
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fe3O4@HKUST-1 and Pd/Fe3O4@HKUST-1 as magnetically recyclable catalysts prepared via conversion from a Cu-based ceramic†
Takashi
Toyao
 ab,
Mark J.
Styles
c,
Tokuichiro
Yago
a,
Muhammad M.
Sadiq
d,
Raffaele
Riccò
ce,
Kiyonori
Suzuki
d,
Yu
Horiuchi
a,
Masahide
Takahashi
a,
Masaya
Matsuoka
*a and
Paolo
Falcaro
*ce
ab,
Mark J.
Styles
c,
Tokuichiro
Yago
a,
Muhammad M.
Sadiq
d,
Raffaele
Riccò
ce,
Kiyonori
Suzuki
d,
Yu
Horiuchi
a,
Masahide
Takahashi
a,
Masaya
Matsuoka
*a and
Paolo
Falcaro
*ce
aDivision of Materials Science & Engineering, Graduate School of Engineering, Osaka Prefecture University, 1-1 Gakuen-cho, Naka-ku, Sakai, Osaka 599-8531, Japan. E-mail: matsumac@chem.osakafu-u.ac.jp
bInstitute for Catalysis, Hokkaido University, N-21, W-10, Sapporo 001-0021, Japan
cCSIRO Manufacturing, Private Bag 10, Clayton South, Victoria 3169, Australia. E-mail: paolo.falcaro@tugraz.at
dDepartment of Materials Science and Engineering, Monash University, Clayton, Victoria 3168, Australia
eGraz University of Technology, Institute of Physical and Theoretical Chemistry, Stremayrgasse 9/Z2, 8010 Graz, Austria
First published on 22nd May 2017
Abstract
Nanocomposites obtained by integrating iron oxide magnetic nanoparticles (Fe3O4) into a metal–organic framework (HKUST-1 or Cu3(BTC)2, BTC = 1,3,5-benzenetricarboxylate) are synthesized through conversion from a composite of a Cu-based ceramic material and Fe3O4. In situ small-angle X-ray scattering (SAXS) and wide-angle X-ray scattering (WAXS) measurements reveal that the presence of Fe3O4 leads to the fast conversion and synthesis of HKUST-1 with small particle sizes. The prepared MOF composite (Fe3O4@HKUST-1) is found to catalyze the one-pot sequential deacetalization–Knoevenagel condensation reaction as a magnetically collectable and recyclable catalyst. In addition, Pd nanoparticles are also incorporated into the material (Pd/Fe3O4@HKUST-1) by addition of a Pd colloidal solution during the conversion of the precursor composite to HKUST-1. The resulting Pd/Fe3O4@HKUST-1 can be utilized for hydrogenation of 1-octene in the liquid phase.
Introduction
Metal–organic frameworks (MOFs), also called porous coordination polymers (PCPs), are a class of crystalline hybrid materials prepared via well-established principles of coordination chemistry using self-assembly of metal ions or metal clusters with organic linkers.1–3 Unlike traditional inorganic porous materials, unlimited possible combinations of such an assembly enables access to reticular structures with tunable porosity and attractive functionalities.4–7 In addition, new synergetic properties have been obtained by incorporating functional species into MOFs, further expanding their potential applications.8–17 While there are a large number of reports describing different methods for successfully combining exogenous species into MOFs,18–21 the encapsulation of nanoparticles is particularly promising due to the innovative functional properties that cannot be obtained from the parent MOFs alone.22–26 Typically, the encapsulation of nanoparticles is achieved through the introduction of precursors using solution impregnation, gas-phase infiltration or a solid-phase method, and subsequent conversion into the corresponding functional components inside the frameworks.27–29 However, in this two-step approach, free nanoparticles are often present which need to be removed from the system. This is generally achieved by a gravimetric separation step, exploiting the different sedimentation rate between the nanoparticles and the MOF crystals, before separating the magnetic composite from the pure MOF.25 Additionally, particle size and morphology are limited by the cavities of the MOFs.30,31 Moreover, the framework can potentially be degraded during nanoparticle formation.30,31In an attempt to overcome this difficulty, approaches for the growth of MOFs around pre-formed functional particles have been successfully proposed.32,33 These approaches involve the addition of preformed functional species into the MOF precursor solution and the subsequent in situ formation of MOF crystals with embedded functional particles. Furthermore, it has been demonstrated that the heterospecies can improve the kinetics of MOF formation,34–36 as nano- and micro-particles can act as crystallization facilitators promoting faster MOF growth. On the other hand, functional nanoparticles tend to aggregate under the acidic environment created by the dissociation of ligands during this solution process.37,38 Therefore, surface modification and the choice of the proper solvent can help the nucleation and growth of the MOFs around the functional components.39,40 An interesting emerging approach that overcomes this issue involves the use of ceramic materials as the metal source for the direct conversion into a MOF composite.41–44 Interestingly, the ceramic materials can act as protecting agents for functional nanoparticles reducing their exposure to harsh conditions.41–44 Furthermore, the use of ceramics as precursors has allowed mild conditions to be used (e.g. water/alcohol at room temperature) with short processing times.45
Here we describe a simple approach to incorporate magnetic nanoparticles into MOFs by taking advantage of the method developed in our previous study, as shown in Fig. 1.46 We previously reported the synthesis of HKUST-1 (also called Cu3(BTC)2, BTC = 1,3,5-benzenetricarboxylate) through conversion from Cu-based ceramic precursors (Cu2(OH)3NO3) at room temperature as an ideal method for the permanent localization46 of the corresponding MOF. In this study, Fe3O4 magnetic nanoparticles have been embedded into the Cu-based ceramic material used as a precursor for the synthesis of repositionable46 HKUST-1 crystals. The Fe3O4@Cu2(OH)3NO3 composite was subsequently converted to Fe3O4-incorporated HKUST-1 (Fe3O4@HKUST-1) through exposure to an alcoholic solution containing 1,3,5-benzenetricarboxylic acid (H3BTC). The conversion of the ceramic into MOFs can be performed at room temperature. The obtained Fe3O4@HKUST-1 has been successfully separated from the HKUST-1 thanks to magnetic localization using a commercial magnet, that enabled the composite to be isolated and removed from the pure MOF crystals.25 With the same approach, the material has been utilized as an easily recyclable heterogeneous catalyst for a one-pot cascade reaction. Moreover, Pd nanoparticles have also been embedded into the HKUST-1 material, to prepare a novel advanced composite based on different nanoparticles encapsulated within the same MOF (i.e. Pd/Fe3O4@HKUST-1). The composite Pd/Fe3O4@HKUST-1 was found to be an active catalyst for the hydrogenation of olefin. Thanks to the magnetic component that confers dynamic localization properties, the composite showed promising recyclability properties. The method proposed here can offer a simple one-step approach to incorporate different functional nanoparticles into the same MOF particle.
