
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comparative studies of oxaliplatin-based platinum(IV) complexes in different in vitro and in vivo tumor models†
Simone
Göschl
a,
Ekaterina
Schreiber-Brynzak
a,
Verena
Pichler
ab,
Klaudia
Cseh
a,
Petra
Heffeter
bcd,
Ute
Jungwirth
ce,
Michael A.
Jakupec
 *ab,
Walter
Berger
bcd and
Bernhard K.
Keppler
ab
*ab,
Walter
Berger
bcd and
Bernhard K.
Keppler
ab
aUniversity of Vienna, Institute of Inorganic Chemistry, Waehringer Strasse 42, 1090 Vienna, Austria. E-mail: michael.jakupec@univie.ac.at; Tel: +43 1 4277 52610
bUniversity of Vienna, Research Platform “Translational Cancer Therapy Research”, Vienna, Austria
cMedical University of Vienna, Department of Medicine I, Institute of Cancer Research, Vienna, Austria
dMedical University of Vienna, Comprehensive Cancer Center, Vienna, Austria
eThe Breast Cancer Now Toby Robins Research Centre, The Institute of Cancer Research, London, UK
First published on 16th February 2017
Abstract
Using platinum(IV) prodrugs of clinically established platinum(II) compounds is a strategy to overcome side effects and acquired resistances. We studied four oxaliplatin-derived platinum(IV) complexes with varying axial ligands in various in vitro and in vivo settings. The ability to interfere with DNA (pUC19) in the presence and absence of a reducing agent (ascorbic acid) was investigated in cell-free experiments. Cytotoxicity was compared under normoxic and hypoxic conditions in monolayer cultures and multicellular spheroids of colon carcinoma cell lines. Effects on the cell cycle were investigated by flow cytometry, and the capacity of inducing apoptosis was confirmed by flow cytometry and Western blotting. The anti-cancer activity of one complex was studied in vivo in immunodeficient and immunocompetent mice, and the platinum levels in various organs and the tumor after treatment were quantified. The results demonstrate that modification of the axial ligands can improve the cytotoxic potency. The complexes are able to interfere with plasmid DNA, which is enhanced by co-incubation with a reducing agent, and cause cell cycle perturbations. At higher concentrations, they induce apoptosis, but generate only low levels of reactive oxygen species. Two of the complexes increase the life span of leukemia (L1210) bearing mice, and one showed effects similar to oxaliplatin in a CT26 solid tumor model, despite the low platinum levels in the tumor. As in the case of oxaliplatin, activity in the latter model depends on an intact immune system. These findings show new perspectives for the development of platinum(IV) prodrugs of the anticancer agent oxaliplatin, combining bioreductive properties and immunogenic aspects.
Significance to metallomicsPlatinum(IV)-based prodrugs of the clinically established drug oxaliplatin have been shown to be potent anticancer compounds maintaining some central properties of the parental drug in vitro and in vivo. One of these compounds, bearing axial (4-propylamino)-4-oxobutanoato ligands, was demonstrated to exert strong antitumor effects in a murine CT26 colon cancer model, with a similar dependence on the host's immunocompetence as oxaliplatin. These findings show a new perspective for the development of platinum(IV) complexes as anticancer prodrugs which combine both bioreductive properties and immunogenic aspects. |
Introduction
Although the field of anticancer therapy has been broadened and changed in the last few years, three platinum(II)-based complexes, cisplatin, carboplatin and oxaliplatin, continue to be used worldwide, mostly in combination with a wide range of other drugs (e.g., doxorubicin, etoposide, gemcitabine, paclitaxel, 5-FU etc.). These platinum(II) complexes show high activity against several tumor types and are among the most successful anticancer drugs. However, severe side effects, resulting from incidental reactions with proteins, such as nephrotoxicity, ototoxicity (in the case of cisplatin) and peripheral neurotoxicity, as well as acquired and hardly predictable intrinsic resistances are major drawbacks.1–4Oxaliplatin ((trans-R,R-cyclohexane-1,2-diamine)oxalatoplatinum(II)) is a third-generation platinum drug active against certain intrinsically cisplatin- and carboplatin-resistant tumors and showing different side effects.5 A combination therapy consisting of oxaliplatin, 5-fluorouracil and folinic acid is a standard adjuvant treatment in patients with stage III colon cancer. To improve convenience for the patients (e.g., by oral administration) and to overcome side effects and resistances, platinum(IV) complexes are being developed based on the following considerations: it is widely accepted that platinum(IV) complexes are kinetically more inert, which may be reflected in lower side effects.6 This inertness together with their higher lipophilicity renders them more suitable for oral administration.7,8 Their octahedral configuration, with two additional ligands (compared to platinum(II) analogs), enables wide ligand variation. These binding sites can be used to influence the reduction parameters, kinetic stability and lipophilicity of the complexes for optimization of oral bioavailability, stability in the bloodstream and cellular accumulation, before they are reduced to the more active platinum(II) species by, e.g., small molecules such as glutathione (GSH) or ascorbic acid (AA). A versatile strategy is to combine a known drug, such as oxaliplatin, with axial ligands introducing an additional function.9–13
In solid tumors, hypoxia can lead to chemotherapy14,15 and radiation therapy resistances16,17 and increase tumor invasiveness. Hence, hypoxia is used as a prognostic marker for malignancy and often correlates with a poor treatment outcome.18,19 Hypoxic tumor regions are associated with decreased pH and poor nutrient supply. Moreover, cancer cells under hypoxia show gene expression changes and altered proteomic patterns due to adaptation of survival mechanisms.19,20 In addition, genomic instabilities and accumulation of mutations are found due to increased selective pressure.21 Therefore, hypoxic regions are hotspots of mutations and sources of resistance development. Low oxygen pressure and decreased pH create a reductive milieu that may alter drug penetration and bio-processing of drugs.21 While, in many cases, bioreduction under hypoxia is an unwanted process since it decreases drug stability, platinum(IV) complexes are prodrugs that require reduction to their more active platinum(II) species. Therefore, platinum(IV) drugs might target hypoxia more selectively and may be advantageous in solid tumors with extensive hypoxia. These considerations argue for the use of cellular models of hypoxia in preclinical drug screening. Using platinum(IV) complexes bearing an oxaliplatin core may have an additional advantage, as oxaliplatin is known to induce immunogenic cell death. Cells treated with oxaliplatin interact with T cells and cause them to produce interferon γ.5,22,23
Herein, four oxaliplatin-based platinum(IV) compounds24 (Fig. 1) as well as oxaliplatin and satraplatin as reference substances were investigated. Their ability to interact with DNA in the presence and absence of the reducing agent ascorbic acid was evaluated. The cytotoxicity was determined in three human colon carcinoma cell lines (HCT15, HCT116 and the oxaliplatin-resistant subline HCT116oxR) under normoxic and hypoxic conditions in monolayer cultures as well as in multicellular spheroids. Furthermore, their influence on the cell cycle and their capacity to induce apoptosis were determined by flow cytometry. In vivo studies against leukemia and a solid tumor model in both immunodeficient and immunocompetent mice were performed to clarify whether these platinum(IV) compounds behave similarly to oxaliplatin with regard to immunodependence. The results support the concept of developing platinum(IV) prodrugs of oxaliplatin, adding the aspect of bioreductive activation while evidently maintaining the immunogenicity of the parent drug.
