
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polystyrene-supported bifunctional resorcinarenes as cheap, metal-free and recyclable catalysts for epoxide/CO2 coupling reactions†
T.
Jose
a,
S.
Cañellas
 a,
M. A.
Pericàs
*ab and
A. W.
Kleij
*ac
a,
M. A.
Pericàs
*ab and
A. W.
Kleij
*ac
aInstitute of Chemical Research of Catalonia (ICIQ), the Barcelona Institute of Science and Technology, Av. Països Catalans 16, 43007 – Tarragona, Spain. E-mail: akleij@iciq.es; mapericas@iciq.es
bDepartament de Química Inorgànica i Orgànica, Universitat de Barcelona, 08028 – Barcelona, Spain
cCatalan Institute for Research and Advanced Studies (ICREA), Pg. Lluis Companys 23, 08010 – Barcelona, Spain
First published on 19th October 2017
Abstract
We here report the synthesis of bifunctional catalysts that can be assembled using simple, cheap and accessible building blocks based on resorcinarenes, and their application as efficient, one-component homogeneous catalysts in the coupling of both terminal and internal epoxides with carbon dioxide affording their cyclic carbonate products. Furthermore, a heterogeneous version was also prepared that combines the activity of these bifunctional systems with excellent stability and recycling potential, allowing for a turnover of >1250. This newly prepared organocatalyst obviates the use of any metal, solvent or additives marking it as an attractive catalyst for CO2 valorization.
Introduction
Carbon dioxide (CO2) is an abundant waste material that may be recycled using effective catalysis technologies.1 Key to CO2 conversion is the use of high-energy reactants that provide a thermodynamic driving force, and historically epoxides have been popular coupling partners to afford cyclic organic carbonates (COCs).2 These heterocyclic structures find many useful applications representing precursors for polymeric carbonates,3 being green solvents4 and offering new opportunities in synthetic chemistry.5 The “fixation” of CO2 into COCs has advanced tremendously over the years with a prominent role for (transition) metal-based catalysts.6 The use of efficient metal catalysis has allowed, inter alia, the resolution of challenging conversions of CO2 with oxetanes,7 and disubstituted8 and even trisubstituted oxiranes.9In contrast, organocatalysts have only recently emerged as potential sustainable alternatives to metal-based catalyst systems,10 though the vast majority of the cases still require the assistance of co-catalysts, solvents and/or elevated temperatures (>100 °C). Exceptions to these harsher temperature requirements were only recently reported, and showed that proper catalyst design and using conceptually new and powerful substrate activation strategies are crucial to be able to bridge the gap between metal- and organo-catalysis.11 In this context, particularly the system from Detrembleur, Tassaing and Jerôme (a fluorinated bis-alcohol derivative) stands out as it represents one of the most active homogeneous, hydrogen-bond donor catalysts reported to date for cyclic carbonate synthesis.12 We have recently set out to develop conceptually new and efficient organocatalysts for the coupling of more challenging internal epoxides and CO2. In order to achieve more powerful catalysts, we used the unique oxo-anion stabilization potential of squaramide based catalysts13 and cooperative hydrogen-bonding catalysis based on polyphenols14 including resorcinarene- and pyrogallene-based structures (Fig. 1a).15
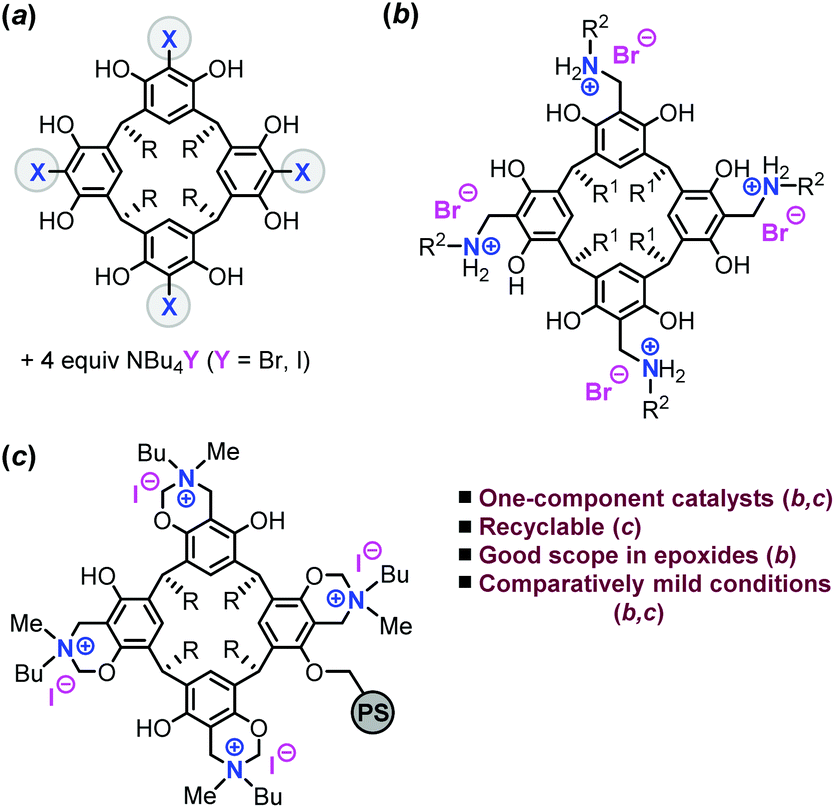 | ||
| Fig. 1 (a) Previously reported binary resorcinarene (X = H) and pyrogallene (X = OH) catalysts, (b) the bifunctional systems developed in this work, and (c) a heterogeneous version of the bifunctional system (PS = polystyrene). | ||
These latter catalysts represent cheap and readily accessible materials comprising of four well-positioned resorcinol or pyrogallol subunits, respectively. These polyphenolic compounds in combination with external (halide-based) nucleophiles showed improved activity for terminal epoxide/CO2 couplings at temperatures as low as 45 °C, and a record-high initial TOF of 488 h−1 at 80 °C.