 | ||
| Fig. 1 Schematic illustrations showing the integration of Fe3O4 nanoparticles into HKUST-1 through conversion from a composite of a Cu-based ceramic material and Fe3O4 (Fe3O4@Cu2(OH)3NO3). (a) Fe3O4@Cu2(OH)3NO3 is obtained by adding aq. NH3 into water containing Cu(NO3)2·3H2O and Fe3O4. (b and c) Fe3O4@Cu2(OH)3NO3 is converted into HKUST-1 material by adding a mixed solution of EtOH and water containing H3BTC at room temperature for 10 min. (d) The prepared composite (Fe3O4@HKUST-1) is collected by immobilizing the composite crystals with a magnet. | ||
Results and discussion
Characterization of materials
Fe3O4@Cu2(OH)3NO3 was prepared from Cu(NO3)2·3H2O and Fe3O4 by exposure to ammonia. Cu2(OH)3NO3 was also prepared for comparison without adding Fe3O4 nanoparticles. Using scanning electron microscopy (SEM), plate-like structures were observed for both Fe3O4@Cu2(OH)3NO3 and Cu2(OH)3NO3 samples, as shown in Fig. 2a and c. Subsequently, the conversion of both samples was carried out by adding a mixed solution of ethanol (EtOH) and water containing H3BTC. After about 10 seconds of reaction time, the color change of the precursor materials from light blue to deep turquoise could be detected visibly, thus indicating the formation of HKUST-1.47 Significant changes in morphology from the Cu-based precursors to the octahedral crystals of HKUST-1 were observed after 10 min of the conversion time (Fig. 2b and d). SEM images at 10 s and 1 min conversion time were also obtained to follow the evolution of the HKUST-1 (see Fig. S1 in the ESI†). The octahedral morphology of the particles suggests the successful formation of HKUST-1 crystals.47 It should be noted that the particle size of the Fe3O4@HKUST-1 composite is smaller than that of HKUST-1 without Fe3O4 nanoparticles. This change in crystal size can be attributed to the Fe3O4 nanoparticles acting as seeding materials and/or crystallization modulators for the formation of HKUST-1 crystals.34–37 Inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis was performed to determine the loading amount of Fe. The results revealed 1.57 wt% of Fe in the Fe3O4@HKUST-1 composite. Transmission electronic microscope (TEM) observations were also made to investigate Fe3O4 nanoparticles in Fe3O4@HKUST-1, as given in Fig. 2e and f. The TEM images demonstrate that Fe3O4@HKUST-1 contains Fe3O4 nanoparticles with an average diameter of 24 nm. The observed particle size matches well with the previous report describing preparation of the Fe3O4 nanoparticles.48 These results suggest that Fe3O4 nanoparticles remained essentially unchanged even after the conversion process and integrated in HKUST-1. | ||
| Fig. 2 SEM images of (a) Fe3O4@Cu2(OH)3NO3, (b) Fe3O4@HKUST-1, (c) Cu2(OH)3NO3 and (d) HKUST-1. (e and f) TEM images of Fe3O4@HKUST-1. | ||
Powder X-ray diffraction (XRD) measurements were carried out in order to ascertain the formation of HKUST-1 crystals, as shown in Fig. 3a. After the conversion to MOF, the measured diffraction patterns matched with previously-reported HKUST-1 diffraction patterns.47 Interestingly, no diffraction peaks corresponding to residual precursors or oxide species were observed in the patterns. Peaks from the Fe3O4 nanoparticles were not observed because of the low loading amount. FT-IR measurements were also performed to characterize the Fe3O4@Cu2(OH)3NO3 and Fe3O4@HKUST-1 (Fig. S2†). In the spectra for Fe3O4@Cu2(OH)3NO3, a wide band attributed to O–H stretching modes was observed in the region of 3550–3200 cm−1.49 The absorption bands at around 877, 1044, 1240, 1296, 1333 and 1416 cm−1 were assigned to vibrational modes of nitrate (NO3−) ions having an interaction with copper hydroxide layers.50,51 The remaining bands at 670 and 772 cm−1 were assigned to the Cu–OH stretching mode.52 After treatments with an alcoholic solution containing H3BTC, the intensity of the absorption bands attributed to O–H stretching modes in the region of 3550–3200 cm−1 decreased significantly. New modes were observed and assigned to the organic molecules used as ligands in the HKUST-1 frameworks (BTC); 729, 760, 940 cm−1: C–CO2 stretching, 1114 cm−1: C–O stretching, 1373, 1449, 1645 cm−1: COO–Cu2 stretching.53–55 These results confirm the successful conversion of the precursor ceramic to HKUST-1. It was also found that the absorption band coming from nitrate ions was present in the Fe3O4@HKUST-1 sample. Thermogravimetric analysis (TGA) curves of Fe3O4@HKUST-1 exhibited weight loss at around 300 °C which is attributable to the framework decomposition while Fe3O4@Cu2(OH)3NO3 showed weight loss at around 250 °C (Fig. S3†). This result supports the formation of HKUST-1 from the ceramic precursor. It was also observed that Fe3O4@HKUST-1 and HKUST-1 showed an almost identical profile, suggesting that incorporation of Fe3O4 in the framework did not affect the thermal stability of the framework material.
