
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Functional metallosupramolecular architectures using 1,2,3-triazole ligands: it's as easy as 1,2,3 “click”
Roan A. S.
Vasdev
,
Dan
Preston
and
James D.
Crowley
 *
*
Department of Chemistry, University of Otago, PO Box 56, Dunedin 9054, Otago, New Zealand. E-mail: jcrowley@chemistry.otago.ac.nz
First published on 19th January 2017
Abstract
Self-assembled metallosupramolecular architectures have become an increasingly popular area of inorganic chemistry. These systems show a range interesting biological, electronic and photophysical properties. Additionally, they display extensive host–guest chemistry that could potentially be exploited for drug delivery and catalysis. To fully realise these types of applications the ability to generate more functionalised metallosupramolecular architectures is required. In this perspective review we examine the exploitation of 1,2,3-triazole ligands, generated using the Cu(I)-catalysed 1,3-cycloaddition of organic azides with terminal alkynes (the CuAAC “click” reaction), for the assembly of discrete functional metallosupramolecular architectures. These “click” ligands have been used to generate metallomacrocycles, cages and helicates. Some of the architectures have shown promise as anti-cancer and anti-bacterial agents while others have been exploited for small molecule activation and catalysis.
 Roan A. S. Vasdev (left), Dan Preston (right) and James D. Crowley (centre) | Roan completed his undergraduate degree majoring in chemistry at the University of Otago. He is currently completing his MSc thesis in the Crowley group. His research interests are self-assembly and supramolecular host–guest interactions. Dan completed his undergraduate degree and honours degree in chemistry at the University of Otago. He is currently working in the Crowley group, writing his doctoral thesis. His research interests are self-assembly and supramolecular host–guest interactions. James obtained his BSc (Hons) (1998) and MSc (2000) from Victoria University of Wellington and completed his PhD (2000–2005) at the University of Chicago under the direction of Prof. Brice Bosnich. In 2005 he moved to Prof. David Leigh's group at the University of Edinburgh, where he was awarded a British Ramsay Memorial Trust Fellowship (2006–2008), to carry out research on molecular machines. He started his independent career at the University of Otago, Department of Chemistry in 2008 and has since moved through the ranks to Associate Professor (2015). His major research interests are in catalysis, self-assembly, molecular recognition and the development of molecular machines. |
Introduction
The past three decades have seen rapid development in the field of metallosupramolecular1 chemistry.2 By exploiting suitably labile metal ions and appropriately designed polydentate ligands a wide range of well-defined descrete multimetallic coordination architectures such as helicates, triangles, squares, rectangles, trigonal prisms, tetrahedra, and other polyhedral cages can be generated (Fig. 1). With such a diverse array of structures accessible, efforts have moved away from simply making new architectures to generating functional systems and exploiting their properties. Metallosupramolecular architectures with interesting biological,3 electronic4 and photophysical5 properties have been developed. Additionally, they have also been extensively used as host molecules for the molecular recognition of a vast array of organic and inorganic guest molecules. This host–guest behaviour of metallosupramolecular systems has been exploited to generate nanoscale reaction flasks and catalysts.2 For the most part these successes have been achieved with polydentate ligands based on classical coordination motifs such as catecholates,6 β-diketonato7 and pyridine (mono- (A), bi- (B) and ter- (C)) units (Fig. 2).8 While these ligand sets have enabled the generation of the wide range of architectures discussed above they are often devoid of other functionality. As efforts become more focussed on the properties and applications of metallosupramolecular systems9 there is a requirement for new methods and/or ligands that allow the ready incorporation of additional functionally into the architectures. The Cu(I)-catalysed 1,3-cycloaddition of organic azides with terminal alkynes (the CuAAC “click” reaction)10 has great potential in this regard due to its reliability, mild reaction conditions and wide substrate scope (Fig. 2). In addition to being an exceedingly useful building block and linking unit in organic chemistry, 1,4-disubstitued-1,2,3-triazoles have been exploited as readily functionalised ligands in coordination chemistry.11 In particular, certain 1,2,3-triazole based ligands (D–F) can be viewed as readily synthesised and functionlised analogues of the very common pyridine (A), bipyridine (B) and terpyridine (C) ligands (Fig. 2b and c).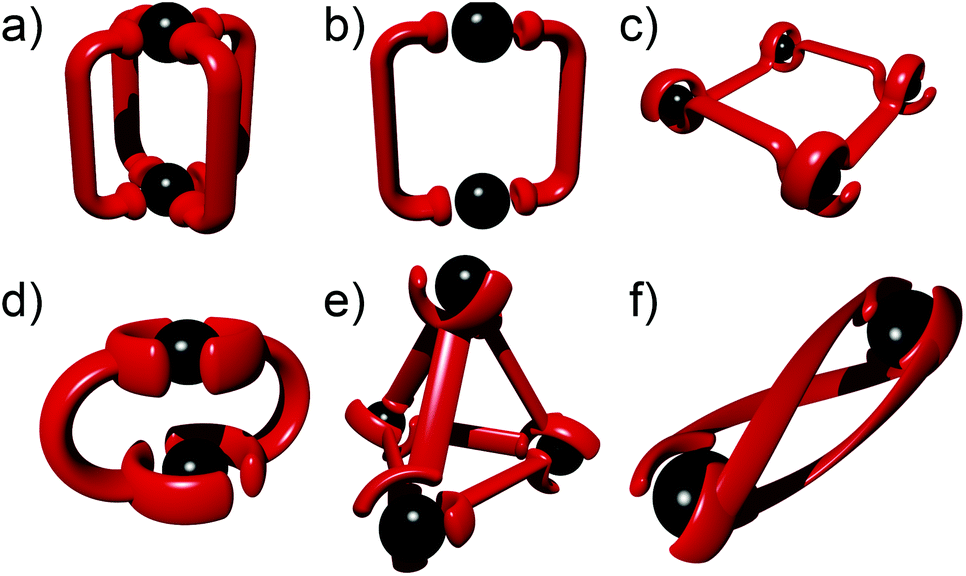 | ||
| Fig. 1 Examples of metallosupramolecular architectures formed from “click” ligands: (a) M2L4 bis-monodentate cage; (b) M2L2 bis-monodentate macrocycle; (c) M4L4 bis-bidentate grid; (d) M2L2 bis-bidentate macrocycle; (e) M4L6 bis-bidentate tetrahedral cage; (f) M2L3 bis-bidentate helicate. | ||
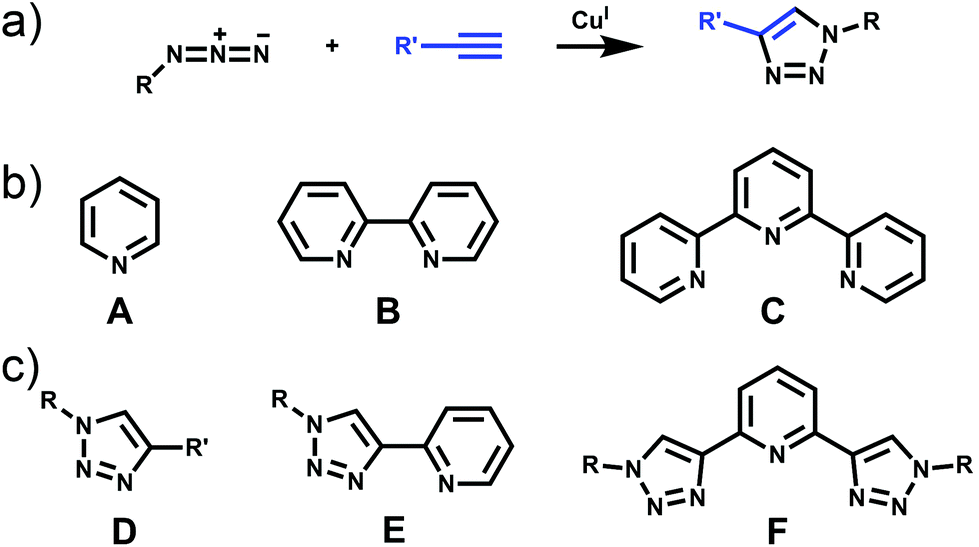 | ||
| Fig. 2 (a) The copper(I) catalysed ‘click’ reaction between an azide and alkyne generating 1,4-disubstitued-1,2,3-triazoles; (b) pyridyl-based chelating units (A–C) and (c) related 1,2,3-triazoles ligands (D–F). | ||
This perspective review will examine the exploitation of 1,2,3-triazole containing “click” ligands for the generation of functional discrete metallosupramolecular architectures. Related “click” ligands have also been used to generate a range of coordination polymers.12 However, as these are not discrete metallosupramolecular architectures they will not be discussed further here. Similary, metallosupramolecular systems that contain uncoordinated “click” derived 1,2,3-triazole units will not be covered.13
Bis-monodentate ligands
Di- and tri-pyridyl ligands have been extensively used for the construction of metallosupramolecular architectures.2 While 1,4-disubstituted-1,2,3-triazoles can potentially display three different N-donor coordination modes (Fig. 3a–c), monodentate coordination through the more electron rich N3 nitrogen atom is most commonly observed (Fig. 3a).11a–d Thus 1,4-disubstituted-1,2,3-triazoles can be viewed as readily functionalised pyridyl surrogates. With this in mind several groups have attempted to use bis-(1,4-disubstituted-1,2,3-triazole) ligands for the formation of a range of metallosupramolecular architectures.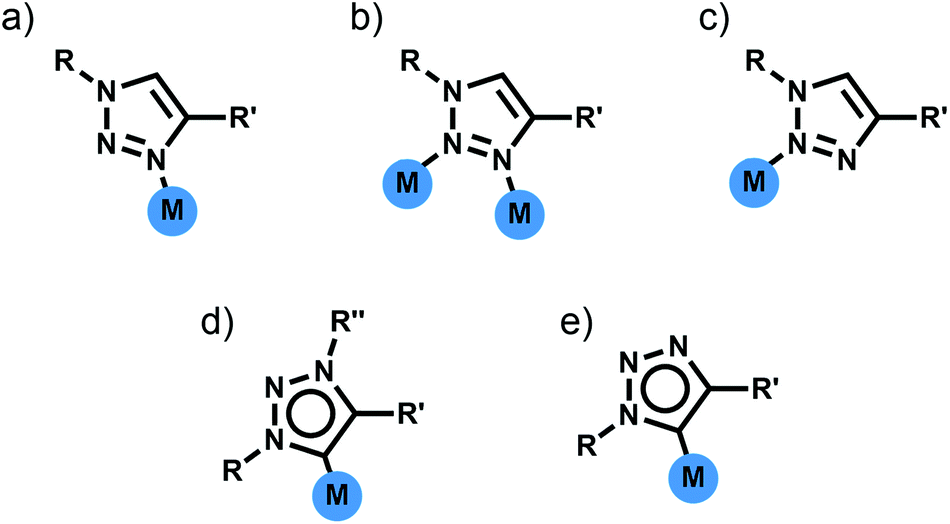 | ||
| Fig. 3 Coordination modes of 1,4-disubstituted-1,2,3-triazole ligands (a) monodentate coordination through the N3 nitrogen atom, (b) bridging through the N3 and N2 nitrogen atoms, (c) monodentate coordination through the N2 nitrogen atom, (d) monodentate coordination through the C5 carbon atom (mesoionic carbene) and (e) monodentate coordination through the C5 carbon atom (carbanionic). | ||
