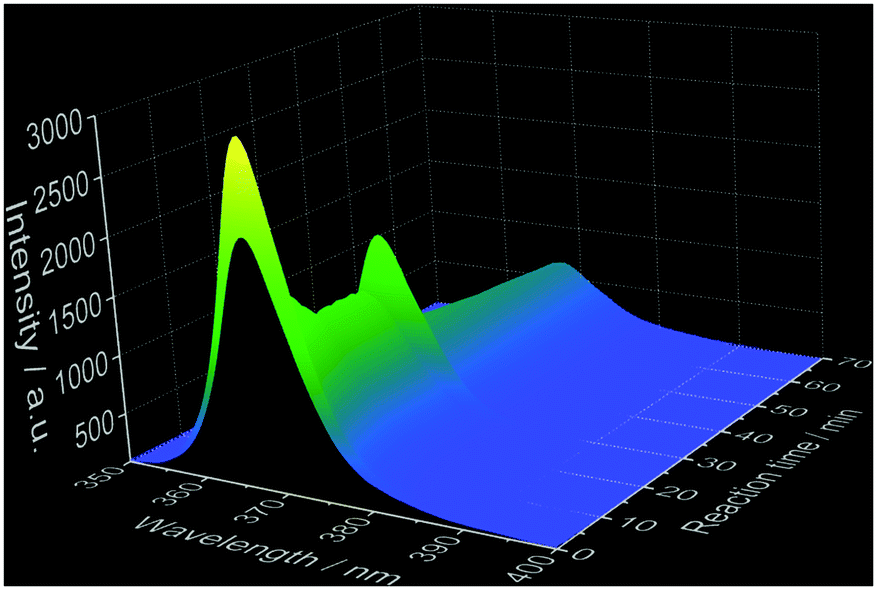
Ex situ characterization methods are helpful for gaining insights into the reaction mechanism, however, they consist only of discrete snapshots and do not deliver continuous information about the processes occurring inside the reaction. For instance, the ex situ XRD patterns presented here (Fig. S6–S8‡) show a time range where the phase transitions can occur, but not the exact time. In addition, the preparation procedure of the samples removed from the reactor for the ex situ characterization such as quenching, washing and drying might influence the product, generating possible divergences between the ex situ results and the actual phenomena occurring during the reactions.2 For this reason, in situ luminescence measurements applying 3% of doping concentration have been carried out for the coordination sensors Ce3+ and Eu3+ (experiments 2 and 6, Table 1) and confirmed by different in situ analysis techniques. The doping concentration of 3% was chosen here due to the non-influence on the calcium phosphate phase transitions in comparison with undoped samples, demonstrated by preliminary ex situ XRD analysis (Fig. S6‡).In general, as for the experiments applying Ce3+ as a coordination sensor (Fig. 1), no luminescence is initially detected upon the presence of pure (NH4)2HPO4 solution in the reactor. On addition of the calcium and cerium solution, a broad emission band between ca. 310 nm and 440 nm with a maximum at 353 nm constantly rises during the first 10 minutes of the reaction, indicating the formation of the product (Fig. 1), identified as a mixture of ACP and Ca5(PO4)3OH by ex situ XRD analysis. This emission range is attributed in the literature to the Ce3+ electronic transition from the lowest 5d state to the ground state levels 2F5/2 and 2F7/2 in, for instance, calcium hydroxylapatite (HAp).38 The non-symmetric shape of the Ce3+ band can be assigned to the simultaneous presence of secondary phases39e.g. ACP. After t = 13 min, the emission intensity starts to decrease and is slightly red shifted, corresponding to the transition to CaHPO4·2H2O indicated by ex situ XRD measurements (Fig. S6b‡).
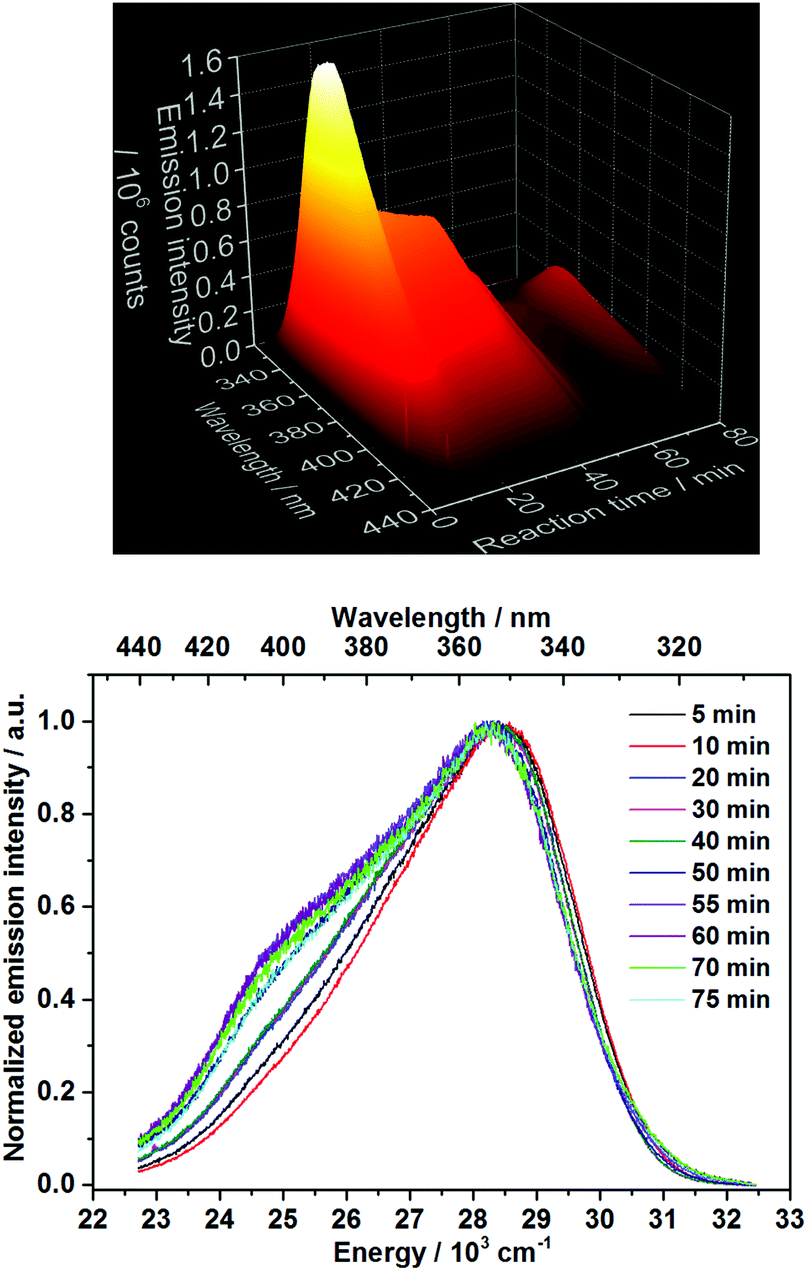 | ||
| Fig. 1 Top: In situ emission spectrum recorded during calcium phosphate phase transitions applying 3% Ce3+ as a coordination sensor (λex = 280 nm). Bottom: Normalized emission spectra for selected reaction times (experiment 2, Table 1). | ||
The decrease of the emission intensity after t = 13 min could be justified by two different explanations. The first possible explanation is the increased quenching effect of the Ln3+ luminescence, caused by the two coordinating water molecules on the doped Ca2+ site on CaHPO4·2H2O.40,41 Within the second possible explanation, since the Ce3+in situ emission spectra are recorded under a constant excitation at 280 nm, the decrease of the emission intensity could be caused by a shift of the excitation spectra during the conversion between different calcium phosphate phases. In order to verify this hypothesis, in situ excitation spectra (λem = 365 nm, experiment 3, Table 1) have been recorded under the same experimental conditions as Fig. 1 (Fig. S13–S15‡). As shown in Fig. S15,‡ the time-dependent profile of the in situ excitation spectra is very similar to the one of the in situ emission spectra and no significant shift in the excitation spectra is observed. Here, the decrease of the intensity of the excitation bands is