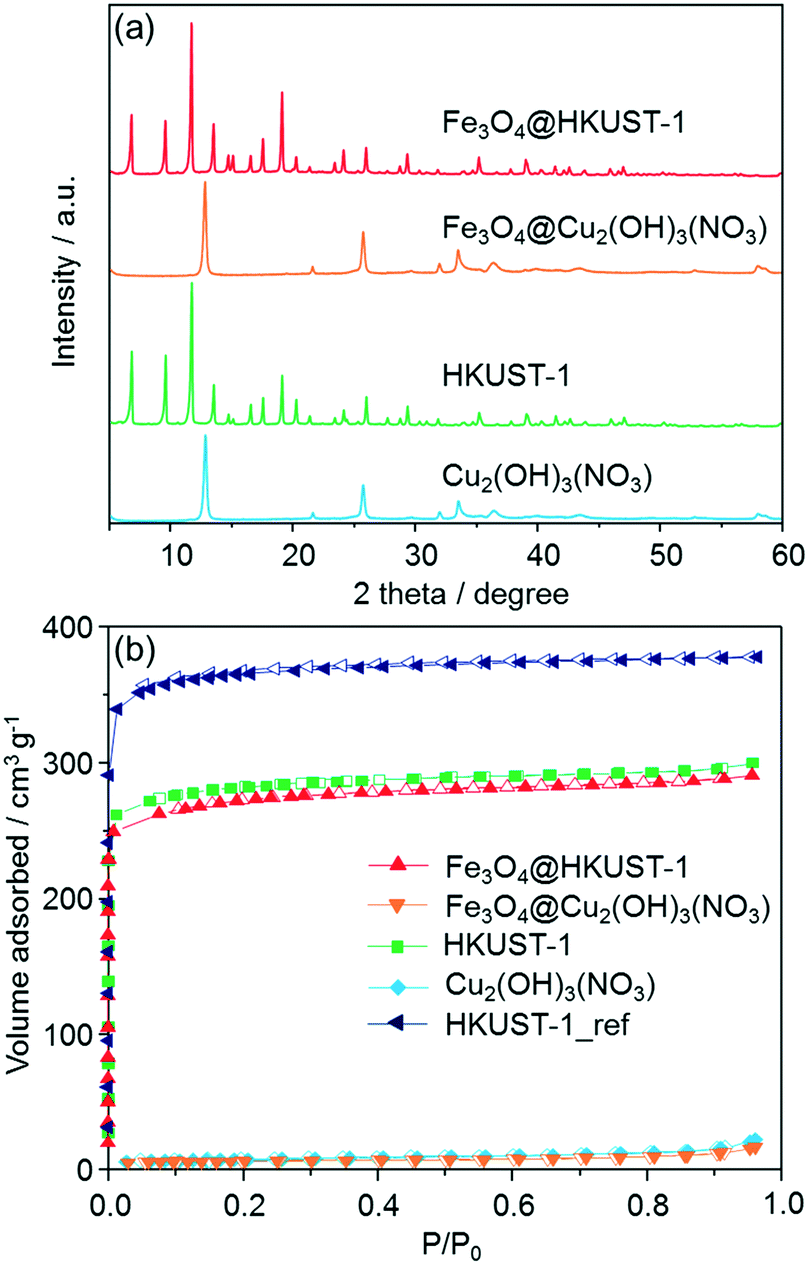 | ||
| Fig. 3 (a) XRD patterns of Fe3O4@HKUST-1, Fe3O4@Cu2(OH)3NO3, HKUST-1 and Cu2(OH)3NO3. The peaks from Fe3O4 nanoparticles cannot be seen in the diffraction patterns of Fe3O4@HKUST-1 and Fe3O4@Cu2(OH)3NO3, most likely due to the low loading amount; (b) N2 adsorption isotherms of Fe3O4@Cu2(OH)3NO3, Fe3O4@HKUST-1, Cu2(OH)3NO3, HKUST-1 and HKUST-1_ref measured at 77 K. Closed and open symbols represent adsorption and desorption isotherms, respectively. | ||
N2 adsorption isotherms are shown in Fig. 3b. Using the BET (Brunauer–Emmett–Teller) method, the specific surface areas of Fe3O4@Cu2(OH)3NO3 and Cu2(OH)3NO3 were measured to be 19 and 23 m2 g−1 respectively, demonstrating that the feedstock ceramic materials have low accessible surface area. After conversion into the MOF, type I isotherms were measured for both Fe3O4@HKUST-1 and HKUST-1, indicating a different microporosity. The specific surface areas of Fe3O4@HKUST-1 and HKUST-1 were determined to be 952 and 986 m2 g−1, respectively. HKUST-1_ref, synthesized using the conventional aqueous room temperature protocol proposed by Bradshaw et al.,56 was found to have the surface area of 1236 m2 g−1. The surface areas of the HKUST-1 materials prepared using the method proposed here were found to be c.a. 20% lower than that of HKUST-1_ref. This result suggests the residual precursor species still exist in the framework, which is consistent with the results obtained by FT-IR measurements. This conclusion is supported by previous studies into converting other Cu-based ceramics into HKUST-1.45,46,57
In order to gain further insights into the growth of the HKUST-1 crystals, in situ small-angle X-ray scattering (SAXS) and wide-angle X-ray scattering (WAXS) measurements were conducted at the Australian Synchrotron. For these measurements, the Cu-based precursor materials were placed in a capillary with the mixed solution of EtOH and water, followed by the controlled addition of the alcoholic solution containing the H3BTC linker using a syringe. Once filled with the solution, the capillary was illuminated by a high-intensity X-ray beam, and the scattered radiation was collected by the two 2D detectors. The obtained 2D scattering patterns have been background-subtracted, radially integrated and plotted as a function of the reaction time. The obtained SAXS/WAXS-based diffraction patterns for the conversion of Fe3O4@Cu2(OH)3NO3 and Cu2(OH)3NO3 are shown in Fig. 4. For the conversion of Fe3O4@Cu2(OH)3NO3 into Fe3O4@HKUST-1, the peaks attributed to the HKUST-1 structure appeared just after the ligand was injected (within 10 seconds). Within 50 seconds, plateaus were found in the intensities of the peaks corresponding to HKUST-1, including two intense peaks that appeared at Q = 0.67 and 0.82 Å−1 arising from the (022) and (222) planes of the HKUST-1 framework, respectively. This result indicates that the conversion process stopped approximately 50 seconds after the injection of the ligand into the capillary containing Fe3O4@Cu2(OH)3NO3 nanopowders. In comparison, the kinetics of the conversion of Cu2(OH)3NO3 into HKUST-1 was slightly slower than that with Fe3O4 nanoparticles and plateaus were found to occur approximately 90 seconds after the injection of the ligand. This suggests that the Fe3O4 nanoparticles induce a faster conversion of the Cu2(OH)3NO3 precursor into HKUST-1. This could be explained by the role of Fe3O4 nanoparticles as crystallization facilitators to promote faster MOF growth relative to the absence of nanoparticles.34–37
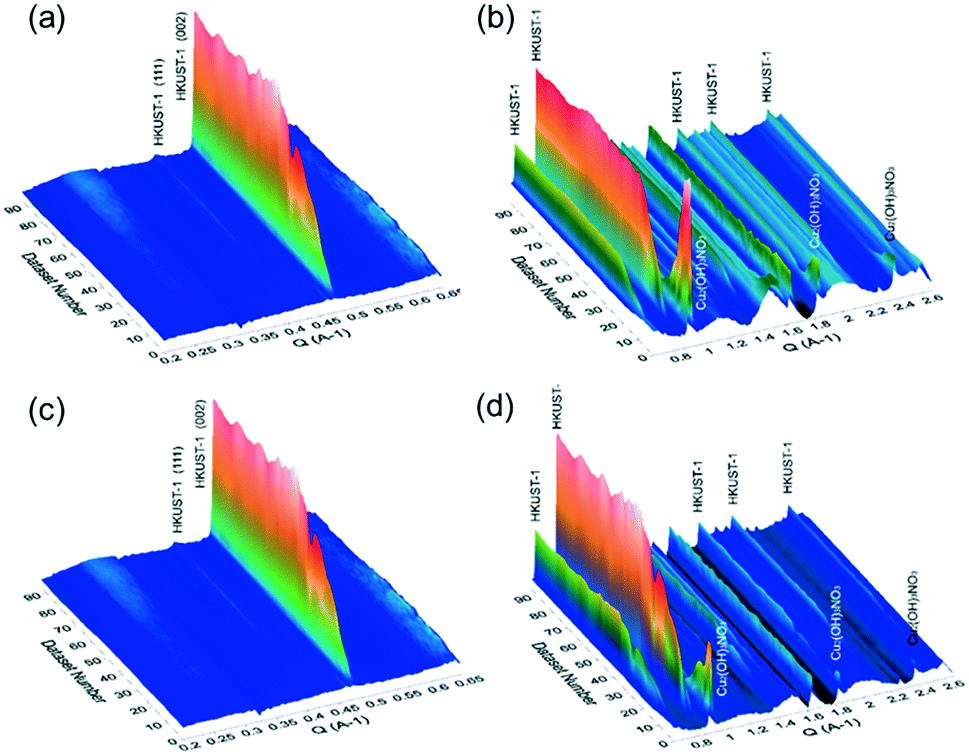 | ||
| Fig. 4 Time series of radially averaged SAXS (a and c) and WAXS (b and d) data. The nucleation and growth of Fe3O4@HKUST-1 (a and b) and HKUST-1 (c and d) at room temperature is observed when the alcoholic solution containing H3BTC is added to the solution containing Cu-based precursors. Dataset number corresponds to SAXS/WAXS images were collected every 3.15 s. The ligand was injected at t = 30 s (dataset 10). | ||
The local structure of Fe species in Fe3O4@HKUST-1 was investigated by Fe K-edge XAFS (XANES and EXAFS) measurements (Fig. 5a and b). The edge position as well as the shape of the XANES spectrum of Fe3O4@HKUST-1 correspond well to those of Fe3O4 used as a reference. Fourier transform of the EXAFS (FT-EXAFS) spectrum (without phase-shift corrections) of Fe3O4@HKUST-1 exhibits two strong peaks corresponding to the Fe–O shell (1.0–2.0 Å) and the Fe–Fe shell (2.5–3.5 Å) which can be seen in the reference Fe3O4 spectrum.58,59 These results indicate that the Fe species in Fe3O4@HKUST-1 exist as Fe3O4 without change during the conversion process.
