
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvances in the preparation of highly selective nanocatalysts for the semi-hydrogenation of alkynes using colloidal approaches
Jorge A.
Delgado
 a,
Olivia
Benkirane
a,
Carmen
Claver
ab,
Daniel
Curulla-Ferré
c and
Cyril
Godard
*ab
a,
Olivia
Benkirane
a,
Carmen
Claver
ab,
Daniel
Curulla-Ferré
c and
Cyril
Godard
*ab
aCentre Tecnològic de la Química, Marcel lí Domingo s/n, Campus Sescelades, 43007 Tarragona, Spain. E-mail: cyril.godard@urv.cat
bDepartament de Química Física i Inorgánica, Universitat Rovira i Virgili, Marcel lí Domingo s/n, Campus Sescelades, 43007 Tarragona, Spain
cTotal Research and Technology Feluy, B-7181 Seneffe, Belgium
First published on 13th June 2017
Abstract
In the last decade, the semi-hydrogenation of alkynes has experienced significant advances in terms of fine control of alkene selectivity and prevention of the over-hydrogenation reaction. Such advances have been possible to a large extent through the progress in colloidal methods for the preparation of metallic nanoparticles. The present review describes the contributions in the field of the selective hydrogenation of alkynes involving the utilization of colloidal methodologies. These approaches permit the fine modulation of several parameters affecting the catalytic performance of the active phase such as the particle size, the bulk and the surface structure and composition. For the transformation of liquid substrates, the nature of the stabilizers, the reducing agents and the metal precursors employed for the synthesis of the catalysts can be tuned to enhance the alkene selectivity. In contrast, in catalytic transformations of gaseous substrates, the presence of adsorbed species at the metal surface usually gives detrimental results while the interplay between the support and the active phase appears to be a more convincing alternative for catalyst tuning.
1. Introduction
One of the current challenges for the industry and the scientific community is the development of not only economical but also environmentally sustainable processes.1 In this context, the use of heterogeneous catalysts offered a key tool to achieve suitable utilization of energy and chemicals.2 Among the most commonly applied catalytic processes, selective hydrogenation of functional groups over heterogeneous catalysts is of paramount importance in various industrial sectors.3 During the last few years, the selective semi-hydrogenation of alkynes into alkenes (Scheme 1) has been the object of particular attention for its relevance in the polymer and fine chemical industries,4–6 for which the development of chemo- and stereoselective catalysts is highly desirable to improve the productivity of the corresponding processes and prevent the over-hydrogenation and/or oligomerisation of the substrates.6,7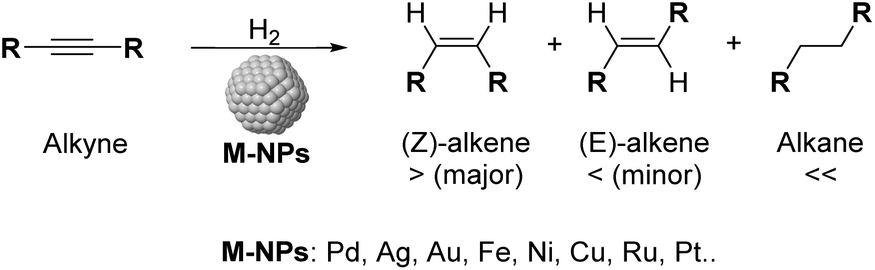 | ||
| Scheme 1 Selective alkyne hydrogenation catalyzed by metal nanoparticles. | ||
Several transition metal catalysts have been reported in the selective semi-hydrogenation of alkynes (e.g. Pd, Ni, Pt, Ag, Rh, Fe).8 Among these metals, those with high hydrogen activation capacity such as Ni or Pt frequently present severe selectivity issues due to their strong over-hydrogenation activity. In contrast, high selectivities to the alkene were reported for metals with low reactivity towards H2 such as Ag or Au, but these metals are hardly competitive in terms of activity.9
Palladium has therefore been identified as the most efficient metal in terms of activity and selectivity, which has been attributed to the stronger adsorption of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C group (relative to C
C group (relative to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) on the Pd surface, thus preventing the over-reduction of the alkene product.10 Palladium based catalysts are generally dispersed on solid carriers such as oxides (Al2O3, TiO2, SiO2), CaCO3 (Lindlar®) and carbon based materials (activated C, graphite, carbon nanofibers or nanotubes).11 Conventionally, the preparation of such supported catalysts has been dominated by impregnation techniques, although these approaches often face difficulties in the fine control of the particle size of the resulting metal NPs and their distribution over the support.11
C) on the Pd surface, thus preventing the over-reduction of the alkene product.10 Palladium based catalysts are generally dispersed on solid carriers such as oxides (Al2O3, TiO2, SiO2), CaCO3 (Lindlar®) and carbon based materials (activated C, graphite, carbon nanofibers or nanotubes).11 Conventionally, the preparation of such supported catalysts has been dominated by impregnation techniques, although these approaches often face difficulties in the fine control of the particle size of the resulting metal NPs and their distribution over the support.11
In this context, colloidal methods offer the possibility to prepare well-defined metallic nanoparticles through a precise control of their size, shape and structure at the nanometric scale.12 The ability to adjust the properties of nanoparticles and compare series of materials prepared in a similar manner has provided, over the last two decades, valuable information on how catalytic systems operate at molecular and atomic levels.13 Moreover, such a control is of particular interest to maximize the available metal surface area and consequently enhance catalyst productivity.
An example of successful application of colloidal chemistry for the preparation of molecularly designed nanocatalysts is the BASF NanoSelect™ Pd catalysts.14,15 This catalyst has been reported as being highly selective in the hydrogenation of alkynes,15 and using DFT calculations, Pérez-Ramírez and co-workers correlated the performance of this catalyst with the accessibility constraints of reagents and products to the metal surface.16 These findings demonstrated that the nanostructure defined at the colloidal preparation stage leads to the production of ultraselective catalysts.
In the following sections, a comprehensive analysis of the literature on the selective hydrogenation of alkynes catalyzed by metal nanoparticles prepared by colloidal methods is presented. This review aims at describing the potential for, and perspectives of, the utilization of colloidal chemistry for the preparation of heterogeneous catalysts for industrial applications and at providing a fundamental understanding.
1.1 Mechanistic and thermodynamic selectivity in the semi-hydrogenation of alkynes
In the partial hydrogenation of alkynes, two types of selectivity to the alkene product have been defined: mechanistic and the thermodynamic selectivity (Scheme 2).17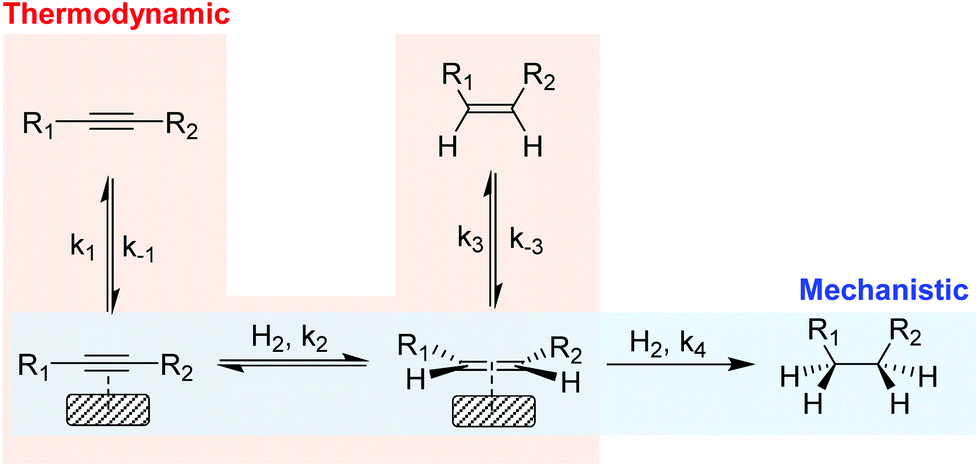 | ||
| Scheme 2 Simplified scheme of the hydrogenation of alkynes illustrating the mechanistic selectivity and thermodynamic selectivity. | ||
Mechanistic selectivity operates when the hydrogenation of the adsorbed alkyne is favored with respect to the adsorbed alkene (k2 ≫ k4).18 In contrast, thermodynamic selectivity consists in the ability of a metal to preferentially adsorb the alkyne in the presence of the alkene (k1/k−1 ≫ k3/k−3), thus suppressing the consecutive hydrogen addition which results in the production of alkane. For most of the metals studied in the literature, however, the alkene hydrogenation rate is faster than that of alkyne hydrogenation19 and consequently, selectivity is usually attributed to the stronger adsorption of the alkyne compared with that of the alkene (thermodynamic selectivity). Under industrial conditions, thermodynamic selectivity is of prime importance due to the high alkene content which favors the competitive adsorption with the alkyne at the metal surface. In addition, studies on Pd, Au, Cu and Ni catalysts identified thermodynamic selectivity as the most important selectivity indicator.20–22
This model offers a basis for the interpretation of the intrinsic selectivity of palladium. However, the selectivity of alkene formation cannot be interpreted without detailed knowledge of the reaction mechanism and the actual surface state of the active Pd sites. Consequently, considerable attention has been paid to the formation of surface intermediates and the role of factors such as dispersion, modifiers and promoters.
1.2 Reaction mechanism
A simplified reaction mechanism for the hydrogenation of alkynes is displayed in Scheme 3. Path a is the sequential hydrogenation of the alkyne, first into the alkene that desorbs and re-adsorbs to be further hydrogenated into alkane. Path b, in turn, is the direct hydrogenation of the alkyne substrate until the alkane without any intermediate desorption/re-adsorption of the alkene. The relevance of the two paths depends on the morphological properties of the catalysts and the reaction parameters. For small and reactive alkynes such as acetylene, an additional route (path c) operates and consists in the oligomerization/polymerization of the substrate to form hydrocarbons (C4–C32). Ranges of 20–40% of C4+ selectivity have been reported for acetylene hydrogenation, thus evidencing the relevance of this reaction path.23–26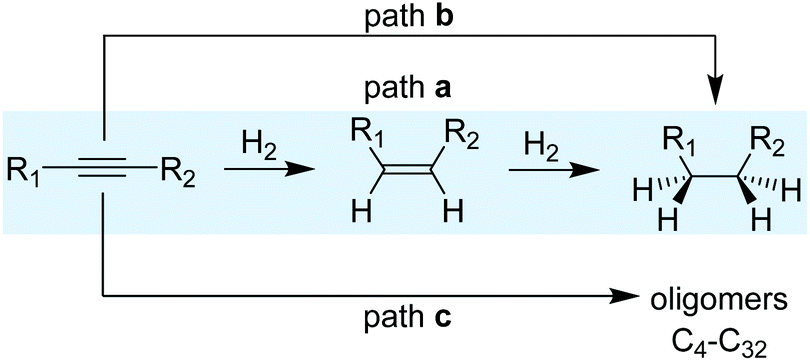 | ||
| Scheme 3 Reaction paths taking place during the alkyne hydrogenation. | ||
For the hydrogenation of acetylene, several intermediates have been postulated based on surface characterization techniques combined, in some cases, with kinetic studies.18,27,28 The reaction starts by H2 dissociation and the adsorption of acetylene on the surface; both steps are exothermic. Then, the sequential addition of H takes place following a Horiuti–Polanyi mechanism.29 For this substrate, despite its higher adsorption enthalpy over palladium surfaces (thermodynamic selectivity), the hydrogenation of ethylene products has been observed even at high acetylene partial pressures.30 This observation suggests the existence of a mixture of sites where acetylene is selectively hydrogenated to ethylene and others where ethylene is hydrogenated even in the presence of acetylene.31
1.3 Descriptors affecting selectivity
Other parameters such as the mixing of the reaction (important for batch or semi-batch systems) need to be considered in order to prevent possible mass-transfer limitations which could affect the activity and/or the selectivity. Kiwi-Minsker and co-workers studied the interactions between mass and reaction kinetics for the three-phase catalytic hydrogenation of 2-methyl-3-butyn-2-ol (MBY) to 2-methyl-3-buten-2-ol (MBE) over a modified Pd/CaCO3 catalyst under solvent free conditions.35 No influence of gas–liquid mass (G–L) transfer was observed for stirring speeds higher than 1500 rpm. Interestingly, the MBE selectivity increased at high stirring rates. Hydrogen liquid–solid (L–S) mass transfer was found to be negligible, whereas the MBY L–S mass transfer became important at high MBY conversions and high hydrogen concentration. These observations thus showed that mass transfer can affect both the activity and the selectivity of a catalyst in alkyne hydrogenation reactions.
The heating method used for the hydrogenation reaction can also have an impact on the catalyst performance. Cravotto and co-workers studied the partial hydrogenation of 2-butyne-1,4-diol in water under microwave irradiation catalyzed by a Pd/boehmite catalyst.36 Despite the low activity usually observed when water is the reaction solvent, the use of microwave irradiation enhanced the conversion while maintaining the alkene selectivity at ca. 97–100%. More recently, the same authors studied the effect of ultrasound on the semi-hydrogenation of phenylacetylene and diphenylacetylene catalyzed by Pd/boehmite.37 Interestingly, the sonication increased the alkene selectivity in both cases and this positive effect of ultrasound was explained by the increased desorption rate of the alkene product from the catalyst surface before over-reduction could occur.
It is generally accepted that the presence of hydrides, either on or below the metal surface, favors the over-hydrogenation and thus the formation of alkanes.32 The low dissociation barrier of H2 and the easy incorporation of hydride into the Pd phase indicate that the entire surface can capture and incorporate H2.38
In the hydrogenation of acetylene, two types of carbon species were reported to modify the selectivity of the catalysts. The effect of subsurface carbon on selectivity was attributed to the improved thermodynamic factor of Pd according to DFT calculations.38,39 In the presence of carbon, a weaker adsorption energy was observed for ethylene in comparison with that on a clean Pd surface (Eads = −0.67 eV vs. −0.82 eV for the clean surface). The formation of such carbides was theoretically proved to be favored on step sites, in agreement with the experimental observations from the group of Kiwi-Minsker, who attributed the formation of a carbide layer to the defect sites present in small particles.40
Furthermore, during the hydrogenation of acetylene, heavy hydrocarbons (C8+) are formed and remain on the catalyst, and are frequently regarded as coke or carbonaceous deposits. Deganello et al. evidenced that the formation of such carbonaceous deposits enhances catalyst selectivity.23 They proposed the existence on the metal surface of two types of sites: some that are accessible to all reagents and others which are poisoned sites coated by carbonaceous deposits and consequently not accessible. These poisoned sites interact with neighboring atoms generating highly hindered sites which become accessible only to acetylene and hydrogen therefore producing ethene selectively.
Moreover, the presence of hydroxyl groups in the alkyne structure was observed to retard the hydrogenation reaction, thus favoring the selectivity control to the alkene.45 Chaudret and co-workers proposed that coordination of the oxygen atom of the alkynol on iron nanoparticles could decrease the hydrogenation rate, and therefore favor the formation of unsaturated species.46 Regarding the stereoselectivity of the hydrogenation of alkynes, the preferential formation of the Z product is usually observed, in agreement with the Horiuti–Polanyi mechanism.29 However, some degree of Z/E isomerisation is sometimes observed depending on the catalytic system and reaction conditions.
2 Strategies for the enhancement of alkene selectivity through colloidal approaches
During the last few decades, the preparation of metal nanocatalysts by colloidal methods has attracted the interest of the scientific community, since they provide a precise control of the size, shape and structure of the resulting nanoparticles.47,48 Moreover, the use of unsupported nanoparticles is also of high interest as they mimic metal surface activation and catalysis at the nanoscale, and can sometimes shed some light on the role of the support during catalysis.49The main parameters that were reported to enhance the alkene selectivity in the semi-hydrogenation of alkynes using colloidal approaches (Fig. 1) are: (1) the properties of the stabilizer employed for the synthesis of the nanocatalysts; (2) the structural properties of the catalysts such as their size and shape and their mono/bimetallic nature; (3) the properties of the support; and (4) catalytic parameters such as additives or use of reducing agents alternative to H2.
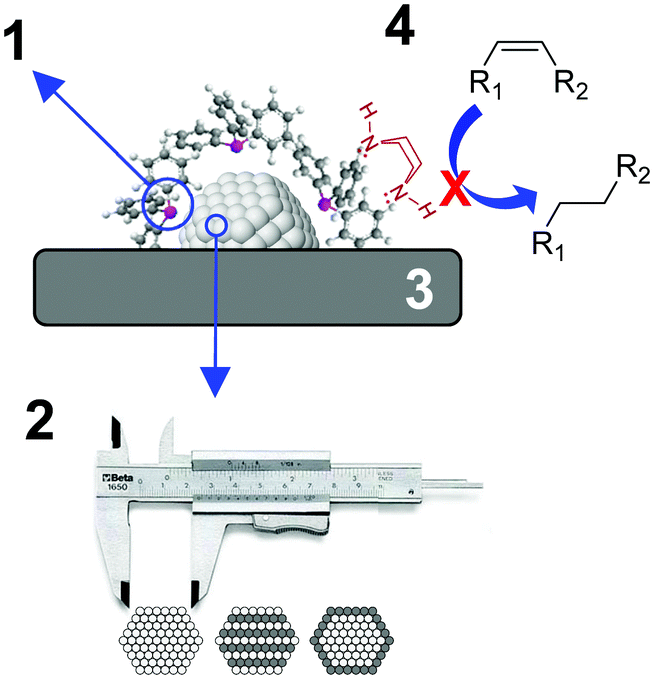 | ||
| Fig. 1 Schematic representation of the main parameters reported to enhance alkene selectivity using colloidal approaches. | ||
In the following sections, an analysis of the results reported on the semi-hydrogenation of alkynes catalyzed by metal nanoparticles is described according to the methodology employed for the enhancement/control of the alkene selectivity.
2.1 Stabilizer
Organic compounds are typically employed during the synthesis of metal nanoparticles to prevent agglomeration of the metallic colloid.48 Compounds such as polymers, dendrimers, surfactants, ionic liquids and ligands have been employed for the stabilization of metal nanoparticles applied in the semi-hydrogenation of alkynes. Depending on the nature of the stabilizer, the stabilization of the metal nanoparticles is either steric (polymers, dendrimers, ligands) or electrostatic (surfactants ionic liquids) and consists in the formation of a layer of the stabilizer at the NP surface preventing their agglomeration.Furthermore, the presence of such compounds at the NP surface can also exert dramatic effects on the catalytic performance of the NPs.47 Even traces of stabilizing agents can induce electronic and steric modifications to the metal NPs that result in important variations in the catalyst activity/selectivity in hydrogenation reactions.50–52
Depending on the phase in which the catalytic reaction takes place (liquid or gas phase), the effect of the stabilizer can be more or less marked. In the liquid phase, it is generally considered that stabilizers are relatively flexible, and that most of the active sites remain accessible to the substrates. In contrast, when the catalytic reactions are performed in the gas phase, the catalysts are generally dried, resulting in the collapse of the stabilizer over the NP surface and consequently, in the blockage of the active sites. For instance, after isolation of PdNPs stabilized by hexadecyl(2-hydroxyethyl)dimethyl ammonium dihydrogenphosphate (HHDMA), no CO uptake could be observed in chemisorption measurements due to the presence of the stabilizer.14
Surfactants. Surfactants are frequently used for the stabilization of metal nanoparticles in aqueous media since they are generally constituted by a polar group able to generate an electronic double layer and a lipophilic side chain that also provides steric repulsion between particles.47
For liquid phase reactions, a clear example that evidences the effect of the stabilizer on the performance of the hydrogenation catalyst is the case of Pd NanoSelect.14,15 This catalyst consists of palladium nanoparticles of 5–8 nm supported on carbon or titanium silicate that exhibit alkene selectivities comparable to those provided by the Lindlar's catalyst in the semi-hydrogenation of a broad range of alkynes and alkynols (Fig. 2a).15 The palladium nanoparticles are prepared by a chemical reduction method employing HHDMA as a reducing agent and stabilizer. Based on deposition experiments onto functionalized surfaces (positive or negative), the authors proposed that the PdNPs are surrounded by a double layer of HHDMA.14 SEM analysis of NanoSelect catalysts revealed a perfect egg–shell distribution of the PdNPs onto the support. Such a distribution was proposed to be responsible for the high selectivity of the NanoSelect catalysts in the semi-hydrogenation of alkynes.