singly related to the same intensity decrease of the respective emission band at 365 nm. Therefore, this hypothesis can be ruled out and the variation of the time-dependent emission intensity can be associated with the attachment and detachment of quenching water molecules within the coordination spheres of the lanthanide-based coordination sensors. The red shift of the Ce3+ emission band upon the formation of CaHPO4·2H2O, could indicate that this compound presents a higher coordination number or a shorter average bond length with the coordinating oxygen ions than in the previously formed ACP phase, since it is not the case for Ca5(PO4)3(OH).42 However, due to the lack of long-range order and Rontgen amorphous character of the ACP phase, very little structural information is available about this phase.43,44 In this context, it is important to note that ex situ XRD analysis shows the formation of CaHPO4·2H2O after t = 10 min (Fig. S6B‡), while in situ measurements locate this phase transition at t = 13 min. As observed in our previous work,45 the sample preparation conditions can lead to the ex situ conversion of the phase mixture to the stable CaHPO4·2H2O compound. Within the in situ luminescence measurements, the Ce3+ emission intensity remains approximately constant at t = 20–40 min, indicating no significant structural change in this time range, in agreement with the respective ex situ XRD patterns (Fig. S6b‡). After t = 40 min, the temperature starts to increase and the emission intensity starts to decrease, caused by thermal quenching effects and confirmed by the consecutive increase of the emission intensity after cooling the system down to room temperature (Fig. 1 and S5‡). At approximately, t = 51 min, the emission intensity slightly increases and decreases again, indicating additional structural changes in the cation environment and therefore a new phase transition by reaching the temperature of ca. 60 °C. This phase transition coincides with the results obtained by ex situ XRD analysis (Fig. S6b‡), showing the conversion from CaHPO4·2H2O to Ca8H2(PO4)6·5H2O between t = 50 and 55 min. The temperature of approximately 60 °C also coincides with the decomposition temperature of CaHPO4·2H2O on the in situ XRD data measured at the DESY synchrotron facility, discussed in detail below. The formation of the new phase is also indicated by the rise of an additional Ce3+ emission band at 24500 cm−1 (Fig. 1), which is not related to the increase of the temperature, since it is still observed after the system was cooled down to room temperature. The rise of additional Ce3+ emission bands is in agreement with the enhancement of crystallographic available Ca2+ doping sites for the coordination sensors, increasing from one Ca2+ site in CaHPO4·2H2O to eight Ca2+ sites on Ca8H2(PO4)6·5H2O.41 The red shift of the Ce3+ emission bands can be explained by the decrease of the average bond lengths between the calcium and coordinating oxygen ions within Ca8H2(PO4)6·5H2O,46 caused by the so-called nephelauxetic effect.39 Moreover, additional structural information can explain the spectroscopic behavior, in which the increase of the emission intensity at t = 51 min during the phase transition occurs most probably due to the decrease of the number of the quenching water molecules on the coordination sphere of the coordination sensor during the conversion from CaHPO4·2H2O to Ca8H2(PO4)6·5H2O, further decreasing afterwards due to thermal quenching effects.