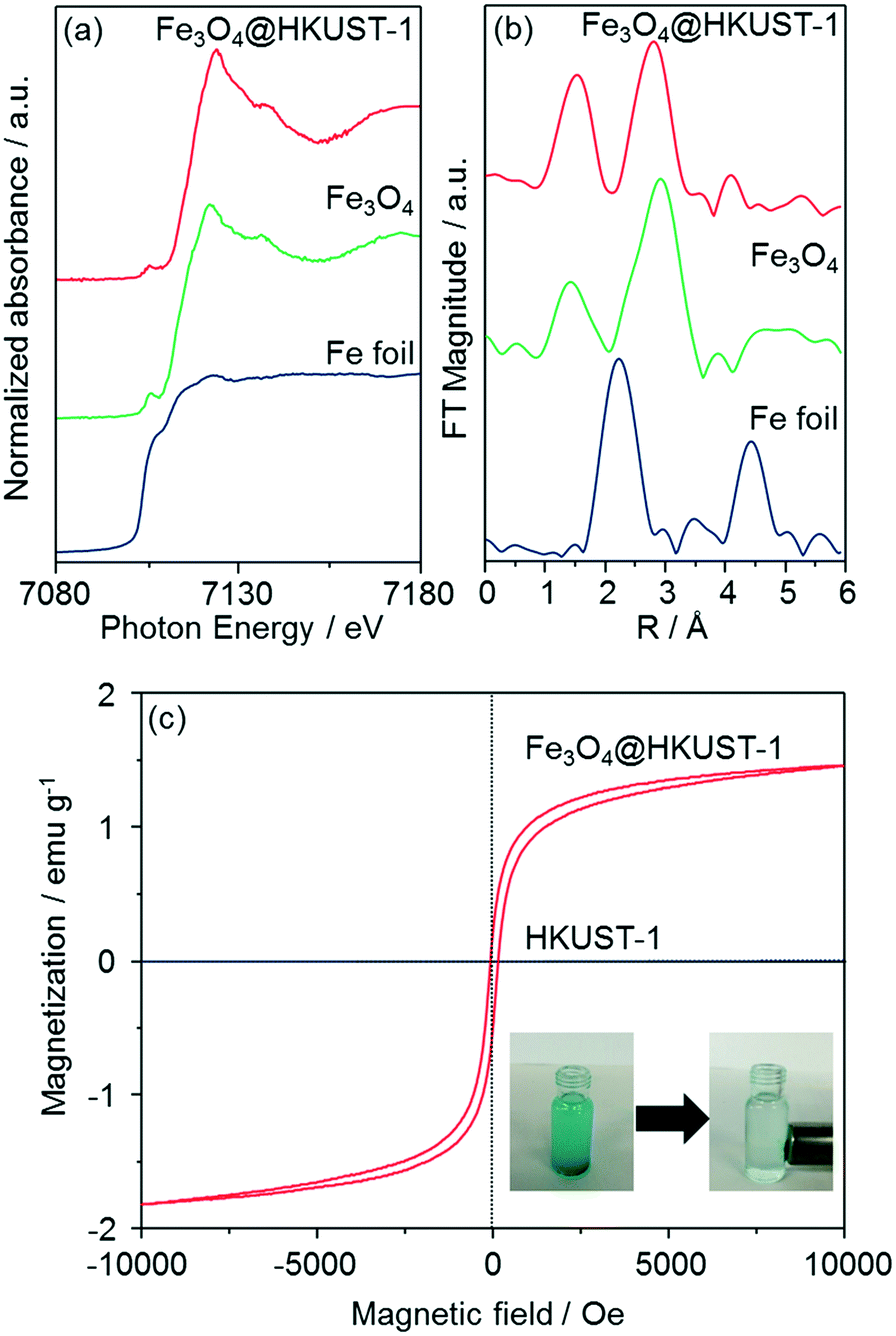 | ||
| Fig. 5 Fe K-edge (a) XANES and (b) FT-EXAFS spectra of Fe3O4, Fe3O4@HKUST-1 and Fe metallic foil. (c) Magnetization curves of Fe3O4@HKUST-1 and HKUST-1, and photographs of Fe3O4@HKUST-1 collected by a magnet. The magnetization measurements were performed at 300 K. | ||
The magnetic properties of Fe3O4@HKUST-1 were measured by a vibrating sample magnetometer with an applied field to investigate the ability of the composite to be used as a recyclable catalyst. Fig. 5c illustrates the room-temperature magnetization curves of Fe3O4@HKUST-1 and HKUST-1 without Fe3O4 nanoparticles. Unlike HKUST-1, Fe3O4@HKUST-1 displayed small hysteresis loops, suggesting that the incorporated Fe3O4 is ferromagnetic.60,61 We also demonstrated that Fe3O4@HKUST-1 is attracted to a magnet (inset of Fig. 5c) and, as a result, it can be easily separated after use as a heterogeneous catalyst.
Catalytic study (one-pot reaction)
Potential catalytic properties of Fe3O4@HKUST-1 were investigated through a one-pot sequential cascade reaction. One-pot reactions have drawn much attention as an attractive synthetic concept for improving overall process efficiency and reducing production wastes.62–66 Among them, we performed a sequential deacetalization–Knoevenagel condensation reaction. It is well-known that deacetalization reactions are catalysed by Brønsted acidic sites67,68 and Knoevenagel condensation reactions are catalysed by base sites.69,70 Also, Knoevenagel condensation reactions are catalysed by Lewis acid.71 Since MOFs contain Brønsted acidic sites and Lewis acidic sites due to carboxylic acid moieties (−COOH groups) and coordinatively unsaturated metal sites,72–75 HKUST-1 is, in principle, a suitable candidate for one-pot sequential cascade. To further investigate the potential of HKUST-1 as a catalyst, deacetalization and Knoevenagel condensation were individually performed and the catalytic activities of Fe3O4@HKUST-1 were tested. Fig. S4† shows the results of deacetalization of benzaldehyde dimethylacetal and Knoevenagel condensation of benzaldehyde with malononitrile. In the presence of Fe3O4@HKUST-1, the reaction efficiently proceeded to give benzaldehyde. Moreover, it was found from the inspection of the time course for Knoevenagel condensation shown in Fig. S5b† that Fe3O4@HKUST-1 also catalyzed the reaction, producing the corresponding benzylidene–malononitrile from benzaldehyde and malononitrile. These findings suggest that Fe3O4@HKUST-1 possesses both Brønsted acidic and Lewis acidic sites to produce benzylidene–malononitrile via two-step deacetalization and Knoevenagel condensation process.Next, Fe3O4@HKUST-1 was applied to one-pot sequential deacetalization and Knoevenagel condensation from benzaldehyde dimethylacetal and malononitrile to produce benzylidenemalononitrile. The reaction was carried out at 363 K using 1,4-dioxane as a solvent. Fig. 6 shows time course of the one-pot reaction. Benzylidenemalononitrile (3, square) was efficiently generated from benzaldehyde dimethylacetal (1, circle) via a pathway involving the initial formation of benzaldehyde (2, diamond), and that the yield of 3 reached 99% after a 5 h reaction time. It was also confirmed that the reaction did not take place in the absence of a catalyst (entry 11 in Table 1). For comparison purposes, other materials including conventional catalysts were applied for this one-pot reaction. The results are summarized in Table 1. The reaction was hardly promoted over Fe3O4 (entry 2), indicating that HKUST-1 played an important role in the progression of the reaction. While Fe3O4@HKUST-1 gave 99% of the final product, HKUST-1 made from Cu2(OH)3NO3 without Fe3O4 and HKUST-1_ref made by the conventional method gave 94% and 67% yields, respectively. This difference in the catalytic activity could be explained by taking into account the different particle size of the materials (See Fig. 2 and Fig. S5 in the ESI† for the particle size of HKUST-1_ref). Since HKUST-1 is a microporous material with ∼1 nm-size channels,47 which can restrict facile substrate diffusion within the particles, a catalyst with smaller particle size is thought to have more easily accessible surface area and better substrate diffusion.76 Additionally, by synthesizing the MOF from the ceramic material, a higher density of coordinatively unsaturated metal sites (defect sites) can be expected. The combination of the two causes would explain the high catalytic activity compared to HKUST-1 without Fe3O4 and HKUST-1_ref. Fe3O4@Cu2(OH)3NO3 as well as Cu(NO3)2·3H2O and H3BTC lead to only low yields under the same condition. The use of conventional heterogeneous catalysts such as MgO, Al2O3 and SiO2 showed reduced performance relative to Fe3O4@HKUST-1. These results demonstrate that Fe3O4@HKUST-1 served as an effective bifunctional catalyst for the sequential deacetalization and Knoevenagel condensation reaction.