We previously reported the successful immobilization of pyrogallol onto a bifunctional polystyrene support and application as a recyclable catalyst for cyclic carbonate synthesis.16 Inspired by our former results, we imagined that the immobilization of amine-functionalized resorcinarenes would offer powerful bifunctional catalysts (Fig. 1c) thus amplifying their potential towards metal-free, recyclable systems for COC formation. In this work, we present a simple and effective approach towards the preparation of modular bifunctional catalysts for COC synthesis, the immobilization onto a polystyrene support and the recycling features of this heterogeneous catalyst. These bifunctional, one-component metal-free catalysts show promise towards the coupling of various terminal and internal epoxides, and thus unite favourable activity, good recycling potential and sustainability features.
Results and discussion
Bifunctional catalyst synthesis
Various resorcin[4]arene precursors (1–4) with different alkyl chain lengths at the lower rim (R1), the tetrabenzoxazine derivatives 5–11 and the bifunctional resorcinarene salts 12–19 were prepared according to previous reported procedures (Scheme 1).15,17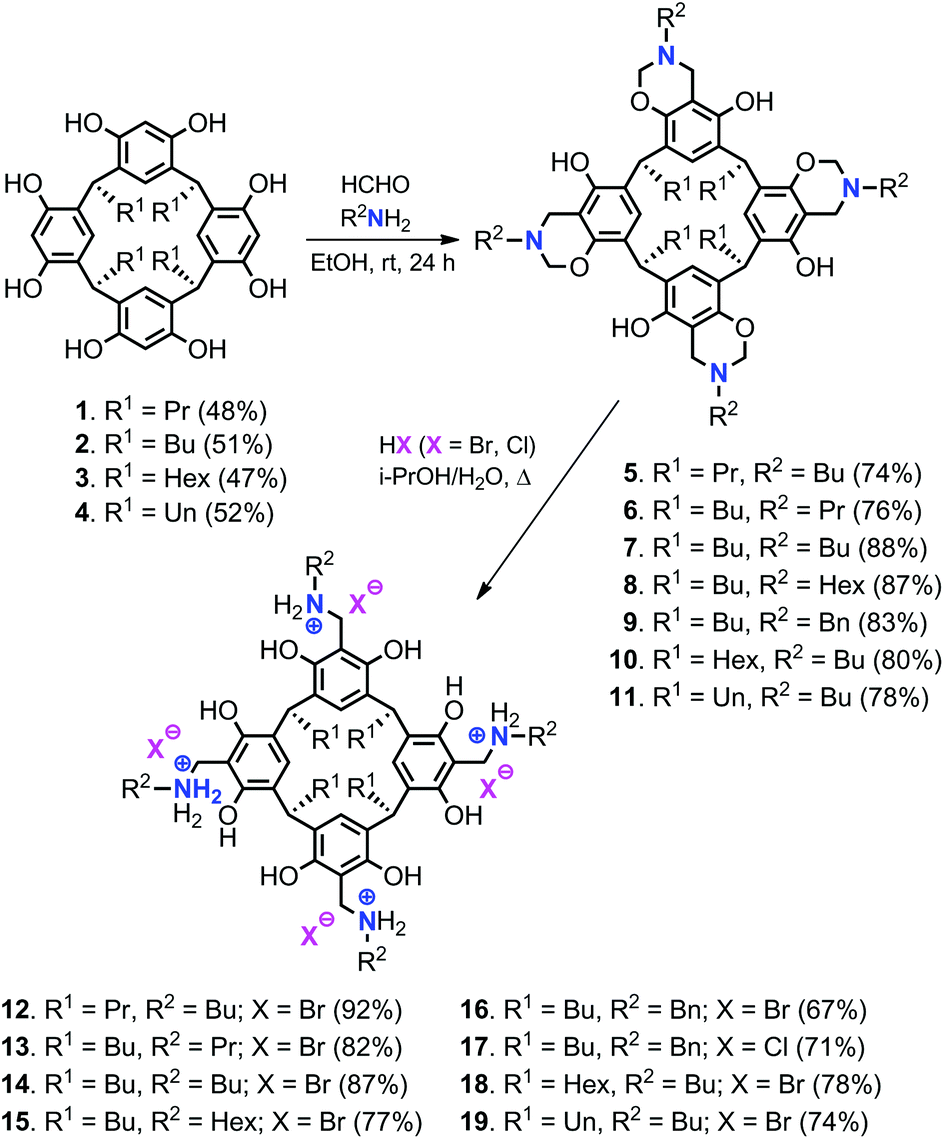 | ||
| Scheme 1 Synthesis of bifunctional resorcinarene derivatives and their isolated yields. | ||
From these precursors 1–4, the bifunctional resorcinarene structures 12–19 were obtained in two steps. First, various tetrabenzoxazines (5–11) were prepared by the condensation reaction of readily available resorcinarenes with various aliphatic primary amines and formaldehyde under ambient conditions. The intermediates 5–11 were then treated with strong aqueous acids (HCl, HBr) to afford the ring-opened benzyl ammonium functionalized resorcinarenes 12–19. The latter compounds were fully characterized by 1H/13C NMR, IR and elemental analysis prior to their use in the catalytic studies (see ESI† for details). Importantly, high-pressure NMR analysis (80 °C, 0.5 MPa of CO2, 18 h) of the bifunctional system 14 did not show any decomposition, a feature of high importance in catalytic applications (vide infra).