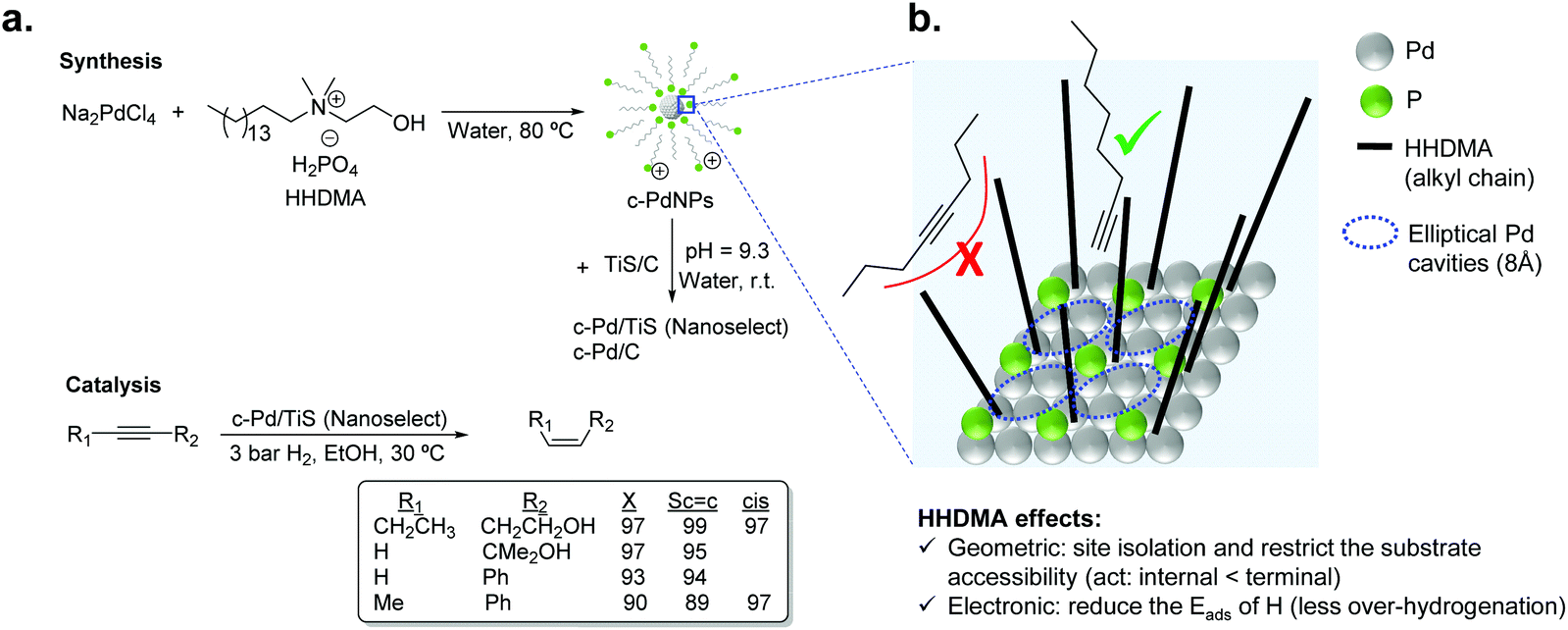 | ||
| Fig. 2 (a) Synthesis of the NanoSelect Pd catalysts and hydrogenation of alkynes and alkynols. (b) Geometric and electronic effects of HHDMA over the surface of PdNPs. | ||
Pérez-Ramírez and co-workers investigated the selectivity patterns and accessibility constraints of NanoSelect Pd catalysts using experimental and theoretical approaches.16 According to DFT calculations, HHDMA is adsorbed rigidly over the Pd surface and such an adsorption reduces both the number of active sites (geometric effect) and the hydride coverage at the Pd surface (electronic effect) (Fig. 2b). This explains the intrinsic alkene selectivity of the NanoSelect Pd catalysts. However, under high H2 pressures, the adsorption of H in the subsurface is relatively easy and over-hydrogenation cannot be avoided. In addition, the authors correlated the hydrogenation performance of a series of Pd-based catalysts with structural descriptors, and concluded that the rigidly adsorbed ligands restrict the accessibility of the substrate to the active sites of the catalyst (forming cavities with elliptical shape), providing exceptional performance for the hydrogenation of terminal alkynes but low activity for disubstituted and bulky alkynes (Fig. 2b). These findings demonstrated that the surface structure determined during the preparation of the colloidal nanoparticles leads to the production of very selective catalysts.16
In a subsequent report, the same authors addressed the impact of the stabilizer content on the catalytic behavior of HHDMA stabilized PdNPs supported on titanium silicate.53 For this purpose, a series of Pd-HHDMA/TiSi2O6 catalysts with distinct HHDMA content (0.3–16.8 wt%) were prepared while maintaining the average particle size (5 nm) and Pd content (0.3 wt%) unchanged. Interestingly, an increase in the catalytic activity at higher ligand content was observed in the semi-hydrogenation of 1-hexyne. DFT calculations indicated that the ligand changes its adsorption configuration as a function of the HHDMA concentration. At low coverage, the organic layer lies almost flat on the surface of the PdNPs blocking a large number of metal sites while a high HHDMA coverage favors an extended three-dimensional configuration of the alkyl chain, and consequently, a lower fraction of Pd sites is blocked by the organic layer. Witte and co-workers also reported the semi-hydrogenation of 3-hexyn-1-ol catalyzed by PdNPs using chiral phosphate stabilizers.54 Small differences were observed in terms of activity and alkene selectivity using these chiral stabilizers in comparison with using HHDMA.
Polymers. Polymeric compounds are frequently applied in the preparation of metal nanoparticles since they can provide steric stabilization to the metal colloid. For this type of stabilization, the polymer, which contains coordinating groups, can prevent M-NP aggregation by providing a protective layer around the NPs through its extended molecular structure.
In an early study, Hirai and Yakura prepared a series of PVP stabilized PdNPs (of ca. 2 nm) using polymers of distinct molecular weights (Mw from 6000 to 574![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) and investigated the thickness of the adsorbed layer of the polymer.51 They observed that the layer of the polymer at the surface of the PdNPs increased from 3.8 to 15.6 nm on increasing the Mw from 6000 to 574000. For NPs stabilized by PVP with Mw = 175000, the authors proposed that the protecting PVP molecules present a conformation constituted by a sequence of many loops (segments in the solution and the surface) and trains (segments in contact with the surface) in an alternating order with a long tail at both ends (Fig. 3). In catalysis, the activity of these NPs in the hydrogenation of cyclooctadiene depended more on the specific surface area of the PdNPs than on the adsorbed layer thickness.
000) and investigated the thickness of the adsorbed layer of the polymer.51 They observed that the layer of the polymer at the surface of the PdNPs increased from 3.8 to 15.6 nm on increasing the Mw from 6000 to 574000. For NPs stabilized by PVP with Mw = 175000, the authors proposed that the protecting PVP molecules present a conformation constituted by a sequence of many loops (segments in the solution and the surface) and trains (segments in contact with the surface) in an alternating order with a long tail at both ends (Fig. 3). In catalysis, the activity of these NPs in the hydrogenation of cyclooctadiene depended more on the specific surface area of the PdNPs than on the adsorbed layer thickness.
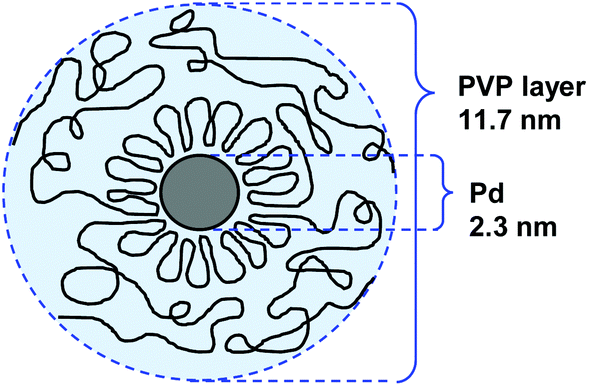 | ||
| Fig. 3 Conformation model of the protecting PVP layer in a suspension of PdNPs. Preparation conditions: PVP Mw = 170000, PVP/Pd = 4.51 | ||
In 2010, Evangelisti et al. reported the application of PVP stabilized PdNPs in the hydrogenation of a series of aliphatic alkynes.50 Excellent alkene selectivities were obtained in the hydrogenation of 1-hexyne, 2-hexyne and 3-hexyne (from 96 to 99% at 95% conversion). For the case of internal alkynes, the authors ascribed the observed high stereoselectivity towards the Z isomer (ca. 99% at 95% of alkyne conversion) to the steric constriction of the PVP surrounding the PdNPs. Interestingly, when 3-hexyne-1-ol was the substrate, a similar performance was observed independently of the Pd/PVP ratio used (>96% of selectivity at 98% of alkyne conversion when using either 99 or 90 wt% PVP).
Similarly, Kiwi-Minsker and co-workers tested the effect of the presence of additional PVP in the selective hydrogenation of alkynols catalyzed by PdNPs of 2.6 nm.8 The NPs were initially synthesized using PVP/Pd = 10. The results obtained in catalysis using these NPs were compared with those obtained when an extra amount of PVP was added to the initial catalyst. No difference in terms of activity or selectivity was observed, suggesting that the surface of the NPs was already saturated by the polymer. These results are in agreement with the observed negligible effect of the PVP/Pd ratio (in the range of 2.5–20) on the Suzuki reaction catalyzed by PdNPs.55
The use of RuNPs stabilized by PEG2000 as catalysts for the semi-hydrogenation of alkynes was reported in biphasic media.56 The RuNPs of ca. 1.6 nm were synthesized by hydrogen reduction (Fig. 4) and were active in the hydrogenation of methyl propiolate. The selectivity to the alkene product was observed to be highly dependent on the reaction conditions (temperature and pressure). Excellent conversions and selectivities were also obtained in the semi-hydrogenation of other terminal alkynes including substituted phenylacetylenes and linear alkynes using this catalyst. It is noteworthy that although Ru is active in the hydrogenation of phenyl groups, no hydrogenation of the arene was observed under the conditions used. This observation could be attributed to the protective effect of the PEG stabilizer preventing the planar orientation of the phenyl ring over the NP surface.
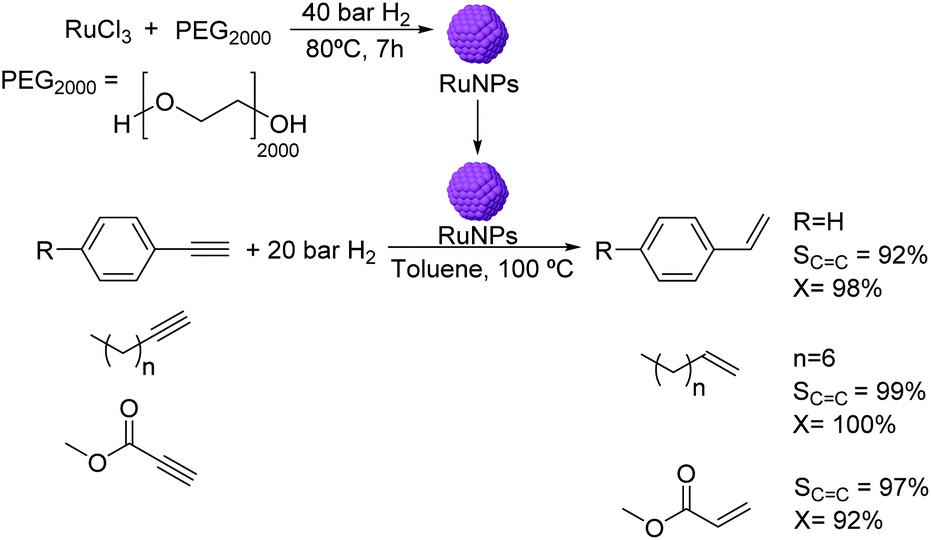 | ||
| Fig. 4 Synthesis of RuNPs stabilized by PEG and catalytic hydrogenation of terminal alkynes. | ||
Jones and co-workers reported the application of PdNPs supported on an aminopolymer–silica composite for the selective hydrogenation of alkynes.57 The palladium nanoparticles were generated through reduction (using H2 or NaBH4) of Pd(II) precursors previously loaded onto a mesoporous silica material functionalized with branched poly(ethyleneimine) polymers (PEI). The PEI served as both a stabilizer of the PdNPs and a linker to the solid support. Interestingly, the authors observed that the rate of over-hydrogenation of phenylacetylene could be significantly reduced by increasing the porosity of the support and the molecular weight of the polymer. The beneficial effect of the aminopolymer on the alkene selectivity was attributed to the efficient competition of the amine groups with the alkenes for the surface palladium sites. Additionally, they observed that the branched PEI of large molecular weight had a stronger effect on the selectivity than the PEI of lower molecular weight and speculated that the branched polymer could cover and passivate the PdNPs more effectively. Moreover, analysis of the used catalysts by TEM, and ICP indicated the excellent stability, recoverability, and recyclability of this catalytic system.
Micelles. The use of amphiphilic compounds permits the formation of micelles which serve as hosts for metallic nanoparticles. Depending on the structural characteristics of the amphiphilic compound (generally co-polymers), the metallic precursor and the polarity of the solvent, the core of the micelle can offer a hydrophilic or hydrophobic environment to the embedded M-NPs.
Kiwi-Minsker reported the selective partial hydrogenation of 2-butyne-1,4-diol catalyzed by PdNPs stabilized in the micelle core of poly(ethylene-oxide)-block-poly-2-vinylpyridine (PEO-b-P2VP) (Fig. 5).58 The amphiphilic character of the block co-polymer allowed the formation of micelles constituted by pyridine units in the core stabilizing the PdNPs and PEO chains in the shell responsible for its dispersion in water. Selectivity towards the alkenol product reached nearly 100% at up to 94% conversion, although beyond this conversion limit, the over-reduced product was formed. The reaction was carried out in the presence of a base (KOH, pH of 13.4) to avoid the acid-catalyzed isomerization of the products. The authors ascribed the high selectivity of this catalyst to the modification of the Pd nanoparticle surface with the 2-vinylpyridine units in the core of the micelle. This system exhibited stable activity and selectivity and could be recycled and reused several times with a simple ultra-filtration procedure.
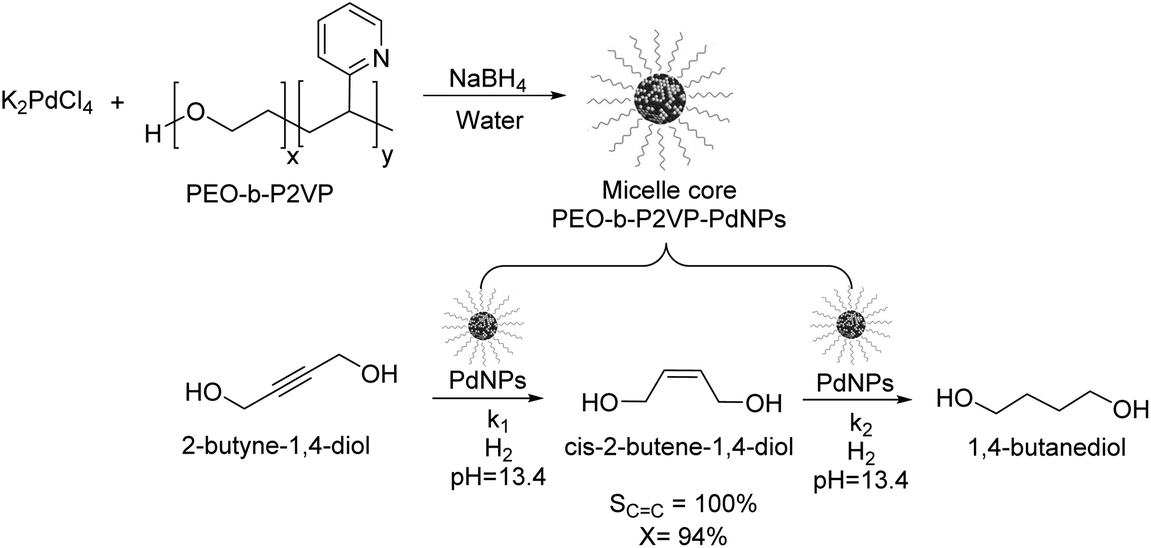 | ||
| Fig. 5 Synthesis of PdNPs stabilized in the micelle core of poly(ethylene-oxide)-block-poly-2-vinylpyridine (PEO-b-P2VP) and their application in the catalytic hydrogenation of 2-butyne-1,4-diol. | ||
By employing a similar supramolecular-micelle approach, Kaneda and coworkers described the stabilization of PdNPs using poly(amidoamine) (PAMAM) dendrons which have a pyridine core and alkyl end groups assembled into spherical micelle-like structures.59 Fine tuning of the Pd nanoparticle size ranging from 3 to 10 nm was achieved by adjusting the alkyl chain length and the structure of the dendrons. The resulting NPs could catalyze the highly selective semi-hydrogenation of dienes and acetylenes. In the hydrogenation of 1-phenyl-1-propyne, 98% selectivity to the Z-alkene product was obtained at 96% conversion. The authors attributed the high alkene selectivity to the ligand effect of the pyridine core of the dendrons.
Ionic liquids. Ionic liquids have also been used as solvents and/or stabilizers for the preparation of M-NPs applied in the semi-hydrogenation of alkynes. Their amphiphilic nature makes them excellent solvents for a wide variety of metallic precursors and organic compounds and since they usually contain polar groups (e.g. the nitrogenated imidazolium moiety), they can interact with the metal surface and stabilize M-NPs. Depending on the miscibility properties of the IL, they can also permit the recycling of catalytic systems by simple solvent extraction processes.60
Dupont and coworkers reported the partial hydrogenation of terminal alkynes using ionic liquid stabilized PdNPs.61 The thermal treatment of Pd(OAc)2 in 1-butyronitrile-3-methylimidazolium-N-bis(trifluoromethanesulfonyl)imide [(BCN)MI·NTf2] resulted in the formation of stable and monodispersed PdNPs exhibiting a diameter of ca. 7.3 nm (Fig. 6). Using these nanocatalysts, phenylacetylene was hydrogenated with 95% selectivity to styrene at 97% conversion and demonstrated the possibility to recycle the catalyst up to 4 times without the loss of activity or selectivity. In these experiments, the hydrogen pressure was observed to be critical to achieve high selectivity. The authors attributed the high selectivity of these NPs to the electronic modification induced by the coordination of functionalized nitrile ionic liquids to the metal surface.
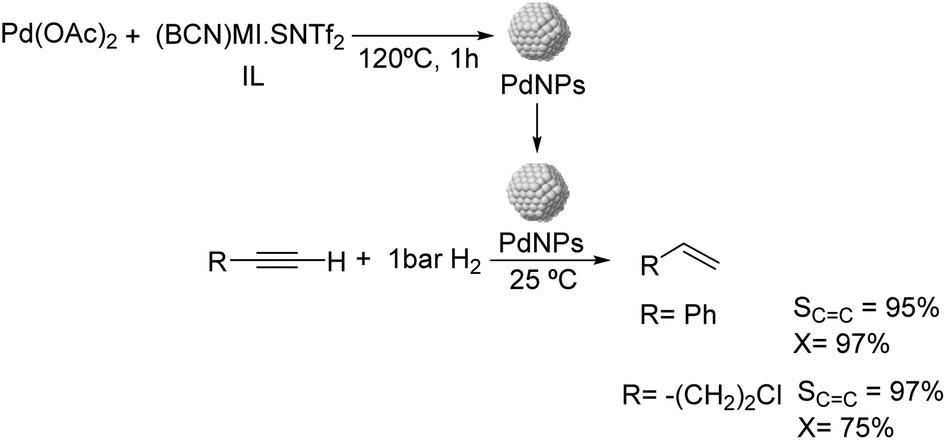 | ||
| Fig. 6 Synthesis of PdNPs stabilized by ILs and catalytic hydrogenation of terminal alkynes. | ||
More recently, the same group reported the preparation of PdNPs stabilized by ILs and imidazolium salts bearing pyridine or phosphine functionalities (Fig. 7) and their application in the semi-hydrogenation of 2-pentyne.62 PdNPs of ca. 3.5 nm were prepared by reduction of [Pd(acac)(COD)]BF4 (acac = acetylacetonate; COD = 1,5-cyclooctadiene), dissolved in n-butyl-3-methylimidazolium tetrafluoroborate (BMI·BF4) in the presence of stoichiometric amounts of the ligands and the use of hydrogen as the reducing agent. Interestingly, the PdNPs formed in the presence of a diphenylphosphine ligand exhibited a particle size remarkably smaller in comparison with the rest of ligands tested (1 nm vs. 3.5 nm). The formation of very small nanoparticles using similar phosphines containing aryl groups as stabilizers had previously been observed by other authors.63 The hydrogenation of 2-pentyne in the presence of any of these P- and N-ligands afforded a significant improvement in the selectivity towards cis-2-pentene in comparison with the reference NPs prepared in the absence of a ligand (up to 87% vs. 20% at ca. 95% of conversion), thus evidencing the positive effect of the ligands on the selectivity. Concerning the nature of the ligand, those presenting π-accepting character provided the highest alkene selectivity. This trend was evident for the series of phosphine ligands, where the alkene selectivity seemed to be inversely correlated with the sigma donating character of the phosphine: diisopropyl < dicyclohexyl < diphenyl. In terms of catalyst recyclability, the authors demonstrated that the ionic-liquid catalytic system could be reused up to 5 times without relevant loss of activity.