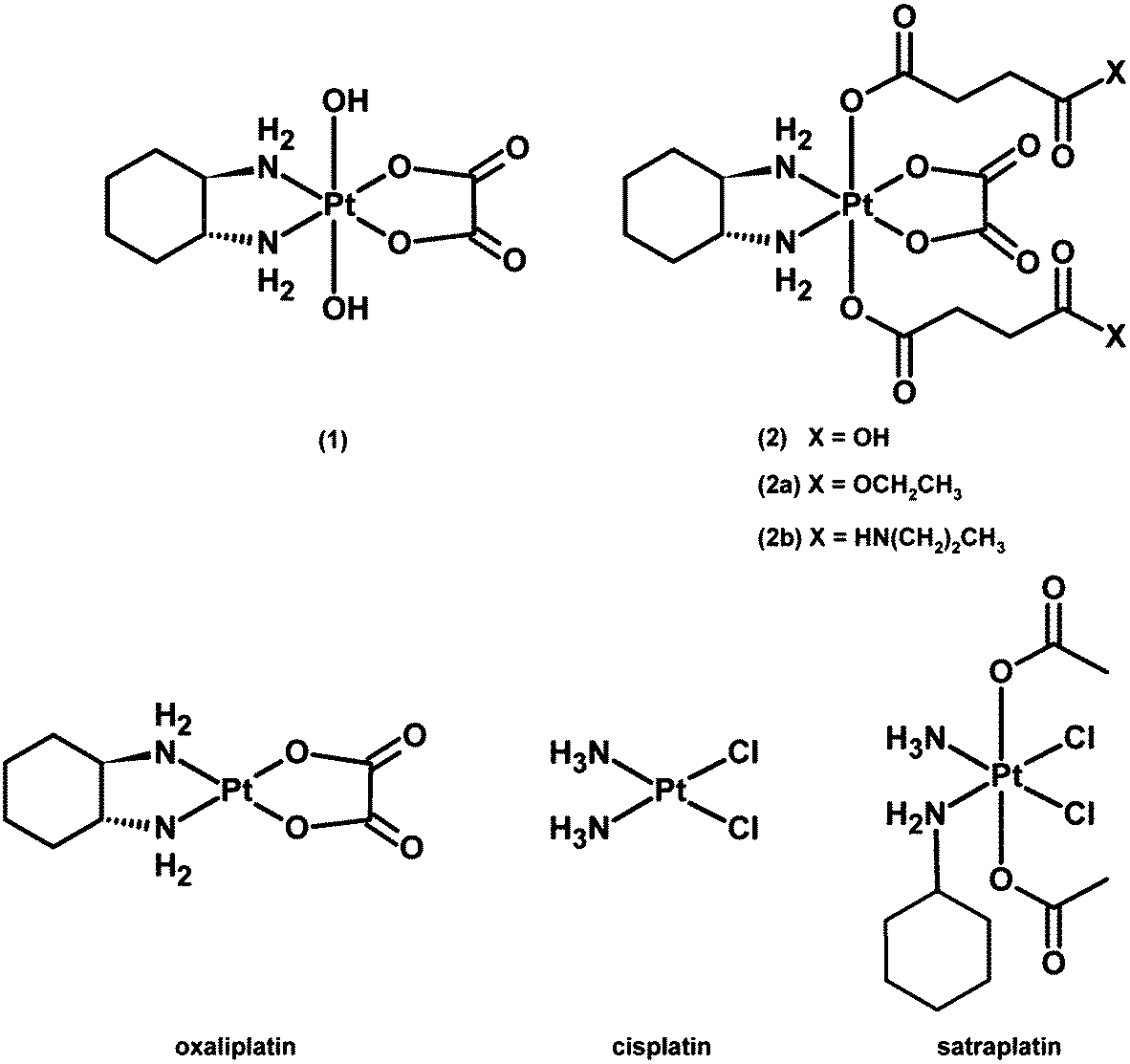 | ||
| Fig. 1 Chemical structures of platinum(IV) complexes and reference compounds under investigation. | ||
Results and discussion
Electrophoretic dsDNA plasmid assay
It is widely accepted that the main target of platinum complexes is DNA.5,7,25 To obtain a first insight into a compound's capacity for interacting with DNA, the cell-free electrophoretic dsDNA plasmid assay is a straightforward method to use. All interfering effects of medium constituents and influences of cellular accumulation or defense mechanisms can be excluded, and the direct impact of equimolar concentrations of the compounds in the absence and presence of ascorbic acid on pUC19 plasmid dsDNA can be examined. To exclude the direct effects of ascorbic acid on DNA, some control samples were treated with 500 μM ascorbic acid alone for 8 h (C8h+AA), yielding electropherograms comparable to untreated controls (except for somewhat intensified bands corresponding to an open circular plasmid, which might suggest a certain incidence of single-strand breaks; these bands also seem to intensify with time in samples treated with, e.g., 2 + AA, or 2b + AA; but none of these would prevent the observation of any impact the platinum complexes might have on the supercoiled form). As shown in Fig. 2, cis-diamminedichloridoplatinum(II) (cisplatin, 50 μM) interacts with plasmid DNA within 15 min, causing total untwisting of the supercoiled form to the open circular form after 30 min, indicated by the maximum DNA band shift, followed by counter-coiling after 1 h. For oxaliplatin, bearing a chelating oxalate ligand instead of chlorides as the leaving group, the interaction with DNA is slowed down and pronounced untwisting of the plasmid starts only after 4 h. Remarkably, the DNA band shift patterns caused by platinum(IV) compounds are more diverse than expected. While complexes 1 and 2 and satraplatin (all 50 μM) have no effect on pUC19 without the reducing agent, 2b interacts slightly with DNA after 6–8 h, and 2a starts to retard DNA migration already after 4 h, similar to oxaliplatin. 2b shows only a minor influence on pUC19, no matter whether it was incubated with or without ascorbic acid. In contrast, addition of a reducing agent strongly increases the effects of 2a and satraplatin, as retardation of the supercoiled plasmid has started already after 2 h. This pattern for satraplatin was already reported by Pichler et al.;262a totally untwists the plasmid after 4 h, but does not start to counter-coil DNA within 8 h. Overall, the electrophoretic dsDNA plasmid assay confirmed that platinum(II) species are more prone to interact with DNA than their platinum(IV) analogs, but showed that these differences might be smaller than expected in some cases.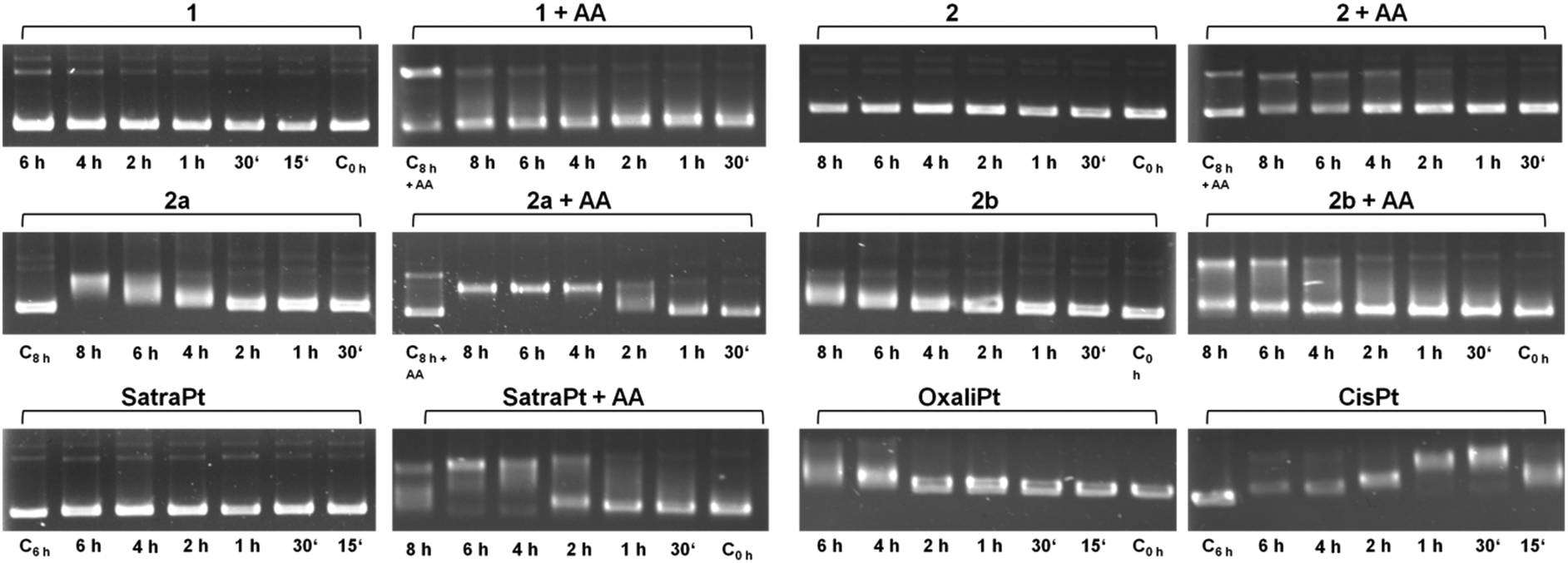 | ||
| Fig. 2 Electropherograms of dsDNA plasmid pUC19 incubated with 50 μM of platinum(IV) compounds 1, 2, 2a, 2b or satraplatin (in the absence or presence of 500 μM ascorbic acid, AA), oxaliplatin or cisplatin for different incubation times at 37 °C in TE buffer (pH = 7.8). | ||
Recently, a series of publications dealt with the rates of reduction of platinum(IV) complexes with different ligand spheres and physiologically available reducing agents such as glutathione and ascorbic acid.26–34 According to the literature, tetracarboxylato platinum(IV) complexes have very slow reduction kinetics, conclusively explainable by the low ability of carboxylates and amines to function as electron transfer ligands. Hydroxido ligands should increase the electron transfer rate and therefore distinctly increase the reduction rate, resulting in a half-life of around 2.5 h for complex 1 as reported by Zhang et al.28 The observed DNA interaction pattern of 2b correlates very well with the published rate of reduction of tetracarboxylato platinum(IV) compounds in phosphate buffered solution. However, the pattern of 1 differs from the expected behavior, as fast reduction rates should facilitate the interaction of the reduced platinum species with DNA, whereas no interaction was observed within the tested 8 h timeframe. As a consequence, the reduction patterns of 1 and 2b under co-incubation with ascorbic acid or glutathione were measured over at least 8 h by 1H NMR (ESI†). No significant reduction of 1 and 2b was observed at room temperature with a molar ratio of compound![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :reductant of 1:2 within 8 h. Increasing the temperature to 37 °C and the amount of reductant to a 10-fold excess still did not result in the reduction of compound 1 when incubated with glutathione, whereas ascorbic acid gave rise to a minor amount of reduced platinum(II) species.
:reductant of 1:2 within 8 h. Increasing the temperature to 37 °C and the amount of reductant to a 10-fold excess still did not result in the reduction of compound 1 when incubated with glutathione, whereas ascorbic acid gave rise to a minor amount of reduced platinum(II) species.
These cell-free experiments demonstrate that the substances under investigation are reducible with ascorbic acid but may require a timeframe of several hours for detectable effects. As reported recently by Pichler et al.26 and Göschl et al.,35 co-incubation of rapidly reduced platinum(IV) complexes with ascorbic acid may even decrease the cytotoxic effect, suggesting that extracellular reduction may result in a different cellular accumulation behavior. For this reason, these platinum(IV) compounds were tested in advanced cell culture models mimicking the reductive environment of solid tumors rather than co-incubating with reducing agents in conventional cell culture settings.
Comparative cytotoxicity in different cell culture models
The platinum(IV) compounds 1 and 2b and reference compounds satraplatin and oxaliplatin were tested in three colon carcinoma cell lines with different levels of drug sensitivity. HCT116 is the most platinum-sensitive cell line in this panel. HCT15 was chosen as it is intrinsically less sensitive than HCT116; and HCT116oxR, a subline with acquired oxaliplatin resistance, shows the lowest sensitivity. To determine the IC50 values in different cellular models and under different oxygenation conditions, the fluorimetric resazurin assay was chosen, which allows staining of the spheroids directly in the treatment medium (Table 1). It is widely accepted that platinum(IV) complexes are activated by reduction.7,26 Hence, the influence of hypoxic conditions on the cytotoxic behavior was one major focus. Hypoxia can be induced in different ways. To compare the effects of normoxia and hypoxia in a 2D adherent monolayer culture, the cells were grown under normal conditions (21% O2) or in a hypoxic chamber (0.5% O2). Secondly, a hypoxic environment was generated in 3D spheroid cultures. To verify the size-dependent generation of hypoxia, HIF-1-alpha (hypoxia-inducible factor 1-alpha) expression and the extent of necrosis (in spheroids with diameters of about 150 and 400 μm) were investigated by using specific antibodies and propidium iodide (PI) staining, respectively. The results confirm the generation of hypoxia, as spheroids with a diameter of >400 μm show a distinct HIF-1-alpha expression already 30–40 μm beneath the surface (Fig. 3B), while spheroids with about 150 μm diameter show no evidence for HIF-1-alpha expression (Fig. 3A). Additionally, PI staining of the necrotic core is highly positive only in spheroids with a size of about 400 μm but not in the smaller (150 μm) ones (Fig. 3A). In conclusion, spheroids with a diameter of about 400 μm were considered highly appropriate for the testing of platinum(IV) compounds for correlation with their reduction properties.| 2D culture – normoxic | |||||||