Catalysis studies
The catalyst screening phase was carried out using 1,2-epoxyhexane as benchmark substrate (S1) and bifunctional catalysts 12–19 under various reaction conditions. Based on previous use of bifunctional Zn-based catalysts for the same reaction,18 we chose to start the screening at 80 °C for 18 h using a mild pressure of 0.5 MPa (5 bar) under solvent-free conditions, see Table 1 (entries 1–11).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | Amount (mol%) | Temp. (°C) | t (h) | P (MPa) | Yieldb (%) |
| a General conditions: 8.3 mmol 1,2-epoxyhexane, 80 °C, 18 h, p(CO2)° = 5 bar, neat. Note that the amount of catalyst is on a molar basis. b Yield and selectivity of epoxide were determined by 1H NMR (CDCl3) using mesitylene as an internal standard, selectivity towards the cyclic carbonate was >99% in all cases. | ||||||
| 1 | — | — | 80 | 18 | 0.5 | 0 |
| 2 | 2 | 1.0 | 80 | 18 | 0.5 | 6 |
| 3 | 7 | 1.0 | 80 | 18 | 0.5 | 21 |
| 4 | 12 | 1.0 | 80 | 18 | 0.5 | 86 |
| 5 | 13 | 1.0 | 80 | 18 | 0.5 | 92 |
| 6 | 14 | 1.0 | 80 | 18 | 0.5 | >99 |
| 7 | 15 | 1.0 | 80 | 18 | 0.5 | >99 |
| 8 | 16 | 1.0 | 80 | 18 | 0.5 | 81 |
| 9 | 17 | 1.0 | 80 | 18 | 0.5 | 19 |
| 10 | 18 | 1.0 | 80 | 18 | 0.5 | >99 |
| 11 | 19 | 1.0 | 80 | 18 | 0.5 | 82 |
| 12 | 14 | 0.8 | 80 | 18 | 0.5 | 80 |
| 13 | 14 | 1.2 | 80 | 18 | 0.5 | >99 |
| 14 | 14 | 1.0 | 40 | 18 | 0.5 | 57 |
| 15 | 14 | 1.0 | 40 | 6 | 0.5 | 21 |
| 16 | 14 | 1.0 | 60 | 18 | 0.5 | 86 |
| 17 | 14 | 1.0 | 60 | 6 | 0.5 | 42 |
| 18 | 14 | 1.0 | 80 | 18 | 0.1 | 84 |
| 19 | 14 | 1.0 | 80 | 6 | 0.5 | 53 |
| 20 | 14 | 1.0 | 80 | 12 | 0.5 | 85 |
| 21 | 14 | 1.0 | 80 | 15 | 0.5 | 91 |
| 22 | 20 | 4.0 | 80 | 18 | 0.5 | 34 |
| 23 | 2 + 20 | 1.0, 4.0 | 80 | 18 | 0.5 | 94 |

As expected no conversion was noted in the absence of any (catalytic) additive (entry 1), while the catalytic performance of the parent resorcinarene 2 and the tetrabenzoxazine derivative 7 was poor (entries 2 and 3) leading only to very low yields of product COC-1. We then turned our focus on the bifunctional catalysts 12–19 (entries 4–11) and examined their potential to mediate the formation of COC-1 from S1 and CO2. As can be judged from the catalytic data, bifunctional 14, 15 and 18 gave quantitative yield of COC-1 under these conditions whereas the other systems showed inferior activities. The lowest activity was noted for 17 that has chloride anions incorporated, and therefore the low potential of this catalyst to mediate COC formation is not surprising as chloride is a weaker nucleophile/poorer leaving group than bromide. Since compound 14 displays the highest comparative overall yield in the two-step preparation, we decided to use 14 (with two butyl-substituents) to further investigate the influence of catalyst loading, reaction temperature and pressure on the yield of COC-1 (entries 12–21).19
First, the amount of 14 was varied from 0.8 to 1.2 mol% (cf., entries 6 versus 12 and 13) showing that a loading of 0.8 mol% of 14 still gave an appreciable yield (80%) of CC-1, whereas higher loadings than 1.0 mol% do not represent any advantage. Lower reaction temperatures were then evaluated (entries 14–17) and we were pleased to find that at 40 °C still a moderately high yield of 57% could be attained after 18 h, while at 60 °C the yield of COC-1 was already high (86%).