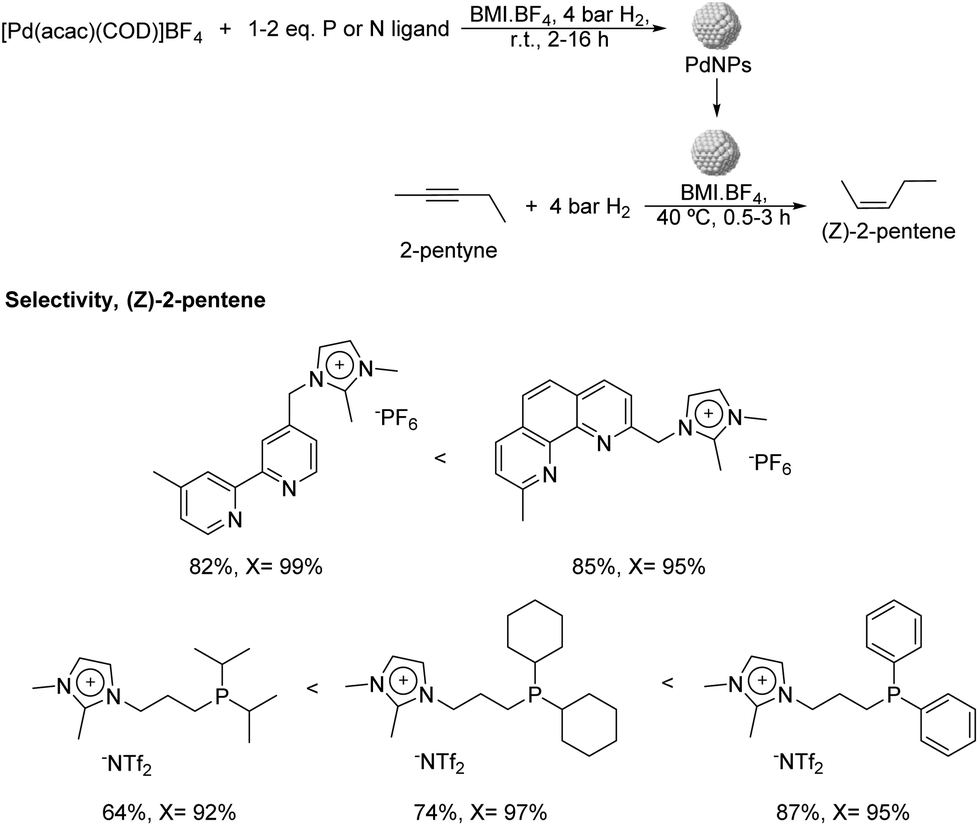 | ||
| Fig. 7 Synthesis of PdNPs stabilized by imidazolium salts bearing P- or N-functional groups and their application in the catalytic hydrogenation of 2-pentyne. | ||
Using a similar approach, the semi-hydrogenation of diphenylacetylene and its derivatives was reported using FeNPs stabilized by ionic liquids.64 These nanocatalysts (ca. 5 nm) were formed by reduction of FeCl3 with EtMgCl (Fig. 8) in the presence of a nitrile-functionalized IL or acetonitrile. The selectivities to the corresponding alkenes ranged from 20% to 98% depending on the substituents (96% for diphenylacetylene at conversion >90%). It is noteworthy that low selectivities towards the alkene were obtained when the phenyl ring of the substrate included electron-withdrawing groups such as –CO2Me or –F (74 and 82%); this effect was even more accentuated for PhCCCO2Me (40%). These results demonstrated the importance of an electron rich triple bond in the substrate to reach high alkene selectivities. Moreover, excellent Z-stereoselectivities were achieved (>92%) although they also resulted in being sensitive to the electronic properties of the substrates. In addition, the biphasic solvent system allowed the facile separation and re-use of the catalyst.
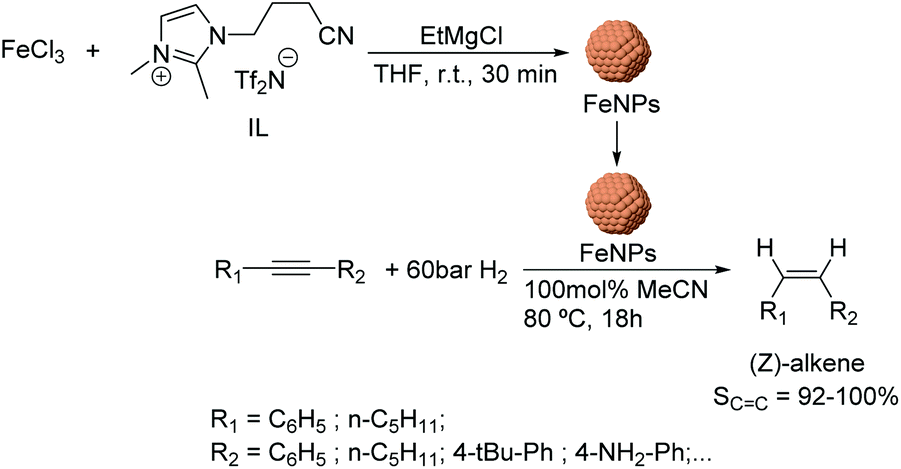 | ||
| Fig. 8 Synthesis of FeNPs stabilized by ILs and catalytic hydrogenation of internal alkynes. | ||
More recently, the preparation of a highly selective Pd/IL/MOF catalyst was reported for the semi-hydrogenation of alkynes.65 Initially the metal–organic framework template based on Cu was formed in the presence of an ionic liquid (1,1,3,3-tetramethylguanidinium trifluoroacetate, TMGT) and a palladium precursor (K2PdCl4), which was subsequently reduced with L-ascorbic acid to form PdNPs of ca. 2 nm. In this case, the role of the IL was to stabilize the PdNPs and act as a linker between these and the MOF support (Fig. 9). This composite catalyst was tested in the semi-hydrogenation of a series of alkynes displaying excellent selectivities (>99%) at >99% conversion. The authors proposed that interactions between the N atoms of the IL and the PdNP surface were responsible for the high selectivity. Moreover, this catalytic system could be recycled up to 4 times without the loss of activity or selectivity. However, after the 5th run, some Pd leaching was detected.
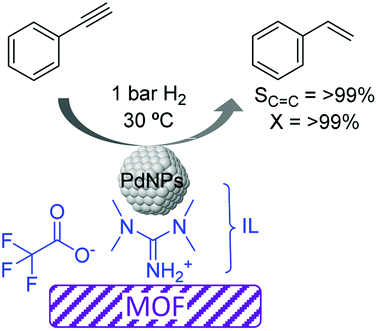 | ||
| Fig. 9 Semi-hydrogenation of alkynes catalyzed by Pd nanoparticles supported by a metal–organic framework embedded in an ionic liquid microphase. | ||
Ligands. Some ligands frequently used in coordination chemistry have also been employed as stabilizers of metal nanoparticles.66,67 Traditionally, the use of ligands has been combined with organometallic precursors provided with olefinic compounds that could be hydrogenated to provide a bare metal atom which would condense in the reaction medium thus resulting in the NPs.67 The presence of the stabilizing ligand in sub-stoichiometric amounts with respect to the metal prevents the agglomeration of the formed nanoclusters.
Much before the popularization of the concept of nanoparticles, Caubère and coworkers introduced the preparation of new versatile reducing agents called complex reducing agents (CRA).68 These reducing agents were prepared by reaction of transition metal salts with reducing systems such as LiAlH4, NaBH4, NaH, among others, and consisted of non pyrophobic black suspensions which, in the presence of hydrogen, could catalyze selective hydrogenation reactions. According to the chemical reduction performed with these reagents, the authors postulated the intervention of transition metal hydrides as the reactive part of the CRA.69 These authors reported the preparation of the very active and highly specific nickel catalyst Nic for alkene and alkyne hydrogenations.70 The designated Nic catalyst was prepared by chemical reduction of Ni(OAc)2 using NaH in t-AmOH under a nitrogen atmosphere (Fig. 10). The Nic catalyzed the hydrogenation of terminal alkynes such as 1-octyne and phenylacetylene providing alkene selectivities up to 90% at full conversion while 96 and 98% selectivities were achieved for 2-hexyne and 3-hexyne, respectively.
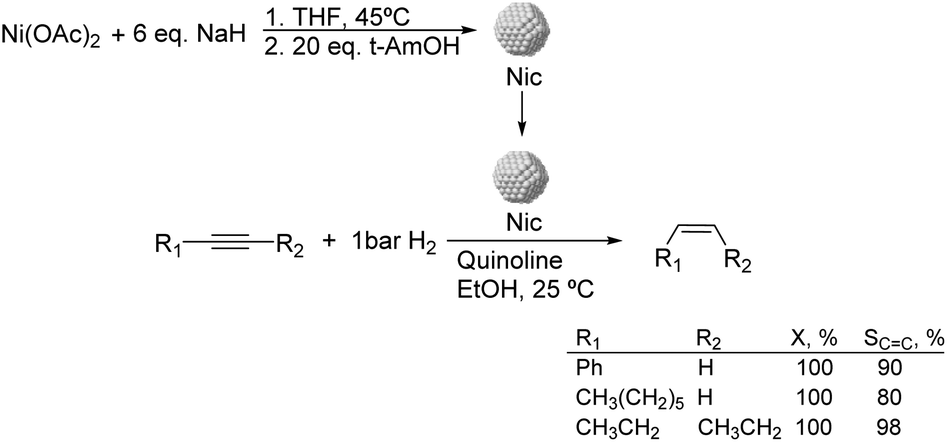 | ||
| Fig. 10 Synthesis of Nic catalyst and its application in the catalytic hydrogenation of alkynes. | ||
Later, the same group reported that the Nic catalyst consisted of slightly magnetic particles of ca. 8–9 nm according to XRD and electron microscopy. In addition, this material contained 60–70 wt% of nickel depending on the alkoxide used during the preparation, which suggested the presence of alkoxides stabilizing the NiNPs. The influence of the nature of the alcohol used during the preparation of the Nic catalyst was also evaluated. Interestingly, when t-BuOH was used, the initial rate of hydrogenation of phenylacetylene was slightly higher than that when i-PrOH and t-AmOH were employed. The effect of the alkyne structure in hydrogenation using the Nic catalyst was also studied.71 For terminal alkynes, the reduction in the substrate is slower than the subsequent alkene hydrogenation, while the opposite occurs for internal alkynes. A similar observation correlating the hydrogenation rates of terminal alkynes and alkenes had previously been reported.41 In the complete hydrogenation of the mono- and disubstituted alkynes 1-octyne and 2-hexyne, alkane formation was observed earlier for the terminal alkyne than with the disubstituted one.71 This was attributed to the over-hydrogenation of the produced 1-alkene without a desorption step. Interestingly, for this Nic catalyst, isomerization of the produced alkene only began when the alkyne had been consumed completely. Moreover, upon addition of quinoline during the hydrogenation of 2-hexyne, both the over-hydrogenation and the cis–trans isomerization reactions of the alkene were notably retarded. In a subsequent report, the same authors reported the application of the Nic catalyst in the hydrogenation of a series of terminal and internal alkynes and alkynols.72 In all cases, the reaction was completed after the consumption of 1–1.2 eq. of hydrogen, resulting in alkene selectivities of 80–90% for terminal alkynes/alkynols while values higher than 95% were usually obtained for internal substrates.
Caubere and coworkers extended their studies on complex reducing agents (CRA) using Pd catalysts. Following the method used for the synthesis of Nic, a colloidal Pd catalyst was synthesized by the reduction of Pd(OAc)2 by NaH in the presence of 2-methylbutan-2-ol (t-AmOH) (Fig. 11).73 This catalyst was active in the self-terminating semi-hydrogenation of 1-heptyne and 1-octyne with an olefin selectivity of 98% for both substrates at complete conversion. However, the presence of N-donor compounds as catalyst modifiers during catalysis was required to avoid over reduction of the substrates and among the series, quinoline provided the highest selectivities. The key role of the alkoxide in the control of the reactivity of this catalyst was demonstrated when a similar Pd catalyst synthesized in the absence of t-AmOH was tested giving full over-hydrogenation of the alkyne under the same conditions.
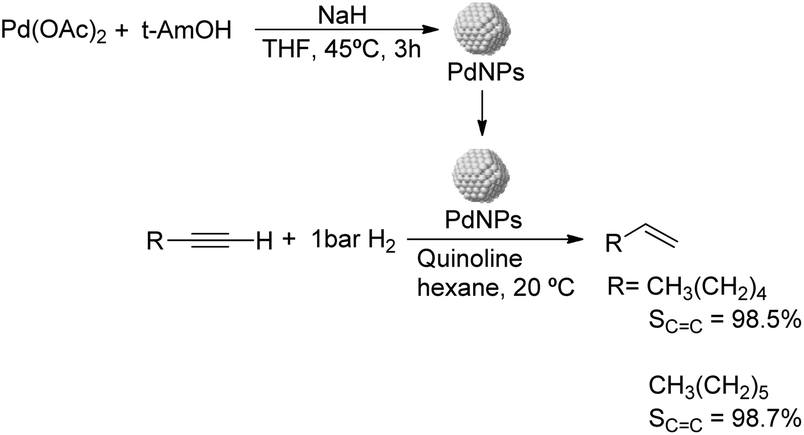 | ||
| Fig. 11 Synthesis of PdNPs reported by Caubere and co-workers and subsequent catalytic hydrogenation of terminal alkynes. | ||
Chaudret and co-workers also reported the use of FeNPs as catalysts for selective hydrogenation reactions.46 These NPs were synthesized by the reduction of an organometallic iron precursor under hydrogen (Fig. 12). Since no additional stabilizer was used, it was proposed that HN[Si(CH3)3]2 (HMDS) released during the decomposition of the metal precursor acts as a stabilizing agent for the FeNPs. These nanocatalysts were very active in the hydrogenation of terminal and internal alkynes. However, over-reduction occurred for most of the tested substrates and only when 3-hydroxyphenylacetylene was tested as a substrate, was a significant selectivity to 3-vinylphenol (88%) obtained.
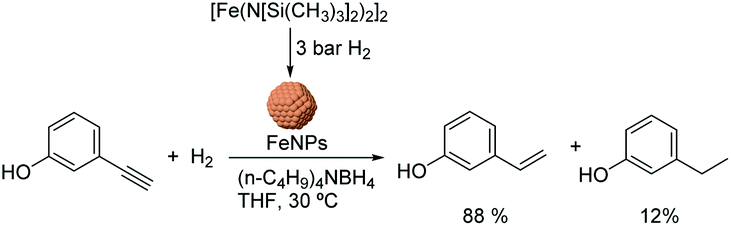 | ||
| Fig. 12 Synthesis of FeNPs via hydrogenation of an organometallic precursor and their application in the hydrogenation of 3-hydroxyphenylacetylene. | ||
Recently, Godard and co-workers reported a versatile method for the preparation of efficient and reusable Pd nanocatalysts immobilized on paper for the selective hydrogenation of alkynes.74 In this report, the Pd nanoparticles were synthesized by a one-pot method involving the decomposition of Pd2(dba)3 (dba = dibenzylideneacetone) under H2 in the presence of tricyclohexylphosphine (PCy3) and multi-walled carbon nanotubes (MWCNTs). Subsequently, a suspension of the Pd/MWCNTs was impregnated in the paper carrier by a simple painting process. The resulting paper based catalyst was active and highly selective in the hydrogenation of a series of alkynes in the presence of quinoline and could be reused up to 5 times without loss of activity or selectivity.
De Vries and coworkers applied FeNPs in the hydrogenation of a series of alkynes with good to excellent conversion under mild conditions.45 These FeNPs were prepared via reduction of iron salts with alkyl Grignards or alkyl lithiums in THF. The resulting FeNPs catalyzed the selective hydrogenation of 1-hexyne and 1-octyne under 10 bar H2 resulting in selectivity to the alkene of ca. 60% at 100% conversion. However, under 20 bar H2 and a longer reaction time (15 h instead 0.5 h), the substrate was completely over-reduced. On the basis of the detection of iron, magnesium and chloride by EDX analysis, the authors proposed that the FeNPs were stabilized by MgCl2 and the solvent, THF.
000), polyquaternium-10, and sodium carboxymethyl cellulose (CMC, Mw: 90000) (Fig. 13).75 The PdNPs stabilized by sodium carboxymethyl cellulose (CMC) in water were the most efficient catalysts in this process with higher selectivity and conversion (50% selectivity at 100% conversion), than those reported for conventional supported Pd catalysts (11% selectivity at 93% conversion for Pd-Ti/SiO2). In this study, the selectivity was highly influenced by the H2/C2H2 ratio and the reaction temperature. The highest ethylene selectivities were obtained when acetylene-rich streams (H2/C2H2 = 1.5) and low temperatures were employed.
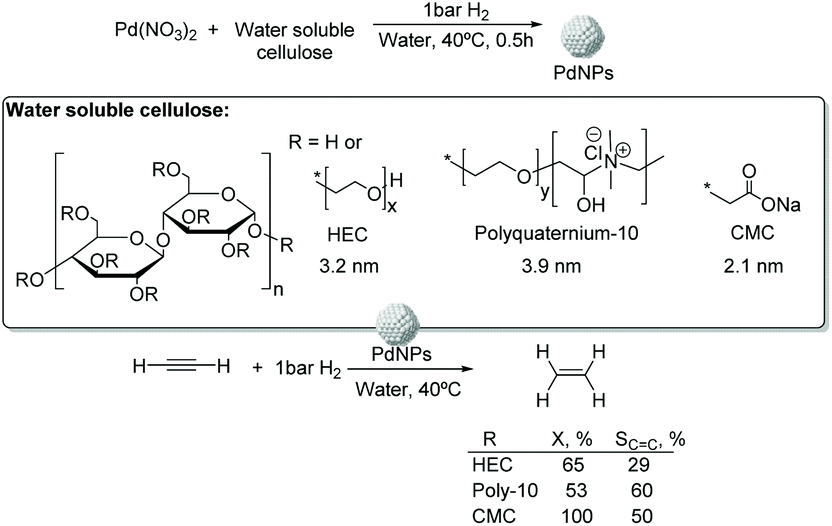 | ||
| Fig. 13 Synthesis of PdNPs stabilized by water soluble cellulose and catalytic hydrogenation of acetylene. | ||
To date only a few studies have been reported on the hydrogenation of acetylene using supported catalysts prepared through colloidal chemistry, and these reports mainly deal with the effect of the stabilizer on the activity of these species. Kiwi-Minsker and co-workers investigated the influence of the organic stabilizer and the reducing agent on the performance of PdNPs supported on carbon nanofibers in the hydrogenation of acetylene.76 The NPs were prepared using sodium di-2-ethylhexylsulfosuccinate (AOT), polyvinylalcohol (PVA), and polyvinyl pyrrolidone (PVP) as stabilizers and various reducing agents (hydrazine hydrate, ascorbic acid, n-butanol, ethanol and methanol). The authors observed a strong decrease in the hydrogenation rate for the NPs stabilized by sodium di-2-ethylhexylsulfosuccinate in comparison with those obtained with the catalysts stabilized by polyvinylpyrrolidone or polyvinylalcohol. These results were attributed to the geometric and electronic effects induced by the stabilisers. In contrast, in terms of alkene selectivity, a similar product distribution was obtained over PdNPs with a similar crystal size, independently of the stabilizer used. Interestingly, when the polymeric stabilizer was removed by a combined ozone/H2 treatment, the ethylene selectivity was negatively affected thus demonstrating the positive effect of the stabilizer in catalysis.