|---|---|---|---|---|---|---|---|
| IC50 [μM] | RF | ||||||
| HCT15 | HCT116 | HCT116oxR | HCT15/HCT116 | p | HCT116oxR/HCT116 | p | |
| 1 | 136 ± 25 | 54 ± 13 | 562 ± 25 | 2.5 | * | 10 | *** |
| 2 | 148 ± 13 | 69 ± 9 | >1000 | 2.1 | ** | >14 | **** |
| 2a | 33 ± 3 | 13 ± 1 | 352 ± 39 | 2.6 | *** | 27 | **** |
| 2b | 83 ± 18 | 47 ± 7 | 371 ± 2 | 1.8 | ns | 7.9 | **** |
| OxaliPt | 1.0 ± 0.2 | 0.55 ± 0.01 | 28 ± 1 | 1.9 | ns | 52 | *** |
| SatraPt | 4.7 ± 0.1 | 1.8 ± 0.1 | 5.8 ± 1.0 | 2.7 | **** | 3.3 | * |
| 2D culture – hypoxic | RF | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| IC50 [μM] | Hypoxic/normoxic | ||||||||
| HCT15 | HCT116 | HCT116oxR | HCT15 | p | HCT116 | p | HCT116oxR | p | |
| 1 | 156 ± 47 | 79 ± 8 | 565 ± 82 | 1.2 | ns | 1.5 | * | 1.0 | ns |
| 2 | 132 ± 8 | 68 ± 15 | >2000 | 0.89 | ns | 0.98 | ns | ∼2 | |
| 2a | 33 ± 7 | 15 ± 2 | 352 ± 75 | 1.0 | ns | 1.1 | ns | 1.0 | ns |
| 2b | 36 ± 4 | 18 ± 4 | 434 ± 58 | 0.44 | * | 0.38 | ** | 1.2 | ns |
| OxaliPt | 1.1 ± 0.3 | 0.91 ± 0.16 | 95 ± 12 | 1.1 | ns | 1.7 | ns | 3.4 | ** |
| SatraPt | 1.5 ± 0.4 | 2.6 ± 0.4 | 4.6 ± 1.0 | 0.33 | **** | 1.5 | ns | 0.80 | ns |
| 3D culture | RF | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| IC50 [μM] | 3D/2D | ||||||||
| HCT15 | HCT116 | HCT116oxR | HCT15 | p | HCT116 | p | HCT116oxR | p | |
| 1 | 185 ± 39 | 136 ± 29 | >1000 | 1.4 | ns | 2.5 | * | >1.8 | ** |
| 2 | 789 ± 21 | 457 ± 46 | >1000 | 5.3 | **** | 6.6 | ** | ||
| 2a | 354 ± 28 | 262 ± 37 | >1000 | 11 | ** | 20 | ** | >2.8 | **** |
| 2b | >1000 | 114 ±26 | >1000 | >12 | *** | 2.4 | * | >2.7 | **** |
| OxaliPt | 66 ± 7 | 9.3 ± 2.7 | 367 ± 98 | 64 | ** | 17 | * | 13 | ** |
| SatraPt | 21 ± 5 | 15 ± 4 | 14 ± 2 | 4.5 | * | 8.6 | ** | 2.5 | ** |
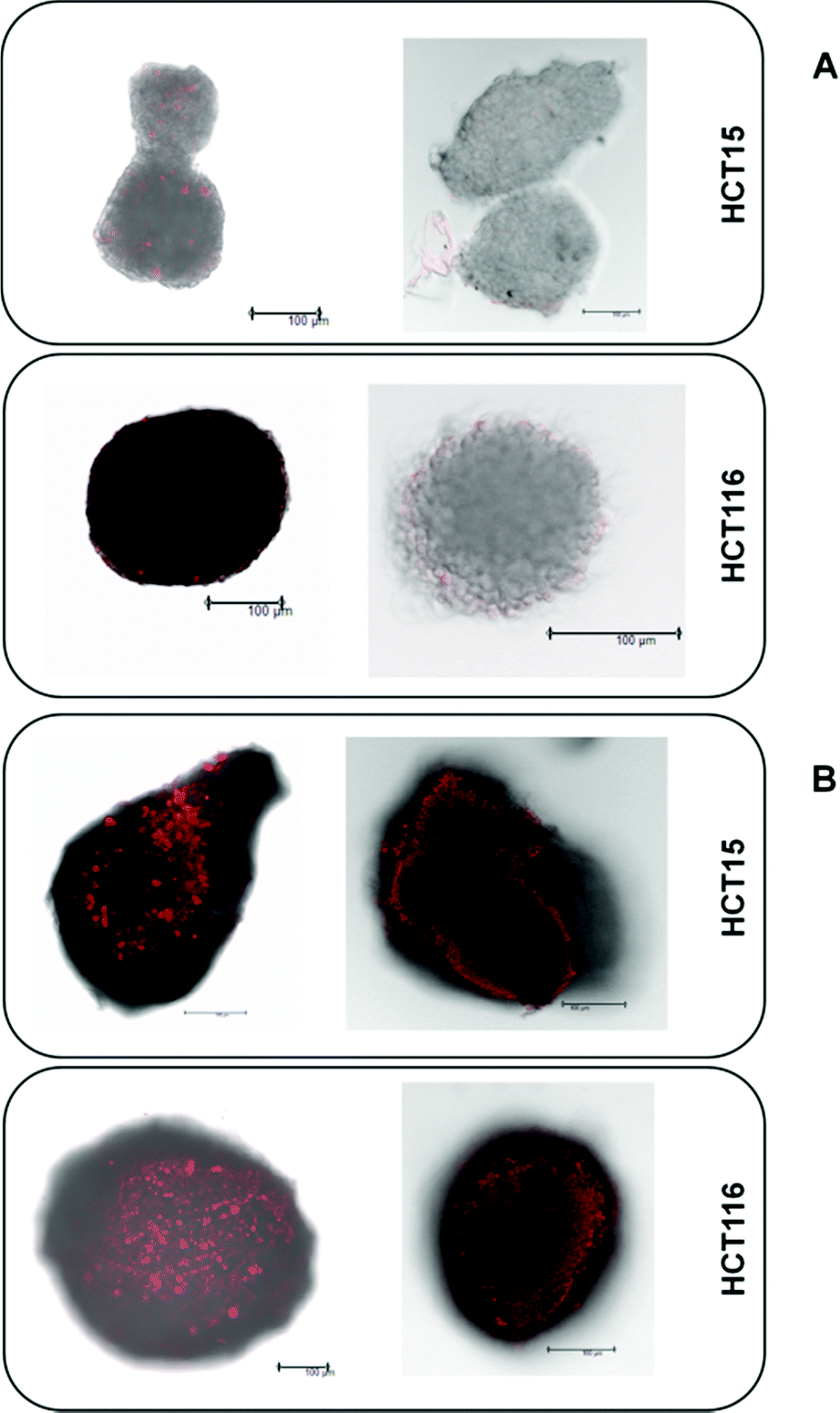 | ||
| Fig. 3 PI staining of the necrotic core (left) and immunohistochemical detection of HIF1α expression (right) in HCT15 and HCT116 spheroids with about 150 μm (A) and 400 μm (B) diameter (scale bars: 100 μm). | ||
Under normoxic conditions, all tested compounds are about twice as active in HCT116 than in HCT15 cells and 3–52 times (the latter in the case of oxaliplatin) more active than in HCT116oxR cells, based on IC50 values. Although HCT116oxR cells are less resistant to all platinum(IV) complexes than to oxaliplatin, none of them could fully overcome the resistance. Even satraplatin, lacking the oxaliplatin core structure, shows similar activity in HCT116oxR as in HCT15 cells. Compounds 2b and 2a are about 2–4 times more active than 2, respectively, in HCT15 cells. 2a is the most active platinum(IV) compound bearing an oxaliplatin moiety in all tested cell lines followed by 2b. Cytotoxicity increases in the following order in HCT15 and HCT116 cells: 2 ≤ 1 < 2b < 2a < satraplatin < oxaliplatin. In HCT116oxR cells, the order is similar, just with satraplatin being about five times more active than oxaliplatin.
A similar cytotoxicity pattern can be observed under hypoxic conditions (1 ≤ 2 < 2a ≈ 2b < satraplatin < oxaliplatin in HCT15 and HCT116 cells, whereas in HCT116oxR 1 is more active than 2 and satraplatin is more active than oxaliplatin). Induction of hypoxia in a monolayer culture seems to have minor effects on cytotoxicity, as IC50 values in hypoxic and non-hypoxic monolayer cultures are for most complexes and cell lines comparable. The only exceptions are satraplatin in HCT15 cells, in which it is 3 times (p < 0.0001) more active than under normoxia, and 2b, which is more than twice as active in HCT15 (p < 0.05) and HCT116 (p < 0.01) cells under hypoxia. For oxaliplatin, a slight decrease in activity in HCT116 and a significant one (p < 0.001) in HCT116oxR cells were noticed. Thus, the expected significant activation of the tested platinum(IV) complexes under hypoxic conditions in a 2D cell culture setting could only partially be observed.
In hypoxic spheroids, however, all tested compounds show a significantly decreased cytotoxicity compared to normoxic monolayer culture in all cell lines. While compound 1 is only weakly affected, the most prominent drop in activity was found for 2a and oxaliplatin (Fig. 4). 2a is up to 20 times and oxaliplatin even 13–64 times less active in spheroids than in the corresponding monolayer cultures. 2b is tremendously less active (>12 times) in HCT15 spheroids but is only about twice less active in HCT116 spheroids than in 2D culture. Such a pattern in HCT116 spheroids was already shown for satraplatin and oxaliplatin previously.36,37
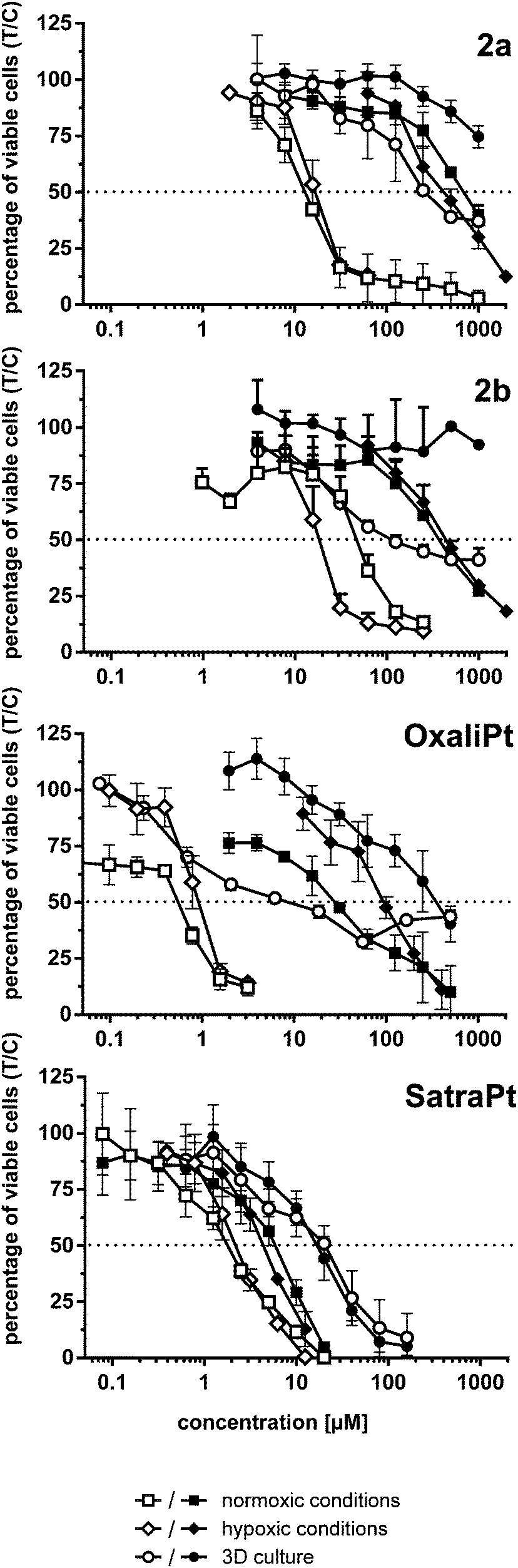 | ||
| Fig. 4 Cytotoxicity (concentration–effect curves) of complexes 2a, and 2b, oxaliplatin and satraplatin in HCT116 (white symbols) and HCT116oxR cells (black symbols) under different conditions, obtained by the fluorimetric resazurin assay after continuous exposure for 96 h. | ||
The lack of activation by reduction of the tested platinum(IV) compounds might be explained by counteractive characteristics of spheroids such as impaired drug penetration and inhomogeneity in drug distribution throughout the spheroid possibly overshadowing the former effect. Theoretically, hypoxia per se might be expected to lead to a decrease in oxidative capacity, without necessarily increasing the reductive capacity significantly, which would influence net rates of reduction only if reduction was reversible (which is not the case for platinum(IV) complexes, though). However, evident gradients of redox potentials within spheroids (such as those demonstrated previously by us38) were proposed to reflect alterations in the cellular metabolism (e.g., activation of glycolysis providing more reducing equivalents of NADPH) associated with hypoxia.39
Annexin V/PI assay
When cells undergo apoptosis, phosphatidylserines (PS) are translocated from the inner leaflet of the lipid bilayer of the plasma membrane to the outer one. Thereupon, annexin V can specifically bind to PS,40 which serves as a marker for all apoptotic cells, whereas the DNA-intercalating stain PI can only penetrate into cells with damaged cell membranes, such as necrotic or late apoptotic cells. Annexin V–FITC/PI double staining thus enables the differentiation of viable (AV−/PI−), early apoptotic (AV+/PI−), late apoptotic (AV+/PI+) or necrotic (AV−/PI+) cells.As shown in Fig. 5, satraplatin is able to induce apoptosis at the highest concentrations in all tested cell lines already after 24 h, whereas oxaliplatin shows no activity in HCT116oxR cells. In accordance with the cytotoxicity data above, all other compounds are less active or inactive in HCT116oxR cells. Furthermore, no or only minor apoptosis induction is detectable in HCT116oxR and HCT15 cells after 24 h. In HCT116 cells, 2a induces apoptosis to a higher extent than 2b or 2, and increasingly with time. When the incubation time is increased to 48 h, all tested compounds are still least active in HCT116oxR cells but demonstrate prominent effects in HCT15 and HCT116 cells. While an increase in the early apoptotic cell fraction is detected in HCT15 cells, more HCT116 cells are already in a late apoptotic phase after the same incubation time with the same concentrations (Fig. 5). Overall, this is in line with the enhanced sensitivity of HCT116 cells in comparison to HCT15. When early and late apoptosis are taken together, satraplatin (50 μM) is the most potent of all tested complexes followed by oxaliplatin (50 μM) > 2a > 1 > 2b ≈ 2 (all 2000 μM) in HCT15 cells and oxaliplatin ≈2a > 2b ≈ 1 > 2 in HCT116 cells. Thus, the investigated platinum(IV) complexes and oxaliplatin are able to induce apoptosis in a concentration- (Fig. S11–S16, ESI†) and time-dependent manner in HCT15 and HCT116 cells, whereas the highly resistant cell line HCT116oxR is less affected by all complexes bearing an oxaliplatin moiety.