Further lowering the pressure from 0.5 MPa to 0.1 MPa (i.e., 1 bar) showed only a small decrease in the yield of the COC product to 84% (entry 18). When following the kinetics in time (entries 19–21 versus 6), it can be observed that catalyst 14 produces already a high yield of COC-1 (85%) after 12 h. These combined observations (vide infra) help to establish that the bifunctionality of 14 allows for the coupling of 1,2-epoxyhexane (S1) and CO2 under comparatively mild reaction conditions. To show the advantage of having the resorcinarene and nucleophile merged into one structure, we also compared bifunctional catalyst 14 with a binary analogue (2 + 20) and with the onium salt itself; the results (entries 6, 22 and 23) indeed show a much higher efficiency for the bifunctional system compared to the use of 20 only. While both bifunctional and binary catalysts 14 and 2 + 20 show similar yields for COC-1, the former obviously has better recycling potential (vide infra).
When considering the kinetic data from entries 14–21 it seems that low temperatures of 40 °C provide relatively slow conversions after 6 and 18 h with molecular turnover frequencies of 3 to 4 h−1. This implies that the activation barrier for the carbonate synthesis mediated by bifunctional catalyst 14 should be at least 24 kcal mol−1, in line with previous data reported for other homogeneous catalysts used for cyclic carbonate synthesis and conversions.20
Substrate scope phase
After having determined the optimized conditions for high substrate conversion of terminal epoxides (Table 1, entry 6: 80 °C, 1.0 mol% of 14, 0.5 MPa, 18 h), we decided to investigate the scope and limitations of the epoxide/CO2 coupling process and the results of these investigations are summarized in Table 2.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Sub. | COC | R1 | R2 | Conv.b (%) | Yieldc (%) |
| a Typical conditions: 8.3 mmol of epoxide, 1.0 mol% of 14, 80 °C, 18 h, p(CO2)° = 5 bar, neat. Note that the amount of catalyst is on a molar basis. b Conversion determined by 1H NMR (CDCl3); selectivity towards the COC > 99% unless stated otherwise in brackets. c Isolated yield after chromatographic purification. d Reactions carried out at 100 °C for 64 h. e Reactions carried out 100 °C for 64 h with 3.0 mol% of 14. | ||||||
| 1 | S1 | 1 |

|
H | >99 | 93 |
| 2 | S2 | 2 |
|
H | >99 | 97 |
| 3 | S3 | 3 |

|
H | >99 | 81 |
| 4 | S4 | 4 |

|
H | >99 | 88 |
| 5 | S5 | 5 |

|
H | >99 | 95 |
| 6 | S6 | 6 |

|
H | >99 | 91 |
| 7 | S7 | 7 |

|
H | >99 | 85 |
| 8 | S8 | 8 |

|
H | >99 | 94 |
| 9 | S9 | 9 |

|
H | >99 | 80 |
| 10d | S10 | 10 |

|
54 (69) | 29 | |
| 11d | S11 | 11 |

|
63 (76) | 41 | |
| 12d | S12 | 12 |
|
39 (86) | 30 | |
| 13e | S10 | 10 |

|
65 (68) | 41 | |
| 14e | S11 | 11 |

|
95 (76) | 69 | |
| 15e | S12 | 12 |
|
71 (86) | 59 | |
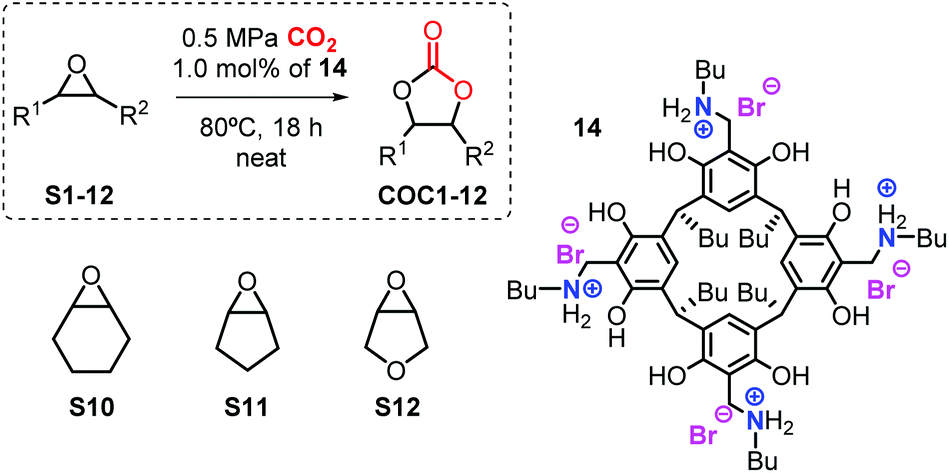