Metallocycles
[2 + 2] Metallocycles have been formed from the combination of bis-triazole ligands with Ag(I) or Zn(II) ions. Two types of isomeric bis-triazole ligands have been examined, these were generated from either a bis-alkyne (giving triazole rings C-bound to the linker Fig. 4a) or a bis-azide (giving N-bound triazole rings, Fig. 4b).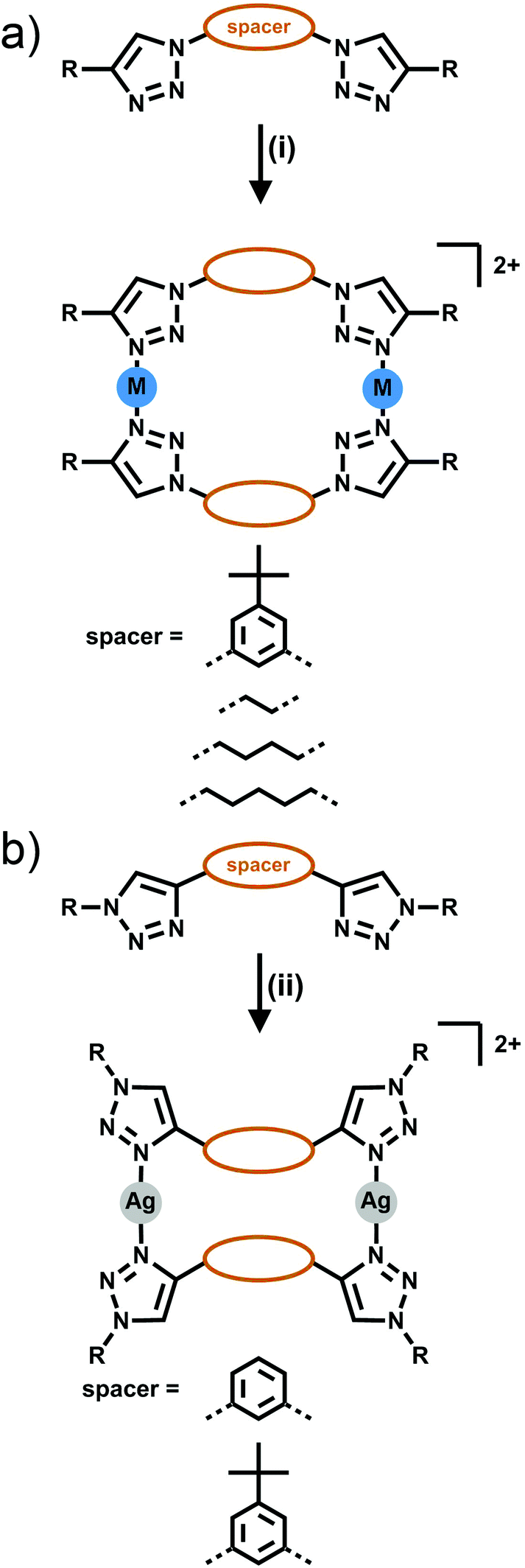 | ||
| Fig. 4 Formation of [2 + 2] bis-monodentate macrocyles either from (a) the dialkyne (C-bound) or (b) diazide (N-bound) linker. Conditions: (i) Ag(CF3SO3), acetone; (ii) AgSbF6, acetone. | ||
In early work Gower and Crowley reported the synthesis of two [2 + 2] Ag(I) metallocycles using 1,3-phenyl-linked bis-triazoles substituted with either benzyl or pentafluorobenzyl groups.141H and DOSY nuclear magnetic resonance (NMR) spectroscopy and electrospray ionisation mass spectrometry (ESI-MS) data were consistent with the quantitative formation of [2 + 2] Ag(I) metallocycles in solution and the pentafluorobenzyl substituted system was successfully crystallised as the anticipated metallo-macrocycle (Fig. 5a). Coordination was through the N3 nitrogen atoms of the triazole units, with a 158° N–Ag–N angle. The oxygen of a diethyl ether or acetone solvent molecule coordinates as well, in a position roughly bisecting the N–Ag–N angle.
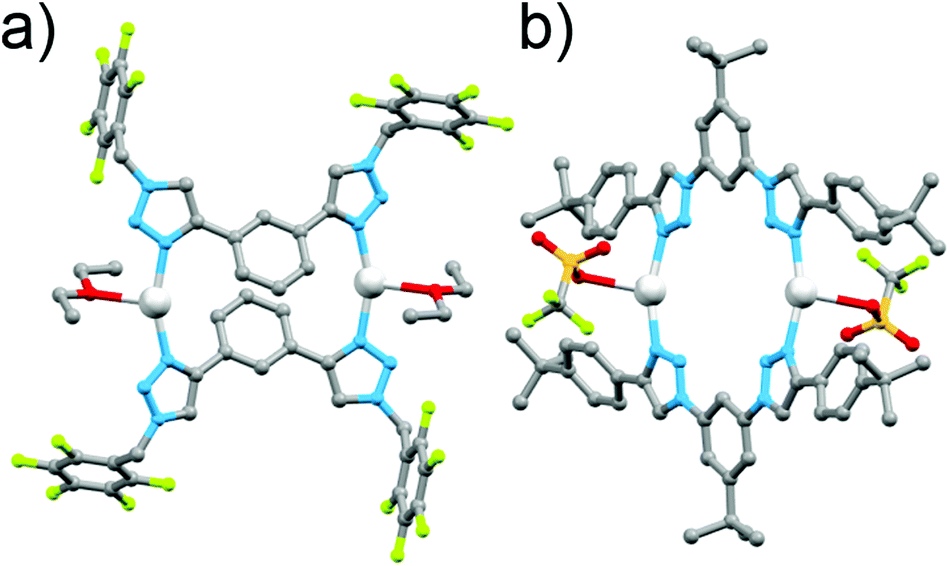 | ||
| Fig. 5 Molecular structures of the [2 + 2] Ag(I) bis-triazole macrocycles: (a) Crowley and Gower's complex and (b) White and Beer's complex. | ||
In a similar vein, White and Beer have reported four bis-monodentate ligands with a 1,3-(5-tert-butyl)phenyl core.15 The triazole units are either N1 or C4 bound, and feature an alkyl or aryl substituent. The C4-connected ligands gave cluster-like compounds consisting of four Ag(I) cations, and four ligands, with three triazole nitrogen bonds to each silver(I) centre, and the fourth coordination bond at each metal ion to either water or a bridging PF2O2 anion, while with Cu(I) a similar [Cu3L4]3+ cluster was formed. The N1-bound triazole ligands gave [2 + 2] macrocycles with Ag(I), coordinating through the N3 nitrogen, with a coordination environment similar to that reported by Crowley and Gower, with a N–Ag–N angle of 158°, and a triflate anion binding through an oxygen atom in a roughly perpendicular position to the N–Ag–N angle (Fig. 5b). 1H NMR studies of these systems were consistent with the presence of the architectures in solution.
Hor and coworkers have also reported a [2 + 2] macrocycle using Zn(II) ions. The zinc(II) ions adopt a tetrahedral coordination geometry and are coordinated to two N3 triazole nitrogen atoms and two chloride anions.16 The ligand possessed bis-(4-(2-pyridylthiomethyl)triazolyl) groups with a pentyl linker: the ethyl- and butyl-linked ligands instead formed dinuclear [(ZnCl2)2L] species with each metal ion bound to both a triazole and chelating pyridine. The authors have only studied the solid state behaviour of these macrocycles thus it is not clear if the macrocyclic structure is maintained in solution.
Metallocycles with carbene ligands
In addition to acting as N-donor ligands 1,4-disubstitued-1,2,3-triazoles can also display monodentate C-donor coordination modes (Fig. 3d and e). In particular there has been considerable recent interest on the use of N3 alkylated 1,3,4-trisubstitued-1,2,3-triazolium salts for the generation of mesoionic 1,3,4-trisubstituted-1,2,3-triazol-5-ylidene ligands.17Bis-1,2,3-triazol-5-ylidene carbene ligands in conjunction with linear Ag(I) or Au(I) ions have been used to assemble a range of metallo-macrocycles. Crudden and coworkers have accessed a [2 + 2] Ag(I) macrocycle of this sort from a bis-1,3(1-aryl-1,2,3-triazol-4-yl)benzene ligand while en route to a bis-rhodium(I) complex (Fig. 6a).18 Frutos and de la Torre have generated an equilibrium mixture of the mononuclear [AgL]+ and [2 + 2] macrocyclic [Ag2L2]+ complexes when using a 1,4-xylyl linked steroid substituted bis-1,2,3-triazol-5-ylidene ligand. Interestingly, the analogous 1,3-xylyl linked system cleanly forms the mononuclear [AgL]+ complex.19 As part of efforts to develop Au(I) 1,2,3-triazol-5-ylidene catalysts, Crowley and coworkers, synthesised a di-Au(I) [2 + 2] macrocycle with a 1,3-phenyl linked bis-1,2,3-triazol-5-ylidene ligand.20 The system was characterised using 1H and DOSY NMR spectroscopy and ESI-MS. Bis-methylated triazolium salts have also been used in the formation of larger metallo-macrocycles; Schubert and coworkers have employed a 2,6-pyridyl linked ligand with dimesityl substituents to form a [Ag4L4]4+ complex. The macrocycles was characterised in solution using 1H and DOSY NMR spectroscopy and ESI-MS and density functional theory (DFT) calculations suggest adopts a square-type structure with the Ag(I) ions in linear coordination geometry,21 while a [Ag8L4]8+ structure based on a tetratopic ligand has also been crystallographically identified by Sessler and coworkers (Fig. 6b).22
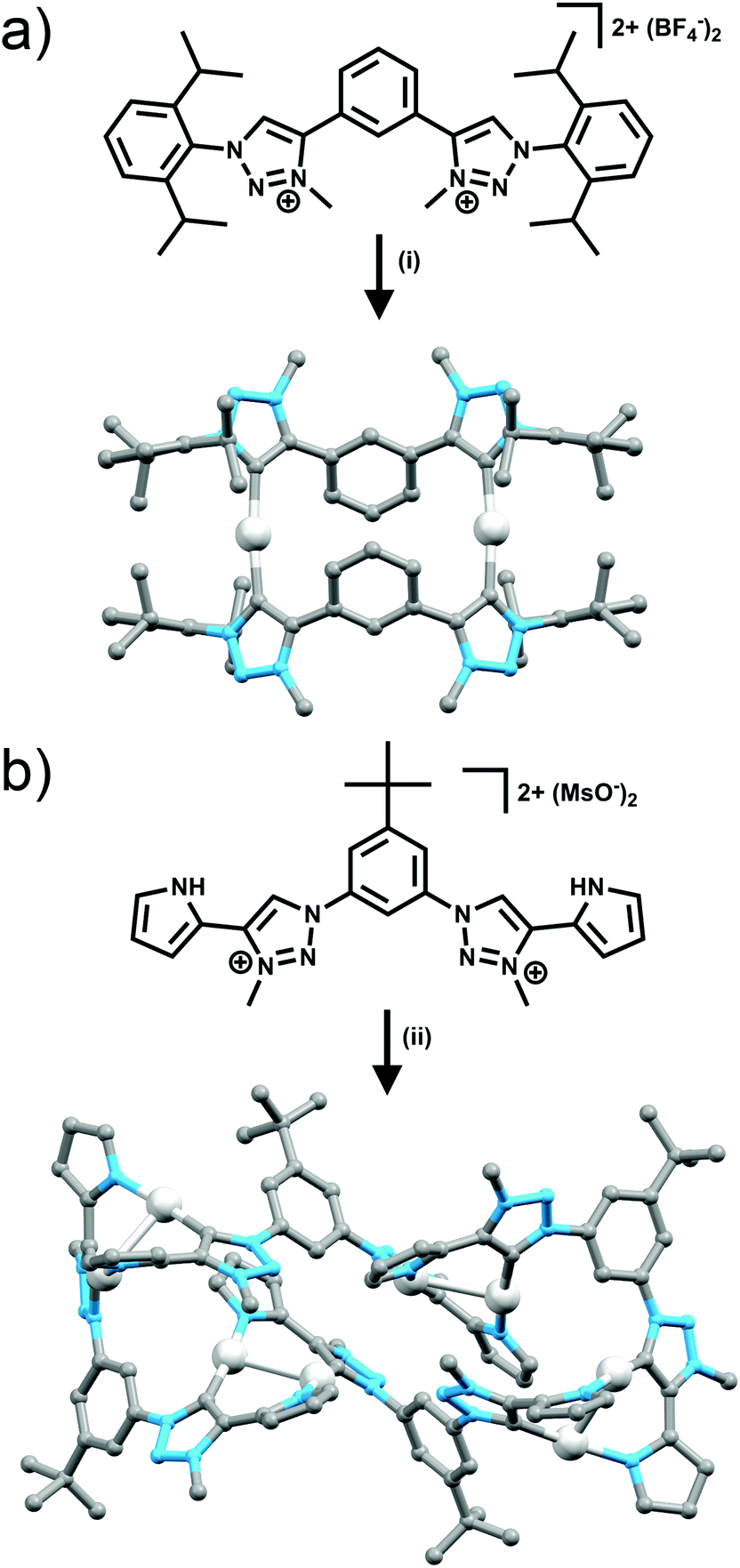 | ||
| Fig. 6 (a) Bis-monodentate and (b) bis-bidentate mesoionic 1,2,3-triazol-5-ylidene ligands with crystal structures obtained upon complexation with Ag(I). Hydrogen atoms and counterions have been omitted for clarity. Conditions: (i) Ag2O, KBr, MeCN; (ii) Ag2O, TBACl, DCM/MeCN. | ||
Helicates
Building on their previous Ag(I) metallocycle work Crowley and co-workers exploited the functional group tolerance of the CuAAC “click” reaction to synthesise a structurally diverse family of bis-triazole ligands. Reaction of a suitable dialkyne with two equivalents of azide, gave a range of bis-monodentate ligands with the two triazole groups separated by a linker (either propyl, 1,3-phenyl or 1,4-phenyl units), and substituted with a wide array of benzyl, aryl and alkyl groups (Fig. 7a).23 When these ligands were combined with square planar palladium(II) ions, in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ligand to metal ratio, dinuclear quadruply-stranded helicates were formed with the 1,3-phenyl and propyl linked bis-triazoles, while the 1,4-phenyl linked bis-triazole ligand gave polymeric mixtures rather than a discrete product. Formation of most of these [Pd2L4]4+ assemblies was facile, occurring instantaneously at room temperature, but with the hexyl substituted ligand the reaction required heating to avoid kinetic polymeric intermediates en route to the formation of the desired [Pd2L4]4+ assembly (Fig. 7). The systems were characterised using a combination of 1H and DOSY NMR spectroscopy and ESI-MS. The X-ray crystal structures of two of the 1,3-phenyl linked helicates were also obtained (Fig. 7c). The Crowley group subsequently examined the biological activity and stability of the some of the quadruply-stranded