Fig. 2 shows the time-dependent emission spectra applying Eu3+ as coordination sensors. Since these measurements have been carried out in solution, a strong quenching effect is observed, caused by the non-radiative depopulation of the excited states of Eu3+ due to the vibrational energy transfer involving the high energy vibrations of the OH oscillator from the H2O solvent molecules.47 This quenching effect results in the low intensity of the Eu3+ emission and consequent enhanced loss of resolution, differing therefore from previously reported emission spectra of Eu3+ doped ACP48 and Ca5(PO4)3OH.49–51 Additional causes for deviations in comparison with luminescence spectra of Eu3+-doped calcium phosphates reported in the literature are different synthesis methods applied and therefore different particle sizes, doping concentrations, measurement temperatures and excitation wavelengths. As mentioned above, no spectral data about the luminescence properties of Eu3+ doped CaHPO4·2H2O and Ca8H2(PO4)6·5H2O is available in the literature so far for comparison. The intensity of the 5D0 → 7FJ (J = 1–4) Eu3+ transitions shows the same behavior as for the Ce3+ experiments. Hence, the continuous increase of the intensity in the first 10 minutes (Fig. 3) indicates the formation of a solid material, assigned to a mixture of ACP and Ca5(PO4)3OH by ex situ XRD (Fig. S6b‡) followed by a decrease of intensity at t ≈ 13 min, indicating the conversion to CaHPO4·2H2O, most probably caused by the quenching effect of the two coordinating water molecules. Also for the Eu3+ coordination sensor, the intensity remains constant up to approximately t = 51 min, reaching the temperature of 60 °C, when an additional intensity oscillation indicates the additional phase transition to Ca8H2(PO4)6·5H2O.
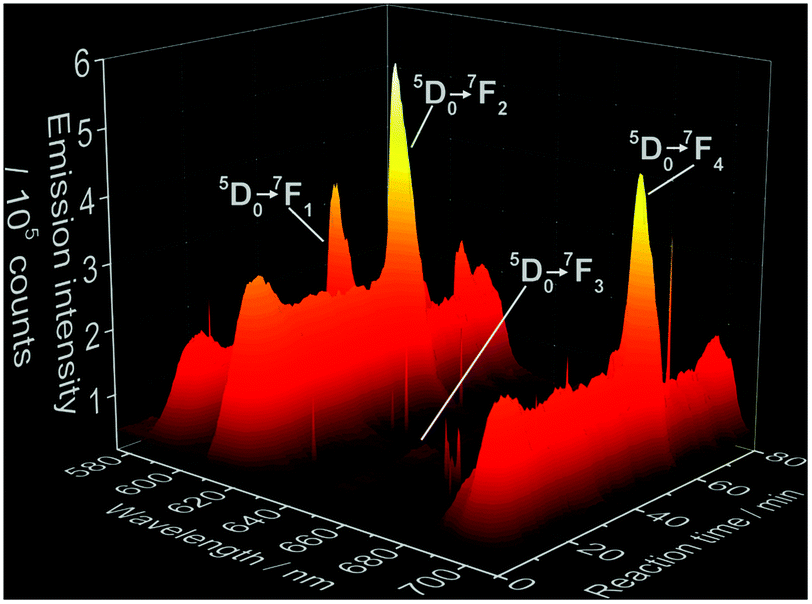 | ||
| Fig. 2 Time-dependence of 5D0 → 7FJ (J = 1–4) electronic transitions of Eu3+ during the formation of doped calcium phosphate (λex = 395 nm, experiment 5, Table 1). Sharper peaks parallel to the Eu3+ emission bands are assigned to measurement artifacts originated by the CCD detector. | ||
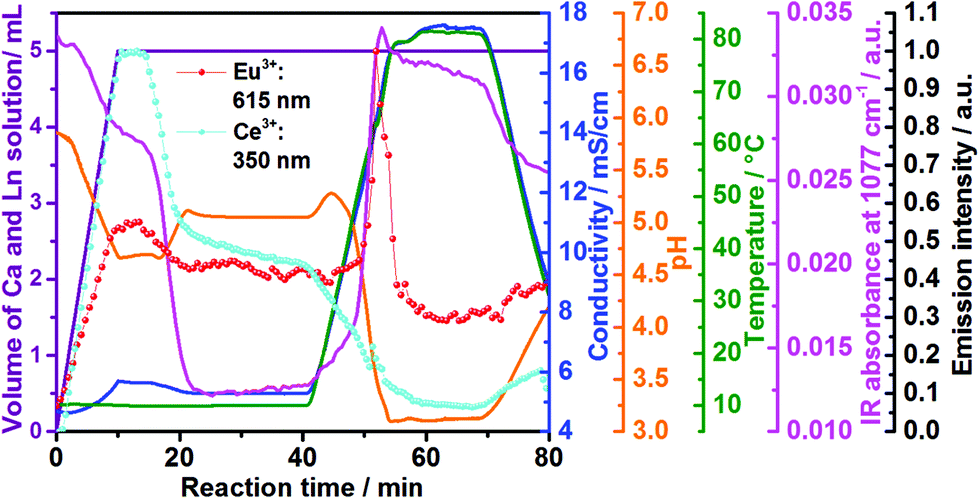 | ||
| Fig. 3 Time dependence of the emission intensity of Ce3+ (light blue curve) and Eu3+ (red curve) doped calcium phosphate, in situ ion conductivity (dark blue curve), in situ pH (orange curve) and IR intensity at 1077 cm−1 (pink curve) in comparison with the volume of the Ca2+ and Ln3+ solution (violett curve) to the reactor containing aqueous (NH4)2HPO4 (experiments 2 and 6, Table 1). | ||
Fig. 3 shows in addition the confirmation of the structural changes detected by in situ luminescence measurements discussed above by comparison with in situ pH value and ion conductivity measurements as well as in situ infrared spectroscopy. Up to t = 10 min, the conductivity increases during the addition of extra ions during the introduction of the solution containing calcium and cerium or europium nitrate. This solution addition also causes the decrease of the pH, due to the acidity of the nitrate solution. Initially, Ca5(PO4)3OH and ACP (Ca9(PO4)6)11,42 formed, according to the ex situ XRD measurements (Fig. S6b‡). The higher pH of the initial phosphate solution inside the reactor (pH = 5.85, Fig. 3, orange curve) can cause the deprotonation of the phosphate anions, stabilizing the PO43− species and therefore, the Ca5(PO4)3OH and ACP phases.11 Upon the addition of more acidic nitrate solution and consequent decrease of the pH value, the reflections assigned to Ca5(PO4)3OH become more clear and with further decrease of the pH value, the phosphate ions are partially protonated,11 resulting in the conversion from Ca5(PO4)3OH to CaHPO4·2H2O. This conversion also causes a slight decrease in the conductivity due to the uptake of the H+ ions for CaHPO4·2H2O formation. Similarly to in situ luminescence measurements, in situ pH and ion conductivity