Other studies have addressed the importance of removing the stabilizer once the nanoparticles are immobilized on the support and prior to the catalysis, in order to maximize the number of accessible active sites. The preparation of poly(vinylpyrrolidone) (PVP)-stabilized Pd nanocubes, their deposition on a carbon-based support, and their subsequent treatment with UV-ozone (UVO) was reported.77 This process allowed complete cleaning of the catalyst surface without affecting the morphology of the particles. The removal of the PVP displayed a positive effect on the selective hydrogenation of acetylene, with a 4-fold increase in the activity after the treatment of the catalyst.
In summary, the selection of the appropriate stabilizer for the preparation of colloidal nanoparticles is crucial to obtain selective catalysts for the hydrogenation of alkynes and should be done based on the substrate that is to be transformed. For instance, gas–solid reactions are highly sensitive to the presence of residues (even in trace amounts) at the catalyst surface, and therefore require the use of cleaning procedures. In this sense, stabilizers containing only C, H, O, and N (e.g. PVP, PVA) are ideal for gas–solid reactions since their decomposition products are generally volatile compounds which do not block the active sites. In contrast, liquid phase reactions permit the modulation of the reactivity of the metallic NPs through the electronic or steric properties of the stabilizers. A common strategy to improve the selectivity in the semi-hydrogenation of alkynes is the use of stabilizers containing nitrogen functionalities such as amines, pyridines or nitrile groups.
2.2 Role of support
In 2012, a comprehensive discussion on the role of the support at the meso/microlevel in the selective hydrogenation of alkynes was reported by Kiwi-Minsker and co-workers.11 Although the immediate role of the support is to dilute the active phase and facilitate the handling of the catalyst, the selection of the appropriate support also represents a powerful tool to influence the properties of the catalyst. Indeed, the support provides some control of the NPs size, metal distribution, and stability of the particles (Fig. 14).78 Furthermore, the performance of a catalyst can be modified by the chemical and electronic properties of the carrier via metal–support interactions while its structural properties affect the diffusion and adsorption of the reactants/products.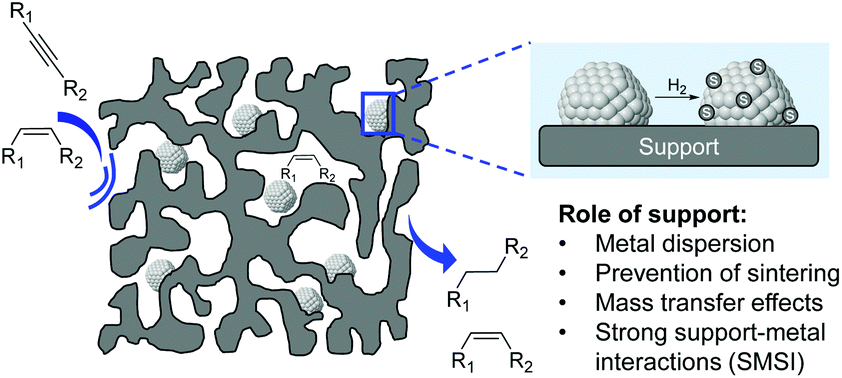 | ||
| Fig. 14 Role of the support in the performance of supported metal nanoparticles. | ||
The impact of the support will also depend on the thermal treatments, including activation steps, to which the catalytic material will be subjected prior to the hydrogenation. For instance, when reducible oxides are used as supports, their partial reduction during the thermal treatment may result in strong metal–support interactions (SMSI) which can cause site blocking via migration of partially reduced oxide species from the support or the formation of new bimetallic phases.79,80 The surface characteristics and adsorption properties of a carrier can also be modified by the incorporation of functionalities in pre-treatment steps. For instance, the treatment of carbon materials with reagents such as ozone, carbon dioxide HNO3, HCl, H2O2, NaOH, etc. can modify their surface by introducing oxygen-containing groups.81
In the following paragraphs, the impact of the support on the performance of nanocatalysts in the selective hydrogenation of alkynes is discussed with particular focus on the recent results involving the use of colloidal techniques for the preparation of the active phase.
High alkenol selectivity (95% at full conversion) was reported in the hydrogenation of 2-methyl-3-buten-2-ol (MBE) over a structured catalyst of Pd/ZnO on sintered metal fibers (SMFs).82 The structured catalyst was prepared by deposition of a colloidal suspension of 7 nm PdNPs over ZnO/SMF followed by reduction in hydrogen at 773 K. Based on the comparison with a Pd/Al2O3 catalyst, the high alkenol selectivity observed for the ZnO supported material was ascribed to the alloyed structure of the active phase, Pd/PdZn, formed during the high-temperature treatment under hydrogen (Fig. 15).
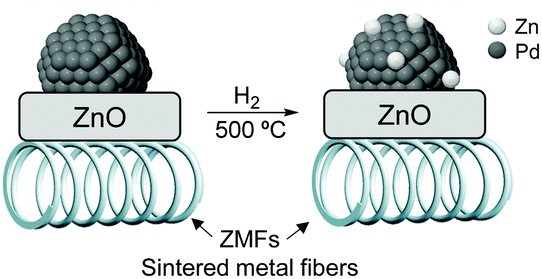 | ||
| Fig. 15 Strong metal–support interactions (SMSI) in the Pd/ZnO/SMF catalyst formed during the activation treatment. | ||
Over the last few years, several studies have been dedicated to the preparation of colloidal NPs which incorporate materials frequently used as supports (e.g. iron oxides). In those cases, the purpose of such incorporation is usually the transmission of magnetic properties to the active phase as a strategy for recycling the colloidal catalyst. For instance, the preparation of magnetically recoverable Pd catalysts was reported via thermal decomposition of palladium acetylacetonate in the presence of iron oxide nanoparticles.83 In the semi-hydrogenation of 2-methyl-3-butyn-2-ol, these catalysts were more efficient than similar PdNPs prepared in the absence of iron oxide NPs (96% selectivity at 95% conversion vs. 100% sel. at 6% conversion) (Fig. 16). The composition of the iron oxide NPs was also shown to influence the catalytic performance of the PdNPs since those prepared from iron species in a lower oxidation state were the most active. According to the authors, electron transfer from the iron oxide NPs to the Pd surface could accelerate the reduction of the alkyne.
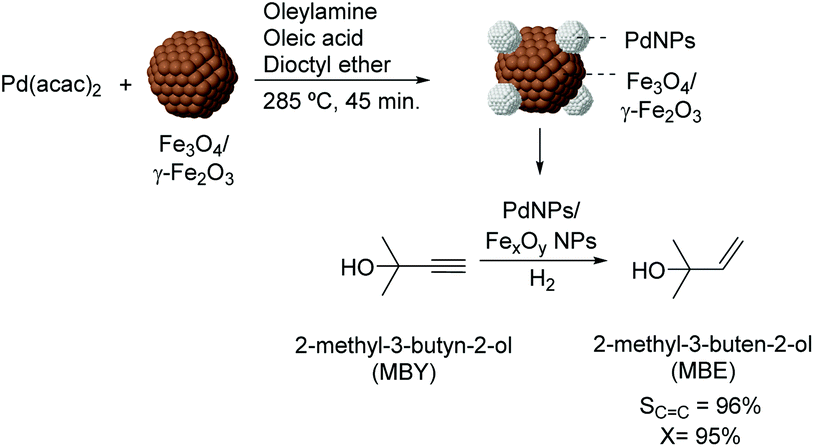 | ||
| Fig. 16 Synthesis of PdNPs/iron oxide NPs and their application in the selective hydrogenation of 2-methyl-3-butyn-2-ol (MBY). | ||
The selective hydrogenation of 3-butyn-1-ol catalyzed by recoverable PdNPs deposited on a colloidal magnetic support (silica-coated magnetite functionalized with amido groups, FFSiNH2) was also recently reported by Philippot and coworkers.84 This catalyst was prepared by decomposition of Pd2(dba)3 over the magnetic support and provided high selectivity in the semi-hydrogenation of 3-butyn-1-ol and benzyl-propargylamines under mild conditions and short reaction times (82% selectivity to 3-buten-1-ol at 98% conversion) (Fig. 17).
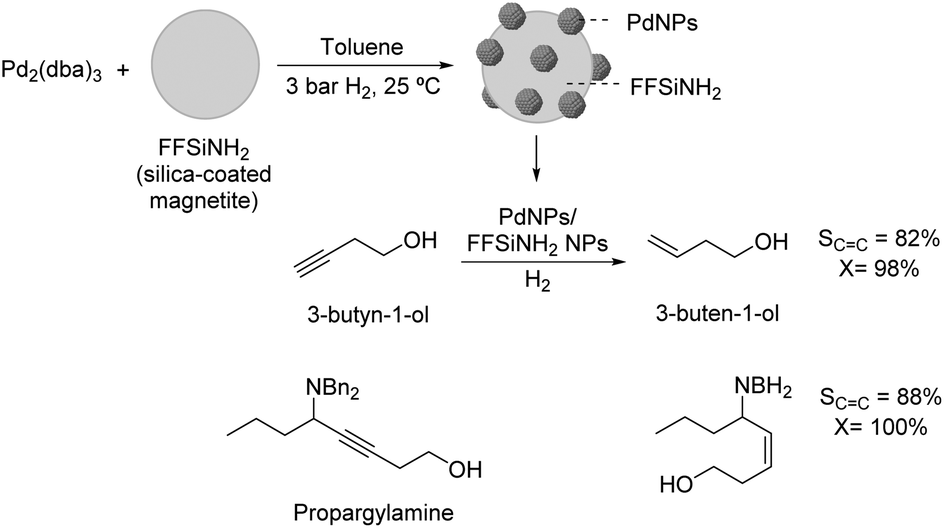 | ||
| Fig. 17 Synthesis of PdNPs/silica-coated magnetite NPs and their application in the selective hydrogenation of 3-butyn-1-ol and benzyl-propargylamines. | ||
Another example of a very selective catalyst for the semi-hydrogenation of alkynes is the multi-walled carbon nanotube (MWCNT) supported Fe3O4–Cu2O–Pd nanocomposite.85 This catalyst was prepared by initial deposition Fe3O4 and CuNPs onto the MWCNTs followed by in situ galvanic replacement between Cu and PdCl42+. Interestingly, this nanocomposite was active only in the hydrogenation of terminal alkynes (internal alkynes were not hydrogenated) leading to the formation of the corresponding alkenes without any over-hydrogenation. The authors demonstrated that Cu2O played a critical role in the selectivity since the absence of this material resulted in the loss of selectivity. However no explanation for such behavior was provided. This catalyst displayed unprecedented selectivity for the semi-hydrogenation of terminal alkynes with self-terminating properties.
The utilization of Au86,87 and CeO233,88 based catalysts in hydrogenation reactions has become popular in recent years. Although cerium oxide is a highly selective catalyst for the partial hydrogenation of alkynes, the high operating conditions required (temperature and pressure, even with promoted versions89) have hampered the practical utilization of this oxide.34 One example combining the synergies of both Au and CeO2 for the semi-hydrogenation of alkynes was reported by Kaneda and co-workers.33 The Au@CeO2 NPs were prepared using a redox-coprecipitation method, where the Au nanoparticle core and the nanoporous shell of CeO2 are simultaneously formed in one step. In the hydrogenation of phenylacetylene at room temperature under 30 bar of H2, the Au@CeO2 catalyst provided 99% selectivity to styrene at full conversion of the substrate, even after prolonged reaction times. The authors propose that the core@shell structure of this catalyst plays a crucial role in reaching such selectivity by maximizing interfacial sites (Fig. 18).
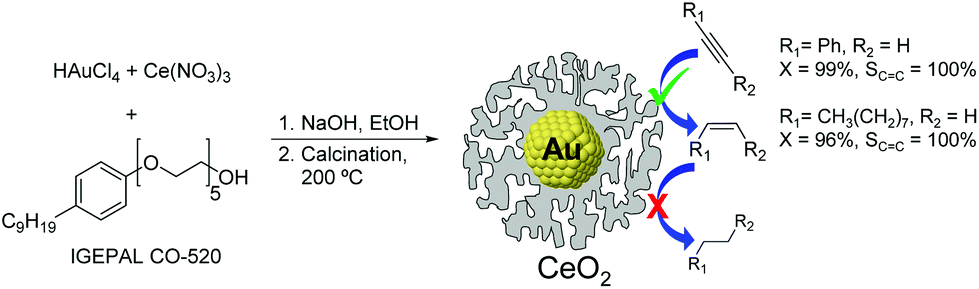 | ||
| Fig. 18 Preparation of Au@CeO2 NPs by a simultaneous coprecipitation method and selective hydrogenation of alkynes with prevented over-hydrogenation. | ||
The encapsulation of the active phase into the solid support is also a rational strategy to enhance the selectivity of catalysts in the semi-hydrogenation of alkynes. In a very interesting contribution, Bal'zhinimaev and co-workers reported that the preparation of Pd nanoclusters of 1–2 nm confined in fiberglass bulk permitted the selective hydrogenation of acetylene in the presence of a large excess of ethylene.90 This fact was explained by the ability of glass to absorb predominantly readily polarizable molecules such as acetylene but not ethylene. In addition, this catalyst was characterized by low oligomerization susceptibility thus providing high resistance towards deactivation under the reaction conditions. This is a clear example of how encapsulation or confinement of the active phase can dramatically impact on the alkene selectivity and prevent side reactions. This type of approach could be very promising for the selective hydrogenation processes of gas substrates.
2.3 Particle size and shape of the metal NPs
The effect of particle size and shape on the catalytic properties of metallic nanoparticles is an important issue in heterogeneous catalysis.91 The particle size of the NPs affects their surface to volume ratio and the coordination numbers of the surface atoms, and therefore their reactivity. Large particles possess mainly large crystal planes, whereas small particles display mainly edge and corner atoms.92 In general, low coordinated sites (located at edges and corners) would strengthen the adsorption of the reagents in comparison with crystal planes. Similarly, the shape of the metal nanoparticles also conditions the fraction of the different types of surface atoms.92Kiwi-Minsker and co-workers studied the structure sensitivity of unsupported PdNPs with diameters in the range 6–14 nm in the selective hydrogenation of 1-hexyne (Fig. 19).93 A 15-fold TOF increase was observed when the diameter of the particle increased from 11 to 14 nm (antipathetic size-sensitivity) while similar TOF values were obtained in the range 6–11 nm. For particles larger than 14 nm, the observed TOF approached the value of the Pd black catalyst. However, the selectivity towards 1-hexene (96.5% at 85% conversion of 1-hexyne) did not vary with the particle size. It was therefore concluded that for 1-hexyne, the selectivity of this reaction was size independent. Similar behaviors in TOF and selectivity were observed in the semi-hydrogenation of 2-methyl-3-butyn-2-ol (MBY) using these catalysts.94 In a complementary study on the semi-hydrogenation of MBY, the evaluation of PVP stabilized PdNPs in the range of 2–9 nm evidenced similar trends as a function of the particle size: higher specific activity (TOF) over larger Pd NPs and constant selectivity independently of the particle size.8 The authors correlated the observed size effect on activity with the fraction of Pdplane active sites being more abundant in the larger NPs.
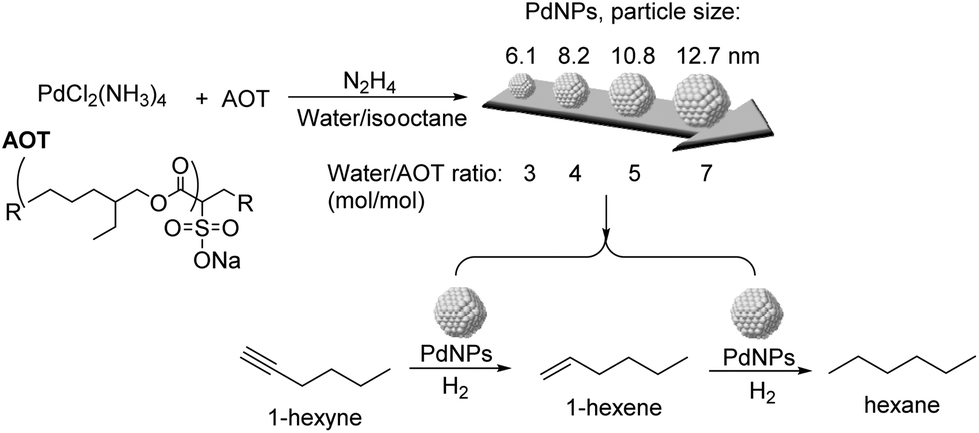 | ||
| Fig. 19 Synthesis of PdNPs of different sizes and their application in the selective hydrogenation of 1-hexyne. | ||
Witte et al. studied the effect of the metal crystallite size in the hydrogenation of 3-hexyn-1-ol using PdNPs between 5–25 nm prepared by the NanoSelect method.14 Interestingly, when the smallest PdNPs were used, the TOF decreased and the alkene selectivity improved.
Zhou and co-workers studied the size effect on the selective hydrogenation of phenylacetylene catalyzed by Pd NPs (4–12 nm) supported on α-Al2O3.95 The catalysts were prepared by a two-step protocol. The first step consisted in the chemical reduction of Pd2+ by NaBH4 in the presence of PVP, followed by the deposition of the metal colloid onto α-Al2O3. In catalysis, an increase in specific activity and selectivity to styrene was observed when increasing the particle size from 3.8 to 6.6 nm. Further increase of the particle size from 6.6 to 12.1 nm resulted in a slight increase in specific activity while the styrene selectivity decreased gradually. The antipathetic size-sensitivity observed in this report is in the same line as that in studies performed using other substrates such as 1-hexyne and 2-methyl-3-butyn-2-ol. This behavior can be associated with the increase in the fraction of low index planes (faces) in larger nanoparticles considering that such sites (Pd100) are more active in the hydrogenation of alkynes.
For acetylene, Kiwi-Minsker and co-workers studied the particle size effects of PdNPs in the range of 8–13 nm for supported Pd/CNF/SMF (carbon nanofibers/sintered metal fibers) nanocatalysts.40 Similarly to what was observed for liquid substrates, the turn-over-frequency was found to increase with the particle size up to a critical diameter of 11 nm, above which the specific activity was size-independent. However, for this substrate, the initial selectivity to ethylene was found to be size-independent. This size effect below 11 nm was suggested to be “geometric” in nature due to the size-sensitive formation of a Pd–Cx phase.96 Therefore, for smaller particles, the C/Pd ratio is higher and carbonaceous deposits block most of the active Pd surface. In a more recent study on the selective hydrogenation of acetylene, Kiwi-Minsker reported that similarly, a decrease in PdNP size from 8 to 2 nm results in a concomitant decrease in activity and increase in selectivity to the ethylene product.76 This observation was linked to site isolation by carbon deposits, which is important over small PdNPs.
The particle shape can also have an impact on the performance of M-NPs in catalysis. For instance, the effect of the shape of nanocatalysts in the selective hydrogenation of 2-methyl-3-butyn-2-ol (MBY) was studied by employing PVP stabilized Pd nanocubes (6 and 18 nm), octahedra (31 nm), and cuboctahedra (5.5 nm).97 Based on the observed activity and selectivity two types of active sites were proposed for this reaction: those on the planes where the semi-hydrogenation of MBY to 2-methyl-3-buten-2-ol (MBE) takes place and those at the edges that are responsible for the over-hydrogenation (Fig. 20). The lower coordination number of the edge atoms could explain the increased adsorption strength of the alkene, thus favoring the over-hydrogenation reaction. By considering the statistics of surface atoms for nanocrystals with different shapes and sizes, the optimal catalyst in terms of productivity and selectivity to MBE was predicted to be cubes of ca. 3.5 nm. Previously, the same authors reported the use of nanohexagons and nanospheres in the same catalytic transformation and the highest activity was obtained using particles with the highest amount of (111) planes.98
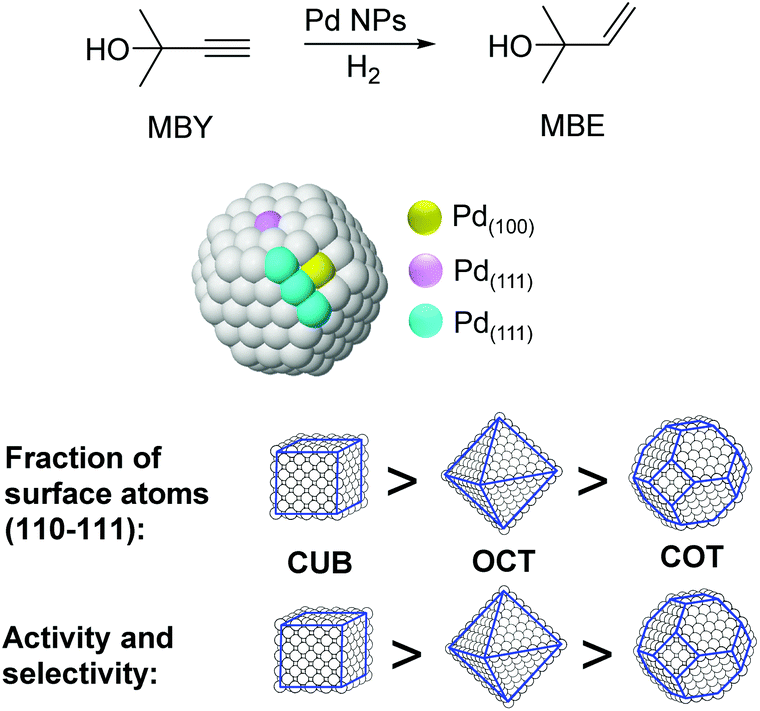 | ||
| Fig. 20 Effect of the palladium nanocrystal shape on the selective hydrogenation of 2-methyl-3-butyn-2-ol (MBY). | ||
However, an analogous study on the effect of the shape/size in the semi-hydrogenation of acetylene indicated that the shape of nanoparticles does not influence the catalyst selectivity, while the activity decreases in the order Pdoctahedra > Pdcuboctahedra > Pdcubes.25 The authors concluded that for acetylene, the octahedron represents the optimal shape for these catalysts. Concerning the optimal particle size for this substrate, calculations predicted that Pd octahedra of 3 nm would display the maximum catalyst activity.