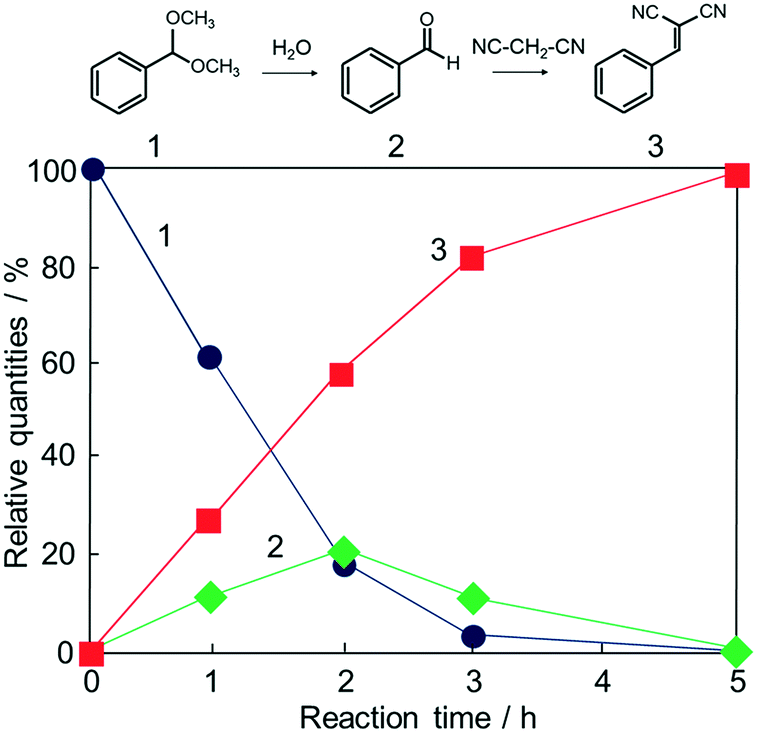 | ||
| Fig. 6 Time course of the one-pot deacetalization–Knoevenagel condensation reaction over Fe3O4@HKUST-1; benzaldehyde dimethylacetal (1, ●), benzaldehyde (2, ◆), benzylidenemalononitrile (3, ■). | ||
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Conversion (%) | Yield (%) | |
| 2 | 3 | |||
| a Reaction conditions: benzaldehyde dimethylacetal (1 mmol), malononitrile (3 mmol), 1,4-dioxane (4 mL), catalyst (50 mg), 363 K, 5 h. | ||||
| 1 | Fe3O4@HKUST-1 | 100 | 1 | 99 |
| 2 | Fe3O4 | 13 | 7 | 6 |
| 3 | HKUST-1 | 100 | 6 | 94 |
| 4 | HKUST-1_ref | 77 | 10 | 67 |
| 5 | Fe3O4@Cu2(OH)3NO3 | 19 | 5 | 14 |
| 6 | Cu(NO3)2·3H2O | 42 | 15 | 27 |
| 7 | H3BTC | 14 | 10 | 2 |
| 8 | MgO | 33 | 15 | 18 |
| 9 | Al2O3 | 38 | 16 | 22 |
| 10 | SiO2 | 8 | 4 | 4 |
| 11 | No catalyst | 2 | 0 | 2 |

Subsequently, recyclability tests using Fe3O4@HKUST-1 were performed. After a 5 h reaction, the Fe3O4@HKUST-1 catalyst was collected using a magnet, washed with 1,4-dioxane, dried at 313 K in air and then reused for the next catalytic test. As shown in Fig. 7a, Fe3O4@HKUST-1 could be reused at least 5 times with the retention of high catalytic activity and selectivity. The stability of Fe3O4@HKUST-1 was investigated by XRD measurements before and after the reaction (Fig. 7b). The diffraction pattern corresponding to the HKUST-1 structure was maintained after the reaction, indicating that Fe3O4@HKUST-1 possesses good resistance to degradation during one-pot reaction cascades. Furthermore, a leaching test was performed, as shown in Fig. S6.† Specifically, we observed that removal of the catalyst following a 1 h period caused the reaction to cease, indicating that the catalyst has heterogeneous nature.
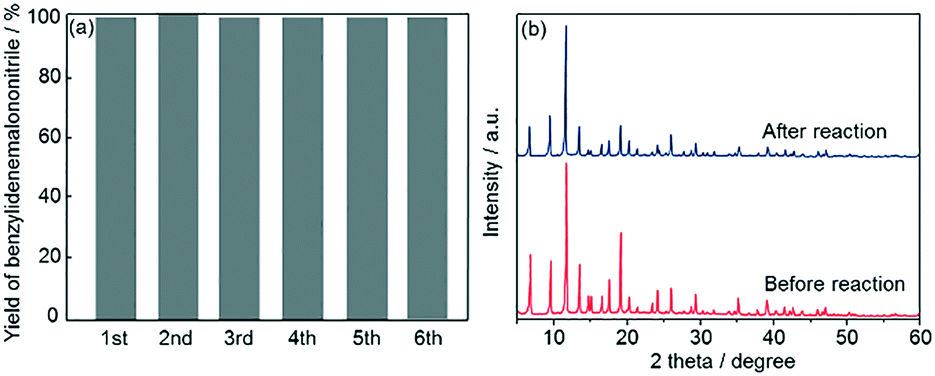 | ||
| Fig. 7 (a) Reusability study of Fe3O4@HKUST-1 for the one-pot deacetalization–Knoevenagel condensation reaction. (b) XRD patterns of Fe3O4@HKUST-1 before and after the catalytic reaction. | ||
Catalytic study (hydrogenation of olefin)
To further exploit the versatility of the current magnetic MOF nanocomposite, Pd nanoparticles were added into the solution containing Fe3O4@Cu2(OH)3NO3, then the conversion into MOF was promoted with the addition of the linker solution. The formation of HKUST-1 was confirmed by XRD analysis (Fig. S7†) performed on the magnetically collected and washed powders. An ICP-OES analysis was carried out to determine the loading amount of Pd. The result revealed that the magnetic composite contained 0.32 wt% of Pd. In order to clarify the state of Pd in the material, XAFS measurements were performed and are shown in Fig. S8.† The edge position and the shape of the XANES spectrum of Pd/Fe3O4@HKUST-1 are similar to those of the Pd metallic foil used as a reference. The FT-EXAFS spectrum (without phase-shift corrections) of the magnetic composite shows one peak corresponding to neighbouring Pd atoms (2.0–3.0 Å). Other peaks arising from potential Pd oxides were not observed in the spectrum. These data indicate that Pd species in Pd/Fe3O4@HKUST-1 system mainly exist in a metal state without a significant change during the conversion process. The composite was investigated using TEM to further confirm the presence of Pd particles in the material (Fig. S9†). The results clearly show 10–20 nm Pd nanoparticles located within the material. The prepared Pd/Fe3O4@HKUST-1 composite has been utilized for hydrogenation reaction of 1-octene to octane in the liquid phase. While no reaction occurred when using Fe3O4@HKUST-1, the presence of Pd/Fe3O4@HKUST-1 successfully produced octane from 1-octene.Fig. 8 shows the time course of the reaction. The yield of octane was 98% within 3 h reaction time. These results indicate that Pd nanoparticles act as an effective catalyst for this hydrogenation reaction while the pure Fe3O4@HKUST-1 does not show appreciable catalytic activity. In order to check the reusability and stability of Pd/Fe3O4@HKUST-1, recycling tests and XRD measurements were performed (Fig. S10†). Pd/Fe3O4@HKUST-1 could be recycled at least 5 times without significant activity loss. Moreover, XRD patterns of the catalyst did not significantly change after the catalytic reaction. As in the sequential deacetalization–Knoevenagel condensation reaction, a leaching test further confirmed that there is no contribution from the leached species (Fig. S11†). Furtheremore, ICP-OES analysis was carried out for the reaction solution after the reaction using Pd/Fe3O4@HKUST-1. The obtained result showed that Pd and Fe species in the solution were below the detection limit (ca. 10 ppm). These results indicate the durability of the Pd/Fe3O4@HKUST-1 as a multifunctional material for catalytic applications. However, it should be noted here that slight morphological change in the catalysts might be caused during the reactions and this could influence the catalytic activity and selectivity77,78 although such changes in the catalysis were not observed for the reactions employed in this study. Compared to previous studies describing the synthesis of complex MOF composites prepared by multistep processes,79,80 the novel conversion method developed in this study is facile and enables the incorporation of different functional nanoparticles, without significant changes81,82 or the need for elaborate structures.79,80,83
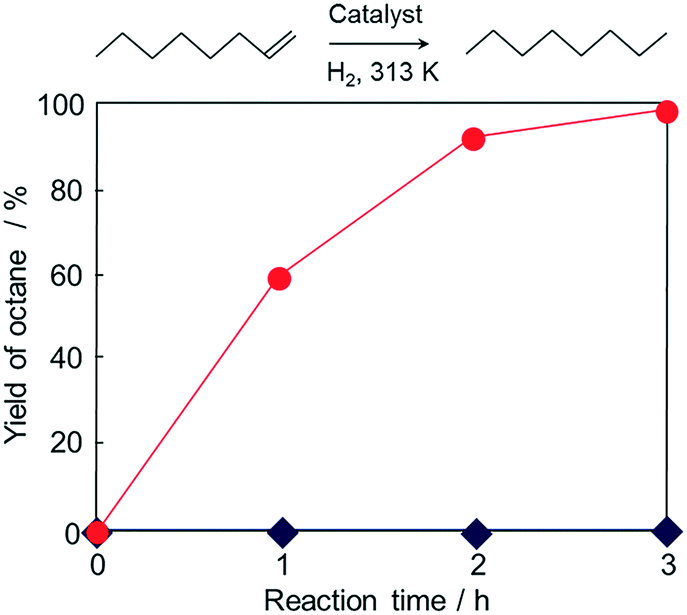 | ||
| Fig. 8 Time course of hydrogenation of 1-octene over Pd/Fe3O4@HKUST-1 (●) and Fe3O4@HKUST-1 (◆). | ||
Experimental details
Materials and methods
Ethanol (EtOH), Cu(NO3)2·3H2O, FeSO4·7H2O and 1,3,5-benzenetricarboxylic acid (H3BTC) were purchased from Acros Organics. Citric acid was obtained from Aldrich Chemical Co. 30% aq. Ammonia was provided by Chem-Supply. 1,4-Dioxane, NaOH, PdCl2, polyvinylpyrrolidone (PVP) K30 and NaBH4 were supplied by Nacalai Tesque Inc. Benzaldehyde dimethylacetal, benzaldehyde, malononitrile, 1-octene and n-decane were purchased from Tokyo Chemical Industry Co., Ltd. MgO was obtained from Kishida Chemical Co., Ltd. Al2O3 was purchased from Evonik. All materials were used as received without purification.Materials preparation