remain nearly constant between t = 20–40 min, indicating no significant structural changes and the stabilization of the CaHPO4·2H2O phase (Fig. S6b‡). After t = 40 min, the large oscillations on pH values and ion conductivity are strongly influenced by the temperature, masking additional structural changes. Also interesting are the changes in the IR absorption bands during the calcium phosphate phase transitions (Fig. 3 and 4), displayed in detail in the ESI (Fig. S16‡). Fig. 3, for instance, shows the time-dependence of the IR absorption band at 1077 cm−1, assigned to the δPOH vibrations within the (NH4)2HPO4 solution.52 Initially, the intensity of this band decreases due to the uptake of phosphate ions from the solution to form ACP and Ca5(PO4)3OH, after deprotonation. The decrease of this IR band is also caused by the uptake of HPO42− ions for the formation of CaHPO4·2H2O and Ca8H2(PO4)6·5H2O (Ca8(HPO4)2(PO4)4·5H2O).
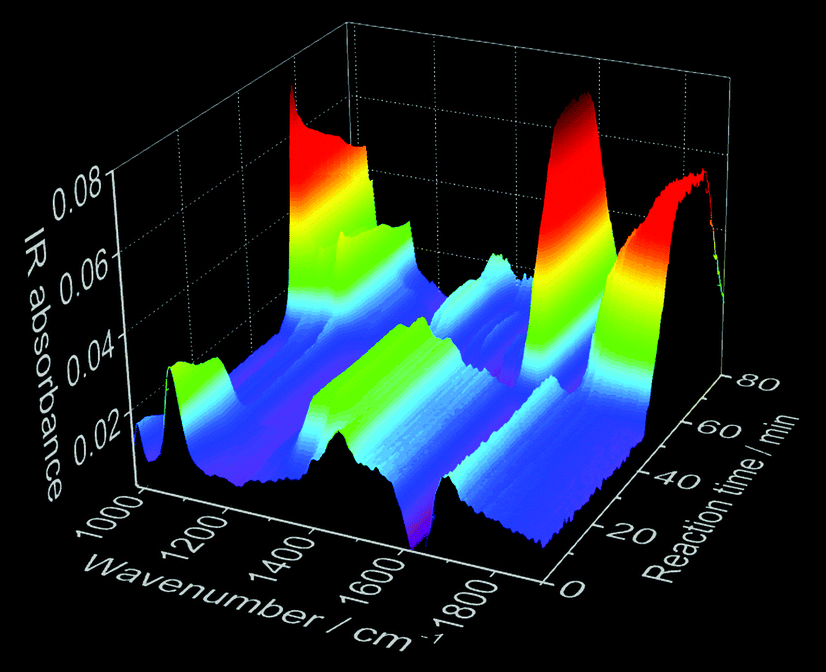 | ||
| Fig. 4 Time-dependence of in situ IR spectroscopy measurements during the synthesis of Ce3+-doped calcium phosphate (experiment 2, Table 1). | ||
3.3. In situ X-ray diffraction and light transmission measurements
As mentioned above, since samples analyzed ex situ can be influenced by the preparation procedure, in situ XRD measurements at the P02.153 and P0854 beamlines at DESY in Hamburg, Germany, have been additionally carried out (Fig. S17–S21‡). In general for all in situ experiments, an increase of the background at low 2θ angles is observed upon the addition of the solution containing calcium and europium or cerium nitrate to the (NH4)2HPO4 solution. This background increase is assigned to the formation of an amorphous phase,55 coinciding with the formation of the combined ACP and Ca5(PO4)3OH phases demonstrated by the ex situ XRD measurements displayed in Fig. S6.‡ Most probably, the reflections assigned to Ca5(PO4)3OH have not been detected within the in situ XRD patterns, because (i) its degree of crystallinity is rather low, (ii) the crystallite size is too small or (iii) this phase is generated only during the ex situ preparation of the samples for the XRD measurements. Even though the broad reflections of the Ca5(PO4)3OH phase in Fig. S6‡ indicate the validation of hypotheses (i) and (ii), additional experiments are necessary to verify these theories. After the rise of the ACP background, this signal starts to decrease, while the reflections of CaHPO4·2H2O (Fig. S17‡) start to increase. At approximately 63 °C, CaHPO4·2H2O starts to decompose, simultaneously with the increase of the Ca8H2(PO4)6·5H2O reflections (Fig. S17‡).According to Engelke et al.,5 different mechanisms can govern the transformation between different phases during the formation of solid materials in solution. These mechanisms are, for instance, (i) a direct solid–solid transition, (ii) the first phase completely dissolves before the emerging phase nucleates and crystallizes from the solution and (iii) the formation of the single phase consists of completely separated processes. Fig. S18‡ shows, for example, that the intensity of the ACP XRD signal is highly correlated with the intensity of the (0,2,0) reflection of the CaHPO4·2H2O phase. Hence, the onset of the decay of the ACP occurs simultaneously with the onset of the crystallization of the CaHPO4·2H2O phase, indicating that CaHPO4·2H2O grows and the intensity of these reflections increases at the cost of the ACP phase. Therefore, the hypothesis (iii) can be ruled out. If hypothesis (ii) were true, the ACP signal would partially or completely disappear before the onset of the CaHPO4·2H2O crystallization and the curves of the normalized reflection intensities of these two phases would not intersect. A similar behaviour can be observed comparing the correlation of the intensities of the reflections assigned to the CaHPO4·2H2O and Ca8H2(PO4)6·5H2O phases. The normalized reflection intensities of the respective transitions show, however, intersections at 0.5 and 0.6 (Fig. S18‡), indicating that the respective conversions from ACP to CaHPO4·2H2O and from CaHPO4·2H2O to Ca8H2(PO4)6·5H2O are rather governed by solid–solid phase transitions, in agreement with hypothesis (i).56