helicates.24 Competition experiments against biological nucleophiles found that despite very similar donor strengths between benzyl-, (2-(2-methoxyethoxy)ethoxy)-, and hexyl-substituted bis-triazole ligands, the hexyl-substituted assembly (Fig. 7b) was completely inert to nucleophilic decomposition whilst the other helicates were rapidly broken down. It was postulated that the hydrophobic hexyl chains clustered over the palladium(II) centres sterically protecting the metal ions and preventing ligand exchange via associative interchange leading to the enhanced stability in the presence of nucleophiles. The robust hexyl-substituted helicate was also the only compound in the series to show any appreciable cytotoxicity. The hexyl-substituted system was found to be cytotoxic against a range of human cell lines, possessing low micromolar IC50 values (3–8 μM) against all of the cell lines examined. Unfortunately, the compound showed no selectivity for cancer cells. Preliminary mechanistic investigations with the helicate revealed that it induced cell death within minutes, accompanied by a loss of cellular membrane integrity.
:1 ligand to metal ratio, dinuclear quadruply-stranded helicates were formed with the 1,3-phenyl and propyl linked bis-triazoles, while the 1,4-phenyl linked bis-triazole ligand gave polymeric mixtures rather than a discrete product. Formation of most of these [Pd2L4]4+ assemblies was facile, occurring instantaneously at room temperature, but with the hexyl substituted ligand the reaction required heating to avoid kinetic polymeric intermediates en route to the formation of the desired [Pd2L4]4+ assembly (Fig. 7). The systems were characterised using a combination of 1H and DOSY NMR spectroscopy and ESI-MS. The X-ray crystal structures of two of the 1,3-phenyl linked helicates were also obtained (Fig. 7c). The Crowley group subsequently examined the biological activity and stability of the some of the quadruply-stranded helicates.24 Competition experiments against biological nucleophiles found that despite very similar donor strengths between benzyl-, (2-(2-methoxyethoxy)ethoxy)-, and hexyl-substituted bis-triazole ligands, the hexyl-substituted assembly (Fig. 7b) was completely inert to nucleophilic decomposition whilst the other helicates were rapidly broken down. It was postulated that the hydrophobic hexyl chains clustered over the palladium(II) centres sterically protecting the metal ions and preventing ligand exchange via associative interchange leading to the enhanced stability in the presence of nucleophiles. The robust hexyl-substituted helicate was also the only compound in the series to show any appreciable cytotoxicity. The hexyl-substituted system was found to be cytotoxic against a range of human cell lines, possessing low micromolar IC50 values (3–8 μM) against all of the cell lines examined. Unfortunately, the compound showed no selectivity for cancer cells. Preliminary mechanistic investigations with the helicate revealed that it induced cell death within minutes, accompanied by a loss of cellular membrane integrity.
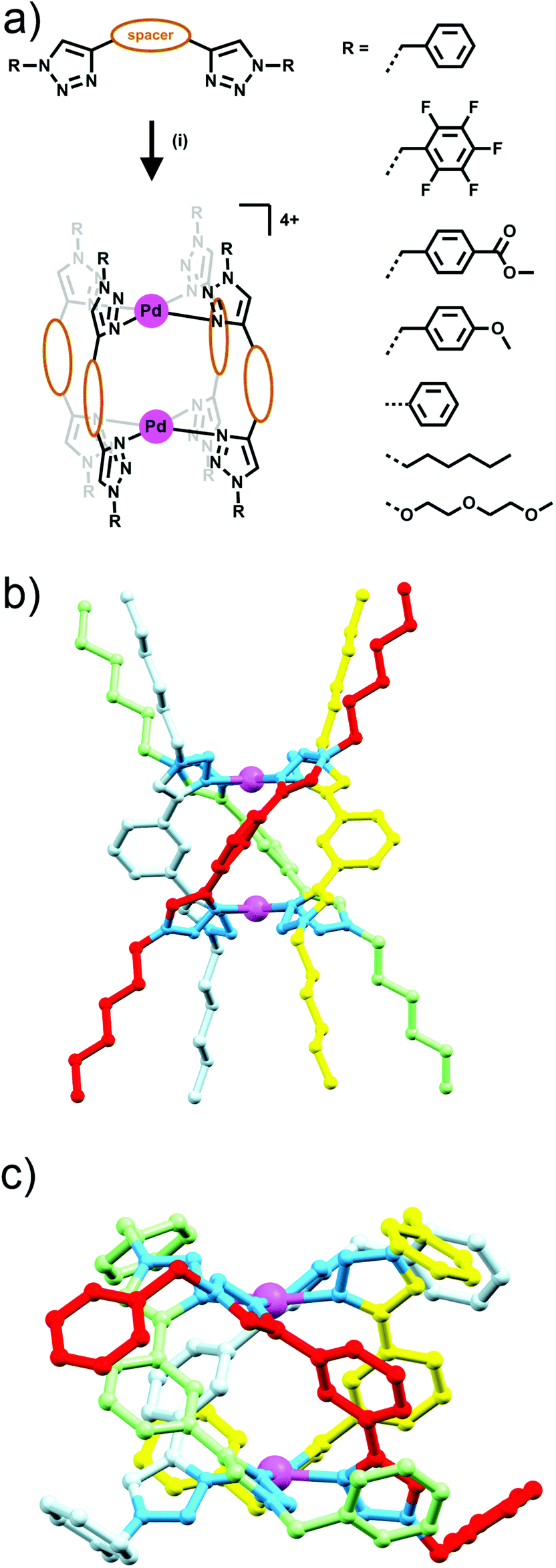 | ||
| Fig. 7 (a) Generic scheme showing the formation of [Pd2L4]4+ helicates from bis-monodentate 1,2,3-triazole ligands, Linker = 1,3-phenyl, 1,4-phenyl or propyl, (i) [Pd(CH3CN)4](BF4)2, CH3CN, RT or 70 °C; (b) MMFF model of the hexyl-substituted [Pd2L4]4+ helicate which exhibited biological activity; (c) X-ray structure of the benzyl-substituted [Pd2L4](BF4)4 helicate. Counterions, solvent molecules and hydrogen atoms have been omitted for clarity. | ||
Bis-bidentate ligands
Bis-bidentate ligand systems based on bipyridine and pyridiylimine ligand motifs have been used to generate a variety of triple-stranded helicates and tetrahedral cages.2 Therefore bis-(2-pyridyl-1,2,3-triazole) ligands should potentially provide access to similar systems. However, an issue with the bis-(2-pyridyl-1,2,3-triazole) ligands is that their synthesis requires the use of potentially explosive diazide intermediates. Bandeen and Crowley exploited the in situ azide formation conditions developed by Fokin and co-workers to safely synthesise a small family of these the bis-(2-pyridyl-1,2,3-triazole) ligands (Fig. 8a). The required diazide are generated in situ from sodium azide and the appropriate dihalide (or ditosylate) then reacted immediately, without isolation, with 2-ethynylpyridine and a copper(I) catalyst to give the bis-(2-pyridyl-1,2,3-triazole) ligands in good to excellent isolated yields (81–94%). | ||
| Fig. 8 (a) Scheme of formation of bis-bidentate pyridyl-triazole ligands, (i) NaN3; (ii) Cu(I); (b) molecular structure of the [2 + 2] Ag(I) metallo-macrocycle generated by Bandeen and Crowley and (c) the molecular structure of the [PtL2]2+ metalloligand (solubilising substituents omitted for clarity) and the heterometallic macrocycles synthesised from it. | ||
Metallocycles
Bandeen and Crowley then used these bis-(2-pyridyl-1,2,3-triazole) ligands to form a series of [2 + 2] metallo-macrocycles, with Ag(I) ions. 1H NMR spectra and ESI-MS data were consistent with the formation the [2 + 2] metallo-macrocycles in solution. Additionally, the disilver complex of the bis-(2-pyridyl-1,2,3-triazole)-1,2-xylene ligand was characterised in the soild state using X-ray crystallography (Fig. 8b). The ligands were coordinated to the Ag(I) cations in the relatively rare square planar geometry with the 2-pyridyl-1,2,3-triazole units adopting a head-to-tail arrangement (Fig. 8b).Subsequently, larger heterometallic macrocycles have been formed, from methylene-bridged bis-(2-pyridyl-1,2,3-triazole) ligands. These systems were synthesised from sodium azide, dichloromethane and the appropriate 2-ethynylpyridine using in situ azide formation conditions. A 2:1 combination of these ligands with platinum(II) ions gave [Pt(L)2]2+ metallo-ligands with vacant peripheral binding sites (Fig. 8c).25 Similar to the previous di-Ag(I) metallo-macrocycle, the Pt(II) complex displayed the expected square planar coordination geometry with the 2-pyridyl-1,2,3-triazole units adopting a head-to-tail arrangement (Fig. 8b). The platinum(II) metallo-ligands could be then combined with other metal ions to generate heterometallic macrocycles. The self-assembly process was studied using 1H and DOSY NMR spectroscopy and ESI-MS. Addition of palladium(II) ions to the platinum(II) metallo-ligands led to the formation of a concentration-dependent equilibrium mixture of tetrameric and hexameric macrocycles. At high concetrations (27 mM) a 5:2 equilibrium mixture of a [Pd3Pt3(L)6]12+ [6 + 6] hexagon and heterometallic [Pd2Pt2(L)4]8+ [4 + 4] square was observed, and at lower concentrations (1.5 mM) essentially only the [4 + 4] square could be detected. The combination of the metallo-ligand with Cu(I) ions resulted in the quantitative, concentration-independent, formation of the [Cu2Pt2(L)4]6+ tetramer (Fig. 8c).