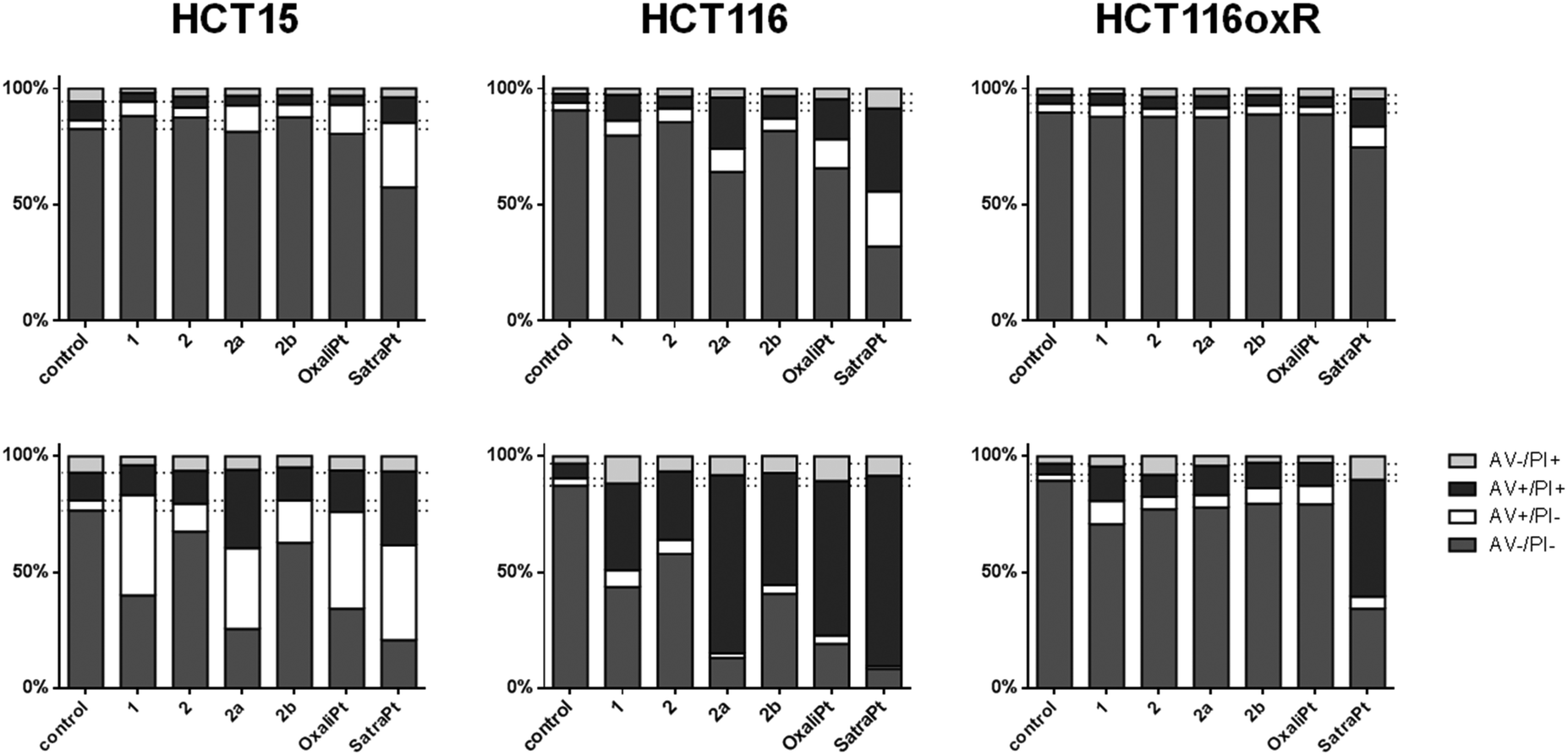 | ||
| Fig. 5 Apoptosis induction by the investigated complexes after 24 h (upper row) and 48 h (lower row) of continuous exposure to either 2000 μM of 1, 2, 2a, 2b or 50 μM of oxaliplatin and satraplatin in HCT15, HCT116 and HCT116oxR cells. Cells were stained with annexin V–FITC/PI and quantified by flow cytometry. Percentages of viable (AV−/PI−), early apoptotic (AV+/PI−), late apoptotic (AV+/PI+) and necrotic (AV−/PI+) cells are means of at least three independent experiments evaluated by FlowJo software. | ||
Western blotting
To investigate in more detail the induction of apoptosis, the cleavage of caspase 7 and PARP was determined by Western blotting. In order to enable comparison with previous results, incubation times of either 24 h or 48 h with similar concentrations (see Fig. 6) were chosen. In accordance with our annexin V studies, satraplatin exhibits the highest activity and is able to induce PARP cleavage in all cell lines at all investigated time points. All compounds are able to induce cleavage of caspase 7 after 24 h (Fig. 6A) or 48 h (Fig. 6B), although it must be kept in mind that considerable amounts of cleaved caspase 7 are found in the untreated controls of every tested cell line. When incubating HCT15 cells with 50 μM oxaliplatin for 48 h, no cleaved caspase 7 is found. Together with the total cleavage of PARP in this sample, this indicates that apoptosis execution is already in an advanced stage at this time point. Overall, HCT116 exhibits the strongest signs for apoptosis induction on the protein level of all cell lines, as 2a, 2b, oxaliplatin and satraplatin induce PARP cleavage after 24 h, whereas no cleaved PARP (except for satraplatin) was observed in HCT116oxR cells. With increasing concentration of 1 and 2b and incubating for 48 h, all compounds are able to induce PARP cleavage in all tested cell lines (Fig. 6C).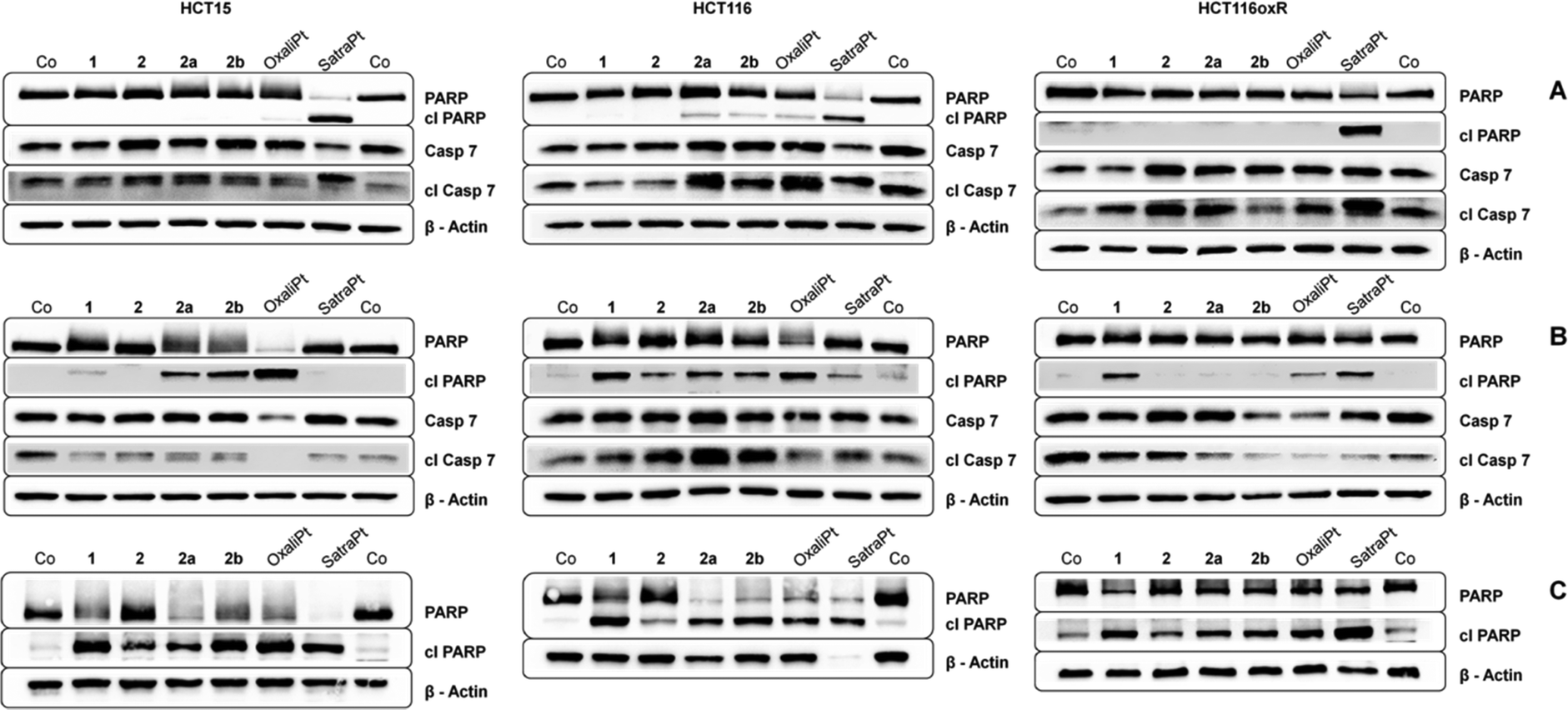 | ||
| Fig. 6 Immunoblots of caspase-induced cleavage of PARP in HCT15, HCT116 and HCT116oxR cells after continuous exposure for (A) 24 h with 2000 μM of 1, 2, 2a, 2b, 50 μM satraplatin and oxaliplatin; (B) 48 h with 1000 μM of 1, 2, 2a, 2b, 10 μM satraplatin and 50 μM oxaliplatin; and (C) 48 h with 2000 μM of 1, 2, 2a, 2b, 50 μM satraplatin and oxaliplatin. Representative blots from at least two independent experiments are depicted. | ||
DCFH-DA assay
In addition to its DNA targeting ability, cisplatin is able to induce reactive oxygen species (ROS).41,42In vitro experiments of Miyajima and coworkers showed that cisplatin is able to firstly generate and secondly enhance ROS levels when GSH depletors are applied.43 Until recently, only very little was known about ROS formation upon treatment with platinum(IV) complexes,26,44 but in experiments using the DCFH-DA assay different platinum(IV) complexes showed the ability to generate ROS in SW480 cells.35 In order to measure the capability of the compounds to produce reactive oxygen species, the oxidation of DCFH-DA to fluorescent DCF was monitored in HCT116 and HCT116oxR cells every 10 min over 150 min fluorimetrically (Fig. 7 and Fig. S17, S18, ESI†).Complexes 1 and 2 and oxaliplatin are not able to generate ROS in the applied concentrations in HCT116 and HCT116oxR cells. At the lowest investigated concentration (1000 μM), 2a did not produce detectable ROS, but at the higher one (2000 μM) a distinct increase up to 1.9 times in comparison to control levels was found (Fig. 7). 2b and satraplatin cause a slight increase in the fluorescence intensity of DCF at lower concentrations in both cell lines. At higher concentrations, both compounds generate ROS levels twice as high as those in the untreated controls. Consequently, these platinum complexes are less active than other platinum(IV) complexes reported previously by, e.g., Pichler et al.26,35 A reason for the reduced potency could be the use of different cell lines. However, an influence of chelating ligands of the oxaliplatin-like core of these platinum(IV) complexes is more likely.