Fe3O4 nanoparticles were prepared according to the method provided by Hui and co-workers.48 In a typical procedure, NaOH (1.60 g, 40.0 mmol) is dissolved, under magnetic stirring, in 190 mL of water, in a 250 mL round-bottomed single-neck flask. Subsequently, NaNO3 (0.170 g, 2.00 mmol) and Na3C6H5O7·2H2O (2.94 g, 10.0 mmol) are added and the mixture heated to 100 °C for 1 h. In another container, FeSO4·7H2O (5.56 g, 20.0 mmol) is dissolved in 10.0 mL of deionized water using an ultrasound bath. The iron(II) solution is added at once into the hydroxide/citrate solution under vigorous stirring (>500 rpm), and the mixture turns to dark brown in few seconds. The flask is kept stirring at 100 °C for an additional hour, then the stirbar removed, and the mixture let to cool down naturally. The magnetic nanoparticles are collected using a commercial N45 magnet, and the supernatant discarded. The magnetic nanoparticles are placed in another vessel and suspended in 100 mL of deionized water, using an ultrasound bath, for 30 min. The material is collected again as described above, and this washing procedure is repeated 3 times. At the end, the magnetic nanoparticles are shaken in 100 mL of acetone, then dried in a rotary evaporator. This water-stripping procedure is repeated twice, then the obtained brown to black material is dried in a vacuum oven (<30 mbar) at 60 °C for at least 3 h. The prepared sample is denoted as Fe3O4@Cu2(OH)3NO3.Fe3O4@HKUST-1 was prepared from Fe3O4@Cu2(OH)3NO3 as follows: Fe3O4@Cu2(OH)3NO3 (97 mg) was added to the mixed solution of 5 mL of EtOH and 2 mL of water containing H3BTC (106 mg) as organic linker. The mixture was stirred for 10 min at room temperature. Then, crystals were separated from the Fe3O4@HKUST-1 composite by the localization of the magnetically responsive composite with a commercial magnet. The resulting powder was washed with water and EtOH, and then collected via centrifugation at 5000 rpm. The collected samples were dried under vacuum.
Pd nanoparticles were prepared by a modified version of the previously reported procedure proposed by Hou and co-workers.84 Typically, PdCl2 (20 mg), PVP (834 mg), and 20 mL of H2O were mixed and stirred at 0 °C in an ice bath. A mixture of NaBH4 (30 mg) and 0.5 mL of H2O was then added in the above solution, which was stirred for 30 min.
Pd/Fe3O4@HKUST-1 was prepared similarly to the synthesis of Fe3O4@HKUST-1 by adding colloidal Pd nanoparticles. Fe3O4@Cu2(OH)3NO3 (97 mg) and 5 mL of the above Pd colloidal solution were added to the mixed solution of 5 mL of EtOH and 2 mL of water containing H3BTC (106 mg) as the organic linker. The mixture was stirred for 10 min at room temperature. Then, the magnetic framework composite crystals were isolated from the pure crystals not containing Fe3O4 particles by magnetic collection. The resulting powder was washed with water and EtOH, and then collected via centrifugation at 5000 rpm. The collected samples were dried under vacuum.
General methods
The surface morphologies of samples were observed using a field emission scanning electron microscope (FE-SEM; Merlin; Carl Zeiss Germany) with a thin iridium film coating. Standard θ–2θ X-ray diffraction (XRD) data were recorded on a Rigaku Smart Lab diffractometer using Cu Kα radiation (λ = 1.5406 Å). Inductively coupled plasma optical emission spectroscopy (ICP-OES) was performed by using Varian 730-ES axial ICP-OES. Nitrogen adsorption–desorption isotherms were collected using a BEL-SORP mini (BEL Japan, Inc.) at 77 K. Fourier transform infrared spectroscopy (FT-IR: ALPHA FT-IR spectrometer, Bruker Optik GmbH) was employed in the ATR configuration. Thermogravimetric analysis (TGA) was carried out using a thermal analyzer (Shimadzu DTG-60), with a heating rate of 10 K min−1 in air. Small-angle X-ray scattering (SAXS) and wide-angle X-ray scattering (WAXS) were performed at the Australian Synchrotron. A capillary with a 1.5 mm diameter was used for the in situ characterization by SAXS and WAXS. Once filled with the solution, the capillary was illuminated by a high-intensity X-ray beam at the beamline, and the scattered radiation was collected by the detector (Pilatus 1 M). An on-axis video camera allowed for parallax free sample viewing and alignment at all times before and during exposure, enabling precise and rapid sample alignment. Transmission electron microscope (TEM) images were recorded using a JEM-2000FX operating under a 200 kV accelerating voltage. XAFS (XANES and EXAFS) spectra were recorded at the BL-01B1 facility of SPring-8 at the Japan Synchrotron Radiation Research Institute (JASRI). Fe K-edge and Pd K-edge XAFS spectra were recorded in the fluorescence mode using a Si(111) double-crystal monochromator at room temperature. Magnetization curves were recorded using a vibrating sample magnetometer (VSM, Riken Denshi Co., Ltd., BHV-50H).Catalytic study
One-pot deacetalization–Knoevenagel condensation reaction was carried out in a 35 mL glass reactor. A solution of benzaldehyde dimethylacetal (1 mmol), malononitrile (5 mmol) and 4 mL of 1,4-dioxane was stirred at 90 °C with 50 mg of the catalysts in powder form. The progression of the reaction was monitored using gas chromatography (Shimadzu GC-14B with a flame ionization detector) equipped with an InertCap®1 capillary column.Reusability of the catalyst was studied as follows. After the first run, the catalyst was collected by a magnet, washed three times with 1,4-dioxane, dried at 40 °C in air and reused for the next run. The above procedure was repeated five times.