The bis-(2-pyridyl-1,2,3-triazole)-1,2-xylene ligand (Fig. 9a) has been employed by Wang and coworkers, in combination with copper(I) iodide (CuI), to form a tetracopper(I) macrocycle, with each pair of copper(I) ions bridged by two iodides (Fig. 9a).26 The tetracopper(I) macrocycle was only characterised in the solid state and unlike related complexes was not luminescent. The [Cu4L2I4] complex was catalytically active, N-(4-phenyl)imidazole was produced in 85% yield using imidazole and iodobenzene under Ullman cross-coupling conditions and 1-benzyl-4-phenyl-1H-1,2,3-triazole was generated in 58% yield using CuAAC “click” cycloaddition reaction conditions. However, the catalytic activity was modest compared to related complexes and the authors attributed this to the insolublity of the [Cu4L2I4] macrocycle.
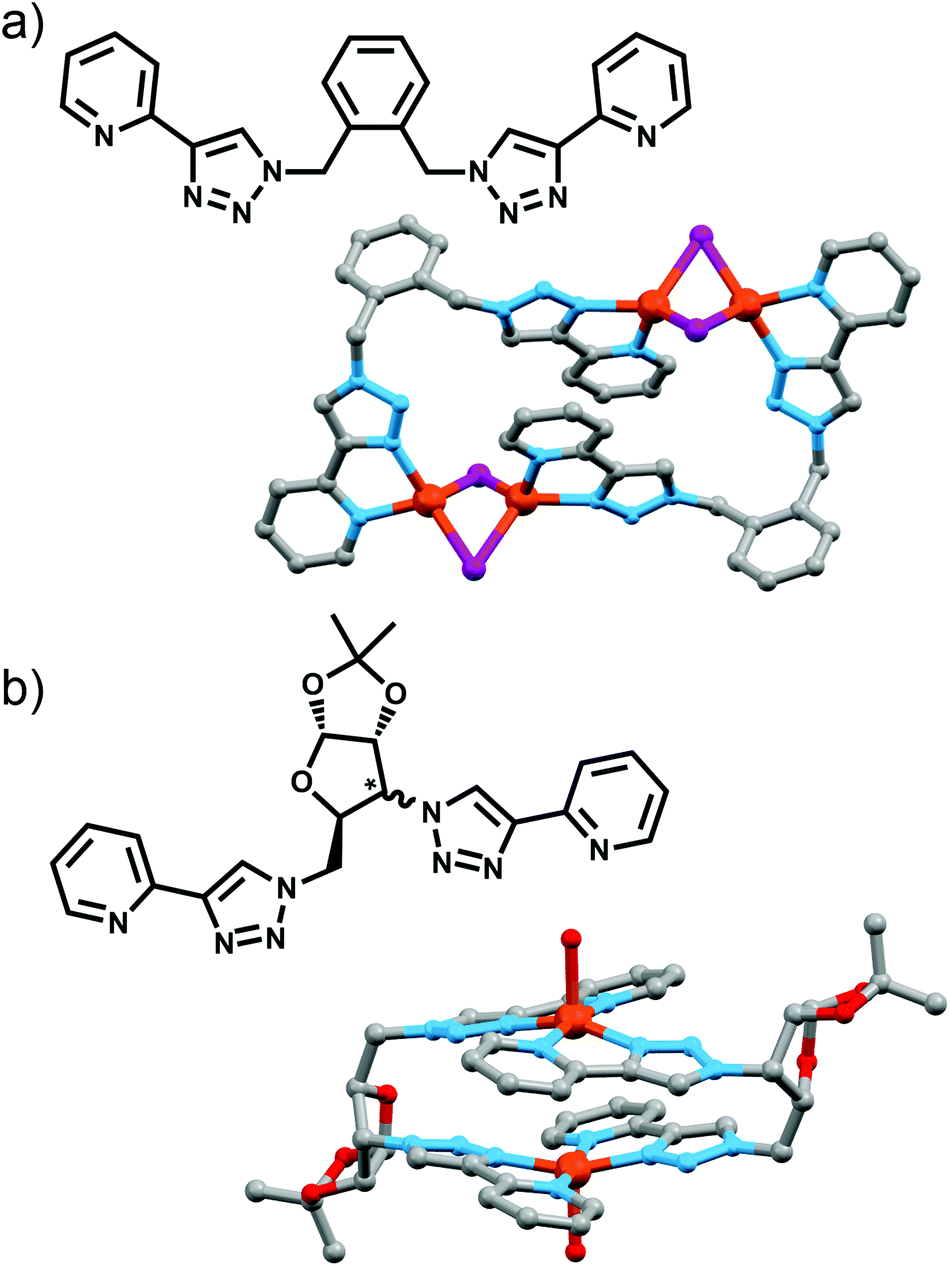 | ||
| Fig. 9 Bis-bidentate ligands and depictions of the molecular structures of their [2 + 2] metallo-macrocycles; (a) Wang's catalytic [Cu4L2I4] complex, and (b) Policar's [Cu2L2]2+ complex from chiral glycoligands (the molecular structure of the xylose-based ligand is shown). | ||
Policar and coworkers utilised a pair of epimeric diazides containing bicyclic furanoses (xylose and ribose) in combination with 2-ethynylpyridine to access two diasteroisomeric bis-(2-pyridyl-1,2,3-triazole) ligands (Fig. 9a).27 These could be combined with Cu(II) ions to give a [2 + 2] macrocycle, with the X-ray crystal structure of the xylose-based compound revealing the copper ion in a distorted square-pyrimidal geometry, with the apical site occupied by a water molecule. Both complexes were chiral, with the chirality originating from the glycoligand, and free ligands exhibited luminescence in ethanol which was quenched upon complex formation.
Maverick and co-workers have synthesised similar [Cu2L2]4+ macrocycles from Cu(II) ions and bis-(2-pyridyl-1,2,3-triazole) ligands, incorporating either a m-xylene (1,3-) or 2,7-dimethylnaphthalene spacer unit.28 The larger cavity of the naphthalene-based metallo-macrocycle enabled binding of DABCO and other guests, with the guest bridging the two internal axial sites of the Cu(II) metal ions, while the xylyl-based system was too small. However, this smaller system (Fig. 10a) was subsequently shown to be capable of selectively extracting carbon dioxide from the air and reducing it to oxalate.29 The mechanism (Fig. 10b) of CO2 uptake proceeds through reduction of the copper atoms from Cu(II) to Cu(I) using sodium ascorbate. During this process, the Cu(II) d–d electronic absorption band disappears and a new strong band at 384 nm becomes present, which is attributed to a metal-to-ligand charge transfer (MLCT) transition. The macrocycle can then selectively extract CO2 from the air. The progression of this process was monitored with electronic absorption spectroscopy, where the disappearance of the MLCT transition band and appearance of the Cu(II) d–d transition were observed. Fourier-transform infrared (FTIR) spectra of the new compound revealed carbonyl stretches at 1670 cm−1, indicating the presence of CO2 or oxalate. Crystallisation of the same complex proved that oxalate was bound within the cavity of the metallo-macrocycle bridging the two copper(II) ions (Fig. 10a). The oxalate could be removed from the complex by addition of either HCl(aq) (8 eq.) or HNO3(aq) (8 eq.) to regenerate the empty dicopper(II) macrocycle. The oxalic acid that was produced in this reaction was characterised by 13C NMR (163.2 ppm) and FTIR (νCO 1668 cm−1). Although this process is reasonably slow (reduction of CO2 was almost complete after 128 h) the Maverick group has been tuning the bis-2-pyridyl-1,2,3-triazole ligand to see if they can increase the reactivity of the macrocycle with CO2, as well as examining alternate methods for removal of oxalate.