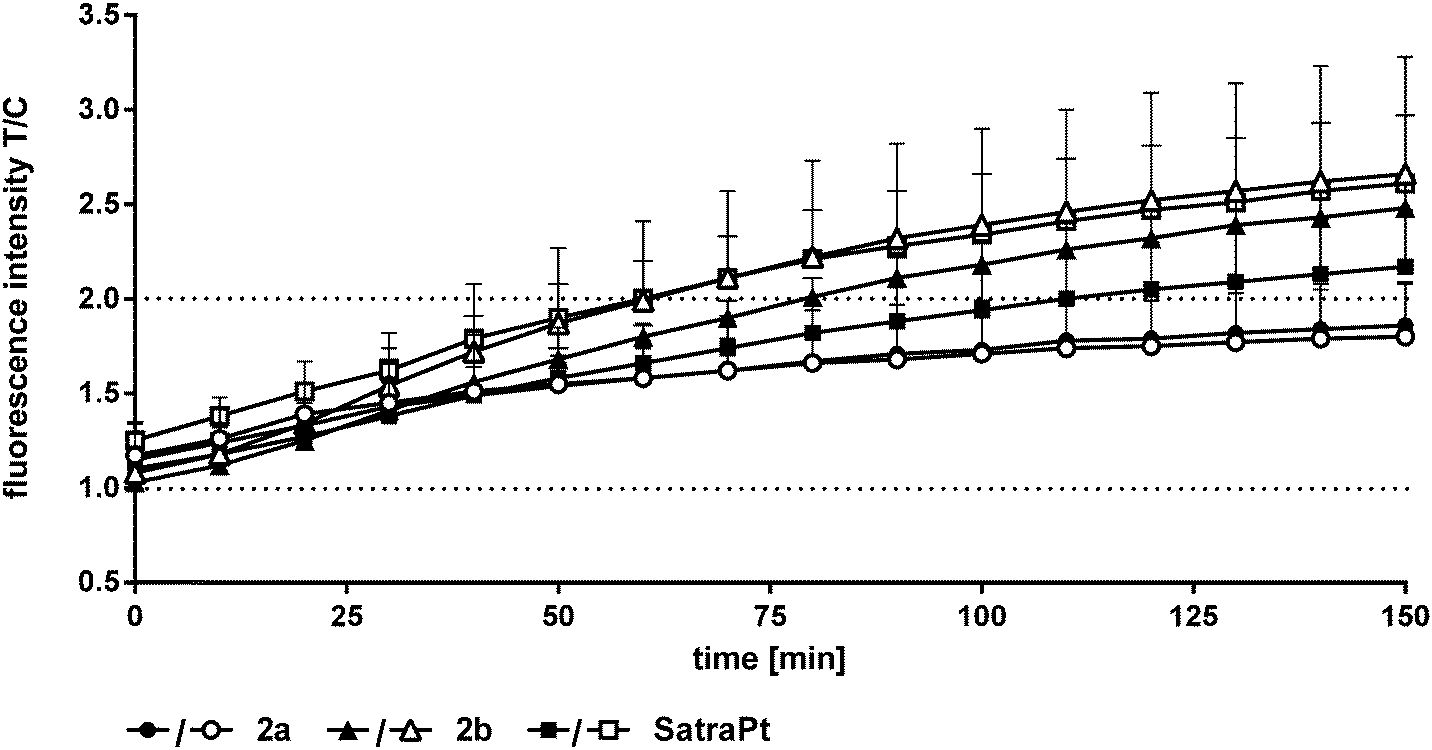 | ||
| Fig. 7 Fluorescence intensities of DCF as a measure of ROS levels in HCT116 (black symbols) and HCT116oxR (white symbols) cells recorded every 10 min during incubation with platinum(IV) complexes (2000 μM) and satraplatin (250 μM) for 2.5 h at 37 °C. | ||
In vivo anticancer activity – antileukemic activity
In order to assess the anticancer activity of 2a and 2b, pilot experiments were performed in the murine L1210 leukemia model with a small number of animals (n = 2 or 3 per group). These experiments were performed in different laboratories (as mentioned in the Experimental section). For experiment 1 (performed at the CRI in Bratislava), 2a was applied on days 1, 2 and 3 after L1210 cell implantation. For experiment 2 (performed at the ICR in Vienna), 2b was administered on days 1, 5 and 9. In both experiments, no long-term survivors without signs of leukemia were observed, but an increase in life span (ILS) was found. For 2a 35% ILS at a dose of 20 mg kg−1 and 24% ILS at 40 mg kg−1 were observed, but at the latter concentration a body weight loss of more than 15% occurred (data not shown), indicating acute toxicity of the substance. A 50% median increase of life span could be detected for 2b at 20 mg kg−1, but at 30 mg kg−1 the substance was toxic (data not shown). These findings suggest that the maximum tolerated dose (MTD) is reached for both compounds at 20 mg kg−1 (Fig. 8).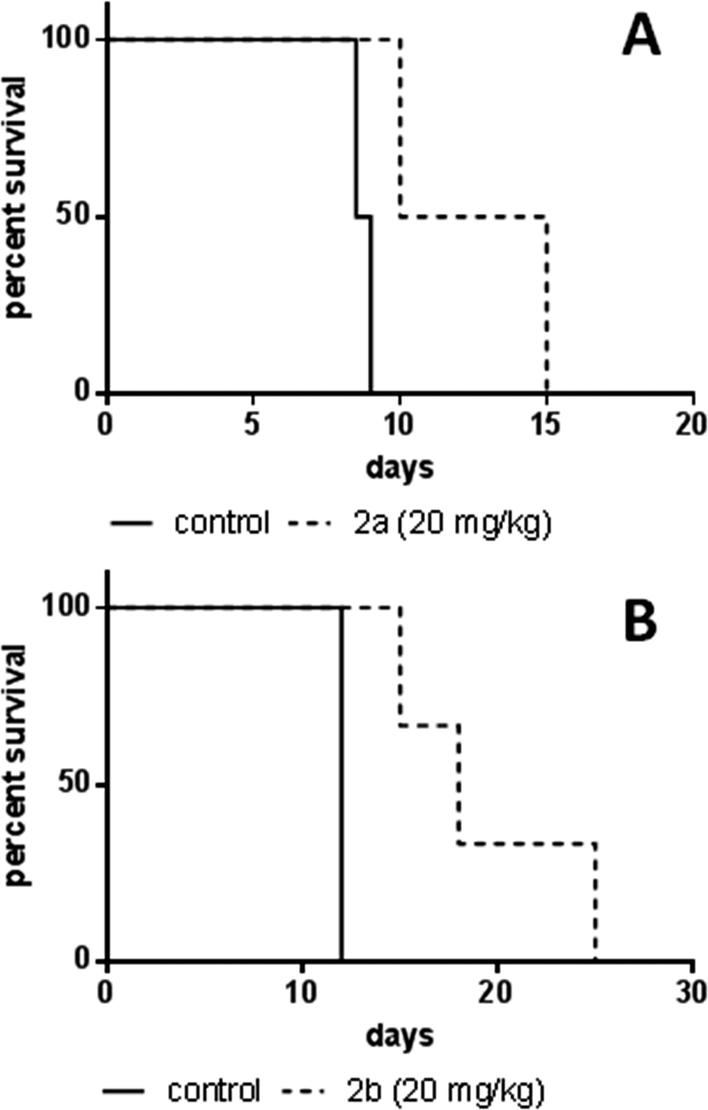 | ||
| Fig. 8 Kaplan–Meier plots of pilot experiments in the L1210 leukemia model. (A) Experiments on 2a were performed at the Cancer Research Institute, Slovak Academy of Sciences, Bratislava. The test compound was dissolved in DMSO and administered intraperitoneally at 20 mg kg−1 in a split-dose regimen on days 1, 2, and 3 after cell implantation (n = 2 per group). Control animals received DMSO i.p. (B) Experiments on 2b were performed at the Institute of Cancer Research, Medical University of Vienna, Austria. The test compound was dissolved in water and administered intraperitoneally at 20 mg kg−1 on days 1, 5, and 9 after cell implantation (n = 3 per group). Control animals received PBS i.p. | ||
In vivo anticancer activity – CT26 solid tumor model
As the anticancer potential of platinum(IV) complexes might be underestimated in a leukemia model being less favorable for reduction to platinum(II), compound 2b was investigated in a CT26 mouse model as well. The efficacy of platinum(IV) compounds may be enhanced due to the specific environment in a solid tumor tissue. It has been reported that the immune system is important for the anticancer activity of oxaliplatin as well as of oxaliplatin analogues.45 This was the reason to compare 2b in immunocompetent BALB/c with immuno-deficient BALB/c SCID mice. 2b was dissolved in water and administered i.p. at a concentration of 20 mg kg−1 on days 4, 7, 11 and 14 after the tumors became palpable. On day 15, the mice were sacrificed and tissues collected for further experiments. In BALB/c SCID mice, no anti-cancer activity was found, as the tumors in the control mice grow as fast as in the treated mice (Fig. 9A). This is in accordance with Jungwirth et al. and previously published data.45 In immunocompetent BALB/c mice, the tumor growth was distinctly retarded in three of the four individuals compared to the controls (Fig. 9B). As in mouse 2 the tumor grew faster than in controls, tumor cells were isolated from the treated tumor as well as from a control mouse. These cells were further investigated for acquired drug resistance against oxaliplatin or 2bin vitro. However, no significant difference in IC50 values in the MTT assay was observed that would indicate acquired drug resistance (Fig. S21, ESI†).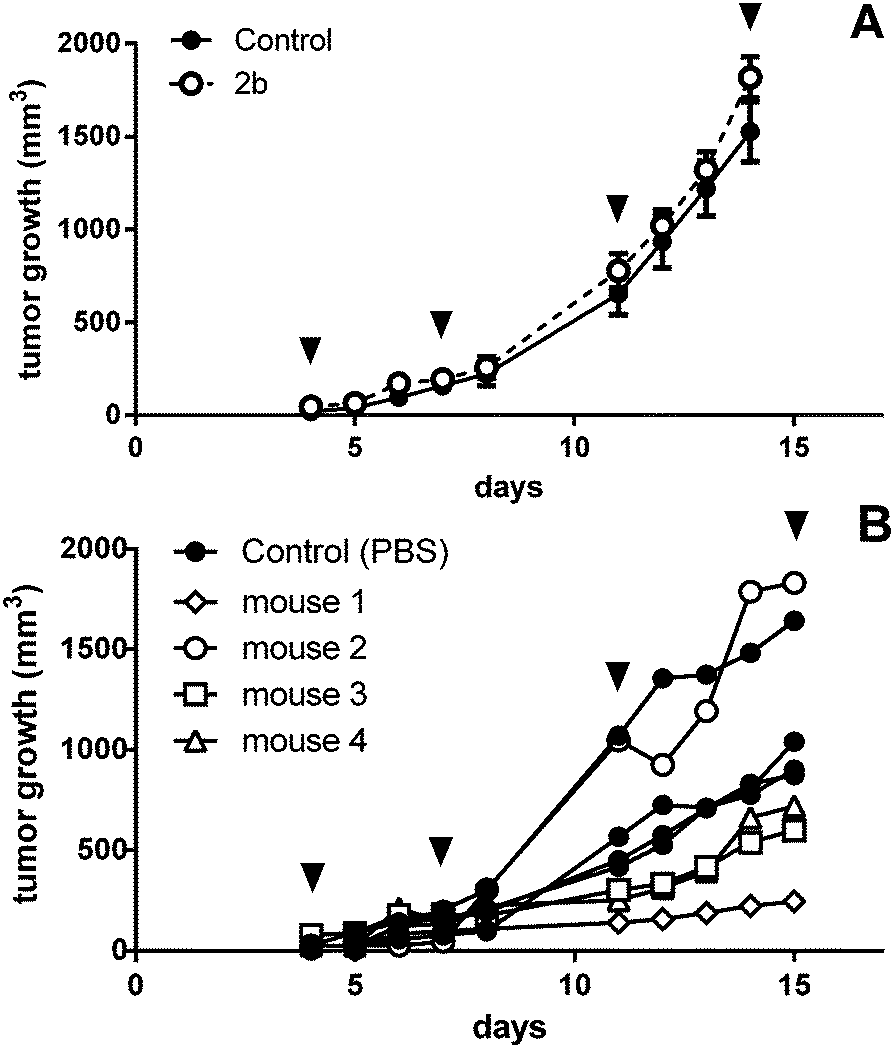 | ||
| Fig. 9 In vivo activity of 2b in male (A) BALB/c SCID (data shown are mean plus SEM; number of animals, 4 per group) and (B) BALB/c mice with subcutaneous CT26 tumors. Animals were treated with 20 mg kg−1 i.p. of 2b on days 4, 7, 11, and 14 (indicated by black arrows). | ||
On day 15, one day after the last treatment with 2b, the mice were sacrificed and organs were collected. The platinum levels in the blood, serum, liver, lungs, kidney, tumors and muscles as a reference were determined by ICP-MS after digestion by using sub-boiled nitric acid and microwave irradiation. To investigate a difference in platinum accumulation in the two investigated mouse models, platinum levels in the organs were compared. In all organs except the liver and kidney, about the same amounts of platinum were observed in BALB/c SCID and BALB/c mice. As expected, the highest accumulation of platinum is found in the liver and kidneys, as these organs are excretory organs and responsible for the detoxification of the body, followed by blood, lungs and serum (Fig. 10). The lowest levels of platinum, which are nearly identical for the immuno-competent and immunodeficient model, were measured in the muscles and tumor. No statistically significant difference in the average platinum levels in the tumor or muscles of BALB/c SCID and BALB/c mice was found. It is noteworthy that mouse 2, which did not respond to the treatment, showed the lowest platinum level compared to the other tested mice (Fig. S20, ESI†). In addition, induction of apoptosis in tumor tissues was determined by H/E staining (Fig. S19, ESI†), but no significant differences were found between treated animals and untreated controls.