The hydrogenation of 1-octene was performed to evaluate the catalytic activity of Pd/Fe3O4@HKUST-1. The catalysts (50 mg), 1-octene (5 mmol), and 10 mL of methanol were introduced into a glass reaction vessel with a reflux condenser. The resulting mixture was bubbled with hydrogen for 15 min and then stirred at 40 °C under hydrogen bubbling (10 mL min−1). The progress of the reaction was monitored by gas chromatography analysis using an internal standard technique (n-decane).
Conclusions
Magnetic Fe3O4 nanoparticles have been successfully incorporated into HKUST-1 through the conversion of a Cu-based ceramic precursor. Fe3O4 played a significant role in the conversion process from ceramic to HKUST-1 with an influence on the kinetics (faster) and the MOF particle size (smaller). It was also revealed by several characterization techniques that the magnetic Fe3O4 particles were incorporated into HKUST-1 without causing significant aggregation. The prepared material (Fe3O4@HKUST-1) has been successfully utilized as a magnetically recyclable heterogeneous catalyst to promote one-pot sequential deacetalization–Knoevenagel condensation reaction. In addition, the results showed that Fe3O4/HKUST-1 exhibits higher catalytic activities than conventional inorganic heterogeneous catalysts (e.g. Al2O3, SiO2, MgO). Pd nanoparticles were also incorporated alongside Fe3O4 within HKUST-1 and successfully used for the hydrogenation of 1-octene in the liquid phase. These findings suggest new possibilities for the encapsulation of different nanoparticles within MOFs demonstrating that multifunctional catalysts can be obtained through the conversion from ceramic composites into MOF composites.Acknowledgements
The present work is supported by “ACCEL Project” from the Japan Science and Technology Agency, by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (No. 25410241 and 15K17903) and Strategic Young Researcher Overseas Visits Program for Accelerating Brain Circulation. Part of this research was undertaken on the SAXS/WAXS beamline at the Australian Synchrotron, Melbourne, Victoria, Australia with the support of Dr. Nigel Kirby and Dr. Adrian Hawley. X-ray absorption measurements were performed at the BL-01B1 facility of SPring-8 at the Japan Synchrotron Radiation Research Institute (JASRI) (No. 2014B1279).References
- S. Furukawa, J. Reboul, S. Diring, K. Sumida and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5700–5734 RSC.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- G. Férey, Chem. Soc. Rev., 2007, 37, 191–214 RSC.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed.
- V. Guillerm, D. Kim, J. F. Eubank, R. Luebke, X. Liu, K. Adil, M. S. Lah and M. Eddaoudi, Chem. Soc. Rev., 2014, 43, 6141–6172 RSC.
- K. Liang, R. Ricco, J. Reboul, S. Furukawa and P. Falcaro, in The Sol-Gel Handbook, ed. D. Levy and M. Zayat, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2015, pp. 471–486 Search PubMed.
- J. D. Evans, C. J. Sumby and C. J. Doonan, Chem. Soc. Rev., 2014, 43, 5933–5951 RSC.
- S. Ou and C.-D. Wu, Inorg. Chem. Front., 2014, 1, 721–734 RSC.
- D. Bradshaw, A. Garai and J. Huo, Chem. Soc. Rev., 2012, 41, 2344–2381 RSC.
- A. Aijaz, A. Karkamkar, Y. J. Choi, N. Tsumori, E. Rönnebro, T. Autrey, H. Shioyama and Q. Xu, J. Am. Chem. Soc., 2012, 134, 13926–13929 CrossRef CAS PubMed.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- M. L. Foo, R. Matsuda and S. Kitagawa, Chem. Mater., 2014, 26, 310–322 CrossRef CAS.
- K. Liang, R. Ricco, C. M. Doherty, M. J. Styles, S. Bell, N. Kirby, S. Mudie, D. Haylock, A. J. Hill, C. J. Doonan and P. Falcaro, Nat. Commun., 2015, 6, 7240 CrossRef CAS PubMed.
- T. Toyao, M. Saito, S. Dohshi, K. Mochizuki, M. Iwata, H. Higashimura, Y. Horiuchi and M. Matsuoka, Res. Chem. Intermed., 2016, 42, 7679–7688 CrossRef CAS.
- P. Falcaro, R. Ricco, A. Yazdi, I. Imaz, S. Furukawa, D. Maspoch, R. Ameloot, J. D. Evans and C. J. Doonan, Coord. Chem. Rev., 2016, 307, 237–254 CrossRef CAS.
- C. M. Doherty, E. Knystautas, D. Buso, L. Villanova, K. Konstas, A. J. Hill, M. Takahashi and P. Falcaro, J. Mater. Chem., 2012, 22, 11470–11474 RSC.
- P. Falcaro, R. Ricco, C. M. Doherty, K. Liang, A. J. Hill and M. J. Styles, Chem. Soc. Rev., 2014, 43, 5513–5560 RSC.
- M. Kim, J. F. Cahill, Y. Su, K. A. Prather and S. M. Cohen, Chem. Sci., 2012, 3, 126–130 RSC.
- P. Falcaro and S. Furukawa, Angew. Chem., Int. Ed., 2012, 51, 8431–8433 CrossRef CAS PubMed.
- C. J. Doonan, W. Morris, H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 9492–9493 CrossRef CAS PubMed.
- Y. Liu and Z. Tang, Adv. Mater., 2013, 25, 5819–5825 CrossRef CAS PubMed.
- W. Zhang, G. Lu, S. Li, Y. Liu, H. Xu, C. Cui, W. Yan, Y. Yang and F. Huo, Chem. Commun., 2014, 50, 4296–4298 RSC.
- H. Zhang, S. Qi, X. Niu, J. Hu, C. Ren, H. Chen and X. Chen, Catal. Sci. Technol., 2014, 4, 3013–3024 CAS.
- R. Ricco, L. Malfatti, M. Takahashi, A. J. Hill and P. Falcaro, J. Mater. Chem. A, 2013, 1, 13033–13045 CAS.
- P. Falcaro, F. Normandin, M. Takahashi, P. Scopece, H. Amenitsch, S. Costacurta, C. M. Doherty, J. S. Laird, M. D. H. Lay, F. Lisi, A. J. Hill and D. Buso, Adv. Mater., 2011, 23, 3901–3906 CrossRef CAS PubMed.
- M. Meilikhov, K. Yusenko, D. Esken, S. Turner, G. Van Tendeloo and R. A. Fischer, Eur. J. Inorg. Chem., 2010, 2010, 3701–3714 CrossRef.
- M. Müller, S. Hermes, K. Kähler, M. W. E. van den Berg, M. Muhler and R. A. Fischer, Chem. Mater., 2008, 20, 4576–4587 CrossRef.