All terminal epoxides (S1–S9) were conveniently coupled with CO2 to afford the cyclic carbonates COC1–9 in excellent chemo-selectivity (>99%) and isolated yields of 80–97% (entries 1–9). Several substitutions and functional groups such as ethers, free alcohols, halogens and double bonds were unaffected by the catalytic procedure. In order to further challenge the catalytic protocol, we chose three internal epoxides (viz., cyclohexene oxide, cyclopentene oxide, and a related tetrahydrofuran oxide; entries 10–15). In these latter conversions, a higher reaction temperature (100 °C) and longer reaction time (64 h) were required to achieve sufficient turnover. The analysis of the crude reaction mixtures in these cases revealed, however, that the overall chemo-selectivity towards the COC targets was negatively affected, and significant amount of diol by-product was observed in all three cases (entries 10–12). We therefore attempted to dry the catalysts and reagents prior to use, though this had no observable effect on the outcome of these transformations. By further increasing the catalyst loading of 14 to 3 mol% (entries 13–15) we finally achieved improved yields for cyclic carbonates COC10–12 (41–69%). All isolated carbonates were characterized by 1H/13C NMR and IR spectroscopy, and these data are summarized in the ESI.†
Immobilization onto a polystyrene support and recycling studies
In order to extend the potential of these cavitand-based systems such as 14 and to be able to recycle such compounds after formation of the COC product, we decided to support tetrabenzoxazine 7 on a Merrifield resin by simple etherification chemistry (Scheme 2).21 The polystyrene-supported tetrabenzoxazine derivative 21 was obtained in 52% yield after treatment with the polystyrene resin (degree of functionalization: f = 1.09 mmol g−1) and 21 (f = 0.26 mmol g−1; fmax = 0.50 mmol g−1) was analysed by elemental analysis and IR spectroscopy (ESI†). Then 21 was reacted with MeI in CH3CN to obtain the tetra-butylmethyl ammonium iodide supported catalyst 22 (f = 0.23 mmol g−1; fmax = 0.25 mmol g−1, ESI† for details) in 92% yield. The reasoning for choosing 21 as starting point to prepare a supported catalyst is two-fold. First, the benzoxazines are rather stable compounds and the N-alkylation process is known. Second, the introduction of methylammonium iodide groups was thought to be not only practical but this would also add a higher stability to the final catalyst structure 22 while preserving the presence of a nucleophilic halide.22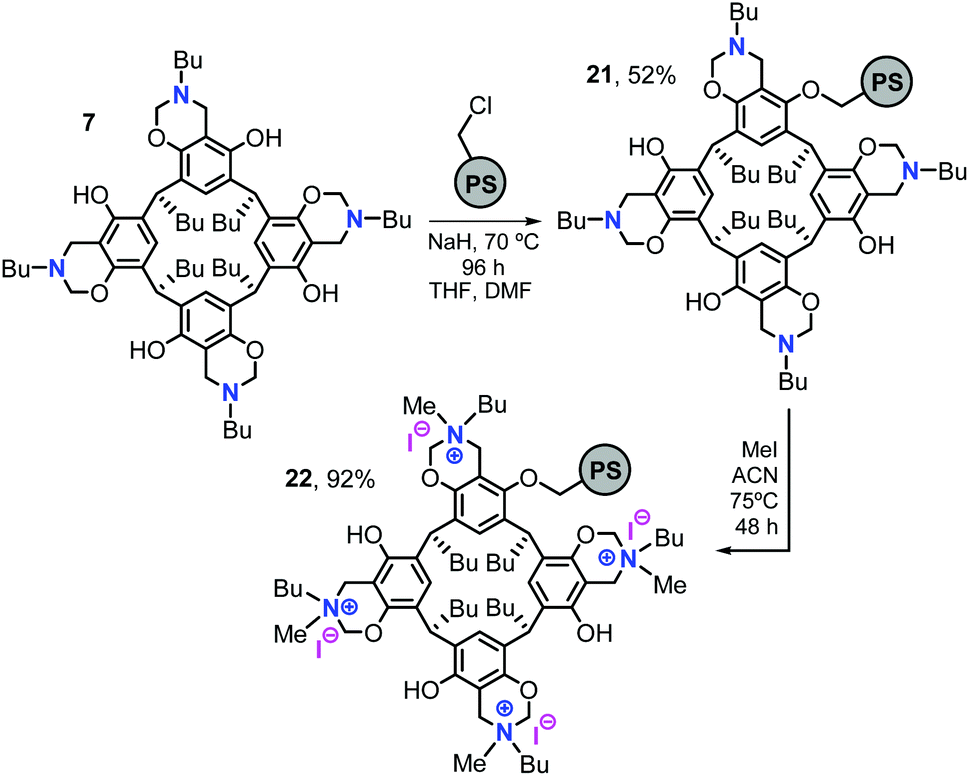 | ||
| Scheme 2 Synthesis of the bifunctional polystyrene-supported catalyst 22. | ||
Heterogeneous bifunctional catalyst 22 was then applied in the coupling of S1 and CO2 under the same optimized conditions as reported for 14 (80 °C, 0.5 MPa CO2 pressure, 18 h) using 0.89 mol% of 22 (Table S3† and Fig. 2). All the starting materials (Merrifield resin with chloromethyl functionalities, polystyrene-supported tetrabenzoxazine derivative 21 and MeI) were first tested under the same conditions to determine any background conversion. As expected, no to low catalytic activity was observed (see Table S1 of the ESI†) with the use of 21 giving only a relatively low yield (25%) of COC-1.