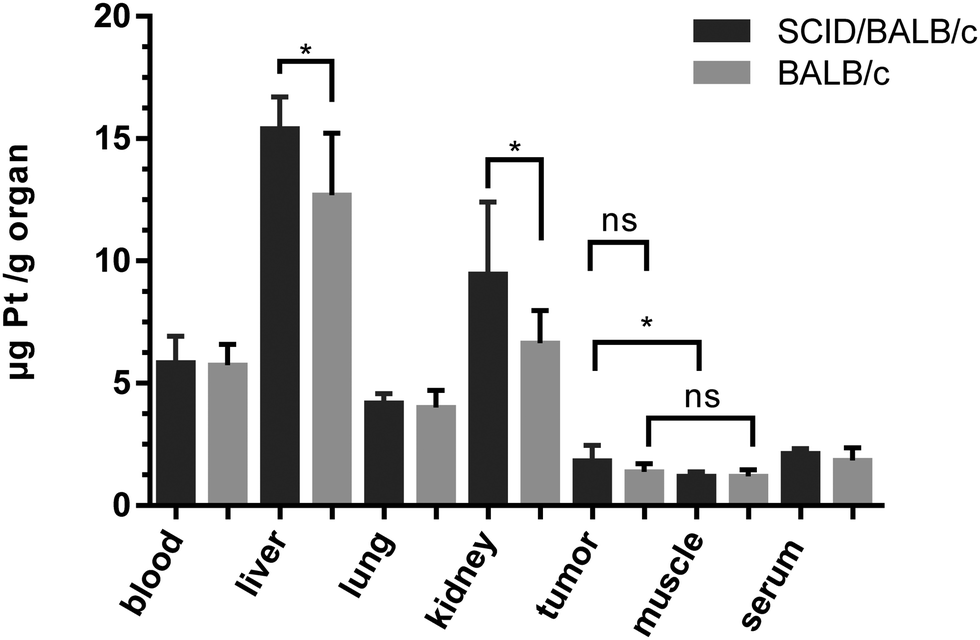 | ||
| Fig. 10 Average platinum concentrations (from treatment with 2b) in mouse tissues collected at day 15, quantified by ICP-MS. | ||
These results suggest that the oxaliplatin-based platinum(IV) complex 2b is processed in vivo to an active metabolite similar to oxaliplatin. The potential anti-cancer activity may be underestimated in immunodeficient mouse models. In fact, compound 2b seems to be strongly affected by the immune system. The extent and mode of action of 2bin vivo should be studied in more detail in further experiments such as investigations with regard to the immune response.
Conclusion
In this study, platinum(IV) prodrugs of the clinically relevant platinum(II)-based complex oxaliplatin were investigated in different in vitro and in vivo studies. The main focus was on effects caused by a reductive milieu and by the immune system.Remarkably, not all platinum(IV) complexes require the presence of a reducing agent to start interacting with DNA as proven in a cell-free assay. In different colon carcinoma cell lines, most of the tested compounds show IC50 values in the medium to high micromolar range. Reducing the oxygen partial pressure to establish a more reducing environment does not increase the cytotoxicity of these complexes. In multicellular spheroids with hypoxic cores, the cytotoxicity of the platinum(IV) complexes is even decreased, but to a lower degree than that of oxaliplatin. Additional investigations are necessary to clarify if this inactivation is due to poor drug penetration and accumulation. It seems that hypoxia, independent of the induction method, is not sufficient to activate these platinum(IV) complexes to a considerable extent. Nevertheless, these complexes induce cell cycle perturbations consistent with DNA-damaging activity (see ESI†) and finally lead to apoptosis in all tested cell lines. Unlike other platinum(IV) complexes, these complexes generate no or only little amounts of ROS in HTC116 and HCT116oxR cells. As 2a and 2b are the most promising of the compounds investigated here, in vivo studies in L1210 leukemia-bearing mice were performed, showing that both complexes are able to increase the life span of the mice distinctly. CT26 tumors in immune-competent BALB/c and immune-deficient SCID/BALB/c mice were treated with 2b to assess the activity in solid tumors. Activity seems to be highly dependent on the immune system, since only immunocompetent mice responded to treatment, while platinum levels in the tumor tissue are similar in both mouse strains and relatively low compared to other organs. These results support the assumption that the platinum(IV) complex is metabolized to oxaliplatin or a platinum(II) species structurally resembling oxaliplatin and induces an immune response.23 Whether these complexes are able to interact with T cells and induce the production of interferon γ like oxaliplatin remains to be investigated.
In conclusion, we demonstrate the high potency of platinum(IV) complexes based on clinically relevant platinum(II) drugs. They show all relevant properties such as DNA interactions, cytotoxicity, apoptosis induction and in vivo activity in addition to the advantages of the kinetically higher stability and lipophilicity that may qualify them for oral administration. Unlike complexes with a cisplatin or related core, reduction parameters hardly affect the cytotoxic potency of the oxaliplatin derivatives applied here. However, the high impact of the immune system on the activity of a platinum(IV)-based oxaliplatin prodrug could be demonstrated in vivo.
Experimental section
Compounds
All compounds were synthesized at the Institute of Inorganic Chemistry, University of Vienna, as described previously.24Rate of reduction
The reactivity of complexes 1 and 2b with cellular reductants was investigated by 1H NMR spectroscopy. Freshly prepared stock solutions of each compound (1 mM) as well as 2 mM or 10 mM ascorbic acid, or 2 mM glutathione (reduced) were prepared in 20 mM phosphate buffer (in D2O, pD = 7.51). Reaction mixtures of compound:reductant (1:2 or 1:10 molar ratio) were mixed directly before measurement, and spectra were measured at ambient temperature or 37 °C over a period of at least 14 h. 1H NMR spectra were recorded with a Bruker Avance III 500 MHz instrument at 500.32 MHz. The solvent residual peaks for 1H were used as internal references.
Electrophoretic dsDNA plasmid assay
300 ng of pUC19 dsDNA (2686 bp) plasmid were incubated with 50 μM of the test compounds alone or in the presence of 500 μM ascorbic acid in a 0.1× TE buffer (pH 8) for different time intervals (30 min to 8 h) at 37 °C. Electrophoresis was carried out in agarose gel 1% w/v for 90 min at 90 V in 1× TBE buffer. Ethidium bromide (EtBr) staining was performed in 1× TBE (0.75 μg mL−1) buffer for 20 min. Images were taken by the GelDoc-It Imaging System Fusion Fx7, Vilber Lourmat Deutschland GmbH.Cell lines and cell culture conditions
For cytotoxicity tests, three different human colon cancer cell lines were used: HCT15 and HCT116 were purchased from the ATCC (American Type Culture Collection). HCT116oxR was established by one of the authors (U. J.) at the Institute of Cancer Research, Department of Medicine I, Medical University Vienna, Austria, as described in ref. 46. Every 3–4 passages, the cells were treated with 10 μM oxaliplatin for 3 days to ensure maintenance of the resistance. All cell culture media and reagents were purchased from Sigma-Aldrich Austria and all cell culture materials such as dishes, plates and flasks from StarLab Germany unless indicated otherwise. Cells were grown as adherent monolayer cultures in 75 cm2 culture flasks in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen) and 4 mM L-glutamine without antibiotics at 37 °C under a humid atmosphere containing 5% CO2 and 95% air. The murine colon cancer cell line CT26 and murine leukemia L1210 (both from ATCC) were grown in DMEM/F12 or RMPI 1640 medium, respectively, supplemented with 10% heat-inactivated fetal bovine serum.Spheroid culture
For spheroid production, HCT15, HCT116, and HCT116oxR cells were harvested from culture flasks by trypsinization and seeded in RPMI 1640 medium (supplemented with 10% heat-inactivated fetal bovine serum and 4 mM L-glutamine) in non-cell culture treated round bottom 96-well plates (Nunclon SpheraTM) at densities of 5 × 103 (HCT15), 3.5 × 103 (HCT116), and 2.5 × 103 (HCT116oxR) viable cells per well 7 days prior to treatment. To minimize evaporation, the outermost wells were not used for spheroid production and filled with 200 μL of PBS instead. The cultures were maintained at 37 °C in a humidified atmosphere containing 95% air and 5% CO2.Cytotoxicity tests
For tests with normoxic monolayers, HCT15, HCT116, and HCT116oxR cells were harvested from the culture flasks by trypsinization and seeded in RPMI 1640 medium in 96-well microculture plates (StarLab) 24 h prior to treatment at densities of 2.5 × 103 (HCT15), 1.5 × 103 (HCT116), or 6 × 103 (HCT116oxR) viable cells per well, respectively. For tests in 3D cultures, spheroids with the required diameter were treated directly in the round-bottom culture plates used for their production.For both monolayer and spheroid treatments, stock solutions of the test compounds were prepared in RPMI 1640 medium and diluted stepwise to obtain a serial dilution. 100 μL of dilution were added to each well, and the plates were incubated for 96 h at 37 °C and 5% CO2. A fresh solution of 440 μM (≈110 μg mL−1) resazurin sodium salt (Sigma-Aldrich) in PBS was prepared and 20 μL of it were added to each well. The plates were stained at 37 °C under 5% CO2 for 4 h (monolayer experiments) or overnight (spheroid experiments). The fluorimetric measurement of the formed resorufin was performed with a Synergy HT reader (BioTek) (excitation wavelength 530 nm; emission wavelength 590 nm).
For tests in hypoxic monolayer cultures, cells were harvested by trypsinization and seeded with different densities (vide supra) in RPMI 1640 medium in 96-well plates in volumes of 100 μL per well 24 h prior to treatment under 0.5% oxygen and 5% CO2 in a C-Chamber with ProOx oxygen/carbon dioxide controllers (Biospherix). The cells were exposed and stained, and fluorescence was measured as described above.