In view of the distinct conclusions reported on the effect of the particle size/shape of Pd nanocrystals on catalyst performance,79 it can be assumed that these effects are sensitive to the substrate under study and therefore, cannot be generalized. Moreover, since the size and shape are closely related parameters, studies in this area need to consider the intrinsic correlation between them.
2.4 Dilution of the active phase: introduction of modifiers
The introduction of modifiers is a widely applied strategy for enhancing the catalyst performance in the selective hydrogenation of alkynes. A modifier is defined as a component of a catalyst that improves the performance of the active substance without necessarily having any significant activity on its own. Some of the more commonly applied modifiers are metals with lower affinity for hydrogen (e.g. Ag, Au, Cu, etc.) than the highly active Pd phase. The introduction of a second metal thus results in the dilution of the active phase. The impact of such a dilution can be classified according to geometric and electronic effects. The geometric effect is attributed to the creation of suitable ensembles of unique character or to the blockage of unselective active sites in order to enhance the selectivity towards the alkene. The electronic effect concerns the ability of a modifier to affect the electronic properties of the active metal in order to enhance the thermodynamic selectivity and/or to suppress the formation of (sub)surface species. A clear example of an electronic effect is the introduction of Zn as modifiers into Ni structures to form a NiZn alloy, which is a promising catalyst for alkyne hydrogenation.10,99 Nørskov and co-workers demonstrated that a catalyst constituted by 25Pd75Zn was more selective in the selective hydrogenation of acetylene than purely Ni or Pd catalysts and even superior to a 25Pd75Ag alloy, commonly used in industrial formulations.99Based on kinetic Monte Carlo simulations for the selective hydrogenation of acetylene, Mei et al. reported that alloying the palladium surface with Ag results in both geometric and electronic effects which weaken the binding energies of acetylene and hydrogen to the surface.100 The presence of Ag at the surface weakens the binding strengths of all surface intermediates and therefore increases their rates of desorption as well as their rates of hydrogenation. The weaker interactions and the decrease of Pd sites control the availability of hydrogen on the surface. This, together with the weaker ethylene binding to the surface, promotes its desorption over its hydrogenation.
One of the classic examples of modified catalysts in alkyne hydrogenation is the Lindlar catalyst which consists of 5 wt% palladium supported on porous calcium carbonate treated with lead and quinoline as modifiers. These two modifiers influence specific parameters which control the selectivity to the alkene: Pb enhances the thermodynamic factor and hinders the formation of hydrides, while quinoline prevents the over-hydrogenation reaction by competition with the alkene for the Pd sites.101
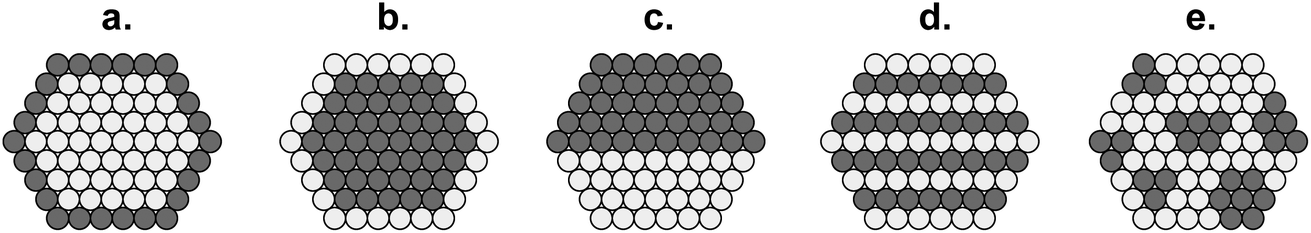 | ||
| Fig. 21 Cross-sections of bimetallic particles: (a) core@shell; (b) inverted core@shell; (c) cluster-in-cluster; (d) alloy with ordered metal atoms; and (e) nanoalloy with randomly disordered metal atoms.105 | ||
In a comprehensive study, Lopez and Vargas-Fuentes performed a series of Density Functional Theory (DFT) calculations that aimed at illustrating the effect of the presence of a second metal on the properties of Pd in the selective hydrogenation of acetylene in ethylene rich mixtures.10 In terms of adsorption properties, this study indicated that a stronger adsorption takes place on Pd-only sites than on mixed sites and that acetylene is always adsorbed more strongly than ethylene. Metals such as Bi improved the thermodynamic selectivity while others such as Cu and Au stabilized the adsorption of both the substrate and alkene product with respect to Pd. Concerning the formation of hydrides, all the promoters studied reduced the availability of the subsurface hydride (β-PdH phase) and as such, should reduce the over-hydrogenation reaction. Among the promoters that were studied, Bi and Sn showed the strongest effect. A similar outcome was also observed for Ag@Pd core@shell structures.106 Regarding carbide formation, the results indicated that metals such as Sn, Pb, and Bi are very unlikely to suffer surface carburization. Concerning oligomer formation, Sn, Pb and Bi efficiently isolated Pd sites, thus inhibiting oligomerization at the surface. In contrast, promoters such as Ag, Zn, and Ga are more prone to suffer oligomerization issues due to electronic effects and for this reason relatively large amounts of these promoters are necessary to achieve site isolation.
In addition to the synthesis methodology, it has been observed that the architecture of bimetallic NPs can be modified by thermal treatment in the presence of H2, O2 or Ar.107
In the following sections, the reported application of bimetallic NPs in the selective hydrogenation of alkynes will be described according to the type of substrate used in catalysis and the metal combinations.
Liquid substrates. PdAg. Kiwi-Minsker and co-workers investigated the role of a second metal using colloidal Pd and bimetallic Pd–Ag and Pd–Cu NPs as catalysts in the selective hydrogenation of dehydroisophytol (DIP) into isophytol (IP).108 The bimetallic NPs were prepared using Pd/M ratios of 1.5–5 (M = Ag or Cu) by chemical reduction on pre-formed Pd colloids stabilized by PVP (Fig. 22). The bimetallic Pd–Ag NPs adopted a core (Pd)–shell (Ag) structure whereas a mixed alloy was obtained for Pd–Cu nanocrystals. In catalysis, a significant increase in selectivity from 91 up to 97% (at 99% conversion) to the target alkenol was observed after incorporation of the second metal. This result was attributed to the dilution of the surface Pd-sites by Ag or Cu and modification of the electronic properties of Pd. Interestingly, the alkenol selectivity was not affected by the amount of modifier used for both Pd–Ag and Pd–Cu systems.
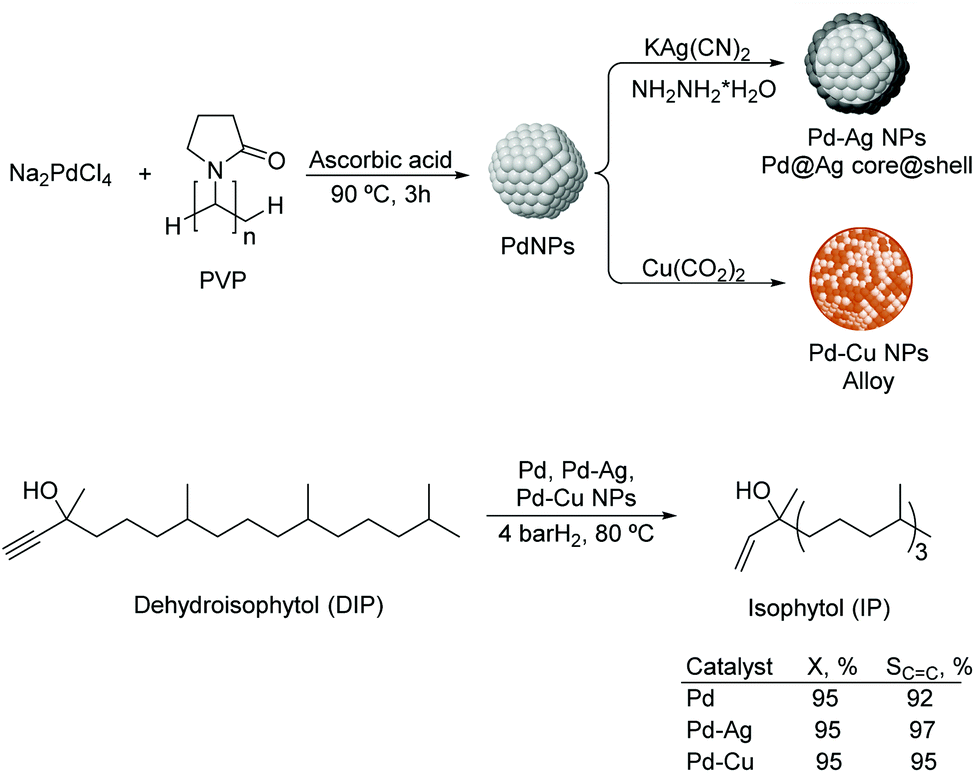 | ||
| Fig. 22 Synthesis of Pd, Pd–Ag and Pd–Cu bimetallic NPs, and their application in the selective hydrogenation of dehydroisophytol. | ||
The selective hydrogenation of 3-hexyl-1-ol was investigated using bimetallic AgPd nanoparticles synthesized via galvanic exchange reactions between AgNP seeds and K2PdCl4 in the presence of PVP as a stabilizer (Fig. 23) using Ag/Pd ratios between 0:1 to 6:1.109 All bimetallic catalysts were highly selective for Z-3-hexen-1-ol (>99%) and within the series of tested catalysts, the highest TOF was achieved with the NPs prepared using a Ag/Pd ratio of 2:1 (935 h−1 compared with 570 h−1 for the 0:1 catalyst). EXAFS analysis indicated that the structure of the catalysts can be described as a cluster-in-cluster.
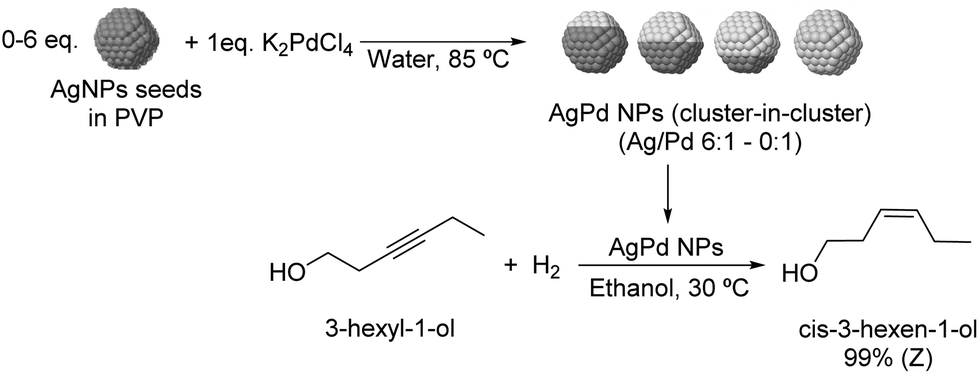 | ||
| Fig. 23 Synthesis of PdAg cluster-in-cluster NPs and their application in the selective catalytic hydrogenation of 3-hexyl-1-ol. | ||
Very recently, the preparation of a series of core@shell Pd@Ag NPs of ca. 26 nm with a variable Ag content (9–33 wt% Ag) was reported. These nanoparticles were prepared by deposition of a silver layer over Pd seeds by reduction of AgNO3 using ascorbic acid in water (Fig. 24).110 In catalysis, excellent alkene selectivity was obtained in the semi-hydrogenation of 1-octyne under mild conditions using the Pd@Ag catalyst of 16.7 wt% Ag (>99% selectivity at >99% conversion at r.t. and 1 bar H2). With this catalyst, the over-hydrogenation of the alkene was prevented even after extended reaction times following the full consumption of the alkyne. In contrast, catalysts with a lower silver loading (<16.7%) provided some degree of over-hydrogenation. The authors attributed the high activity and selectivity displayed by these catalysts to the complementary relationship between Pd and Ag derived from the core–shell arrangement. On the one hand, the Pd core enhanced the activity of the Ag-shell by activating the hydrogen. On the other hand, the Ag shell served as the surface for the semi-hydrogenation of the alkyne and reduced the intrinsically low alkene-selectivity of Pd by inhibiting the contact between Pd and the alkenes.
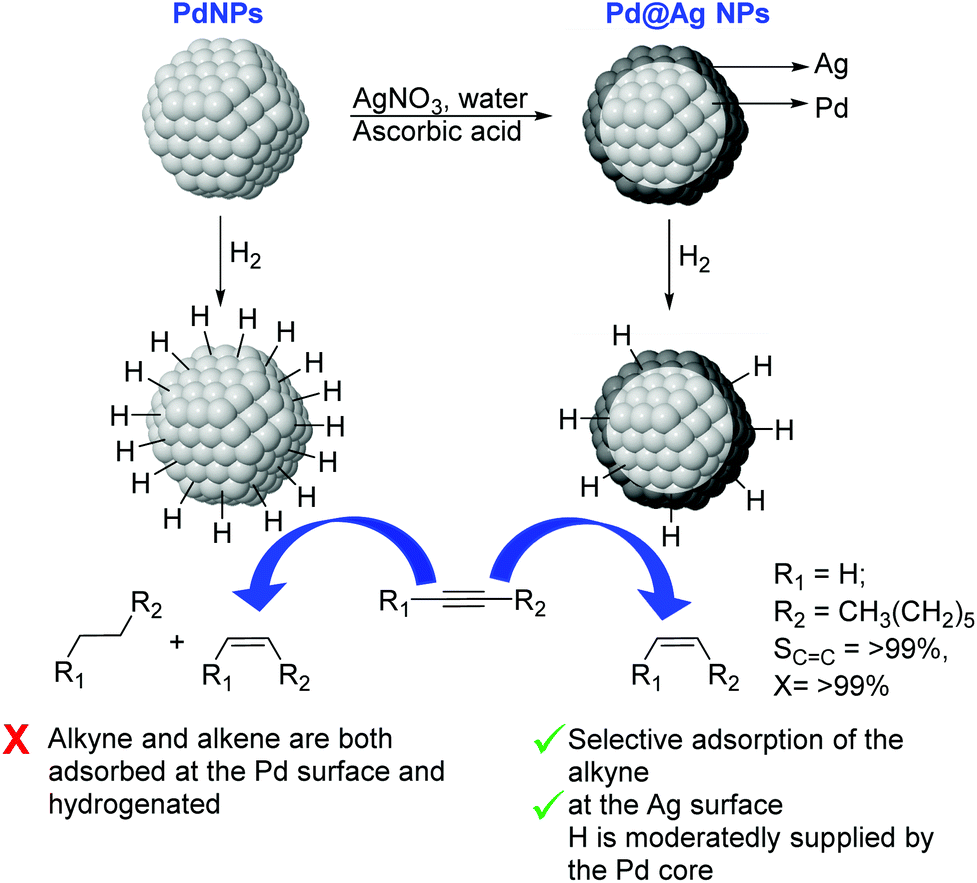 | ||
| Fig. 24 Synthesis of Pd@Ag core@shell NPs and selective hydrogenation of alkynes. | ||
The preparation of a series of core–shell PdAg NPs (Pd:Ag ratios of 40:60, 50:50, and 60:40) was achieved by co-reduction of Pd–Ag salts (using NaBH4 as the reducing agent) in the presence of poly(propylene imine) dendrimers covalently bound to the surface of a silica polyamine composite.111 In this approach, the poly(propylene imine) dendrimers play the role of stabilizers of the NPs. Based on XPS analysis, the authors proposed a core–shell structure of the PdAg NPs where silver dominates the outer shell. The evaluation of the catalytic performance of these composite materials in the selective hydrogenation of phenylacetylene, 1- and 4-octyne revealed moderate alkene selectivity even before full conversion of the alkyne (50–90%) and important isomerization activity. The relatively strong catalytic conditions employed in this study (80 °C and 10 bar H2) probably negatively affected the performance of these bimetallic catalysts in terms of selectivity. However, the immobilization approach employed permitted the successful recycling of the catalysts up to 7 times without loss of activity.
PdAu. One of the earliest studies dealing with the semi-hydrogenation of alkynols was reported by Bronstein and co-workers in 1999. In this report, the catalytic properties of palladium and bimetallic PdAu, PdPt, and PdZn nanoparticles formed in block polystyrene poly-4-vinylpyridine (PS-b-P4VP) micelles were studied in the hydrogenation of dehydrolinalool (DHL) (Fig. 25).103,112 High alkene selectivity (99.8% at 100% conversion) was achieved for all these micellar catalysts. The excellent selectivity was ascribed to the chemical modification of the nanoparticle surface with pyridine units of the block-copolymer. In addition, the second metal (Au, Pt, or Zn) acted as both a geometric and an electronic modifier of the Pd-NPs.
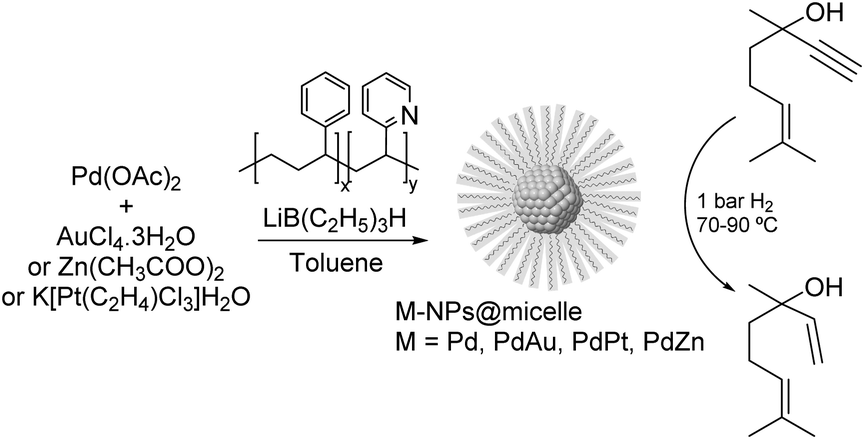 | ||
| Fig. 25 Synthesis of Pd, PdAu, PdPt and PdZn NPs and their application in the selective hydrogenation of dehydrolinalool (DHL). | ||
Very recently, the preparation of a supported PdAu/CNT catalyst containing PdAu NPs of 10 nm and a gold content of ca. 73 wt% was reported.113 The NPs were prepared by simultaneous reduction of Pd(NO3)2 and HAuCl4·3H2O using hydrogen bubbling over an ethanol solution. The bimetallic PdAu catalyst exhibited intermediate activity while the alkene selectivity was increased when compared to monometallic reference catalysts of Pd and Au.
Other PdX nanocatalysts. The preparation of palladium–rhodium–phosphorus alloy nanoparticles and their application in the selective hydrogenation of alkynes under mild conditions were reported.114 The monodispersed PdRhP nanoparticles were prepared via co-reduction of Pd(acac)2 and Rh(acac)3, by employing trioctylphosphine (TOP) as the stabilizer. According to ICP analysis, the atom composition of this amorphous alloy was Pd34Rh39P27. The resulting alloy was active in the semi-hydrogenation of 1-ethynyl-4-nitrobenzene and 1-bromo-4-ethynylbenzene under 1 bar H2 at 35 °C resulting in selectivity towards the alkene of 99% and 92% respectively at 100% conversion (Fig. 26). These excellent results were ascribed to the intrinsic nature of the amorphous alloy with a modified geometric/electronic structure affecting the adsorption of reactants, thus controlling the selectivity.