 | ||
| Fig. 2 Recycling studies performed with supported catalyst 22 in the coupling of 1,2-epoxyhexane (S1) and CO2 to afford cyclic carbonate COC-1. Yields were determined by 1H NMR (CDCl3) using mesitylene as internal standard after collecting an aliquot of the crude reaction mixture. | ||
Contrary to the starting materials required for the synthesis of 22, the latter was significantly more active. Fig. 2 demonstrates that the bifunctional catalyst shows a quantitative yield (>99%, selectivity towards COC-1 was >99%) in the first four runs for the cyclic carbonate product. After each run, the catalyst was recycled and washed thoroughly with diethyl ether, then dried, weighed and subsequently applied in a subsequent run. During this catalyst isolation process, we noted a slight loss of material with the amount of 22 in the fourth run (third recycle) being reduced to 0.80 mol%. Since in the fourth run still a quantitative yield of COC-1 was achieved, the results give the impression that 0.80 mol% is the minimal amount required for quantitative conversion of S1/CO2 into COC-1 under these experimental conditions.
Upon further recycling of 22, the activity decreased slightly as shown in the Fig. 2 and the catalyst was used in 12 runs without a dramatic loss in catalytic efficiency. To get further insight into the loss of the activity we compared the mass of catalyst obtained after the ninth run (expressed in 0.69 mol% relative to the epoxide substrate) with the required amount for quantitative conversion of S1 under the experimental conditions (0.80 mol%) which is a mass loss of 14%. Interestingly, about 15% loss in conversion was noted between the fourth and the ninth run, which seem to corroborate well with the idea that most of the loss in activity is caused by the washing and isolation of 22 prior to the next run and not be any decomposition process of the resorcinarene backbone. After the twelfth run, we isolated the catalyst and subjected it to elemental analysis: both the N- and I-content had decreased throughout the entire recycling campaign from 1.26 → 0.91% and 12.28 → 3.86%, respectively (ESI† for more details). This seems to suggest that some degree of retro-Mentschutkin reactions had taken place as observed previously with other bifunctional catalysts in cyclic carbonate synthesis.23 These analytical data clearly show that the catalyst structure is affected in due course with significant loss of the nucleophile. Nonetheless, the overall activity of the original catalyst 22 is not dramatically influenced, and after 12 runs, an appreciable total turnover of 1252 could be achieved, which is comparatively a very good result for an organocatalyst in the area of cyclic carbonate synthesis.
The advantages and the enhancement of the catalytic activity in these bifunctional systems (both homogeneous and heterogeneous) is believed to be the result of a high local concentration of phenol sites (cf., hydrogen bond activation of the epoxide) and the presence of ammonium halide groups (providing the nucleophilic sites for epoxide ring opening). The presence of all these groups and their synergistic involvement in the conversion of CO2 and epoxides towards cyclic carbonate formation are aspects that were studied in detail in our previous work.13–16 However, the current work represents a unique bifunctional one-component homogenous catalyst that is able to perform well without the addition of any co-catalyst or solvent. The system is also easily immobilized onto polystyrene supports through simple transformations affording higher substrate turnover numbers.
Conclusions
Here we report on easily accessible, cheap and efficient bifunctional catalysts for the formation of cyclic carbonates from various epoxides and CO2 in excellent yields and solvent-free conditions. The starting materials are modular in nature allowing for the optimization of the catalytic activity, and providing an easy entry towards heterogeneous versions of bifunctional catalysts for carbonate synthesis. The presented recycling studies help to establish that also a heterogenized bifunctional catalyst can display attractive activity under comparatively mild reaction conditions with a high substrate turnover number.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank ICREA, the CERCA Program/Generalitat de Catalunya, the Spanish Ministerio de Economía y Competitividad (MINECO: CTQ-2014-60419-R, CTQ2015-69136-R and Severo Ochoa Excellence Accreditation 2014–2018, SEV-2013-0319) for financial support. S. C. thanks MINECO for a Severo Ochoa FPI fellowship, and T. J. acknowledges the EU for co-financing a postdoctoral position through the COFUND programme. We also thank Dr Noemí Cabello for the MS studies.Notes and references
- For some recent reviews: (a) B. Yu and L.-N. He, ChemSusChem, 2015, 8, 52 CrossRef CAS PubMed; (b) Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed; (c) Q.-W. Song, Z.-H. Zhou and L.-N. He, Green Chem., 2017, 19, 3707 RSC; (d) N. Kielland, C. J. Whiteoak and A. W. Kleij, Adv. Synth. Catal., 2013, 355, 2115 CrossRef CAS; (e) F. Manjolinho, M. Arndt, K. Gooßen and L. J. Gooßen, ACS Catal., 2012, 2, 2014 CrossRef CAS; (f) M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709 CrossRef CAS PubMed; (g) J. Klankermayer, S. Wesselbaum, K. Beydoun and W. Leitner, Angew. Chem., Int. Ed., 2016, 55, 7296 CrossRef CAS PubMed; (h) C. Maeda, Y. Miyazaki and T. Ema, Catal. Sci. Technol., 2014, 4, 1482 RSC.