Confocal microscopy
For hypoxia staining, 30 spheroids were transferred into 1.5 mL microreaction tubes and washed with phosphate buffered saline (PBS, Sigma-Aldrich). Subsequently, samples were fixed with PBS containing 4% paraformaldehyde (Sigma-Aldrich) and 1% Triton X-100 (Sigma-Aldrich) for 3 h at 4 °C and washed with PBS (three times for 10 min). An ascending methanol series was used for dehydration at 4 °C in PBS (25%, 50%, 75%, and 95% for 30 min each and 100% for 3 h) and the reverse methanol series was used for rehydration. The samples were washed with PBS (three times for 10 min). The samples were blocked and permeabilized with PBST (0.1% Triton X-100 in PBS) containing 3% bovine serum albumin (Sigma-Aldrich) overnight at 4 °C, and subsequently washed twice for 15 min with PBST and incubated for 48 h at 4 °C with HIF1α antibody (Abcam, Cambridge, UK) diluted 1:400 in PBST. After washing four times for 30 min with PBST, the secondary antibody – Alexa Flour 647 (Cell Signaling, Frankfurt am Main, Germany) – diluted 1:1000 in PBST was added for 24 h at 4 °C. The samples were finally washed four times for 30 min in PBST and investigated with a confocal microscope.
For staining of necrotic cores, viable spheroids were transferred into a Falcon non-tissue culture treated u-bottom 96-well plate, one spheroid per well in a total volume of 40 μL MEM (supplemented with 10% heat-inactivated fetal bovine serum (Life Technologies), 1% glutamine (Sigma-Aldrich), 1% sodium pyruvate (Sigma-Aldrich), and 1% non-essential amino acids (Sigma-Aldrich)). 160 μL of propidium iodide (Sigma-Aldrich) diluted in MEM to a final concentration of 1 μg mL−1 was added per well. The samples were incubated for 8 h at 37 °C and 5% CO2 prior to image acquisition with a confocal laser scanning microscope.
Confocal imaging was performed with a CLSM from Leica (Leica SP5). It was equipped with 2.5×, 10× and 20× multi-immersion objectives for low magnification, and for high magnification with 40× and 60× oil, water and glycerol objectives. Tonal range adjustment of the micrographs was carried out in Adobe Photoshop CS 4.
Apoptosis induction – Annexin V/PI assay
Cell death induction was analyzed by flow cytometry using FITC-conjugated annexin V (BioVision, USA) and propidium iodide (PI, Fluka) double staining. Cells were seeded 24 h prior to treatment into 48-well plates at densities of 6 × 104 cells (for 24 h) and 4 × 104 cells (for 48 h) and incubated in 250 μL per well in RPMI 1640 medium. The medium was removed and the cells were exposed to different concentrations of the substances in volumes of 200 μL per well for 24 h and 48 h (37 °C, 5% CO2). After incubation, the cells were gently trypsinized and harvested, centrifuged (300 g, 3 min), and the supernatant was discarded. The cells were resuspended with annexin V/FITC (0.25 μg mL−1) in binding buffer (10 mM HEPES/NaOH pH 7.4, 140 mM NaCl, 2.5 mM CaCl2) and incubated at 37 °C for 15 min. PI (1 μg mL−1) was added shortly before measurement. The stained cells were analyzed with a Millipore Guava easyCyte™ 8HT flow cytometer using InCyte software. The resulting dot plots were quantified by FlowJo software (TreeStar). At least three independent experiments were conducted with 5 × 103 cells per analysis.Apoptosis induction – Western blotting
Cells were harvested by trypsinization and 6 × 105 cells per dish were seeded into a cell culture Petri dish (6 cm diameter) in 6 mL of RPMI 1640 medium, and after incubation for 48 h the cells were allowed to settle and resume exponential growth. The medium was removed and the cells were exposed to the test compounds for 24 h or 48 h. Thereafter, the medium was removed again, the cells were washed with PBS and total cell lysates were prepared by lysis with RIPA buffer containing protease inhibitor cocktails (P8340, Sigma-Aldrich). Identical amounts (determined with the Micro BCA™ protein assay kit following manufacturer's instructions, purchased from Thermo Scientific) of total proteins were separated by SDS-PAGE (9–12%) and electrophoretically transferred onto a PVDF (Immobilon® Transfer Membrane, Millipore) membrane by using a semi-dry blotter (Peqlab, Erlangen, Germany). The membrane was blocked with 5% w/v BSA in Tris-buffered saline/Tween 20 overnight at 4 °C. All antibodies were purchased from Cell Signaling and used according to the manufacturer's instructions. The anti-β-actin antibody was used as a loading control. Horseradish peroxidase-coupled secondary antibodies were appropriately diluted and incubated for 1 h at room temperature and detected by chemiluminescence using the Pierce SuperSignal chemiluminescence substrate (Thermo Fisher Scientific, Inc., Rockford, IL, USA) and the GelDoc-It Imaging System Fusion Fx7 (Vilber Lourmat Deutschland GmbH).ROS generation
HCT116 and HCT116oxR cells were harvested and 2.5 × 104 cells per well were seeded in RPMI 1640 medium into a 96-well plate in volumes of 100 μL per well. In the first 24 h, the cells were allowed to settle and resume exponential growth. The cells were washed with Hanks' balanced salt solution (HBSS, Sigma-Aldrich, containing 1% FCS) and incubated with 25 μM DCFH-DA (2′,7′-dichlorofluorescein diacetate, Sigma-Aldrich) in HBSS for 45 min at 37 °C and 5% CO2. After staining, the cells were washed with HBSS and incubated with the compounds at different concentrations in 160 μL HBSS. ROS production was measured online with the Synergy HT reader (Ex 480 nm/Em 516 nm) over 2.5 h. The results are given relative to the untreated control. As a positive assay control, 200 μM tert-butyl hydroperoxide was used.Antileukemic activity in vivo
Animal experiments were performed in accordance with the European Community Guidelines for the use of experimental animals in the animal facility at the Cancer Research Institute, Slovak Academy of Sciences, Bratislava, Slovak Republic. L1210 murine leukemia cells (1 × 105) were injected in a volume of 0.5 mL into DBA/2J mice (weighing 18–22 g) intraperitoneally on day 0. The test compound was dissolved in DMSO and immediately administered intraperitoneally in a volume of 50 μL per mouse per day in a split-dose regimen (days 1, 2, and 3 after implantation of cells) to groups of two animals per dose. The toxicity of the test compounds was monitored by daily observation of the animals and registration of their body weight. The therapeutic efficacy of the test compounds was monitored by recording the lengths of survival of the experimental mice compared to vehicle controls.At the Institute of Cancer Research, Medical University of Vienna, L1210 murine leukemia cells (1 × 105) were injected in a volume of 0.2 mL into male DBA/2J mice (weighing 19–25 g) intraperitoneally on day 0. The test compound was dissolved in water and immediately administered intraperitoneally in a volume of 100 μL per mouse on days 1, 5, and 9 after implantation of cells. The toxicity of the test compounds was monitored by daily observation of animals and registration of their body weight. The therapeutic efficacy of the test compounds was monitored by recording the lengths of survival of the experimental mice compared to vehicle controls.
Anticancer activity against CT26 colon cancer cells in vivo
Murine CT26 cells (5 × 105) were injected subcutaneously into the right flank of male BALB/c or CB-17scid/scid (BALB/c SCID) mice and therapy was started when tumor nodules were palpable (day 4). The mice were treated with 2b (i.p. 20 mg kg−1 in H2O at days 4, 7, 11, and 14). Control mice received PBS i.p. The animals were monitored for distress development every day and the tumor size was assessed regularly by caliper measurement. The mice were anesthetized 24 h after the last drug application (day 15) and tissues were collected. Samples for ICP-MS measurements were stored at −20 °C. Samples for histological evaluation were formalin-fixed and paraffin-embedded. H/E stains were performed by using standard procedures. For the re-establishment of a CT26 subline after treatment with 2b, a tumor piece was mechanically minced in medium and the supernatant further diluted in culture medium containing 10% FCS.Platinum quantification in mouse tissues by ICP-MS
Digestion of mouse organs was performed as described previously.38 The organs were cut and digested in sub-boiled nitric acid by using the microwave system Discover SP-D (CEM Microwave Technology, Germany) at a temperature of 200 °C (ramp. time 4 min, hold 6 min) with maximal power of 300 W. The samples were further diluted with MilliQ water (final HNO3 concentration <3%) and platinum concentrations were quantified by ICP-MS.Statistical analysis
The results were expressed as mean ± SD. Statistical calculations were performed by using GraphPad Prism 6. Statistical significance levels with p < 0.001, p < 0.01 or p < 0.05 were obtained by the t test with Welch's correction, whenever indicated by ***, ** or *, respectively.Conflict of interests
The authors declare no conflict of interests.Abbreviations
| AA | Ascorbic acid |
| AV | Annexin V |
| casp 7 | Caspase 7 |
| DNA | Deoxyribonucleic acid |
| FCS | Fetal calf serum |
| GSH | Glutathione |
| HBSS | Hanks' balanced salt solution |
| HSF | Hypotonic fluorochrome solution |
| ICP-MS | Inductively coupled plasma mass spectrometry |
| MEM | Minimum essential medium |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NMR | Nuclear magnetic resonance |
| oc | Open circular |
| PARP | Poly(ADP-ribose)polymerase |
| PBS | Dulbecco's phosphate-buffered saline |
| PI | Propidium iodide |
| RF | Resistance factor |
| PVDF | Polyvinylidene difluoride |
| RIPA | Radioimmunoprecipitation assay buffer |
| ROS | Reactive oxygen species |
| RPMI | Roswell Park Memorial Institute |
| sc | Supercoiled |
| SD | Standard deviation |
| SDS-PAGE | Sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| TBE | Tris-borate-EDTA buffer |
| TE | Tris-EDTA buffer. |
Acknowledgements
The authors thank Markus Galanski for providing compounds and expertise. S. G. wants to thank S. Theiner, M. Klose and L. Bamonti for ICP-MS measurements and V. Somoza for providing the Fusion Fx7 detection system. The in vivo experiments were performed in the course of P26603, a project funded by the Austrian Science Fund (FWF). Many thanks to Gerhard Zeitler for his devoted animal care and Sushilla van Schoonhoven who helped with the H&E staining.References
- J. Reedijk, Increased understanding of platinum anticancer chemistry, Pure Appl. Chem., 2011, 83, 1709–1719 CrossRef CAS.
- N. J. Wheate, S. Walker, G. E. Craig and R. Oun, The status of platinum anticancer drugs in the clinic and in clinical trials, Dalton Trans., 2010, 39, 8113–8127 RSC.
- L. Kelland, The resurgence of platinum-based cancer chemotherapy, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- Y. Jung and S. J. Lippard, Direct cellular responses to platinum-induced DNA damage, Chem. Rev., 2007, 107, 1387–1407 CrossRef CAS PubMed.
- R. K. Mehmood, Review of cisplatin and oxaliplatin in current immunogenic and monoclonal antibody treatments, Oncol. Rev., 2014, 8, 256 Search PubMed.
- J. X. Ong, S. Q. Yap, D. Y. Q. Wong, C. F. Chin and W. H. Ang, Platinum(IV) carboxylate prodrug complexes as versatile platforms for targeted chemotherapy, Chim. Int. J. Chem., 2015, 69, 100–103 CrossRef CAS PubMed.
- M. Galanski, M. A. Jakupec and B. K. Keppler, Update of the preclinical situation of anticancer platinum complexes: novel design strategies and innovative analytical approaches, Curr. Med. Chem., 2005, 12, 2075–2094 CrossRef CAS PubMed.
- M. D. Hall and T. W. Hambley, Platinum(IV) antitumour compounds: their bioinorganic chemistry, Coord. Chem. Rev., 2002, 232, 49–67 CrossRef CAS.