Fig. S18‡ shows the normalized intensity of XRD intensities (λ = 0.4959 Å) at 0.5732° 2θ (green curve), assigned to the amorphous phase,55 1.5142° 2θ (pink curve), assigned to the (1,0,0) reflection of Ca8H2(PO4)6·5H2O46 (Fig. S17‡) and 3.7471° 2θ (violet curve), assigned to the (0,2,0) reflection of CaHPO4·2H2O41 (Fig. S19‡), measured in situ during the synthesis of Eu3+-doped calcium phosphate at the DESY P08 beamline (experiment 9, Table 1). This diagram shows the initial formation of amorphous calcium phosphate, which converts at t = 7.5–13.5 min to CaHPO4·2H2O, growing further up to t = 22 min. When the temperature is increased to 63 °C, the intensity of the reflection assigned to CaHPO4·2H2O starts to decrease, upon the increase of the reflection assigned to the Ca8H2(PO4)6·5H2O phase. In this context, it is important to note that the increase of the intensity of the reflection at 1.5142° 2θ (pink curve) at t = 0–14 min occurs due to the overlap with the broad signal of the amorphous phase and not because of a premature formation of Ca8H2(PO4)6·5H2O. As explained within section 2.2, slight shifts in the time range of the phase transitions in comparison with experiments carried out with Setup A might be assigned to the adaptation of the experimental conditions, necessary for performing these experiments at the synchrotron facility.
The formation of the amorphous phase at t = 1 min is also detected by the simultaneous increase of the intensity of the Eu3+ 5D0 → 7F2 electronic transition at 613 nm. The changes in the intensity ratio between the 5D0 → 7F2 and 5D0 → 7F1 transitions indicate changes in the symmetry around the cation sites during the formation of the amorphous phase (Fig. S19‡).
Similar to Fig. S18,‡Fig. 5 shows the initial formation of ACP during the synthesis of Ce3+-doped calcium phosphate, which converts at t = 9–14.5 min to CaHPO4·2H2O (Fig. S21‡), growing further up to approximately t = 23 min. Similar to that indicated in Fig. S6,‡ a possible explanation for the longer stabilization of the amorphous phase on the measurements in Fig. 5 (7% Ln3+) than the measurements in Fig. S18‡ (5% Ln3+) could be the higher amount of coordination sensors. Because the differences in ionic radii and charge between Ln3+ and Ca2+ ions, doping trivalent lanthanides within calcium phosphate could increase the disorder within the solid material, delaying crystallization. When the temperature is increased to ca. 60 °C, the intensity of the reflection assigned to CaHPO4·2H2O decreases, upon the increase of the reflection assigned to the Ca8H2(PO4)6·5H2O phase (Fig. S20‡). As previously mentioned in Fig. S18,‡ the increase of the intensity of the reflection at 0.6347° 2θ (pink curve) at t = 0–20 min occurs due to an overlap with the broad signal of the amorphous phase and not because of a premature formation of Ca8H2(PO4)6·5H2O (Fig. S21‡). The intensity of the reflection assigned to Ca8H2(PO4)6·5H2O reaches its maximum at the maximum temperature of 100 °C and decreases afterward turning the heating system off at t = 28.5 min, which could be caused by (i) a decrease of crystallinity or (ii) the partial dissolution of the product. Hence, the simultaneously performed in situ measurements of light transmission through the reaction solution offer additional insights into this open question and is discussed in detail in Fig. 6. At t = 55 min, a 25% NH3 solution was added to the reactor in order to evaluate the effect of the increase of the pH in this reaction stage. However, the addition of NH3 has not shown a significant influence on the calcium phosphate phase transitions.