- For earlier reviews on this subject: (a) A.-A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951 CrossRef CAS PubMed; (b) T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS PubMed; (c) B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554 CrossRef PubMed; (d) T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312 RSC.
- See for some examples: (a) D. J. Darensbourg, A. I. Moncada and S.-H. Wei, Macromolecules, 2011, 44, 2568 CrossRef CAS; (b) M. Helou, O. Miserque, J.-M. Brusson, J.-F. Carpentier and S. Guillaume, Chem. – Eur. J., 2010, 16, 13805 CrossRef CAS PubMed.
- (a) H. L. Parker, J. Sherwood, A. J. Hunt and J. H. Clark, ACS Sustainable Chem. Eng., 2014, 2, 1739 CrossRef CAS; (b) M. North, F. Pizzato and P. Villuendas, ChemSusChem, 2009, 2, 862 CrossRef CAS PubMed.
- (a) W. Guo, L. Martínez-Rodríguez, E. Martín, E. C. Escudero-Adán and A. W. Kleij, Angew. Chem., Int. Ed., 2016, 55, 11037 CrossRef CAS PubMed; (b) A. Cai, W. Guo, L. Martínez-Rodríguez and A. W. Kleij, J. Am. Chem. Soc., 2016, 138, 14194 CrossRef CAS PubMed.
- For recent reviews on this topic: (a) J. W. Comerford, I. D. V. Ingram, M. North and X. Wu, Green Chem., 2015, 17, 1966 RSC; (b) C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353 CrossRef; (c) J. E. Gómez and A. W. Kleij, Curr. Opin. Green Sustainable Chem., 2017, 3, 55 CrossRef; (d) P. P. Pescarmona and M. Taherimehr, Catal. Sci. Technol., 2012, 2, 2169 RSC.
- (a) B. R. Buckley, A. P. Patel and K. G. Upul Wijayantha, Eur. J. Org. Chem., 2015, 474–478 CrossRef CAS PubMed; (b) D. J. Darensbourg, A. Horn Jr. and A. I. Moncada, Green Chem., 2010, 12, 1376 RSC; (c) J. Rintjema, W. Guo, E. Martin, E. C. Escudero-Adán and A. W. Kleij, Chem. – Eur. J., 2015, 21, 10754 CrossRef CAS PubMed.
- Selected recent examples: (a) J. A. Castro-Osma, K. Lamb and M. North, ACS Catal., 2016, 6, 5012 CrossRef CAS; (b) F. Della Monica, S. V. C. Vummaleti, A. Buonerba, A. De Nisi, M. Monari, S. Milione, A. Grassi, L. Cavallo and C. Capacchione, Adv. Synth. Catal., 2016, 358, 3231 CrossRef; (c) V. Laserna, G. Fiorani, C. J. Whiteoak, E. Martin, E. C. Escudero-Adán and A. W. Kleij, Angew. Chem., Int. Ed., 2014, 53, 10416 CrossRef CAS PubMed; (d) C. Maeda, J. Shimonishi, R. Miyazaki, J.-Y. Hasegawa and T. Ema, Chem. – Eur. J., 2016, 22, 6556 CrossRef CAS PubMed.
- (a) G. Fiorani, M. Stuck, C. Martín, M. Martínez-Belmonte, E. Martin, E. C. Escudero-Adán and A. W. Kleij, ChemSusChem, 2016, 9, 1304 CrossRef CAS PubMed; (b) J. Martínez, J. Fernández-Baeza, L. F. Sánchez-Barba, J. A. Castro-Osma, A. Lara-Sánchez and A. Otero, ChemSusChem, 2017, 10, 2886 CrossRef PubMed; (c) V. Laserna, E. Martin, E. C. Escudero-Adán and A. W. Kleij, ACS Catal., 2017, 7, 5478 CrossRef CAS.
- For recent reviews: (a) M. Alves, B. Grignard, R. Mereau, C. Jerome, T. Tassaing and C. Detrembleur, Catal. Sci. Technol., 2017, 7, 2651 RSC; (b) M. Cokoja, M. E. Wilhelm, M. H. Anthofer, W. A. Herrmann and F. E. Kühn, ChemSusChem, 2015, 8, 2436 CrossRef CAS PubMed; (c) Q. He, J. W. O'Brien, K. A. Kitselman, L. E. Tompkins, G. C. T. Curtis and F. M. Kerton, Catal. Sci. Technol., 2014, 4, 1513 RSC; (d) G. Fiorani, W. Guo and A. W. Kleij, Green Chem., 2015, 17, 1375 RSC.