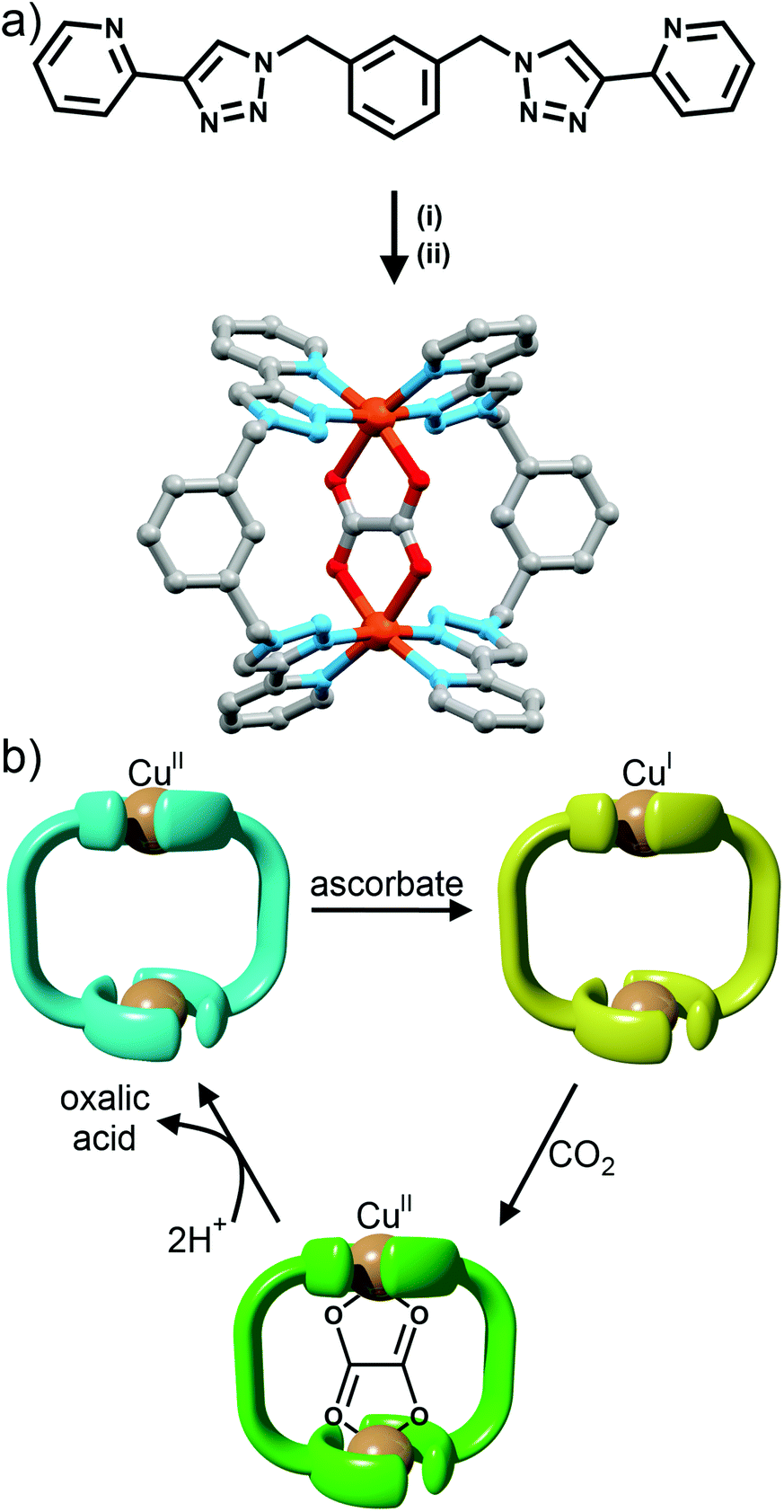 | ||
| Fig. 10 (a) Molecular structure of Maverick's dicopper(II) metallocycle, (i) Cu(I); (ii) NH4PF6; and (b) a cartoon representation of the reduction of CO2 with the metallocycle. From top-left: free Cu(II) macrocycle, reduced by ascorbate to free Cu(I) macrocycle; uptake of CO2 oxidises macrocycle back to Cu(II) with oxalate bound inside; addition of acid removes oxalate from macrocycle as oxalic acid to reform free Cu(II) macrocycle. | ||
M2L3 helicate and mesocates and M4L6 tetrahedral cages
The metallo-macrocyles above were generated using metal ions with a preference for forming either four or five coordinate complexes. Metal ions that typically form six coordinate octahedral complexes should enable the formation of M2L3 cylinders (Fig. 11a) or M4L6 tetrahedral cages (Fig. 12) with bis-(2-pyridyl-1,2,3-triazole) ligands.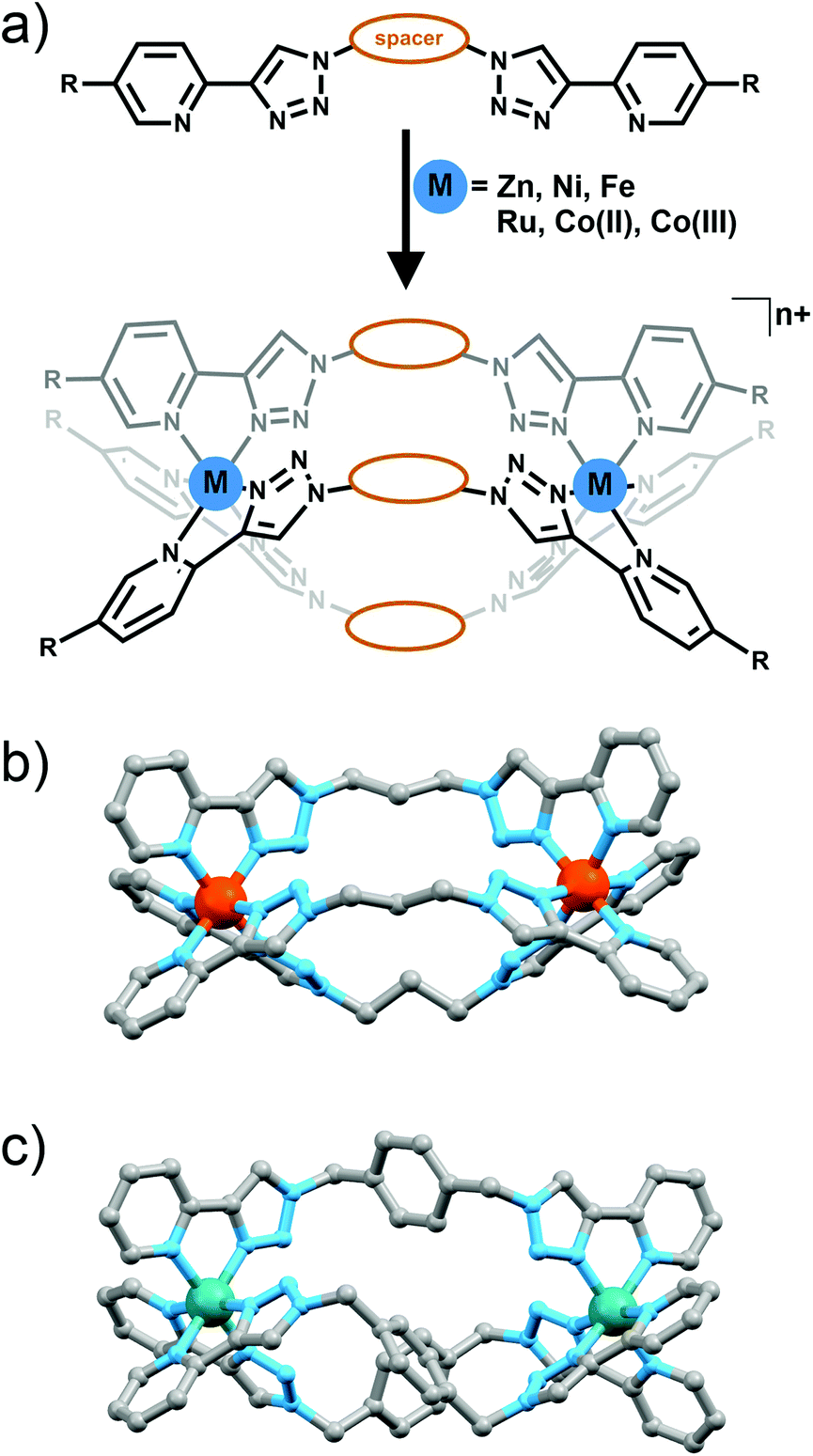 | ||
| Fig. 11 (a) Generic scheme showing formation of [M2L3]n+ cylinders from bis-bidentate 2-pyridyl-1,2,3-triazole ligands. Spacer = variety of alkyl or aryl groups, R = H or alkyloxy chain; (b) crystal structure of one of Petitjean's Fe(II) cylinders and (d) the molecular structure of Crowley and co-workers Ru(II) helicate. Hydrogen atoms, solvent molecules and counterions have been omitted for clarity. | ||
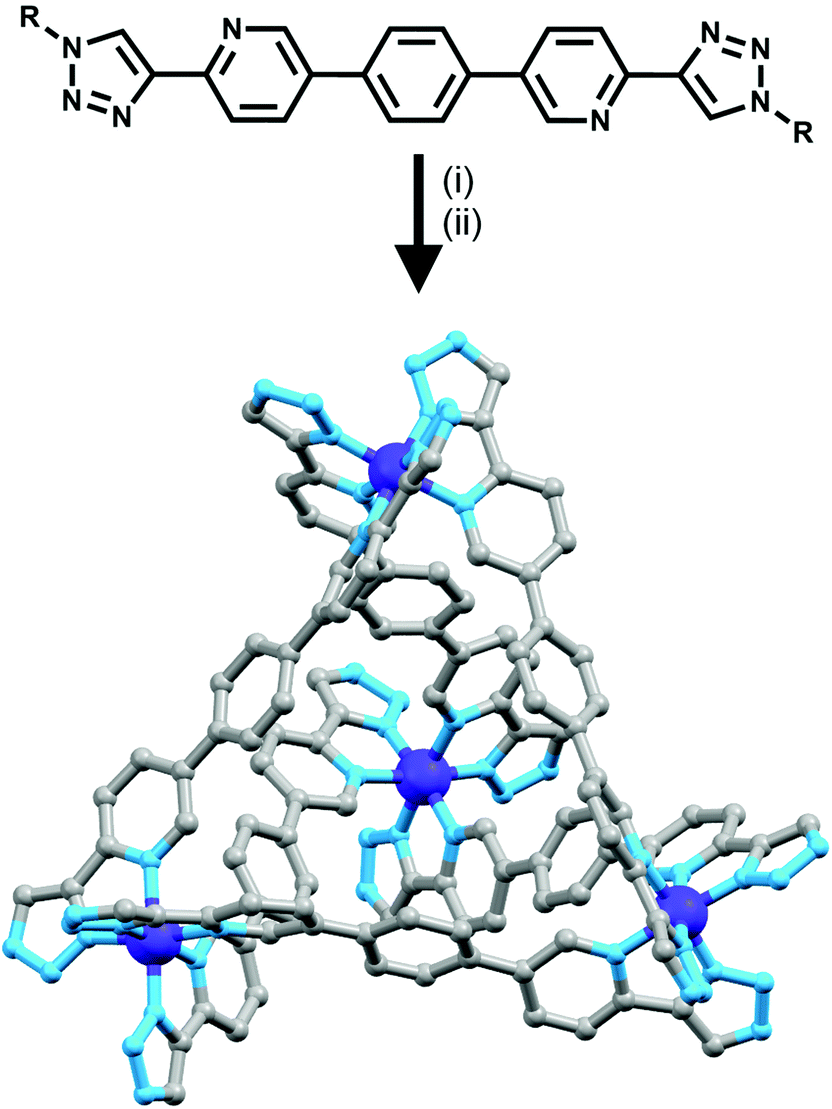 | ||
| Fig. 12 Scheme showing formation of a [Co4L6]12+ tetrahedron. R = adamantane or PEG; (i) Co(II), MeCN; (ii) CAN, MeCN; NH4PF6, H2O, MeCN. Terminal adamantane groups, hydrogen atoms and counterions have been omitted from the crystal structure for clarity. | ||
In a preliminary study Petitjean and co-workers showed that treating bis-(2-pyridyl-1,2,3-triazole) ligands containing 1,3-propyl, 1,4-xylyl or 2,6-dimethylnaphthalene spacers with Fe(II) or Ni(II) ion lead to the formation of M2L3 cylinders (Fig. 11a and b).30 Exploiting the structural diversity provided by the functional group tolerance of the CuAAC reaction Petitjean and co-workers subsequently synthesised a larger family of bis-(2-pyridyl-1,2,3-triazole) ligands and examined their behaviour in the presence of Fe(II), Ni(II) and Zn(II) ions (Fig. 11a).31 The systems were characterised using 1H and 13C NMR and UV/Vis spectroscopy, ESI-MS and vapour pressure osmometry and in some case X-ray crystallography (Fig. 11b). For the most part [M2L3]4+ cylinders were obtained. However as the central spacer unit becomes larger and more flexible other architectures were also observed highlighting the importance of ligand design in these systems. Similar types of [M2L3]n+ cylinder have been shown to follow the so called “odd-even”6b,32 rule where [M2L3]n+ complexes with an even number of carbon units in the spacer form racemic mixture of the ΔΔ and ΛΛ helicates while systems with odd numbers of carbons in the spacer lead to ΔΛ mesocates. Some of the “click” cylinders (those with 1,4-xylyl, 2,6-napthlyl and 1,3-propyl spacer units) followed this rule but for compounds featuring the 1,2-ethyl, 1,4-butyl and 1,5-pentyl spacers the rule breaks down. 1H NMR spectra showed that the complex with the long flexible 1,5-pentyl spacer unit formed a 1:2 mixture of helicate and mesocate in solution. The 1,2-ethyl and 1,4-butyl linked complexes also did not lead to the formation of a pure species in solution. In the case of the 1,2-ethyl linked complex the authors attribute this to the proximity of the six non-coordinating nitrogen atoms of the 1,2,3-triazolyl unit generating lone pair––lone pair electronic repulsion. However, electrostatic repulsion due to the close proximity of the two divalent Fe(II) centers could also play a role. Both these factors are destabilizing leading to the formation of other oligomeric species in addition to the desired [Fe2L3]4+ cylinder.