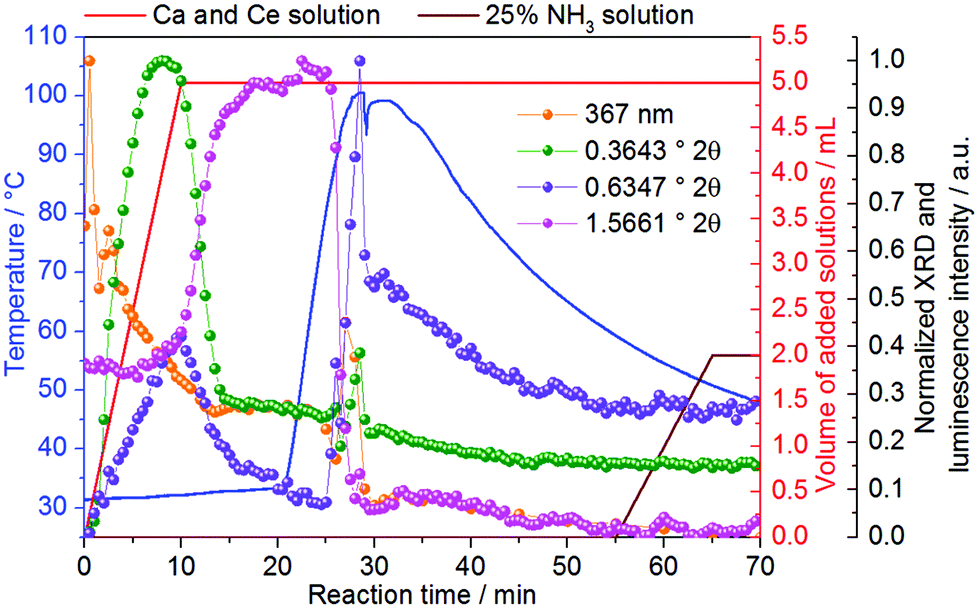 | ||
| Fig. 5 Normalized intensity of XRD reflections (λ = 0.2075 Å) at 0.3643° 2θ (green curve), assigned to the amorphous phase,55 0.6347° 2θ (pink curve), assigned to the (1,0,0) reflection of Ca8H2(PO4)6·5H2O46 (Fig. S20‡) and 1.5661° 2θ (violet curve), assigned to the (0,2,0) reflection of CaHPO4·2H2O41 (Fig. S20‡), measured in situ during the formation of Ce3+-doped calcium phosphate at the DESY P02.1 beamline (experiment 10, Table 1). | ||
 | ||
| Fig. 6 Time-dependent light transmission of a 365 nm light source during the synthesis of Ce3+-doped calcium phosphate (experiment 10, Table 1). | ||
As explained in section 1.2 of the ESI,‡ for these experiments, a 365 nm light source was used for irradiating the reactor, while an optical fiber submersed in the reactor content and connected to a CCD detector, was applied for measuring the intensity of the light source transmitted through the solution during the reaction. Upon the addition of the calcium and cerium nitrate solution to the reactor containing (NH4)2HPO4 (Fig. 6), the intensity of the light source firstly increases, probably due to a brief diluting effect caused by the addition of the second solution to the reactor. The formation of solid material at t = 0.5 min is detected by the strong decrease of the intensity of the light source, caused by the increase of the turbidity of the solution, blocking light transmission.45 The light transmission decreases further up to t ≈ 14 min, during the formation of CaHPO4·2H2O (Fig. 5), and is rather constant while the growth of CaHPO4·2H2O stabilizes. At approximately t = 23 min, the light transmission starts to decrease again, coinciding with the formation of Ca8H2(PO4)6·5H2O (Fig. 5). Even though the intensity of the reflections decreases during the cooling process, the further decrease of the light intensity indicates an increase in the turbidity of the solution, ruling out the hypothesis of product dissolution, discussed in Fig. 5. The oscillation of the intensity of the light source and signals of all other sensors at t = 28.5 min are assigned to the oscillation of the temperature by turning off the heating system.
4. Conclusions
This work introduces the recently developed ILACS26in situ luminescence approach as a new technique for characterizing the phase transitions in calcium phosphate. Calcium phosphate is a major inorganic component of the human body,11 often found in both healthy and pathologic tissues such as bones and teeth, heart calcifications or caries. Here, the ILACS approach explored the coordination sensitive emissive f–d and f–f electronic transitions on, respectively, Ce3+ and Eu3+ as local sensors for delivering information about structural changes around the doped cation sites. Therewith, it was possible to detect the formation of ACP and Ca5(PO4)3OH as well as their subsequent conversion to CaHPO4·2H2O and Ca8H2(PO4)6·5H2O. The detection of these phase transitions has been confirmed by additional characterization methods as in situ measurements of pH value, ion conductivity, IR spectroscopy, besides ex situ and synchrotron-based in situ techniques. Upon addition of the solutions of calcium and europium or cerium nitrates to (NH4)2HPO4, a mixture of ACP and Ca5(PO4)3OH is formed, converted to CaHPO4·2H2O, most probably due to a decrease of the pH. On increasing the temperature, CaHPO4·2H2O is decomposed upon the formation of CaHPO4 with a higher reactant concentration and Ca8H2(PO4)6·5H2O with a lower reactant concentration. Understanding the phase transitions of calcium phosphate is important for, for instance, comprehending and treating diseases.Acknowledgements
The authors thank Prof. Dr W. Bensch, Prof. Dr C. Wickleder and Prof. Dr N. Stock for the applied equipment, L. Mahnke, P. Rönfeldt and S. Leubner for the help with the experiments at the beamlines, Dr N. Pienack for the helpful discussions, M. Radke for the photographs, M. Köppen for the development of the in situ analysis software as well as the DFG (Priority Program 1415, project BE 1653/29-1, project TE 1147/1-1), Daimler and Benz Foundation and MATsynCELL for the financial support. Parts of this research were carried out at PETRA III at DESY, a member of the Helmholtz Association (HGF). We would like to also thank Dr U. Rütt for assistance in using the beamline P08.References
- N. Pienack and W. Bensch, Angew. Chem., Int. Ed., 2011, 50, 2014 CrossRef CAS PubMed and references therein.