- (a) Y. Toda, Y. Komiyama, A. Kikuchi and H. Suga, ACS Catal., 2016, 6, 6906 CrossRef CAS; (b) V. B. Saptal, T. Sasaki, K. Harada, D. Nishio-Hamane and B. M. Bhanage, ChemSusChem, 2016, 9, 644 CrossRef CAS PubMed; (c) S. Sopeña, G. Fiorani, C. Martin and A. W. Kleij, ChemSusChem, 2015, 8, 3248 CrossRef PubMed; (d) J. Wang and Y. Zhang, ACS Catal., 2016, 6, 4871 CrossRef CAS; (e) H. Büttner, J. Steinbauer and T. Werner, ChemSusChem, 2015, 8, 2655 CrossRef PubMed; (f) A. Mirabaud, J.-C. Mulatier, A. Martinez, J.-P. Dutasta and V. Dufaud, ACS Catal., 2015, 5, 6748 CrossRef CAS; (g) M. H. Anthofer, M. Wilhelm, M. Cokoja, M. Drees, W. A. Herrmann and F. E. Kühn, ChemCatChem, 2015, 7, 94 CrossRef CAS; (h) T. Werner and H. Büttner, ChemSusChem, 2014, 7, 3268 CrossRef CAS PubMed.
- S. Gennen, M. Alves, R. Méreau, T. Tassaing, B. Gilbert, C. Detrembleur, C. Jerome and B. Grignard, ChemSusChem, 2015, 8, 1845 CrossRef CAS PubMed.
- S. Sopeña, E. Martín, E. C. Escudero-Adán and A. W. Kleij, ACS Catal., 2017, 7, 3532 CrossRef.
- C. J. Whiteoak, A. Nova, F. Maseras and A. W. Kleij, ChemSusChem, 2012, 5, 2032 CrossRef CAS PubMed . See also ref. 11c.
- L. Martínez-Rodríguez, J. Otalora Garmilla and A. W. Kleij, ChemSusChem, 2016, 9, 749–755 CrossRef PubMed.
- C. J. Whiteoak, A. H. Henseler, C. Ayats, A. W. Kleij and M. A. Pericàs, Green Chem., 2014, 16, 1552 RSC.
- (a) K. Airola, V. Böhmer, E. F. Paulus, K. Rissanen, C. Schmidt, I. Thondorf and W. Vogt, Tetrahedron, 1997, 53, 10709 CrossRef CAS; (b) N. K. Beyeh, A. Ala-Korpi, M. Cetina, A. Valkonen and K. Rissanen, Chem. – Eur. J., 2014, 20, 15144 CrossRef CAS PubMed.
- C. Martín, C. J. Whiteoak, E. Martin, M. Martínez Belmonte, E. C. Escudero-Adán and A. W. Kleij, Catal. Sci. Technol., 2014, 4, 1615 Search PubMed.
- Note that we did try to prepare iodide based bifunctional catalyst systems derived from 5–11; however, during purification of these compounds we found by elemental analyses that the iodide content was consistently significantly higher than expected, and we therefore continued with the bromide-based bifunctional catalysts.
- For illustrative examples please refer to: (a) W. Guo, J. Gónzalez-Fabra, N. A. G. Bandeira, C. Bo and A. W. Kleij, Angew. Chem., Int. Ed., 2015, 54, 11686 CrossRef CAS PubMed; (b) M. Alves, R. Méreau, B. Grignard, C. Detrembleur, C. Jérôme and T. Tassaing, RSC Adv., 2017, 7, 18993–19001 RSC; (c) C. J. Whiteoak, A. Nova, F. Maseras and A. W. Kleij, ChemSusChem, 2012, 5, 2032 CrossRef CAS PubMed. Note that bifunctional catalyst 14 contains four active sites though it may be difficult to assess whether all sites are involved simultaneously. As the heterogeneous system 22 shows comparable conversion kinetics, it may be assumed that the activation barrier for the homogeneous and heterogeneous catalyzed formation of COC-1 is rather similar.
- Part of us have reported the immobilization of various organocatalysts onto similar supports using comparable approaches, see: (a) S. Cañellas, C. Ayats, A. H. Henseler and M. A. Pericàs, ACS Catal., 2017, 7, 1383 CrossRef; (b) L. Clot-Almenara, C. Rodríguez-Escrich, L. Osorio-Planes and M. A. Pericàs, ACS Catal., 2016, 6, 7647 CrossRef CAS; (c) P. Llanes, S. Sayalero, C. Rodríguez-Escrich and M. A. Pericàs, Green Chem., 2016, 18, 3507 RSC; (d) J. Izquierdo and M. A. Pericàs, ACS Catal., 2016, 6, 348 CrossRef CAS.
- Note that the presence of bromide or iodide likely does not bear too much difference in overall activity, as they are known for other systems to mediate the coupling of epoxides and CO2 with similar efficiencies.
- (a) M. North, B. Wang and C. Young, Energy Environ. Sci., 2011, 4, 4163 RSC; (b) See also ref. 16; in this particular case we were able to re-alkylate the catalyst structure with MeI to regenerate the required ammonium iodide groups.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and catalytic procedures, substrate synthesis and full characterization data. See DOI: 10.1039/c7gc02856c |
| This journal is © The Royal Society of Chemistry 2017 |