As many iron(II) triazole complexes display spin-crossover phenomena33 Petitjean and co-workers examined the family of iron(II) cylinders for this behaviour. The authors were unable to obtain a complete spin cross-over in the temperature window investigated (2.5 K to 300 K) and no thermal hysteresis was observed.31
Using an overlapping family of bis-(2-pyridyl-1,2,3-triazole) ligands to Petitjean and co-workers, the Crowley group has synthesised a related family of diiron(II) cylinders. The iron(II) complexes were readily synthesised and characterised using 1H and DOSY NMR spectroscopy and X-ray crystallography. Similar to the findings of Petitjean and co-workers the authors found that the diiron(II) cylinders mostly obey the “odd–even” rule. The exceptions were the 1,6-hexyl and diphenylmethylene linked systems. The diphenylmethylene linked cylinder crystallised as a racemic mixture of the ΔΔ and ΛΛ helicates but 1H NMR spectra indicated that the complex formed mixture of the helicate and mesocate species in solution. The more flexible 1,6-hexyl linked ligand formed both the desired [Fe2L3]4+ cylinder and other oligomers. Inspired by the interesting biological activity of other iron(II) cylinders34 Crowley and co-workers examined the triazole based compounds for antifugal activity against Saccharomyces cerevisiae. Interestingly, the iron(II) cylinders were completely inactive against the yeast. The authors traced the lack of activity back to the stability of the complexes. Unlike related pyridylimine based cylinders the complexes featuring the 2-pyridyl-1,2,3-triazole units were rapidily decomposed in the presence of histidine and DMSO. The authors suggested that the lower stability of the diiron(II) 2-pyridyl-1,2,3-triazole cylinders, compared to the related pyridylimine systems,34 was connected to both the weaker ligand donor strength and the presence of lone pair–lone pair replusions between the non-coordinating nitrogen atoms.
Crowley and co-workers have also generated a related ruthenium(II) helicate using the 1,4-xylyl linked bis-(2-pyridyl-1,2,3-triazole) ligand (Fig. 11c).35 The complex was characterised using 1H and DOSY NMR spectroscopy, electrospray ionisation mass spectrometry and the solid state structure was determined using X-ray crystallography. Efforts to generate a larger family of [Ru2L3]4+ cylinders were unsuccessful, only intractable oligomeric/polymeric species were observed. As expected the diruthenium(II) cylinder proved more inert than the iron(II) complexes and exhibited modest antimicrobial activity against Escherichia coli (MIC = 256 μg mL−1) but was inactive against Staphylococcus aureus.
Building on this work Crowley and co-workers examined the antimicrobial activity properties of some related dicobalt(III) cylinders. Exploiting the assembly-followed-by-oxidation method developed by Lusby and co-workers (vide infra) they assembled a small family of dicobalt(III) cylinders, linked by either a methylene or 1,4-xylyl spacer units featuring either unsubstituted or hexyloxy substituted terminal pyridyl-triazole units. Similar to the diruthenium(II) helicate and unlike the diiron(II) cylinders, the more inert dicobalt(III) complexes were DMSO and water soluble, and stable in D2O in the presence of biological nucleophiles for an extended period of time. Although decomposition was observed in DMSO, this was light-activated and complexes stored in the absence of light the exhibited longer half-lives. Despite displaying excellent kinetic stability none of the cylinders exhibited any antimicrobial activity in either a disk-diffusion or broth dilution assays. The authors have suggested that the lack of observed biological activity is likely connected to the high 6+ charge of the dicobalt(III) cylinders which prevents the molecules from readily passing across the bacterial cell membrane,36 this is similar to what other have observed with related diiridium(III) complexes that are overall 6+ cations.
M4L6 tetrahedral cages
Using rigid bis-(2-pyridyl-1,2,3-triazole) ligands Lusby and co-wokers have successfully erected a series of [M4L6]n+ tetrahedral cage architectures (Fig. 12).37 Their ligand system differs from the previously discussed 2-pyridyl-1,2,3-triazole based structures, the rigid phenyl spacer unit is attached to the pyridyl moieties of the 2-pyridyl-1,2,3-triazole ligands. This allowed the authors to tune the solubility of the ligands by exploiting CuAAC “click” chemistry. The authors developed an assembly-followed-by-oxidation approach to generate a family of [Co4L6]12+ tetrahedral cages. Co(II) ions are labile and allow the rapid assembly of the tetrahedral cages under thermodynamic control. However, while the high lability possessed by the Co(II) ions enables the facile generation of the cages it also allows the rapid decomposition of the cages in the presence of competitive ligands/nucleophiles. Lusby and co-workers were able to turn the labile [Co4L6]8+ tetrahedral cages into kinetically robust [Co4L6]12+ complexes by chemical oxidation with cerium(IV) ammonium nitrate (CAN). Once oxidised the Co(III) ions within the architecture are kinetically inert providing a robust cage system. The authors have gone on to show, through NMR studies, that the [Co4L6]12+ tetrahedra can bind (K = 100–1200 M−1) a wide range of neutral organic molecules such as nitrobenzene and chromanone within the cavity of the cage architecture.Exploiting their [Co4L6]12+ tetrahedral cages as a starting material Lusby and co-workers have also engineered a small family of dicobalt(III) cylinders.38 Using an assembly-followed-by-fixing process (Fig. 13) the aforementioned [Co4L6]12+ tetrahedra were treated with tetrabutylammonium iodide in order to reduce the tetracobalt(III) tetrahedron to a tetracobalt(II) cage and enable the switching to the dicobalt(III) cylinders. The tetracobalt(II) cage complex was then heated at 70 °C and the formation of the entropically favoured paramagnetic dicobalt(II) helicate was confirmed by 1H NMR spectroscopy. Finally, CAN was rapidly added and the dicobalt(III) helicate precipitated out of the acetonitrile solution. This process can be reversed using ambient conditions to reform the cobalt(III) tetrahedron. Once again, tetrabutylammonium iodide is utilised as a reducing agent and then after a period of three days, the formation of the tetracobalt(II) tetrahedron is confirmed via1H NMR spectroscopy. Again, CAN was added, this time slowly (∼5 hours), to reoxidise the tetrahedron to the tetracobalt(III) system. The Lusby group have also explored using photoredox to access the tetrahedron from the helicate. A solution of the dicobalt(III) helicate is mixed with [Ir(ppy)2(dtbbpy)](PF6) (where ppy = 2-phenylpyridine and dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine) and degassed with argon. After irradiating the sample with a standard 42 W lightbulb for 2 days, the conversion from the helicate to tetrahedron was confirmed via1H NMR spectrscopy. A control experiment was also conducted, whereby the sample was kept in the dark. After 5 days, only about 33% of the helicate had been converted, indicating that the illumination catalysed the transformation of the helicate to the tetrahedron. Interestingly, the authors have shown the photoredox method only works for the pyridyl-triazole based ligand systems.