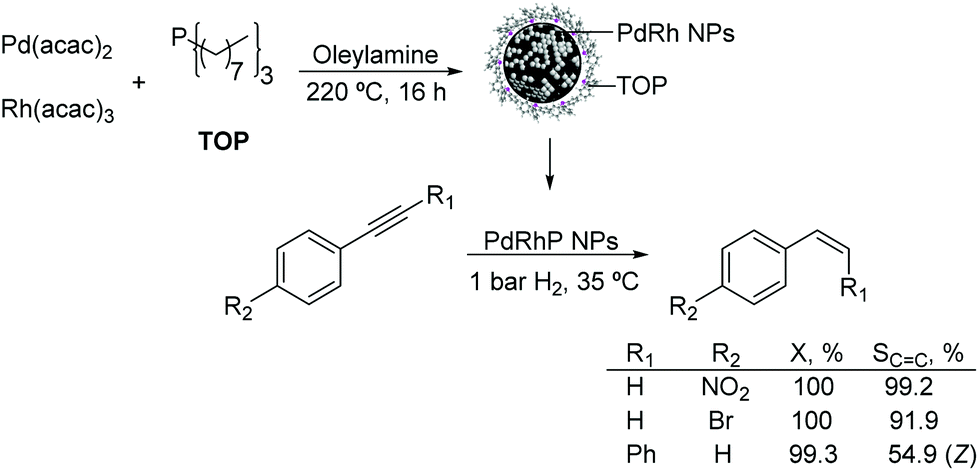 | ||
| Fig. 26 Synthesis of PdRhP amorphous alloy nanoparticles and their application in the selective hydrogenation of p-substituted phenylacetylenes. | ||
In 2014, Tsang and co-workers reported the interstitial modification of palladium nanoparticles with boron atoms.115 The treatment with borane tetrahydrofuran solutions of a previously prepared Pd/C catalyst produces the doping of palladium interstitial lattice sites with boron atoms with good chemical and thermal stability (Fig. 27). DFT calculations suggested that the adsorption properties of palladium are modified by the presence of subsurface boron atoms resulting in an improvement in the alkene selectivity in comparison with that of the pure Pd catalyst. In addition, the occupancy of the interstitial boron atoms can displace the subsurface hydrides usually present at the same sites in Pd and thus reduces over-hydrogenation.116
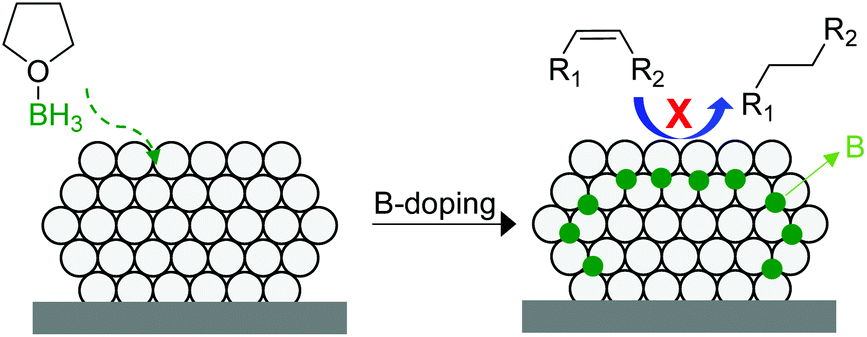 | ||
| Fig. 27 Promotion of a Pd/C catalyst by the doping of palladium interstitial lattice sites with boron atoms. | ||
Other M1–M2 alloys. As previously mentioned, De Vries and coworkers reported the use of FeNPs in the hydrogenation of 1-octyne.45,117 Interestingly, the authors observed the formation of n-octane from the beginning of the reaction, which suggested its formation via direct hydrogenation of 1-octyne without release and re-adsorption of 1-octene.117 With the aim to suppress the hypothetic direct pathway to octane, bimetallic nanoparticles of Fe:Ni (1:1), Fe:Co (1:1) and Fe:Cu (1:1), as well as monometallic Ni, Co and Cu NPs were prepared and tested in catalysis. Ni and CoNPs were more active and selective towards 1-octene than Fe (86 and 76% vs. 70% at full octyne conversion). In contrast, CuNPs were inactive under the tested conditions. In terms of alkene selectivity, the bimetallic Fe:Co and Fe:Ni NPs exhibited intermediate values compared with the corresponding monometallic components.
Although in the selective hydrogenation of alkynes, iron oxide has been mostly employed as a support to assist the catalyst separation, there is one example dealing with its use as the active phase of hydrogenation. Moores and co-workers reported the application of colloidal iron@iron oxide core@shell nanoparticles of ca. 50 nm (Fe@FeO NPs) in the hydrogenation of olefins and alkynes in aqueous media.118,119 The core@shell nanoparticles were synthesized in a water/methanol mixture via a colloidal method using NaBH4 as the reducing agent (Fig. 28) resulting in NPs of ca. 50 nm (core diameter of 44 nm and shell thickness of 6 nm). These NPs were tested in the hydrogenation of several substrates with selectivity towards the reduction of alkene and alkyne functions over carbonyl or aromatic groups. Additionally, they were observed to be stable and magnetically recoverable. The results indicated that the presence of an oxide shell is not an obstacle to activity and does provide protection against oxidation by oxygen and water. For the particular case of alkynes as substrates, the selectivity towards the alkene product in the hydrogenation of decyne was very low (6% at 88% conversion). The authors demonstrated that the Fe@FeO NPs could be magnetically recovered and recycled up to 10 times. Although these NPs did not exhibit remarkable results in terms of alkene selectivity, they constitute a clear example of the beneficial effect that a protective oxide shell can provide on the core of highly sensitive NPs, thus allowing application under non-inert conditions. Based on other iron based catalysts employed in selective hydrogenation of alkynes,45,46,64,117,120 only relevant alkene selectivities were obtained when promoting stabilizers (e.g. nitrogen containing compounds) or additives (e.g. acetonitrile) are used. It is likely that the Fe@FeO NPs in the presence of additives or a promoting stabilizer would display improved alkene selectivities. This report is remarkable because it deals with the simple and practical application of one of the cheapest and most non-toxic metals in hydrogenation reactions.
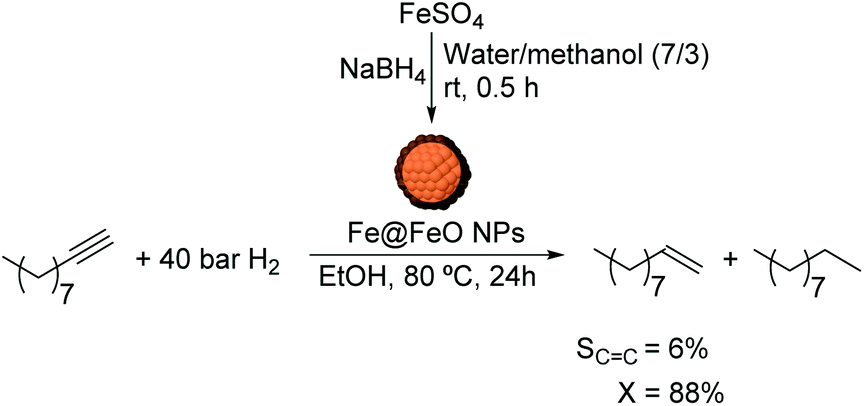 | ||
| Fig. 28 Synthesis of FeNPs and catalytic hydrogenation of 1-decyne. | ||
Acetylene as substrate. For acetylene hydrogenation, the most extended bimetallic approach consists in the doping of PdNPs with a second metal (Ag or Au generally). For Pd@Ag/Au core@shell structures, most reports describe the use of electroless deposition of Ag or Au on PdNPs.104,108,121,122 This method consists in the reduction of the second metal by employing reducing agents which are activated at the Pd surface (e.g. hydrazine), thus providing a controlled deposition of the second metal. In general, the degree of coverage of the second metal can be modulated by the stoichiometry of the Pd and Ag/Au solutions. For instance, Williams and co-workers reported the selective hydrogenation of acetylene in excess ethylene using Ag– and Au–Pd/SiO2 bimetallic catalysts prepared by electroless deposition.123 With this catalyst, the selectivity towards ethylene was enhanced at high Ag or Au coverages on Pd. Similar performance trends for Ag– and Au–Pd/SiO2 suggest that the bimetallic effect for these catalysts is geometric and not electronic in nature. When the same catalytic process was studied using Pd–Ag/SiO2 bimetallic catalysts prepared by galvanic displacement during which controlled loadings of Pd were added to the Ag/SiO2 pre-catalyst, the resulting bimetallic nanoparticles provided lower selectivity to ethene than in the previous case.26
Experimental and computational studies were reported on the effect of the coverage of the palladium phase with a second metal, in the semi-hydrogenation of acetylene. Zhang and co-workers reported that the optimal Pd@Ag and Pd@Au catalysts (in terms of TOF and selectivity) were those with a coverage of 0.92, obtained using a molar ratio of ca. 1:1 during the synthesis.123 In a more comprehensive report, Yang et al. studied the reaction mechanism and the adsorption energies of the structures involved in the acetylene hydrogenation at different Pd sites: 111, 100 and 211 (Fig. 20); as well as the effect of the decoration of a Pd (111) surface with Ag or Au.124 They concluded that Pd (211) is the most active site while Pd (111) is the most selective site for partial hydrogenation. For Pd (111) decorated with Cu, Ag and Au, the selectivity for the hydrogenation of acetylene to ethylene is enhanced. Adsorption energies of ethylene and activation barriers justify these results. Further increase in selectivity was also observed on alloying Pd (211) surfaces. In these cases, the blocking of the sites at the step edge resulted in significant increases in selectivity to ethylene.
There are a more limited number of studies dealing with the use of alloyed structures for the hydrogenation of acetylene. The preparation of a series of Pd/Ag alloy catalysts by simultaneous reduction of the metal precursors was reported using N2H4 as a reducing agent.125 These catalysts were tested in the semi-hydrogenation of acetylene, and the results showed that the catalyst prepared using a 1:1.5 Pd/Ag molar ratio was the most selective to ethylene. The study of Pd/Ag alloys has also been addressed from computational approaches. The addition of Ag to Pd was observed to increase the electron density of the Pd d-band as a result of charge transfer from Ag to Pd, weakening the adsorption of both ethylene and acetylene on the alloy surface. The electronic and ensemble effects decrease the acetylene and ethylene binding energy by 10–20 kJ mol−1, and 30–40 kJ mol−1, respectively. This should favor both thermodynamic and kinetic considerations as the weaker metal–adsorbate bond strength promotes both desorption and hydrogenation.126
NiBx – nickel borides and other metal borides. One of the first examples of application of unsupported metal colloids for the selective semi-hydrogenation of alkynes was reported using nickel based catalysts in the early 1960s.127,128 The preparation of nickel borides Ni2B was achieved by reduction of nickel acetate using sodium borohydride in water or ethanol. When water was the solvent, the obtained material was observed to be very active in the hydrogenation of olefins under mild conditions.127 In contrast, when ethanol was used, the resulting material (P2–Ni) was less active than P1–Ni but provided good Z-stereoselectivity in the partial hydrogenation of 3-hexyne in the presence of quinoline (98–99% purity of Z-3-hexene, Fig. 29).128 Interestingly, both nickel boride catalysts exhibited a low tendency to isomerize olefins during the hydrogenation reactions.
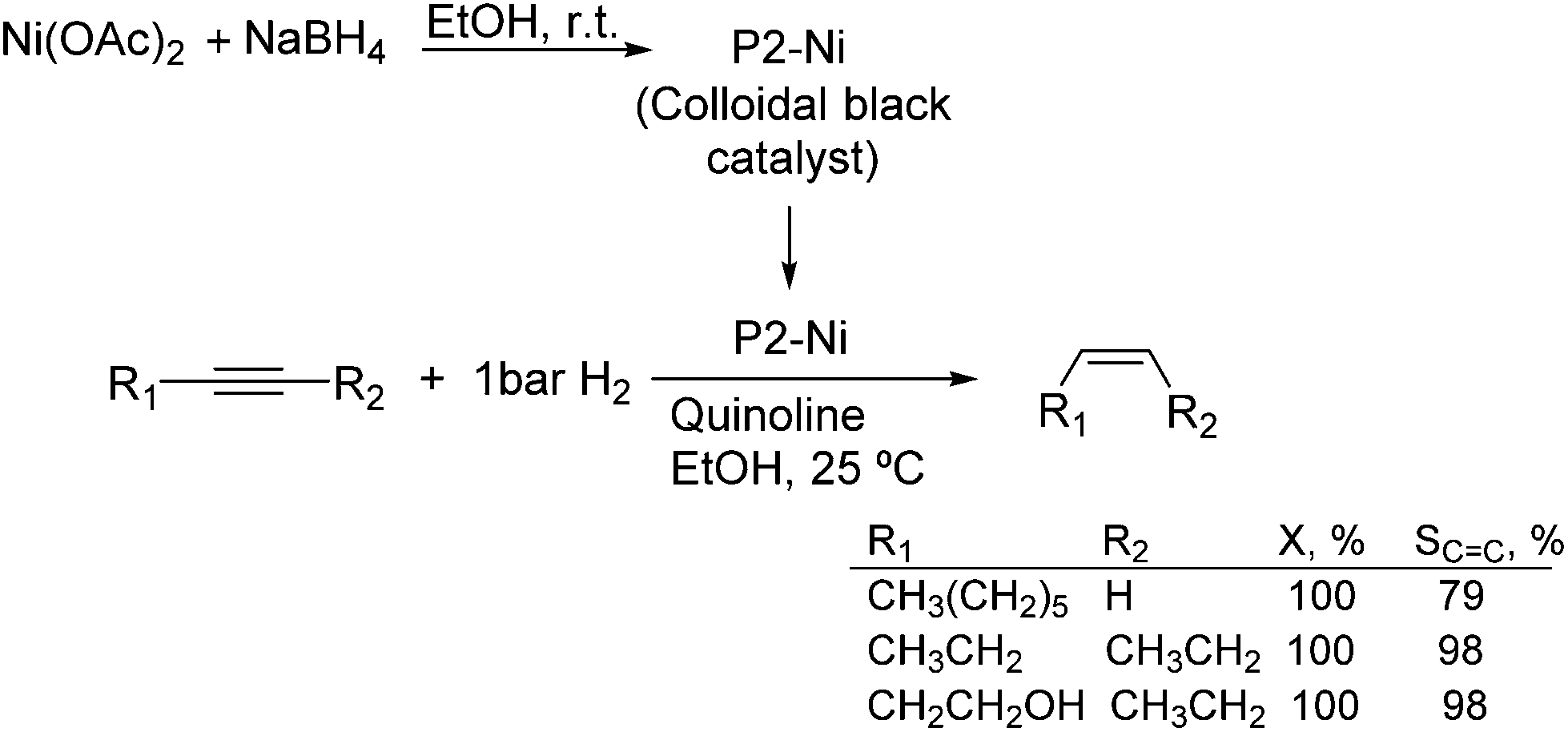 | ||
| Fig. 29 Synthesis of P2–Ni catalyst and catalytic hydrogenation of alkynes. | ||
An analogous method was later reported for the preparation of colloidal Pd, Pt and Rh catalysts.129 Interestingly, if the precipitation was carried out in the presence of high surface area supports such as activated carbon, the resulting catalysts exhibited a greater activity in the hydrogenation of olefins than the corresponding unsupported NPs, which was explained by the increase in the dispersion of the metal catalysts. The hydrogenations of 1-hexyne and 1-octene were examined separately using the carbon supported Pd, Pt and Rh and the P1 and P2–Ni catalysts.41 Interestingly, in all cases, the observed rate of hydrogen uptake was appreciably slower for the alkyne than for the alkene. In the hydrogenation of 1-hexyne, only the supported palladium Pd-BH4/C was highly selective to 1-hexene (97% after consumption of 1 eq. of hydrogen) while Pt-BH4/C and the P1/P2–Ni catalysts provided 75 and 85% of alkene selectivity, respectively.
Via the monitoring of the alkane/alkene ratio during the reaction, the authors proposed that P1–Ni and P2–Ni operate through a mechanism in which hexene and hexane are produced directly from hexyne. In contrast, the results indicated that the Pd catalyst Pd-BH4/C could operate via a combination of two mechanisms: the sequential reduction of the alkyne into the alkene and subsequently to the alkane and the direct reduction of the alkyne into the alkane.
Later, the sensitivity of the P2–Ni catalyst to the substrate substituents was investigated and it was concluded that the substitution favored selectivity since P2–Ni provided much higher selectivity for internal than for terminal alkynes.130 For instance, 3-hexyne was hydrogenated over P2–Ni to yield 3-hexene quantitatively with excellent cis-selectivity (cis:trans > 30:1); in contrast, in the hydrogenation of 1-hexyne, a 1:4:1 ratio of hexane:1-hexene:1-hexyne was measured. Interestingly, during the reaction, the hexane:hexene ratio remained constant while 1-hexyne was present, suggesting that both products were formed via a common chemisorbed intermediate (mechanistic selectivity). The large difference in selectivity between terminal and internal alkynes is explained in terms of the high sensitivity of the catalysts towards the steric hindrance induced in the resulting terminal and internal olefins. The same author also investigated the effect of amines as modifiers of the P2–Ni catalyst.131 The addition of ethylenediamine (2–3 eq. vs. Ni) improved further the stereoselectivity in the hydrogenation of internal alkynes and alkynols to produce the corresponding olefins with Z/E ratios as high as 200:1 at full conversion.
NixP – nickel phosphide. Corma and co-workers reported the chemoselective hydrogenation of terminal alkynes catalyzed by nickel phosphide nanoparticles, which were synthesized through the addition of P4 over NiNPs stabilized by trioctylphosphine in toluene followed by an annealing process at 220 °C.4,5 The Ni2P composite hydrogenated a series of terminal and internal alkenes with good conversion and selectivity. Fig. 30 displays the synthetic procedure of the Ni2P NPs and the catalytic results for the hydrogenation of a series of phenylacetylenes substituted in para positions. The conversion and the alkene selectivity were dependent on the nature of the substituents. For instance, halide substituted substrates exhibited high conversions and selectivities (Fig. 30) compared with those containing a NH2 group. In contrast, the lowest activity and selectivity were observed for the substrate provided with the most electron-withdrawing NO2 group (X = 3%, and SC
C = 43%).
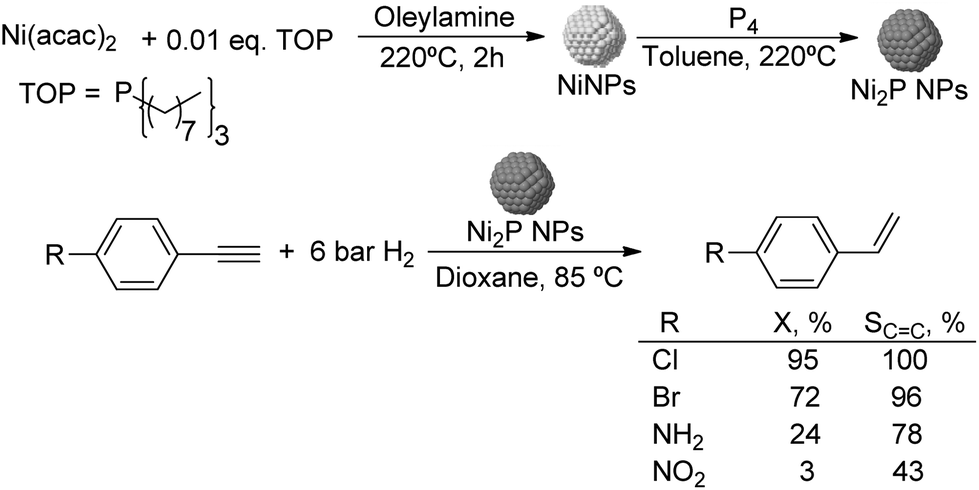 | ||
| Fig. 30 Synthesis of Ni2P NPs stabilized and catalytic hydrogenation of para substituted phenylacetylenes. | ||
In this study, the Ni/P ratio was observed to be critical for selectivity (Fig. 31). With a high Ni/P ratio of 3.5, phosphorus only blocked the most unsaturated sites and/or slightly withdrew electron density from the active Ni sites whereas with a lower Ni/P ratio of 2, the catalyst was significantly less active in alkene hydrogenation, thus reducing the over-hydrogenation and enhancing selectivity.