- H. Terraschke, M. Rothe and P. Lindenberg, Rev. Anal. Chem., 2017 Search PubMed , submitted.
- N. Heidenreich, U. Rütt, M. Köppen, A. K. Inge, A.-C. Dippel, R. Suren and N. Stock, manuscript in preparation.
- E. Antonova, B. Seidlhofer, J. Wang, M. Hinz and W. Bensch, Chem. – Eur. J., 2012, 18, 15316–15322 CrossRef CAS PubMed.
- L. Engelke, M. Schaefer, M. Schur and W. Bensch, Chem. Mater., 2001, 13, 1383 CrossRef CAS.
- R. Kiebach, N. Pienack, M.-E. Ordolff, F. Studt and W. Bensch, Chem. Mater., 2006, 18, 1196 CrossRef CAS.
- R. Kiebach, M. Schaefer, F. Porsch and W. Bensch, Z. Anorg. Allg. Chem., 2005, 631, 369 CrossRef CAS.
- N. Pienack, C. Näther and W. Bensch, Eur. J. Inorg. Chem., 2009, 7, 937 CrossRef.
- T. Ahnfeldt, J. Moellmer, V. Guillerm, R. Staudt, C. Serre and N. Stock, Chem. – Eur. J., 2011, 17, 6462–6468 CrossRef CAS PubMed.
- R. El Osta, M. Feyand, N. Stock, F. Millange and R. I. Walton, Powder Diffr., 2013, 28, S256 CrossRef CAS.
- S. V. Dorozhkin, Materials, 2009, 2, 399 CrossRef CAS and references therein.
- H. Pan, S. Jiang, T. Zhang and R. Tang, Methods Enzymol., 2013, 532, 129 CAS.
- R. Z. LeGeros, J. Dent. Res., 1974, 53, 45 CrossRef CAS PubMed.
- S. Takagi, L. C. Chow and K. Ishikawa, Biomaterials, 1998, 19, 1593 CrossRef CAS PubMed.
- G. Willmann, Adv. Eng. Mater., 1999, 1, 95 CrossRef CAS.
- A. S. Posner and F. Betts, Acc. Chem. Res., 1975, 8, 273 CrossRef CAS.
- D. Gebauer, M. Kellermeier, J. D. Gale, L. Bergström and H. Cölfen, Chem. Soc. Rev., 2014, 43, 2348 RSC.
- S. Bach, V. R. Celinski, M. Dietzsch, M. Panthöfer, R. Bienert, F. Emmerling, J. Schmedt auf der Günne and W. Tremel, J. Am. Chem. Soc., 2015, 137, 2285 CrossRef CAS PubMed.
- X. Yang, B. Xie, L. Wang, Y. Qin, Z. J. Henneman and G. H. Nancollas, CrystEngComm, 2011, 13, 1153 RSC.
- C.-G. Wang, J.-W. Liao, B.-D. Gou, J. Huang, R.-K. Tang, J.-H. Tao, T.-L. Zhang and K. Wang, Cryst. Growth Des., 2009, 9, 2620 CAS.
- G. B. Ramirez-Rodriguez, J. M. Delgado-Lopez and J. Gomez-Morales, CrystEngComm, 2013, 15, 2206 RSC.
- E. I. Dorozhkina and S. V. Dorozhkin, Colloids Surf., A, 2002, 203, 237 CrossRef CAS.
- R. Tang, Z. J. Henneman and G. H. Nancollas, J. Cryst. Growth, 2003, 249, 614 CrossRef CAS.
- J. V. Rau, M. Fosca and V. S. Komlev, Key Eng. Mater., 2013, 541, 115 CrossRef.
- Q. Zhang, Y. Jiang, B.-D. Gou, J. Huang, Y.-X. Gao, J.-T. Zhao, L. Zheng, Y.-D. Zhao, T.-L. Zhang and K. Wang, Cryst. Growth Des., 2015, 15, 2204 CAS.
- H. Terraschke, L. R. Arana, P. Lindenberg and W. Bensch, Analyst, 2016, 141, 2588 RSC.
- H. Terraschke and C. Wickleder, Chem. Rev., 2015, 115, 11352 CrossRef CAS PubMed.
- H. Terraschke, M. Suta, M. Adlung, S. Mammadova, N. Musayeva, R. Jabbarov, M. Nazarov and C. Wickleder, J. Spectrosc., 2015, 2015, 541958 Search PubMed.