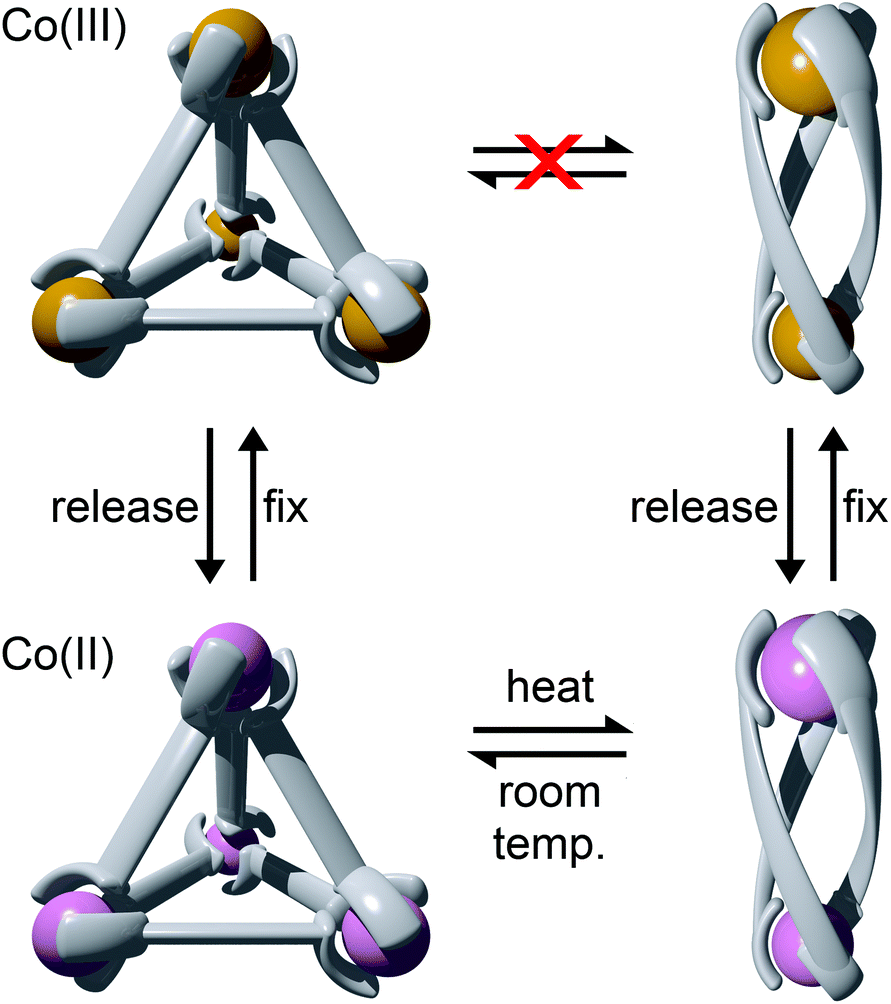 | ||
| Fig. 13 Scheme showing formation of Co(II) and Co(III) tetrahedra and helicates through the assembly-followed-by-fixing process. Conditions: release: tetrabutylammonium iodide; fix: CAN (added quickly to achieve the helicate and slowly to achieve the tetrahedron). | ||
Grids
Schubert et al. have successfully synthesised a small family of [2 + 2] grids (Fig. 14).39 The ligands, complexed to either copper(I) or silver(I), are based on a pyridazine unit, with two terminal 1,2,3-triazole groups. These triazole moieties have either a mesityl or PEG chain as the R group, providing the grids with different physical properties. Synthesis of the ligands followed a simple pathway: bromination of 1,2-dihydro-3,6-pyridazinedione, followed by a Pd(0)-catalyzed Sonogashira cross-coupling and culminating in a CuAAC click reaction. Complexation of the mesityl-substituted ligand with [Cu(CH3CN)4](PF6) resulted in the instantaneous formation of the [2 + 2] copper(I) grid. Combining the same ligand with AgSbF6 and stirring for an hour produced the [2 + 2] silver(I) grid. Characterisation of the grids via various NMR techniques, along with UV/Vis spectroscopy, ESI-MS and sedimentation velocity experiments confirmed the formation and structure of the copper(I) and silver(I) complexes. Flood and co-workers40 have synthesised an analogous [2 + 2] copper(I) grid and examined the self-assembly process in detail using 1H NMR and UV-vis titrations.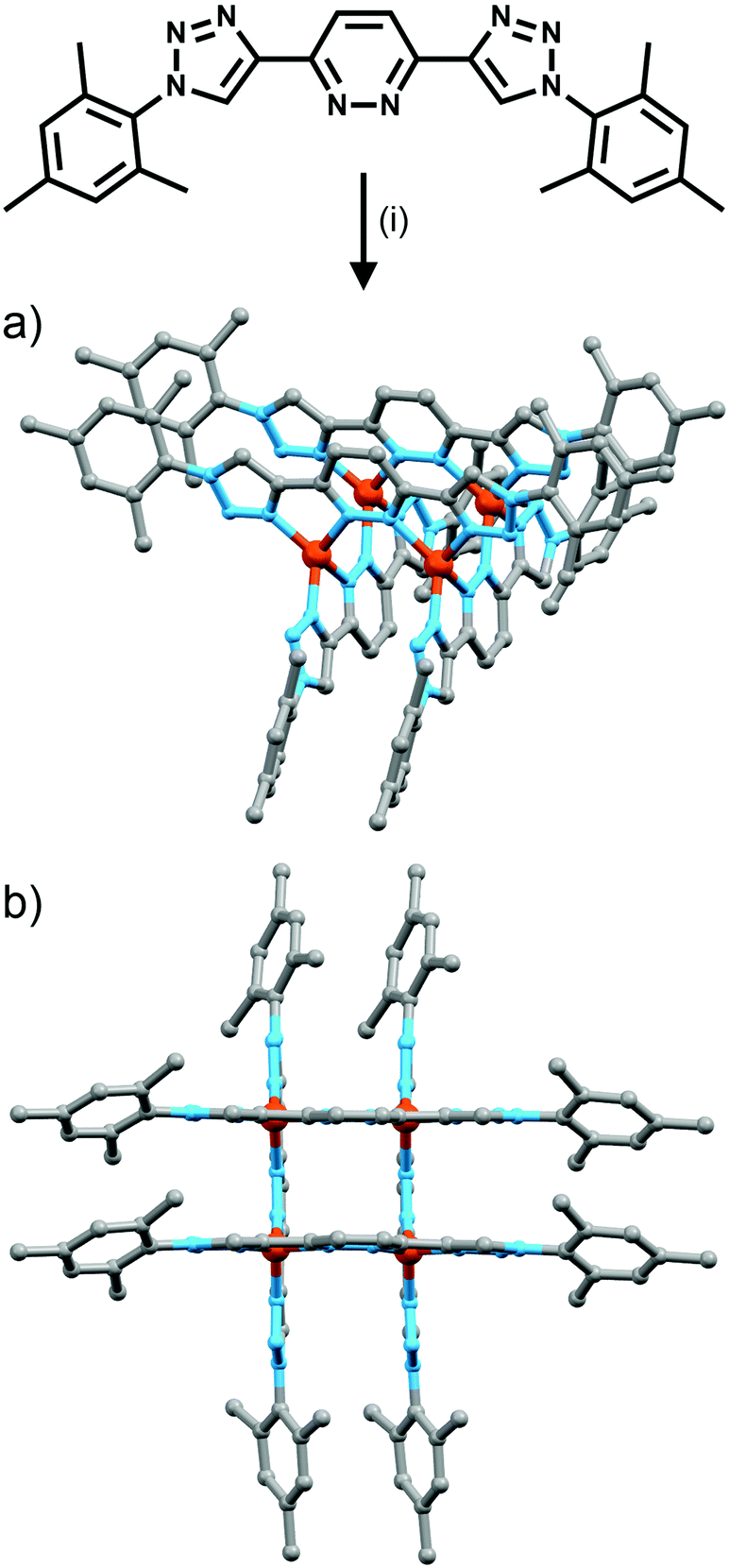 | ||
| Fig. 14 Formation of “click” grids. Conditions: (i) Cu(I), CH2Cl2 (a) side view and (b) top view of the molecular models (MMFF) showing the proposed structure of the copper(I) based grid. | ||
Poly-bidentate ligands
A trinuclear cylinder has been reported by Ghosh and co-workers,41 capable of being switched between being homometallic and heterometallic. Using a tris-bidentate ligand composed of a 2,2′-bipyridine flanked by two 2-pyridyl-1,2,3-triazole units, they have been able to create a cylinder around three iron(II) metal centres which was kinetically inert at room temperature. They then added one equivalent of zinc(II) triflate and with heating at 85 °C, all three iron(II) ions were replaced with zinc(II). However, this complex was kinetically labile at room temperature and hence back-exchanged to the thermodynamically favoured heteronuclear complex (two terminal Fe(II) ions and one central Zn(II) ion). This heteronuclear complex is also accessible from the trizinc(II) compound. Upon addition of iron(II) ions, the cylinder transforms into the thermodynamic product at room temperature.Other systems featuring tris- and tetra-2-pyridyl-1,2,3-triazole binding units have also been developed. Crowley and Bandeen used a 3:2 ratio of Ag(I) ions with a tritopic 1,3,5-methylbenzene-linked tris-(2-pyridyl-1,2,3-triazole) ligand, to form a [Ag3L2]3+ cage.42 A zinc porphyrin ligand with four 2-pyridyl-1,2,3-triazole arms at the peripheries of the porphyrin ring was synthesised by Ballester, Heitz and coworkers (Fig. 15).43 The authors combined the ligand with AgSbF6, forming an [M4L2]4+ cage (Fig. 15). NMR spectroscopy in d6-DMSO showed that a single discrete species was formed, with 1H DOSY NMR experiments revealing that all the protons resonances were diffusing at the same rate, slower than the free ligand, confirming the formation of a larger architecture. The peaks were sharp, indicating non-fluxional behaviour, and a UV-vis titration along with mass spectral analysis confirmed the [Ag4L2]4+ stoichiometry.
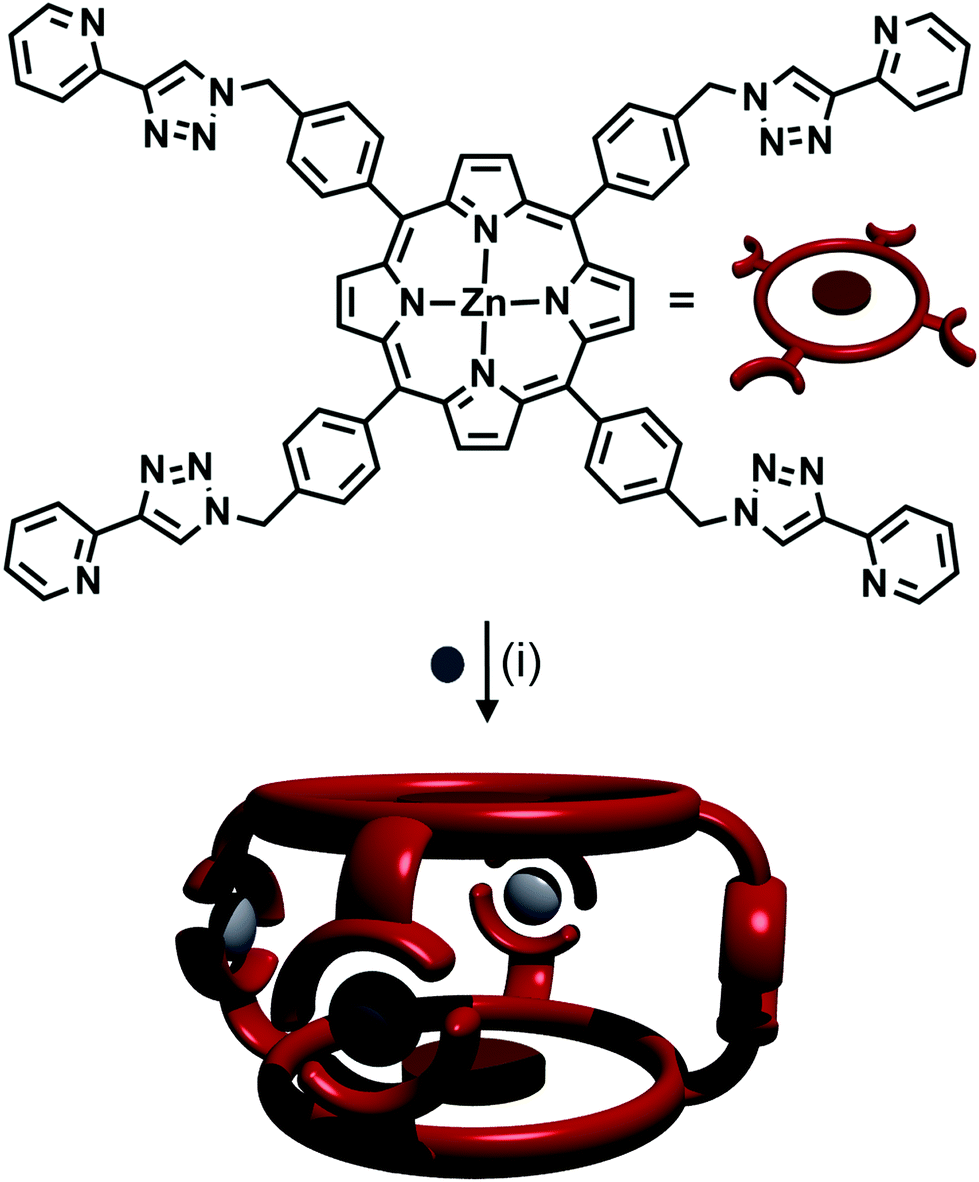 | ||
| Fig. 15 Formation of [Ag4L2]4+ cage from Ag(I) ions and a tetra-pyridyl-triazole zinc porphyrin ligand. Conditions: Ag(I), CH2Cl2/CHCl3. | ||
Tridentate ligand systems
Somewhat surprisingly, especially given the ubiquitous use of terpyridine (terpy, C) ligands in the construction of metallosupramolecular systems, tridentate “click” ligands have not yet been extensively exploited for the generation of discrete metallo-architectures. The coordination chemistry of tridentate “click” ligands (F)44 has been well explored and bis-tridentate “click” ligands have been used to generate metallo-polymers45 and rotaxanes46 and catenanes.47 Thus it seems only matter of time before these types of ligands are used to generate some type of discrete functional metallosupramolecular architecture.Conclusions
The use of “click”-derived ligands in metallosupramolecular chemistry is nascent but steadily growing. There appeared to be little interest in generating discrete metallopsupramolecular architectures with coordinating ‘structural’ 1,2,3-triazoles prior to 2010. But in the intervening six years a considerable amount of work has been done to develop these systems. The reasons for the increasing use of the 1,2,3-triazole containing ligands are not difficult to elucidate. The reliability, mild reaction conditions and wide substrate scope of the CuAAC “click” reaction10 mean a wide range of functionlized ligands with a variety of denticities can be readily accessed. The majority of the “click” ligands systems examined to date are analogues of pyridine, bipyridine and terpyridine ligands. Exploiting the priniciples previously developed with the pyridyl ligands a range of architectures have been generated with the “click” derived systems, including metallomacrocycles, helicates, grids and cages. These results confirm that “click” ligands can be used for the construction of metallosupramolecular systems. Additionally, no there is no doubt that the synthetic ease (under mild conditions) of the “click” reaction could allow the generation of a huge variety of polytopic ligands with tuned linkers and/or donor groups, leading to the generation of large variety of accessible metallosupramolecular assemblies.However, with the principles of metallosupramolecular assembly now well established for simpler unfunctionalised systems, the field is moving towards greater structural complexity due to the desire to generate systems for “real” applications. Thus the ease with which functionality and molecular character of these “click” ligands can be tuned using CuAAC chemistry is a powerful tool. Ligands with enhanced biological, electrochemical and photophysical properties can be readily evisioned, while the facile alteration of ligand character could allow the tuning of cavity size and molecular recognition properties of “click” derived metallosupramolecular systems. As was shown herein suitably designed “click” architectures have already displayed both anti-cancer and anti-bacterial properties. “Click” derived macrocycles and cage systems have shown good host–guest properties and one macrocyclic system has been used as molecular reaction flask for the remediation of CO2. Other systems have been examined for their interesting electronic (spin-crossover) and catalytic properties. Additionally, during the reviewing time of this manuscript a new tetra-2-pyridyl-1,2,3-triazole porphyrin ligand has been used to generate a Fe(II) based molecular cube system which can be exploited as a molecular flask.48 The cube enabled the selective reaction between C60 and anthrance to be carried out. For these reasons, we believe that the popularity of “click”-derived 1,2,3-triazole ligands as components of metallosupramolecular systems will continue to grow, and that the potential applications of the assemblies thus generated will likewise flourish.