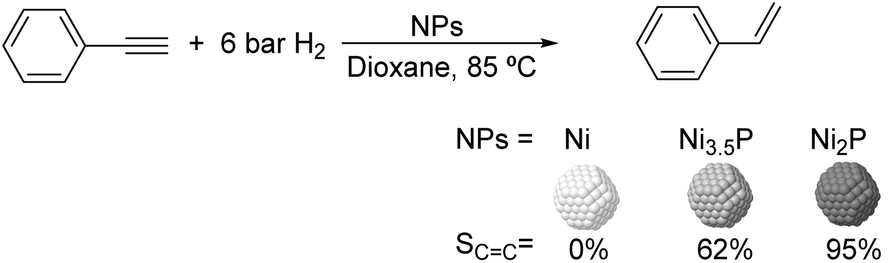 | ||
| Fig. 31 Hydrogenation of phenylacetylene using pure Ni, Ni3.5P and Ni2P nanoparticles as catalysts. Styrene selectivities at 100% conversion. | ||
Similarly, the preparation of monodisperse palladium phosphide nanoparticles possessing diameters of ca. 4.5 nm was reported using a colloidal methodology and these NPs were tested in the selective hydrogenation of alkynes.132 The PdP NPs were synthesised via the chemical reduction of Pd(acac)2 in the presence of triphenylphosphine and trioctylphosphine as phosphorous sources, borane tert-butylamine as the reducing agent and oleylamine as the stabilizer. Analysis of these NPs by XRD and ICP evidenced the alloyed structure of the phosphide with a composition of Pd75P25. These NPs were subsequently supported on activated carbon, annealed at 300 or 400 °C and tested in the selective hydrogenation of alkynes. Although these catalysts displayed a high activity under mild conditions (r.t. and 1 bar H2), the addition of quinoline was generally required to suppress over-hydrogenation.
RhNi. Very recently, the preparation of a bimetallic NP@MOF composite catalyst containing a core constituted of RhNi nanoflowers (NFs) encapsulated in a shell of a metal–organic framework (MOF) was reported.133 RhNi nanoflowers of variable composition (Rh1/3/9Ni1) were prepared by simultaneous reduction of Rh(acac)3 and NiCl2·6H2O salts using formaldehyde as the reducing agent. These NPs served as templates for the subsequent deposition of the MOF via a dealloying process using 2,5-dihydroxyterephthalic acid (DOT) as an organic linker. Both the nanoflowers and the composite materials were tested in the semi-hydrogenation of diphenylacetylene. Poor selectivities towards the cis-product (around 20% at ca. 70% of conversion) were obtained for the bimetallic RhNi nanoflowers, while the NPs encapsulated in the MOFs provided cis selectivity of 60%; however, in the latter case, the alkyne conversion decreased by a factor of 2.
2.5 Additives
Recently, the role of surface modification of Pt and CoPt3 nanoparticles was studied in the selective hydrogenation of 4-octyne.134,135 This work was performed using experimental and computational approaches and showed that the balance between the adsorption energetics of the additives and those of alkynes and alkenes at the surface of NPs define the activity and selectivity of the nanocatalyst (Fig. 32). On the one hand, if the additive presents higher adsorption energies than that of the alkyne substrate, the activity of the catalyst is reduced, which results in lower conversions. On the other hand, when the adsorption energy of the additive at the NP surface is higher than that of the alkenes, the selectivity can be improved. For instance, the addition of primary alkylamines to Pt and CoPt3NPs resulted in a drastic increase in the selectivity of the alkene from 0 to over 90% at 99.9% conversion, indicating that the coverage of the catalyst surface with the alkylamine prevents the hydrogenation of alkenes into alkanes. This study provided new insights into how surface modification affects the activity and/or selectivity of nanocatalysts and constitutes one of the few examples reported on selective alkyne hydrogenation catalyzed by highly active colloidal PtNPs.134,135
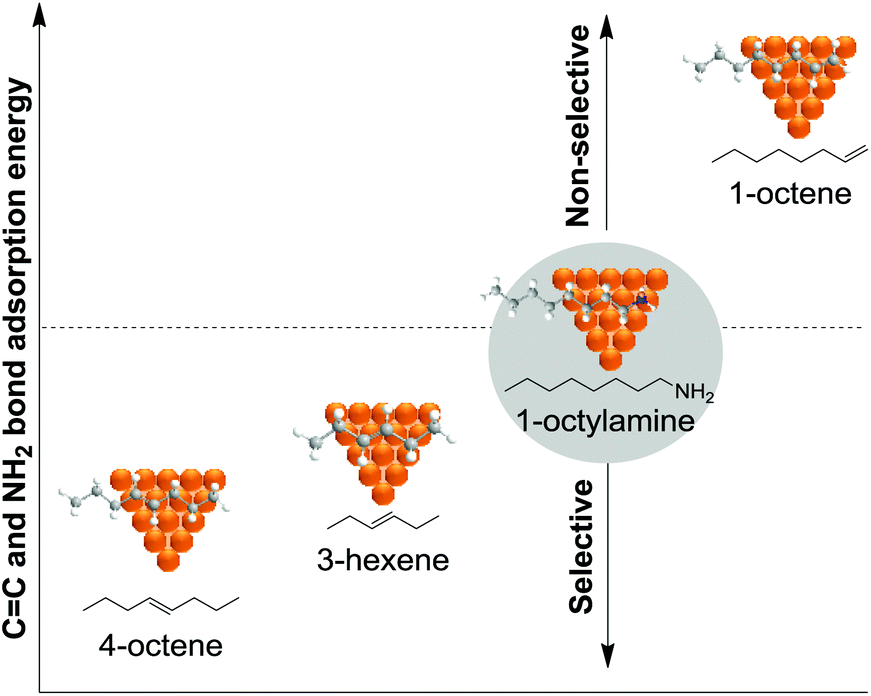 | ||
| Fig. 32 Correlation between CC and NH2 adsorption energy at the surface of a Pt cluster and the selectivity to the corresponding alkenes. | ||
Very recently, Rossi and co-workers reported the promoting effect of N-containing bases on the selective hydrogenation of alkynes catalyzed by AuNPs.86 The addition of stoichiometric amounts of the nitrogenated base into the reaction media resulted in a significant enhancement of activity while maintaining optimal alkene selectivities (e.g. >99% at full conversion of phenylacetylene). This promoting effect was shown to depend on the structure of the base, with piperazine being the one which exerted the most positive effect. Based on experimental data and DFT calculations, the authors proposed that the adsorption of nitrogen-containing bases on gold nanoparticles results in a frustrated Lewis pair interface that activates H2 heterolytically, providing high hydrogenation activity.
Another approach was reported using palladium nanoparticle-tetrabutylammonium borohydride as a catalytic system.7 The PdNPs were prepared by reduction of Pd(OAc)2 by t-ButOK (Fig. 33) in the presence of 4-octyne which, together with the corresponding oligomers formed during the synthesis, played the role of a stabilizer. In the hydrogenation of 1-pentyne and phenylacetylene, selectivities to 1-pentene and styrene of 98 and 94% were obtained at 100% conversion. For disubstituted alkynes, the addition of (n-C4H9)4NBH4 was shown to inhibit the isomerization and the over-reduction of the Z-alkene, which takes place in the presence of the PdNPs alone. (n-C4H9)4NBH4 was proposed to reversibly interact with the surface of the PdNPs thus avoiding the reaction of the Z-alkene.
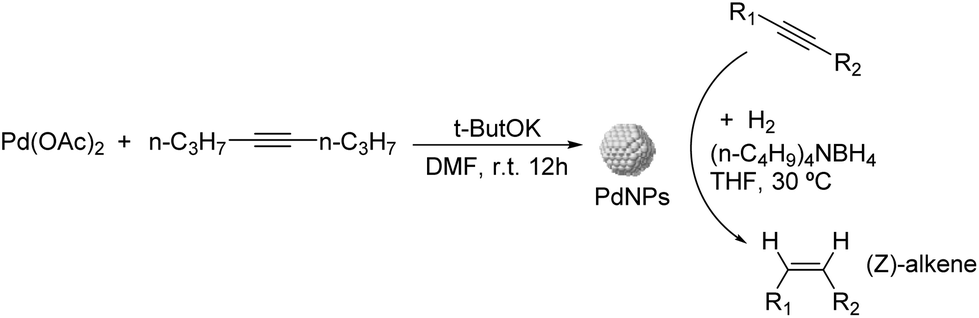 | ||
| Fig. 33 Synthesis of PdNPs stabilized by n-octyne (and its oligomers) applied in the selective hydrogenation of alkynes. | ||
Acetylene. For the selective hydrogenation of acetylene CO is the most common process modifier applied over Pd-based catalysts.79 Based on DTF calculations and experimental results, Bridier et al. reported that CO forms a dense blanket that covers the Pd surface, improves the thermodynamic factor, reduces the amount of available H at the surface and the formation of subsurface H species, and shrinks the size of active ensembles.38 However, CO also behaves as a poison and reduces the number of active sites on the surface, depending on both temperature and pressure.10 For the case of bimetallic preparations (e.g. PdM), an additional effect to be considered when using CO as a process modifier is the possibility to induce segregation of the promoting metal. According to DFT calculations, none of the investigated PdM alloys (M = Cu, Ag, Au, Zn, Ga, Sn, Pb and Bi) exhibited sufficient resistance towards segregation under high CO pressures.10
Other types of compounds such as amines, alkali and sulfur compounds, have also been employed as molecular modifiers.79 Recently, the grafting of nitrogen-containing modifiers (polyvinylpyridine or polyethylenimine) on the palladium based catalysts supported on CNF/SMF was shown to provide a significant increase in ethylene selectivity (up to 93%) where the catalyst shows remarkable stability during 120 h on-stream.76 This behavior was explained in terms of charge-transfer induced by the grafted ligands over the PdNPs.
3. Outlook and perspectives
Current nano-engineering offers a wide spectrum of possibilities for the preparation of highly functionalized nanomaterials with synergistic capabilities which could result in ultra-selective catalysts for the semi-hydrogenation of alkynes. Depending on the topological location of the variation in the active phase, two types of functionalization can be distinguished (Fig. 34): surface functionalization and structural modifications.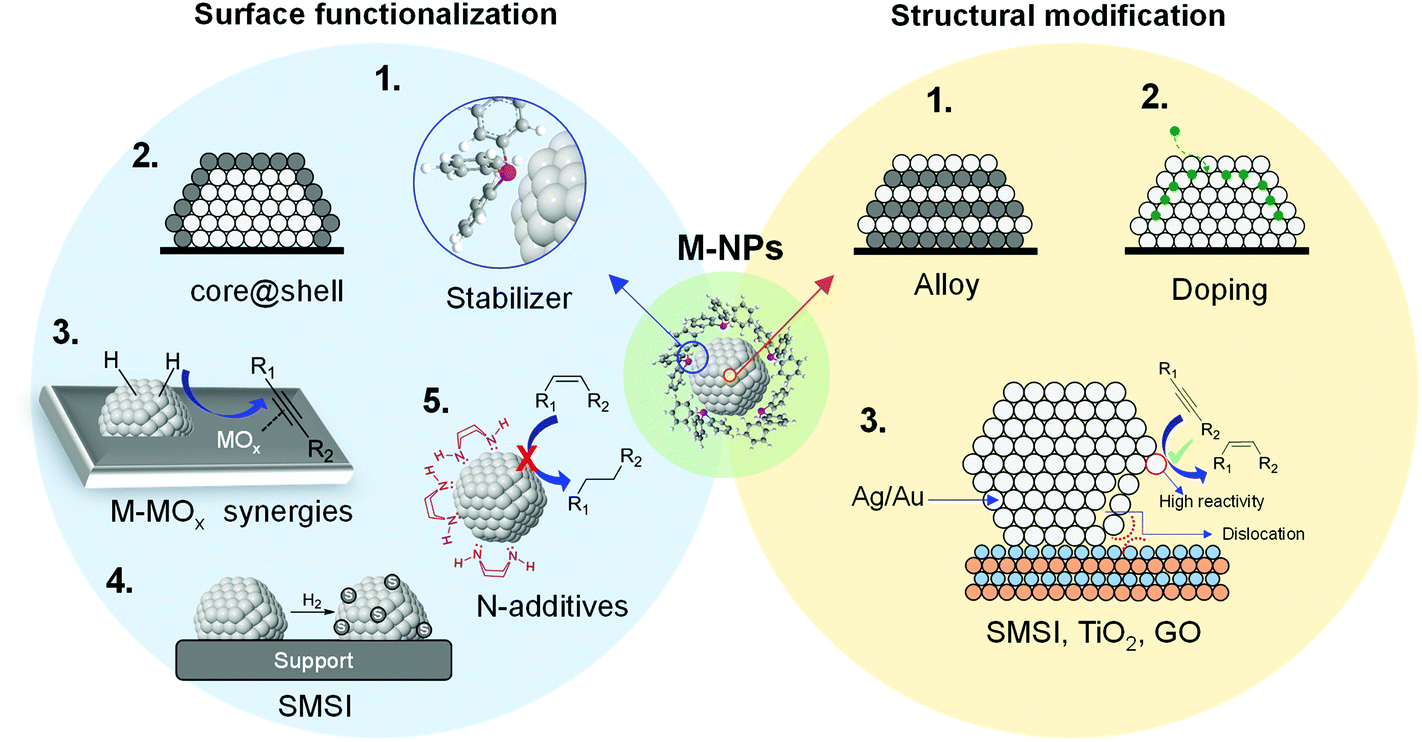 | ||
| Fig. 34 Surface and structural modifications of metal nanoparticles as strategies for the improvement of the catalytic performance in the semi-hydrogenation of alkynes. | ||
Surface functionalization constitutes a site blocking strategy that usually alters the accessibility or the adsorption of the alkene product by exerting geometric or electronic effects on the active phase. For instance, the use of multifunctional stabilizers such as aminopolymers that play the roles of both an NP stabilizer and a surface modifier, improves the alkene selectivity through the competitive adsorption of the amine and of the alkene but does not affect the alkyne adsorption. Similarly, the covering of PdNPs by nitrogen containing ILs favors the alkene selectivity and helps the catalyst recycling. It is noteworthy that in the semi-hydrogenation of acetylene, a positive effect on selectivity was also observed in the presence of stabilizers or additives although the activity is generally reduced. Functionalization of the catalyst surface via the utilization of a support that will provide interactions with the active phase is also of interest. For instance, the cooperation between Pd–Cu2O provided the selective semi-hydrogenation of terminal alkynes. The design of core@shell nanoparticles is also a powerful tool for surface functionalization. For instance, Au@CeO2 core@shell nanocatalysts display self-terminating properties in the semi-hydrogenation of both terminal and internal alkynes. For bimetallic systems, the Pd@Ag core@shell combines the high activity of palladium with the high selectivity of silver, resulting in very efficient catalysts and also exhibits self-terminating properties. In this area, the optimization of the size of Pd@Ag NPs (<20 nm), the preparation of efficient Pd@Au NPs or the use of other metal pairs such as Ni@Ag, or Ni@Au are challenges that are still to be tackled to further improve catalytic performance in this process.
Another promising approach to functionalize the catalyst surface is the use of bifunctional additives during catalysis. For instance, the addition of piperazine enhanced the activity and alkene selectivity more significantly than any other nitrogenated compounds to date in the gold catalyzed hydrogenation of alkynes. The formation of frustrated Lewis pairs at the surface of the NPs that is favored by the structure of piperazine, could explain the catalytic performance obtained with such an additive. Ethylene diamine has also been pointed out as an efficient additive.
Structural catalyst modification consists in the functionalization of the active phase in the sub-surface or the core of the metal NPs in order to alter the adsorption properties of the active phase towards the alkyne/alkene product. For instance, the adsorption properties of palladium can be modified by the presence of subsurface boron atoms that result in an improvement of the alkene selectivity. Alloyed systems remain one of the main strategies investigated in this field. Among the wide variety of bimetallic formulations, NiZn should be highlighted in view of the excellent performance/cost ratio in comparison with the more traditional options based on Pd (PdX, X = Ag, Au, Cu).
Other strategies based on metal-free catalytic systems,136 or the use of alternative reducing agents to hydrogen gas also provided promising results in the selective hydrogenation of alkynes. These approaches exhibit the advantage of a precise control of the alkyne/reducing agent stoichiometry which prevents over-hydrogenation and offers facile handling of the process since no special equipment (reactors or pressurized gases) is required. It is noteworthy that in the reported literature, some catalytic systems involve the in situ production of hydrogen gas,120,137 while others employ transfer hydrogen reagents (e.g. NaBH4, ammonium formate and ethanolamine).138–140
Acknowledgements
The authors are grateful to the Total Research & Technology Feluy, the Ministerio de Economia y Competividad and the Fondo Europeo de Desarrollo Regional FEDER (CTQ2016-75016-R, AEI/FEDER, UE) and the Generalitat de Catalunya (SGR2014) for financial support.References
- A. E. Marteel-Parrish and M. A. Abraham, in Green Chemistry and Engineering, John Wiley & Sons, Inc., 2013, pp. 1–19 Search PubMed.
- C. Descorme, P. Gallezot, C. Geantet and C. George, ChemCatChem, 2012, 4, 1897–1906 CrossRef CAS.
- J. R. H. Ross, in Heterogeneous Catalysis, ed. J. R. H. Ross, Elsevier, Amsterdam, 2012, pp. 1–15 Search PubMed.
- S. Carenco, A. Leyva-Pérez, P. Concepción, C. Boissière, N. Mézailles, C. Sanchez and A. Corma, Nano Today, 2012, 7, 21–28 CrossRef CAS.
- S. Carenco, X. F. Le Goff, J. Shi, L. Roiban, O. Ersen, C. Boissière, C. Sanchez and N. Mézailles, Chem. Mater., 2011, 23, 2270–2277 CrossRef CAS.
- K. L. Planken, B. W. M. Kuipers and A. P. Philipse, Anal. Chem., 2008, 80, 8871 CrossRef CAS PubMed.
- J. Hori, K. Murata, T. Sugai, H. Shinohara, R. Noyori, N. Arai, N. Kurono and T. Ohkuma, Adv. Synth. Catal., 2009, 351, 3143–3149 CrossRef CAS.
- A. Yarulin, I. Yuranov, F. Cárdenas-Lizana, P. Abdulkin and L. Kiwi-Minsker, J. Phys. Chem. C, 2013, 117, 13424–13434 CAS.
- G. Vilé, D. Baudouin, I. N. Remediakis, C. Copéret, N. López and J. Pérez-Ramírez, ChemCatChem, 2013, 5, 3750–3759 CrossRef.
- N. Lopez and C. Vargas-Fuentes, Chem. Commun., 2012, 48, 1379–1391 RSC.
- M. Crespo-Quesada, F. Cárdenas-Lizana, A.-L. Dessimoz and L. Kiwi-Minsker, ACS Catal., 2012, 2, 1773–1786 CrossRef CAS.
- T. W. Odom and M.-P. Pileni, Acc. Chem. Res., 2008, 41, 1565–1565 CrossRef CAS PubMed.
- S. K. Beaumont, Phys. Chem. Chem. Phys., 2014, 16, 5034–5043 RSC.
- P. T. Witte, S. Boland, F. Kirby, R. van Maanen, B. F. Bleeker, D. A. M. de Winter, J. A. Post, J. W. Geus and P. H. Berben, ChemCatChem, 2013, 5, 582–587 CrossRef CAS.
- P. Witte, P. Berben, S. Boland, E. Boymans, D. Vogt, J. Geus and J. Donkervoort, Top. Catal., 2012, 55, 505–511 CrossRef CAS.
- G. Vilé, N. Almora-Barrios, S. Mitchell, N. López and J. Pérez-Ramírez, Chem. – Eur. J, 2014, 20, 5926–5937 CrossRef PubMed.
- L. Börger and H. Cölfen, Prog. Colloid Polym. Sci., 1999, 113, 23 Search PubMed.
- Á. Molnár, A. Sárkány and M. Varga, J. Mol. Catal. A: Chem., 2001, 173, 185–221 CrossRef.
- G. C. Bond, Catalysis by Metals, Academic Press, New York, 1962 Search PubMed.
- H. Cölfen and A. Völkel, Eur. Biophys. J., 2003, 32, 432 CrossRef PubMed.
- T. Vossmeyer, L. Katsikas, M. Giersig, I. G. Popovic, K. Diesner, A. Chemseddine, A. Eychmüller and H. Weller, J. Phys. Chem., 1994, 98, 7665 CrossRef CAS.