- H. Terraschke, M. F. T. Meier, Y. Voss, H. Schönherr and C. Wickleder, J. Cer. Proc. Res., 2015, 16, 59 Search PubMed.
- P. Dorenbos, J. Lumin., 2013, 135, 93 CrossRef CAS.
- K. Binnemans, Coord. Chem. Rev., 2015, 295, 1 CrossRef CAS.
- C. Gorller-Walrand and K. Binnemans, Handb. Phys. Chem. Rare Earths, 1996, 23, 121 CAS.
- G. Jia, P. A. Tanner, C.-K. Duan and J. Dexpert-Ghys, J. Phys. Chem. C, 2010, 114, 2769 CAS.
- G. Blasse and B. C. Grabmaier, in Luminescent Materials, Springer, Berlin, 1994 Search PubMed.
- G. Li, Y. Tian, Y. Zhao and J. Lin, Chem. Soc. Rev., 2015, 44, 8688 RSC.
- R. D. Shannon and C. T. Prewitt, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1969, 25, 925 CrossRef CAS.
- A. Dey, P. H. H. Bomans, F. A. Müller, J. Will, P. M. Frederik, G. de With and N. A. J. M. Sommerdijk, Nat. Mater., 2010, 9, 1010 CrossRef CAS PubMed.
- I. V. Berezovskaya, N. P. Efryushina, E. V. Zubar and V. P. Dotsenko, Distribution and Luminescent Properties of Ce3+ Ions in Nanosized Calcium Hydroxyapatite, Sumy State University, 2012, vol. 1, p. 01PCN24 Search PubMed.
- W. M. Yen, H. Yamamoto and S. Shionoya, in Phosphor Handbook, CRC Press Laser and Optical Science and Technology, Boca Raton, 2006 Search PubMed.
- T. Moon, S.-T. Hwang, D.-R. Jung, D. Son, C. Kim, J. Kim, M. Kang and B. Park, J. Phys. Chem. C, 2007, 111, 4164 CAS.
- C. A. Beevers, Acta Crystallogr., 1958, 11, 273 CrossRef CAS.
- M. I. Kay, R. A. Young and A. S. Posner, Nature, 1964, 204, 1050 CrossRef CAS PubMed.
- L.-W. Du, S. Bian, B.-D. Gou, Y. Jiang, J. Huang, Y.-X. Gao, Y.-D. Zhao, W. Wen, T.-L. Zhang and K. Wang, Cryst. Growth Des., 2013, 13, 3103 CAS.
- S. V. Dorozhkin, Adv. Mater. Sci. Res., 2013, 15, 1 CAS.
- N. Pienack, L. Ruiz Arana, W. Bensch and H. Terraschke, Crystals, 2016, 6, 157/1 CrossRef CAS.
- W. E. Brown, Nature, 1962, 196, 1048 CrossRef CAS.
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. Gareth Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493 RSC.
- F. Chen, Y.-J. Zhu, K.-H. Zhang, J. Wu, K.-W. Wang, Q.-L. Tang and X.-M. Mo, Nanoscale Res. Lett., 2011, 6, 67 CrossRef PubMed.
- R. Ternane, M. Trabelsi-Ayedi, N. Kbir-Ariguib and B. Piriou, J. Lumin., 1999, 81, 165 CrossRef CAS.
- O. A. Graeve, R. Kanakala, A. Madadi, B. C. Williams and K. C. Glass, Biomaterials, 2010, 31, 4259 CrossRef CAS PubMed.
- C. S. Ciobanu, S. L. Iconaru, F. Massuyeau, L. V. Constantin, A. Costescu and D. Predoi, J. Nanomater., 2012, 942801 Search PubMed.
- C. Sun and D. Xue, J. Phys. Chem. C, 2013, 117, 19146 CAS.
- A.-C. Dippel, H.-P. Liermann, J. T. Delitz, P. Walter, H. Schulte-Schrepping, O. Seeck and H. Franz, J. Synchrotron Radiat., 2015, 22, 675 CAS.
- O. H. Seeck, C. Deiter, K. Pflaum, F. Bertam, A. Beerlink, H. Franz, J. Horbach, H. Schulte-Schrepping, B. M. Murphy, M. Greve and O. Magnussen, J. Synchrotron Radiat., 2012, 19, 30 CrossRef CAS PubMed.
- M. Wendt, L. K. Mahnke, N. Heidenreich and W. Bensch, Eur. J. Inorg. Chem., 2016, 5393 CrossRef CAS.
- L. Engelke, M. Schaefer, F. Porsch and W. Bensch, Eur. J. Inorg. Chem., 2003, 3, 506–513 CrossRef.
Footnotes |
| † These results have been partially reported as posters and oral presentations at the conferences “18. Vortragstagung Fachgruppe Festkörperchemie und Materialforschung”, September 19th–21st 2016, Innsbruck, Austria (DOI: 10.1002/zaac.201605095); “2nd joint workshop of MATsynCELL and C3”, November 16th–17th 2015, Hamburg, Germany; “Summer School on Time-resolved and In situ Studies of Materials: Basics and Applications”, August 22nd–29th 2015, Sellin, Island of Rügen, Germany. |
| ‡ Electronic supplementary information (ESI) available: Detailed description of experimental setup, complementary ex situ XRD and SEM measurements as well as additional results of in situ excitation spectra, IR spectroscopy and synchrotron-based XRD experiments. See DOI: 10.1039/c7qi00172j |
| This journal is © the Partner Organisations 2017 |