Acknowledgements
RASV and DP would like to thank the University of Otago for a MSc stipend and a PhD scholarship, respectively.Notes and references
-
(a) P. J. Steel, Chem. N. Z., 2003, 67, 57–60 CAS
; (b) I. Dance, New J. Chem., 2003, 27, 1–2 RSC
- For some selected recent examples see:
(a) H. Li, Z.-J. Yao, D. Liu and G.-X. Jin, Coord. Chem. Rev., 2015, 293-294, 139–157 CrossRef CAS
-
(a) B. Therrien, CrystEngComm, 2015, 17, 484–491 RSC
- V. Croue, S. Goeb and M. Salle, Chem. Commun., 2015, 51, 7275–7289 RSC
-
(a) M. L. Saha, X. Yan and P. J. Stang, Acc. Chem. Res., 2016, 49, 2527–2539 CrossRef CAS PubMed
-
(a) D. L. Caulder and K. N. Raymond, J. Chem. Soc., Dalton Trans., 1999, 1185–1200 RSC
-
(a) J. K. Clegg, F. Li and L. F. Lindoy, Coord. Chem. Rev., 2013, 257, 2536–2550 CrossRef CAS
-
(a) G. R. Newkome, A. K. Patri, E. Holder and U. S. Schubert, Eur. J. Org. Chem., 2004, 235–254 CrossRef CAS
- M. D. Ward and P. R. Raithby, Chem. Soc. Rev., 2013, 42, 1619–1636 RSC
-
(a) M. Meldal and C. W. Tornoe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed
-
(a) B. Schulze and U. S. Schubert, Chem. Soc. Rev., 2014, 43, 2522–2571 RSC
-
(a) L. Jiang, Z. Wang, S.-Q. Bai, X. J. Loh and T. S. A. Hor, Aust. J. Chem., 2016, 69, 645–651 CrossRef CAS
-
(a) A. B. S. Elliott, J. E. M. Lewis, H. van der Salm, C. J. McAdam, J. D. Crowley and K. C. Gordon, Inorg. Chem., 2016, 55, 3440–3447 CrossRef CAS PubMed
- M. L. Gower and J. D. Crowley, Dalton Trans., 2010, 39, 2371–2378 RSC
- N. G. White and P. D. Beer, Supramol. Chem., 2012, 24, 473–480 CrossRef CAS
- L. Jiang, Z. Wang, S.-Q. Bai and T. S. A. Hor, CrystEngComm, 2013, 15, 10451–10458 RSC
-
(a) K. F. Donnelly, A. Petronilho and M. Albrecht, Chem. Commun., 2013, 49, 1145–1159 RSC
- E. C. Keske, O. V. Zenkina, R. Wang and C. M. Crudden, Organometallics, 2012, 31, 456–461 CrossRef CAS
- M. Frutos, M. C. de la Torre and M. A. Sierra, Inorg. Chem., 2015, 54, 11174–11185 CrossRef CAS PubMed
- K. J. Kilpin, U. S. D. Paul, A.-L. Lee and J. D. Crowley, Chem. Commun., 2011, 47, 328–330 RSC
- B. Schulze, D. Escudero, C. Friebe, R. Siebert, H. Görls, U. Köhn, E. Altuntas, A. Baumgaertel, M. D. Hager, A. Winter, B. Dietzek, J. Popp, L. González and U. S. Schubert, Chem. – Eur. J., 2011, 17, 5494–5498 CrossRef CAS PubMed
- J. Cai, X. Yang, K. Arumugam, C. W. Bielawski and J. L. Sessler, Organometallics, 2011, 30, 5033–5037 CrossRef CAS PubMed
-
(a) S. O. Scott, E. L. Gavey, S. J. Lind, K. C. Gordon and J. D. Crowley, Dalton Trans., 2011, 40, 12117–12124 RSC
- S. M. McNeill, D. Preston, J. E. M. Lewis, A. Robert, K. Knerr-Rupp, D. O. Graham, J. R. Wright, G. I. Giles and J. D. Crowley, Dalton Trans., 2015, 44, 11129–11136 RSC
- D. Preston, R. A. J. Tucker, A. L. Garden and J. D. Crowley, Inorg. Chem., 2016, 55, 8928–8934 CrossRef CAS PubMed
- H. Zhao, X. Li, J. Wang, L. Li and R. Wang, ChemPlusChem, 2013, 78, 1491–1502 CrossRef CAS
- L. Garcia, S. Maisonneuve, J. Oudinet-Sin Marcu, R. Guillot, F. Lambert, J. Xie and C. Policar, Inorg. Chem., 2011, 50, 11353–11362 CrossRef CAS PubMed
- U. R. Pokharel, F. R. Fronczek and A. W. Maverick, Dalton Trans., 2013, 42, 14064–14067 RSC
- U. R. Pokharel, F. R. Fronczek and A. W. Maverick, Nat. Commun., 2014, 5, 5883 CrossRef CAS PubMed
- K. A. Stevenson, C. F. C. Melan, O. Fleischel, R. Wang and A. Petitjean, Cryst. Growth Des., 2012, 12, 5169–5173 CAS
- N. Wu, C. F. C. Melan, K. A. Stevenson, O. Fleischel, H. Guo, F. Habib, R. J. Holmberg, M. Murugesu, N. J. Mosey, H. Nierengarten and A. Petitjean, Dalton Trans., 2015, 44, 14991–15005 RSC
- M. Albrecht, Chem. Rev., 2001, 101, 3457–3497 CrossRef CAS PubMed
-
(a) H. L. C. Feltham, A. S. Barltrop and S. Brooker, Coord. Chem. Rev., 2016 DOI:10.1016/j.ccr.2016.10.006
-
(a) R. A. Kaner, S. J. Allison, A. D. Faulkner, R. M. Phillips, D. I. Roper, S. L. Shepherd, D. H. Simpson, N. R. Waterfield and P. Scott, Chem. Sci., 2016, 7, 951–958 RSC
- S. V. Kumar, W. K. C. Lo, H. J. L. Brooks and J. D. Crowley, Inorg. Chim. Acta, 2015, 425, 1–6 CrossRef CAS
- M. Pandrala, F. Li, L. Wallace, P. J. Steel, B. Moore, J. Autschbach, J. G. Collins and F. R. Keene, Aust. J. Chem., 2013, 66, 1065–1073 CrossRef CAS
- P. R. Symmers, M. J. Burke, D. P. August, P. I. T. Thomson, G. S. Nichol, M. R. Warren, C. J. Campbell and P. J. Lusby, Chem. Sci., 2015, 6, 756–760 RSC
- M. J. Burke, G. S. Nichol and P. J. Lusby, J. Am. Chem. Soc., 2016, 138, 9308–9315 CrossRef CAS PubMed
- B. Happ, G. M. Pavlov, E. Altuntas, C. Friebe, M. D. Hager, A. Winter, H. Goerls, W. Guenther and U. S. Schubert, Chem. – Asian J., 2011, 6, 873–880 CrossRef CAS PubMed
- L. E. Manck, C. R. Benson, A. I. Share, H. Park, D. A. Vander Griend and A. H. Flood, Supramol. Chem., 2014, 26, 267–279 CrossRef CAS
- B. Akhuli, L. Cera, B. Jana, S. Saha, C. A. Schalley and P. Ghosh, Inorg. Chem., 2015, 54, 4231–4242 CrossRef CAS PubMed
- J. D. Crowley and P. H. Bandeen, Dalton Trans., 2010, 39, 612–623 RSC
- P. Ballester, M. Claudel, S. Durot, L. Kocher, L. Schoepff and V. Heitz, Chem. – Eur. J., 2015, 21, 15339–15348 CrossRef CAS PubMed
- J. P. Byrne, J. A. Kitchen and T. Gunnlaugsson, Chem. Soc. Rev., 2014, 43, 5302–5325 RSC
-
(a) A. Winter and U. S. Schubert, Chem. Soc. Rev., 2016, 45, 5311–5357 RSC
- J. E. M. Lewis, R. J. Bordoli, M. Denis, C. J. Fletcher, M. Galli, E. A. Neal, E. M. Rochette and S. M. Goldup, Chem. Sci., 2016, 7, 3154–3161 RSC
- J. P. Byrne, S. Blasco, A. B. Aletti, G. Hessman and T. Gunnlaugsson, Angew. Chem., Int. Ed., 2016, 55, 8938–8943 CrossRef CAS PubMed
- W. Brenner, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2017, 139, 75–78 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2017 |