- J. A. Jamison, K. M. Krueger, C. T. Yavuz, J. T. Mayo, D. LeCrone, J. J. Redden and V. L. Colvin, ACS Nano, 2008, 2/2, 311 CrossRef PubMed.
- D. Duca, F. Arena, A. Parmaliana and G. Deganello, Appl. Catal., A, 1998, 172, 207–216 CrossRef CAS.
- J. Osswald, R. Giedigkeit, R. E. Jentoft, M. Armbrüster, F. Girgsdies, K. Kovnir, T. Ressler, Y. Grin and R. Schlögl, J. Catal., 2008, 258, 210–218 CrossRef CAS.
- A. E. Yarulin, R. M. Crespo-Quesada, E. V. Egorova and L. L. Kiwi-Minsker, Kinet. Catal., 2012, 53, 253–261 CrossRef CAS.
- Y. Zhang, W. Diao, J. R. Monnier and C. T. Williams, Catal. Sci. Technol., 2015, 5, 4123–4132 CAS.
- H. Molero, B. F. Bartlett and W. T. Tysoe, J. Catal., 1999, 181, 49–56 CrossRef CAS.
- A. Sandell, A. Beutler, A. Jaworowski, M. Wiklund, K. Heister, R. Nyholm and J. N. Andersen, Surf. Sci., 1998, 415, 411–422 CrossRef CAS.
- I. Horiuti and M. Polanyi, Trans. Faraday Soc., 1934, 30, 1164–1172 RSC.
- W. T. McGown, C. Kemball and D. A. Whan, J. Catal., 1978, 51, 173–184 CrossRef CAS.
- S. Leviness, V. Nair, A. H. Weiss, Z. Schay and L. Guczi, J. Mol. Catal., 1984, 25, 131–140 CrossRef CAS.
- A. Borodziński and G. C. Bond, Catal. Rev., 2006, 48, 91–144 Search PubMed.
- T. Mitsudome, M. Yamamoto, Z. Maeno, T. Mizugaki, K. Jitsukawa and K. Kaneda, J. Am. Chem. Soc., 2015, 137, 13452–13455 CrossRef CAS PubMed.
- G. Vile and J. Perez-Ramirez, Nanoscale, 2014, 6, 13476–13482 RSC.
- A. Bruehwiler, N. Semagina, M. Grasemann, A. Renken, L. Kiwi-Minsker, A. Saaler, H. Lehmann, W. Bonrath and F. Roessler, Ind. Eng. Chem. Res., 2008, 47, 6862–6869 CrossRef CAS.
- Z. Wu, E. Calcio Gaudino, L. Rotolo, J. Medlock, W. Bonrath and G. Cravotto, Chem. Eng. Process.: Process. Intensif., 2016, 110, 220–224 CrossRef CAS.
- Z. Wu, G. Cravotto, E. C. Gaudino, A. Giacomino, J. Medlock and W. Bonrath, Ultrason. Sonochem., 2017, 35(Part B), 664–672 CrossRef CAS PubMed.
- M. García-Mota, B. Bridier, J. Pérez-Ramírez and N. López, J. Catal., 2010, 273, 92–102 CrossRef.
- F. Studt, F. Abild-Pedersen, T. Bligaard, R. Z. Sørensen, C. H. Christensen and J. K. Nørskov, Angew. Chem., Int. Ed., 2008, 47, 9299–9302 CrossRef CAS PubMed.
- M. Ruta, N. Semagina and L. Kiwi-Minsker, J. Phys. Chem. C, 2008, 112, 13635–13641 CAS.
- C. A. Brown, J. Chem. Soc. D, 1970, 139–140 RSC.
- L. E. J. Brus, Chem. Phys., 1984, 80, 4403 CAS.
- F. Delannay, Characterization of Heterogeneous Catalysts, 1984 Search PubMed.
- W. Bae and R. K. Mehra, J. Inorg. Biochem., 1998, 70, 125 CrossRef CAS.
- P.-H. Phua, L. Lefort, J. A. F. Boogers, M. Tristany and J. G. de Vries, Chem. Commun., 2009, 3747–3749 RSC.
- V. Kelsen, B. Wendt, S. Werkmeister, K. Junge, M. Beller and B. Chaudret, Chem. Commun., 2013, 49, 3416–3418 RSC.
- A. Roucoux, J. Schulz and H. Patin, Chem. Rev., 2002, 102, 3757–3778 CrossRef CAS PubMed.
- L. S. Ott and R. G. Finke, Coord. Chem. Rev., 2007, 251, 1075–1100 CrossRef CAS.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS PubMed.
- C. Evangelisti, N. Panziera, A. D'Alessio, L. Bertinetti, M. Botavina and G. Vitulli, J. Catal., 2010, 272, 246–252 CrossRef CAS.
- H. Hirai and N. Yakura, Polym. Adv. Technol., 2001, 12, 724–733 CrossRef CAS.
- M. M. Telkar, C. V. Rode, R. V. Chaudhari, S. S. Joshi and A. M. Nalawade, Appl. Catal., A, 2004, 273, 11–19 CrossRef CAS.
- D. Albani, G. Vile, S. Mitchell, P. T. Witte, N. Almora-Barrios, R. Verel, N. Lopez and J. Perez-Ramirez, Catal. Sci. Technol., 2016, 6, 1621–1631 CAS.
- F. Kirby, C. Moreno-Marrodan, Z. Baán, B. F. Bleeker, P. Barbaro, P. H. Berben and P. T. Witte, ChemCatChem, 2014, 6, 2904–2909 CrossRef CAS.
- Y. Li, E. Boone and M. A. El-Sayed, Langmuir, 2002, 18, 4921–4925 CrossRef CAS.
- M. Niu, Y. Wang, W. Li, J. Jiang and Z. Jin, Catal. Commun., 2013, 38, 77–81 CrossRef CAS.
- W. Long, N. A. Brunelli, S. A. Didas, E. W. Ping and C. W. Jones, ACS Catal., 2013, 3, 1700–1708 CrossRef CAS.
- N. Semagina, E. Joannet, S. Parra, E. Sulman, A. Renken and L. Kiwi-Minsker, Appl. Catal., A, 2005, 280, 141–147 CrossRef CAS.
- T. Mizugaki, M. Murata, S. Fukubayashi, T. Mitsudome, K. Jitsukawa and K. Kaneda, Chem. Commun., 2008, 241–243 RSC.
- H. Olivier-Bourbigou, L. Magna and D. Morvan, Appl. Catal., A, 2010, 373, 1–56 CrossRef CAS.
- R. Venkatesan, M. H. G. Prechtl, J. D. Scholten, R. P. Pezzi, G. Machado and J. Dupont, J. Mater. Chem., 2011, 21, 3030–3036 RSC.
- B. C. Leal, C. S. Consorti, G. Machado and J. Dupont, Catal. Sci. Technol., 2015, 5, 903–909 CAS.
- E. Ramirez, S. Jansat, K. Philippot, P. Lecante, M. Gomez, A. M. Masdeu-Bultó and B. Chaudret, J. Organomet. Chem., 2004, 689, 4601–4610 CrossRef CAS.
- T. N. Gieshoff, A. Welther, M. T. Kessler, M. H. G. Prechtl and A. Jacobi von Wangelin, Chem. Commun., 2014, 50, 2261–2264 RSC.
- L. Peng, J. Zhang, S. Yang, B. Han, X. Sang, C. Liu and G. Yang, Green Chem., 2015, 17, 4178–4182 RSC.
- K. Philippot and B. Chaudret, C. R. Chim., 2003, 6, 1019–1034 CrossRef CAS.
- B. Chaudret, C. R. Phys., 2005, 6, 117–131 CrossRef CAS.
- P. Caubère, Angew. Chem., Int. Ed. Engl., 1983, 22, 599–613 CrossRef.
- J. J. Brunet, L. Mordenti, B. Loubinoux and P. Caubere, Tetrahedron Lett., 1977, 18, 1069–1072 CrossRef.
- J. J. Brunet, P. Gallois and P. Caubere, Tetrahedron Lett., 1977, 18, 3955–3958 CrossRef.
- J. J. Brunet, P. Gallois and P. Caubere, J. Org. Chem., 1980, 45, 1937–1945 CrossRef CAS.
- P. Gallois, J. J. Brunet and P. Caubere, J. Org. Chem., 1980, 45, 1946–1950 CrossRef CAS.
- J. J. Brunet and P. Caubere, J. Org. Chem., 1984, 49, 4058–4060 CrossRef CAS.
- L. Montiel, J. A. Delgado, M. Novell, F. J. Andrade, C. Claver, P. Blondeau and C. Godard, ChemCatChem, 2016, 8, 3041–3044 CrossRef CAS.
- H. Zhang, Y. Yang, W. Dai, D. Yang, S. Lu and Y. Ji, Catal. Sci. Technol., 2012, 2, 1319–1323 CAS.
- D. Lamey, I. Prokopyeva, F. Cárdenas-Lizana and L. Kiwi-Minsker, Catal. Today, 2014, 235, 79–89 CrossRef CAS.
- M. Crespo-Quesada, J.-M. Andanson, A. Yarulin, B. Lim, Y. Xia and L. Kiwi-Minsker, Langmuir, 2011, 27, 7909–7916 CrossRef CAS PubMed.
- D. Uzio and G. Berhault, Catal. Rev., 2010, 52, 106–131 CAS.
- A. Borodziński and G. C. Bond, Catal. Rev., 2008, 50, 379–469 Search PubMed.
- A. Corma, P. Serna, P. Concepción and J. J. Calvino, J. Am. Chem. Soc., 2008, 130, 8748–8753 CrossRef CAS PubMed.
- C. Moreno-Castilla, M. A. Ferro-Garcia, J. P. Joly, I. Bautista-Toledo, F. Carrasco-Marin and J. Rivera-Utrilla, Langmuir, 1995, 11, 4386–4392 CrossRef CAS.
- N. Semagina, M. Grasemann, N. Xanthopoulos, A. Renken and L. Kiwi-Minsker, J. Catal., 2007, 251, 213–222 CrossRef CAS.
- R. Easterday, C. Leonard, O. Sanchez-Felix, Y. Losovyj, M. Pink, B. D. Stein, D. G. Morgan, N. A. Lyubimova, L. Z. Nikoshvili, E. M. Sulman, W. E. Mahmoud, A. A. Al-Ghamdi and L. M. Bronstein, ACS Appl. Mater. Interfaces, 2014, 6, 21652–21660 CAS.
- P. M. Uberman, N. J. S. Costa, K. Philippot, R. C. Carmona, A. A. Dos Santos and L. M. Rossi, Green Chem., 2014, 16, 4566–4574 RSC.
- S. Yang, C. Cao, L. Peng, J. Zhang, B. Han and W. Song, Chem. Commun., 2016, 52, 3627–3630 RSC.
- J. L. Fiorio, N. López and L. M. Rossi, ACS Catal., 2017, 7, 2973–2980 CrossRef CAS.
- L. Shao, X. Huang, D. Teschner and W. Zhang, ACS Catal., 2014, 4, 2369–2373 CrossRef CAS.
- M.-M. Wang, L. He, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, Green Chem., 2011, 13, 602–607 RSC.
- G. Vilé, P. Dähler, J. Vecchietti, M. Baltanás, S. Collins, M. Calatayud, A. Bonivardi and J. Pérez-Ramírez, J. Catal., 2015, 324, 69–78 CrossRef.
- Y. K. Gulyaeva, V. V. Kaichev, V. I. Zaikovskii, A. P. Suknev and B. S. Bal'zhinimaev, Appl. Catal., A, 2015, 506, 197–205 CrossRef CAS.
- G. C. Bond, Chem. Soc. Rev., 1991, 20, 441–475 RSC.
- R. Van Hardeveld and F. Hartog, Surf. Sci., 1969, 15, 189–230 CrossRef CAS.
- N. Semagina, A. Renken and L. Kiwi-Minsker, J. Phys. Chem. C, 2007, 111, 13933–13937 CAS.
- N. Semagina, A. Renken, D. Laub and L. Kiwi-Minsker, J. Catal., 2007, 246, 308–314 CrossRef CAS.
- J. Hu, Z. Zhou, R. Zhang, L. Li and Z. Cheng, J. Mol. Catal. A: Chem., 2014, 381, 61–69 CrossRef CAS.
- D. Teschner, E. Vass, M. Hävecker, S. Zafeiratos, P. Schnörch, H. Sauer, A. Knop-Gericke, R. Schlögl, M. Chamam, A. Wootsch, A. S. Canning, J. J. Gamman, S. D. Jackson, J. McGregor and L. F. Gladden, J. Catal., 2006, 242, 26–37 CrossRef CAS.
- M. Crespo-Quesada, A. Yarulin, M. Jin, Y. Xia and L. Kiwi-Minsker, J. Am. Chem. Soc., 2011, 133, 12787–12794 CrossRef CAS PubMed.
- N. Semagina and L. Kiwi-Minsker, Catal. Lett., 2009, 127, 334–338 CrossRef CAS.
- F. Studt, F. Abild-Pedersen, T. Bligaard, R. Z. Sørensen, C. H. Christensen and J. K. Nørskov, Science, 2008, 320, 1320–1322 CrossRef CAS PubMed.
- D. Mei, M. Neurock and C. M. Smith, J. Catal., 2009, 268, 181–195 CrossRef CAS.
- M. García-Mota, J. Gómez-Díaz, G. Novell-Leruth, C. Vargas-Fuentes, L. Bellarosa, B. Bridier, J. Pérez-Ramírez and N. López, Theor. Chem. Acc., 2011, 128, 663–673 CrossRef.
- T. Naoki, Y. Hu and S. Yiukihide, in Metal Nanoclusters in Catalysis and Materials Science, Elsevier, Amsterdam, 2008, pp. 49–75 Search PubMed.
- L. M. Bronstein, D. M. Chernyshov, I. O. Volkov, M. G. Ezernitskaya, P. M. Valetsky, V. G. Matveeva and E. M. Sulman, J. Catal., 2000, 196, 302–314 CrossRef CAS.
- T. Rieker, A. Hanprasopwattana, A. Datye and P. Hubbard, Langmuir, 1999, 15, 638 CrossRef CAS.
- E. V. Shevchenko, D. V. Talapin, H. Schnablegger, A. Kornowski, O. Festin, P. Svedlindh, M. Haase and H. Weller, J. Am. Chem. Soc., 2003, 125, 9090 CrossRef CAS PubMed.
- N. A. Khan, S. Shaikhutdinov and H.-J. Freund, Catal. Lett., 2006, 108, 159–164 CrossRef CAS.
- B. Marinkovic, R. R. de Avillez, A. Saavedra and F. C. R. Assuncao, Mater. Res., 2001, 4, 71 CrossRef CAS.
- A. Yarulin, I. Yuranov, F. Cárdenas-Lizana, D. T. L. Alexander and L. Kiwi-Minsker, Appl. Catal., A, 2014, 478, 186–193 CrossRef CAS.
- C. F. Calver, P. Dash and R. W. J. Scott, ChemCatChem, 2011, 3, 695–697 CrossRef CAS.
- T. Mitsudome, T. Urayama, K. Yamazaki, Y. Maehara, J. Yamasaki, K. Gohara, Z. Maeno, T. Mizugaki, K. Jitsukawa and K. Kaneda, ACS Catal., 2016, 6, 666–670 CrossRef CAS.
- E. A. Karakhanov, A. L. Maximov, A. V. Zolotukhina, N. Yatmanova and E. Rosenberg, Appl. Organomet. Chem., 2015, 29, 777–784 CrossRef CAS.
- E. Sulman, V. Matveeva, A. Usanov, Y. Kosivtsov, G. Demidenko, L. Bronstein, D. Chernyshov and P. Valetsky, J. Mol. Catal. A: Chem., 1999, 146, 265–269 CrossRef CAS.
- S. Wang, Z. Xin, X. Huang, W. Yu, S. Niu and L. Shao, Phys. Chem. Chem. Phys., 2017, 19, 6164–6168 RSC.
- M. Ren, C. Li, J. Chen, M. Wei and S. Shi, Catal. Sci. Technol., 2014, 4, 1920–1924 CAS.
- C. W. A. Chan, A. H. Mahadi, M. M.-J. Li, E. C. Corbos, C. Tang, G. Jones, W. C. H. Kuo, J. Cookson, C. M. Brown, P. T. Bishop and S. C. E. Tsang, Nat. Commun., 2014, 5, 5787 CrossRef CAS PubMed.
- R. Burch and F. A. Lewis, Trans. Faraday Soc., 1970, 66, 727–735 RSC.
- C. Rangheard, C. de Julian Fernandez, P.-H. Phua, J. Hoorn, L. Lefort and J. G. de Vries, Dalton Trans., 2010, 39, 8464–8471 RSC.
- R. Hudson, A. Riviere, C. M. Cirtiu, K. L. Luska and A. Moores, Chem. Commun., 2012, 48, 3360–3362 RSC.
- C. M. Cirtiu, T. Raychoudhury, S. Ghoshal and A. Moores, Colloids Surf., A, 2011, 390, 95–104 CrossRef CAS.
- B. Huang, T. Wang, Z. Yang, W. Qian, J. Long, G. Zeng and C. Lei, ACS Sustainable Chem. Eng., 2017, 5, 1668–1674 CrossRef CAS.
- P. Scherrer, Nachr. Ges. Wiss. Göttingen, 1918, 2, 98 Search PubMed.
- B. D. Hall, D. Zanchet and D. Ugarte, J. Appl. Crystallogr., 2000, 33, 1335 CrossRef CAS.
- Y. Zhang, W. Diao, C. T. Williams and J. R. Monnier, Appl. Catal., A, 2014, 469, 419–426 CrossRef CAS.
- B. Yang, R. Burch, C. Hardacre, G. Headdock and P. Hu, J. Catal., 2013, 305, 264–276 CrossRef CAS.
- J. I. Langford and A. J. C. Wilson, J. Appl. Crystallogr., 1978, 11, 102 CrossRef CAS.
- A. A. Guzelian, J. E. B. Katari, A. V. Kadavanich, U. Banin, K. Hamd, E. Juban and A. P. Alivisatos, J. Phys. Chem., 1996, 100, 7212 CrossRef CAS.
- C. A. Brown and H. C. Brown, J. Am. Chem. Soc., 1963, 85, 1003–1005 CrossRef CAS.
- H. C. Brown and C. A. Brown, J. Am. Chem. Soc., 1963, 85, 1005–1006 CrossRef CAS.
- H. C. Brown and C. A. Brown, Tetrahedron, 1966, 22(Suppl. 8), 149–164 CrossRef.
- C. A. Brown and V. K. Ahuja, J. Org. Chem., 1973, 38, 2226–2230 CrossRef CAS.
- C. A. Brown and V. K. Ahuja, J. Chem. Soc., Chem. Commun., 1973, 553–554 RSC.
- M. Zhao, Chem. – Asian J., 2016, 11, 461–464 CrossRef CAS PubMed.
- L. Chen, H. Li, W. Zhan, Z. Cao, J. Chen, Q. Jiang, Y. Jiang, Z. Xie, Q. Kuang and L. Zheng, ACS Appl. Mater. Interfaces, 2016, 8, 31059–31066 CAS.
- S. G. Kwon, G. Krylova, A. Sumer, M. M. Schwartz, E. E. Bunel, C. L. Marshall, S. Chattopadhyay, B. Lee, J. Jellinek and E. V. Shevchenko, Nano Lett., 2012, 12, 5382–5388 CrossRef CAS PubMed.
- E. V. Shevchenko, D. V. Talapin, A. L. Rogach, A. Kornowski, M. Haase and H. Weller, J. Am. Chem. Soc., 2002, 124, 11480–11485 CrossRef CAS PubMed.
- Y. Liu, L. Hu, H. Chen and H. Du, Chem. – Eur. J., 2015, 21, 3495–3501 CrossRef CAS PubMed.
- F. Alonso, I. Osante and M. Yus, Tetrahedron, 2007, 63, 93–102 CrossRef CAS.
- S. Liang, G. B. Hammond and B. Xu, Chem. Commun., 2016, 52, 6013–6016 RSC.
- E. D. Slack, C. M. Gabriel and B. H. Lipshutz, Angew. Chem., Int. Ed., 2014, 53, 14051–14054 CrossRef CAS PubMed.
- J.-J. Zhong, Q. Liu, C.-J. Wu, Q.-Y. Meng, X.-W. Gao, Z.-J. Li, B. Chen, C.-H. Tung and L.-Z. Wu, Chem. Commun., 2016, 52, 1800–1803 RSC.
| This journal is © The Royal Society of Chemistry 2017 |