- V. Novohradsky, L. Zerzankova, J. Stepankova, O. Vrana, R. Raveendran, D. Gibson, J. Kasparkova and V. Brabec, Antitumor platinum(IV) derivatives of oxaliplatin with axial valproato ligands, J. Inorg. Biochem., 2014, 140, 72–79 CrossRef CAS PubMed.
- R. Raveendran, J. P. Braude, E. Wexselblatt, V. Novohradsky, O. Stuchlikova, V. Brabec, V. Gandin and D. Gibson, Pt(II) derivatives of cisplatin and oxaliplatin with phenylbutyrate axial ligands are potent cytotoxic agents that act by several mechanisms of action, Chem. Sci., 2016, 7, 2381–2391 RSC.
- N. Margiotta, S. Savino, C. Marzano, C. Pacifico, J. D. Hoeschele, V. Gandin and G. Natile, Cytotoxicity-boosting of kiteplatin by Pt(IV) prodrugs with axial benzoate ligands, J. Inorg. Biochem., 2015, 160, 85–93 CrossRef PubMed.
- W. H. Ang, S. Pilet, R. Scopelliti, F. Bussy, L. Juillerat-Jeanneret and P. J. Dyson, Synthesis and characterization of platinum(IV) anticancer drugs with functionalized aromatic carboxylate ligands: influence of the ligands on drug efficacies and uptake, J. Med. Chem., 2005, 48, 8060–8069 CrossRef CAS PubMed.
- W. H. Ang, I. Khalaila, C. S. Allardyce, L. Juillerat-Jeanneret and P. J. Dyson, Rational design of platinum(IV) compounds to overcome glutathione-S-transferase mediated drug resistance, J. Am. Chem. Soc., 2005, 127, 1382–1383 CrossRef CAS PubMed.
- Z. Dong and J. Wang, Hypoxia selection of death-resistant cells: a role for Bcl-XL, J. Biol. Chem., 2004, 279, 9215–9221 CrossRef CAS PubMed.
- N. Rohwer and T. Cramer, Hypoxia-mediated drug resistance: Novel insights on the functional interaction of HIFs and cell death pathways, Drug Resist. Updates, 2011, 14, 191–201 CrossRef CAS PubMed.
- G. L. Semenza, Intratumoral hypoxia, radiation resistance, and HIF-1, Cancer Cell, 2004, 5, 405–406 CrossRef CAS PubMed.
- C. H. Hsieh, C. P. Wu, H. T. Lee, J. A. Liang, C. Y. Yu and Y. J. Lin, NADPH oxidase subunit 4 mediates cycling hypoxia-promoted radiation resistance in glioblastoma multiforme, Free Radical Biol. Med., 2012, 53, 649–658 CrossRef CAS PubMed.
- M. Milosevic, P. Warde, C. Menard, P. Chung, A. Toi, A. Ishkanian, M. McLean, M. Pintilie, J. Sykes, M. Gospodarowicz, C. Catton, P. R. Hill and R. Bristow, Tumor hypoxia predicts biochemical failure following radiotherapy for clinically localized prostate cancer, Clin. Cancer Res., 2012, 18, 2108–2114 CrossRef CAS PubMed.
- M. Höckel and P. Vaupel, Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects, J. Natl. Cancer Inst., 2001, 93, 266–276 CrossRef.
- P. Vaupel and L. Harrison, Tumor hypoxia: causative factors, compensatory mechanisms, and cellular response, Oncologist, 2004, 9, 4–9 CrossRef PubMed.
- K. R. Luoto, R. Kumareswaran and R. G. Bristow, Tumor hypoxia as a driving force in genetic instability, Genome Integr., 2013, 4, 1–5 CrossRef PubMed.
- T. Alcindor and N. Beauger, Oxaliplatin: a review in the era of molecularly targeted therapy, Curr. Oncol., 2011, 18, 18–25 CAS.
- A. Tesniere, F. Schlemmer, V. Boige, O. Kepp, I. Martins, F. Ghiringhelli, L. Aymeric, M. Michaud, L. Apetoh, L. Barault, J. Mendiboure, J.-P. Pignon, V. Jooste, P. van Endert, M. Ducreux, L. Zitvogel, F. Piard and G. Kroemer, Immunogenic death of colon cancer cells treated with oxaliplatin, Oncogene, 2010, 29, 482–491 CrossRef CAS PubMed.
- M. R. Reithofer, S. M. Valiahdi, M. A. Jakupec, V. B. Arion, A. Egger, M. Galanski and B. K. Keppler, Novel di- and tetracarboxylatoplatinum(IV) complexes, Synthesis, characterization, cytotoxic activity, and DNA platination, J. Med. Chem., 2007, 50, 6692–6699 CrossRef CAS PubMed.
- J. Reedijk, Fast and slow versus strong and weak metal–DNA binding: consequences for anti-cancer activity, Metallomics, 2012, 4, 628–632 RSC.
- V. Pichler, S. Göschl, E. Schreiber-Brynzak, M. A. Jakupec, M. Galanski and B. K. Keppler, Influence of reducing agents on the cytotoxic activity of platinum(IV) complexes: induction of G2/M arrest, apoptosis and oxidative stress in A2780 and cisplatin resistant A2780cis cell lines, Metallomics, 2015, 7, 1078–1090 RSC.
- V. Pichler, S. Göschl, S. M. Meier, A. Roller, M. A. Jakupec, M. Galanski and B. K. Keppler, Bulky (NN)-(di)alkylethane-1,2-diamineplatinum(II) compounds as precursors for generating unsymmetrically substituted platinum(IV) complexes, Inorg. Chem., 2013, 52, 8151–8162 CrossRef CAS PubMed.
- J. Z. Zhang, E. Wexselblatt, T. W. Hambley and D. Gibson, Pt(IV) analogs of oxaliplatin that do not follow the expected correlation between electrochemical reduction potential and rate of reduction by ascorbate, Chem. Commun., 2012, 48, 847–849 RSC.
- E. Wexselblatt and D. Gibson, What do we know about the reduction of Pt(IV) pro-drugs?, J. Inorg. Biochem., 2012, 117, 220–229 CrossRef CAS PubMed.
- H. P. Varbanov, M. A. Jakupec, A. Roller, F. Jensen, M. Galanski and B. K. Keppler, Theoretical investigations and density functional theory based quantitative structure-activity relationships model for novel cytotoxic platinum(IV) complexes, J. Med. Chem., 2013, 56, 330–344 CrossRef CAS PubMed.
- H. P. Varbanov, S. M. Valiahdi, C. R. Kowol, M. A. Jakupec, M. Galanski and B. K. Keppler, Novel tetracarboxylatoplatinum(IV) complexes as carboplatin prodrugs, Dalton Trans., 2012, 41, 14404–14415 RSC.
- M. Sinisi, F. P. Intini and G. Natile, Dependence of the reduction products of platinum(IV) prodrugs upon the configuration of the substrate, bulk of the carrier ligands, and nature of the reducing agent, Inorg. Chem., 2012, 51, 9694–9704 CrossRef CAS PubMed.
- B. R. Hoffmeister, M. Hejl, M. A. Jakupec, M. Galanski and B. K. Keppler, Bis- and tris(carboxylato)platinum(IV) complexes with mixed am(m)ine ligands in the trans position exhibiting exceptionally high cytotoxicity, Eur. J. Inorg. Chem., 2015, 1700–1708 CrossRef CAS.
- D. Höfer, H. P. Varbanov, A. Legin, M. A. Jakupec, A. Roller, M. Galanski and B. K. Keppler, Tetracarboxylatoplatinum(IV) complexes featuring monodentate leaving groups – a rational approach toward exploiting the platinum(IV) prodrug strategy, J. Inorg. Biochem., 2015, 259–271 CrossRef PubMed.
- S. Göschl, H. P. Varbanov, S. Theiner, M. A. Jakupec, M. Galanski and B. K. Keppler, The role of the equatorial ligands for the redox behavior, mode of cellular accumulation and cytotoxicity of platinum(IV) prodrugs, J. Inorg. Biochem., 2016, 160, 264–274 CrossRef PubMed.
- E. Schreiber-Brynzak, E. Klapproth, C. Unger, I. Lichtscheidl-Schultz, S. Göschl, S. Schweighofer, R. Trondl, H. Dolznig, M. A. Jakupec and B. K. Keppler, Three-dimensional and co-culture models for preclinical evaluation of metal-based anticancer drugs, Invest. New Drugs, 2015, 33, 835–847 CrossRef CAS PubMed.
- J. Laurent, C. Frongia, M. Cazales, O. Mondesert, B. Ducommun and V. Lobjois, Multicellular tumor spheroid models to explore cell cycle checkpoints in 3D, BMC Cancer, 2013, 13, 73 CrossRef PubMed.
- E. Schreiber-Brynzak, V. Pichler, P. Heffeter, B. Hanson, S. Theiner, I. Lichtscheidl-Schultz, C. Kornauth, L. Bamonti, V. Dhery, D. Groza, D. Berry, W. Berger, M. Galanski, M. A. Jakupec and B. K. Keppler, Behavior of platinum(IV) complexes in models of tumor hypoxia: cytotoxicity, compound distribution and accumulation, Metallomics, 2016, 8, 422–433 RSC.
- L. E. Jamieson, V. L. Camus, P. O. Bagnaninchi, K. M. Fisher, G. D. Stewart, W. H. Nailon, D. B. McLaren, D. J. Harrison and C. J. Campbell, Targeted SERS nanosensors measure physicochemical gradients and free energy changes in live 3D tumor spheroids, Nanoscale, 2016, 8, 16710–16718 RSC.
- B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, Molecular Biology of the Cell, Garland Science, 2008, p. 1117 Search PubMed.
- T. Ozben, Oxidative stress and apoptosis: impact on cancer therapy, J. Pharm. Sci., 2007, 96, 2181–2196 CrossRef CAS PubMed.
- J. Wang and J. Yi, Cancer cell killing via ROS: to increase or decrease, that is a question, Cancer Biol. Ther., 2008, 7, 1875–1884 CrossRef CAS PubMed.
- A. Miyajima, J. Nakashima, K. Yoshioka, M. Tachibana, H. Tazaki and M. Murai, Role of reactive oxygen species in cis-dichlorodiammineplatinum-induced cytotoxicity on bladder cancer cells, Br. J. Cancer, 1997, 76, 206–210 CrossRef CAS PubMed.
- G. N. Kaluđerović, S. A. Mijatović, B. B. Zmejkovski, M. Z. Bulatović, S. Gómez-Ruiz, M. K. Mojić, D. Steinborn, D. M. Miljković, H. Schmidt, S. D. Stošić-Grujičić, T. J. Sabo and D. D. Maksimović-Ivanić, Platinum(II/IV) complexes containing ethylenediamine-NN′-di-2/3-propionate ester ligands induced caspase-dependent apoptosis in cisplatin-resistant colon cancer cells, Metallomics, 2012, 4, 979–987 RSC.
- U. Jungwirth, D. N. Xanthos, J. Gojo, A. K. Bytzek, W. Körner, P. Heffeter, S. A. Abramkin, M. A. Jakupec, C. G. Hartinger, U. Windberger, M. Galanski, B. K. Keppler and W. Berger, Anticancer activity of methyl-substituted oxaliplatin analogs, Mol. Pharmacol., 2012, 81, 719–728 CrossRef CAS PubMed.
- S. A. Abramkin, U. Jungwirth, S. M. Valiahdi, C. Dworak, L. Habala, K. Meelich, W. Berger, M. A. Jakupec, C. G. Hartinger, A. A. Nazarov, M. Galanski and B. K. Keppler, {(1R,2R,4R)-4-methyl-1,2-cyclohexanediamine}oxalatoplatinum(II): a novel enantiomerically pure oxaliplatin derivative showing improved anticancer activity in vivo, J. Med. Chem., 2010, 53, 7356–7364 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6mt00226a |
| This journal is © The Royal Society of Chemistry 2017 |