- H.-L. Jiang, T. Akita, T. Ishida, M. Haruta and Q. Xu, J. Am. Chem. Soc., 2011, 133, 1304–1306 CrossRef CAS PubMed.
- C. Rösler and R. A. Fischer, CrystEngComm, 2015, 17, 199–217 RSC.
- G. Li, H. Kobayashi, K. Kusada, J. M. Taylor, Y. Kubota, K. Kato, M. Takata, T. Yamamoto, S. Matsumura and H. Kitagawa, Chem. Commun., 2014, 50, 13750–13753 RSC.
- G. Lu, S. Li, Z. Guo, O. K. Farha, B. G. Hauser, X. Qi, Y. Wang, X. Wang, S. Han, X. Liu, J. S. DuChene, H. Zhang, Q. Zhang, X. Chen, J. Ma, S. C. J. Loo, W. D. Wei, Y. Yang, J. T. Hupp and F. Huo, Nat. Chem., 2012, 4, 310–316 CrossRef CAS PubMed.
- K. Sugikawa, Y. Furukawa and K. Sada, Chem. Mater., 2011, 23, 3132–3134 CrossRef CAS.
- C. M. Doherty, D. Buso, A. J. Hill, S. Furukawa, S. Kitagawa and P. Falcaro, Acc. Chem. Res., 2014, 47, 396–405 CrossRef CAS PubMed.
- D. Buso, K. M. Nairn, M. Gimona, A. J. Hill and P. Falcaro, Chem. Mater., 2011, 23, 929–934 CrossRef CAS.
- D. Buso, A. J. Hill, T. Colson, H. J. Whitfield, A. Patelli, P. Scopece, C. M. Doherty and P. Falcaro, Cryst. Growth Des., 2011, 11, 5268–5274 CAS.
- K. Sugikawa, S. Nagata, Y. Furukawa, K. Kokado and K. Sada, Chem. Mater., 2013, 25, 2565–2570 CrossRef CAS.
- T. Tsuruoka, H. Kawasaki, H. Nawafune and K. Akamatsu, ACS Appl. Mater. Interfaces, 2011, 3, 3788–3791 CAS.
- Y. Abdollahian, J. L. Hauser, I. R. Colinas, C. Agustin, A. S. Ichimura and S. R. J. Oliver, Cryst. Growth Des., 2014, 14, 1506–1509 CAS.
- P. Falcaro, A. J. Hill, K. M. Nairn, J. Jasieniak, J. I. Mardel, T. J. Bastow, S. C. Mayo, M. Gimona, D. Gomez, H. J. Whitfield, R. Riccò, A. Patelli, B. Marmiroli, H. Amenitsch, T. Colson, L. Villanova and D. Buso, Nat. Commun., 2011, 2, 237 CrossRef PubMed.
- Y. Mao, J. Li, W. Cao, Y. Ying, P. Hu, Y. Liu, L. Sun, H. Wang, C. Jin and X. Peng, Nat. Commun., 2014, 5, 5532 CrossRef PubMed.
- Y. Liu, W. Zhang, S. Li, C. Cui, J. Wu, H. Chen and F. Huo, Chem. Mater., 2014, 26, 1119–1125 CrossRef CAS.
- J. Reboul, S. Furukawa, N. Horike, M. Tsotsalas, K. Hirai, H. Uehara, M. Kondo, N. Louvain, O. Sakata and S. Kitagawa, Nat. Mater., 2012, 11, 717–723 CrossRef CAS PubMed.
- J. Reboul, K. Yoshida, S. Furukawa and S. Kitagawa, CrystEngComm, 2014, 17, 323–330 RSC.
- G. Majano and J. Pérez-Ramírez, Adv. Mater., 2013, 25, 1052–1057 CrossRef CAS PubMed.
- T. Toyao, K. Liang, K. Okada, R. Ricco, M. J. Styles, Y. Tokudome, Y. Horiuchi, A. J. Hill, M. Takahashi, M. Matsuoka and P. Falcaro, Inorg. Chem. Front., 2015, 2, 434–441 RSC.
- S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed.
- C. Hui, C. Shen, T. Yang, L. Bao, J. Tian, H. Ding, C. Li and H.-J. Gao, J. Phys. Chem. C, 2008, 112, 11336–11339 CAS.
- I. Y. Park, K. Kuroda and C. Kato, Solid State Ionics, 1990, 42, 197–203 CrossRef CAS.
- G. Socrates, Infrared and Raman characteristic group frequencies: tables and charts, Wiley, Chichester, 3rd edn, 2010, as paperback Search PubMed.
- K. Nakamoto, Infrared and Raman spectra of inorganic and coordination compounds, Wiley, Hoboken, N.J, 6th edn, 2009 Search PubMed.
- A. Yin, X. Guo, W.-L. Dai and K. Fan, J. Phys. Chem. C, 2009, 113, 11003–11013 CAS.
- J. L. C. Rowsell and O. M. Yaghi, J. Am. Chem. Soc., 2006, 128, 1304–1315 CrossRef CAS PubMed.
- S. Loera-Serna, L. L. Núñez, J. Flores, R. López-Simeon and H. I. Beltrán, RSC Adv., 2013, 3, 10962–10972 RSC.
- S. Loera-Serna, M. A. Oliver-Tolentino, M. de Lourdes López-Núñez, A. Santana-Cruz, A. Guzmán-Vargas, R. Cabrera-Sierra, H. I. Beltrán and J. Flores, J. Alloys Compd., 2012, 540, 113–120 CrossRef CAS.
- J. Huo, M. Brightwell, S. El Hankari, A. Garai and D. Bradshaw, J. Mater. Chem. A, 2013, 1, 15220–15223 CAS.
- K. Okada, R. Ricco, Y. Tokudome, M. J. Styles, A. J. Hill, M. Takahashi and P. Falcaro, Adv. Funct. Mater., 2014, 24, 1969–1977 CrossRef CAS.
- L. Signorini, L. Pasquini, L. Savini, R. Carboni, F. Boscherini, E. Bonetti, A. Giglia, M. Pedio, N. Mahne and S. Nannarone, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 195423 CrossRef.
- G. Subías, J. García and J. Blasco, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 155103 CrossRef.
- T. Yang, C. Shen, Z. Li, H. Zhang, C. Xiao, S. Chen, Z. Xu, D. Shi, J. Li and H. Gao, J. Phys. Chem. B, 2005, 109, 23233–23236 CrossRef CAS PubMed.
- Z. L. Liu, Y. J. Liu, K. L. Yao, Z. H. Ding, J. Tao and X. Wang, J. Mater. Synth. Process., 2002, 10, 83–87 CrossRef CAS.
- K. Motokura, M. Tada and Y. Iwasawa, J. Am. Chem. Soc., 2009, 131, 7944–7945 CrossRef CAS PubMed.
- S. Shylesh, A. Wagener, A. Seifert, S. Ernst and W. R. Thiel, Angew. Chem., Int. Ed., 2010, 49, 184–187 CrossRef CAS PubMed.
- M. Sasidharan, S. Fujita, M. Ohashi, Y. Goto, K. Nakashima and S. Inagaki, Chem. Commun., 2011, 47, 10422–10424 RSC.
- A. Corma, T. Ródenas and M. J. Sabater, J. Catal., 2011, 279, 319–327 CrossRef CAS.
- T. Toyao, M. Saito, Y. Horiuchi and M. Matsuoka, Catal. Sci. Technol., 2014, 4, 625–628 CAS.
- R. A. Duval, R. L. Allmon and J. R. Lever, J. Med. Chem., 2007, 50, 2144–2156 CrossRef CAS PubMed.
- F. Douelle, A. S. Capes and M. F. Greaney, Org. Lett., 2007, 9, 1931–1934 CrossRef CAS PubMed.
- S. Hasegawa, S. Horike, R. Matsuda, S. Furukawa, K. Mochizuki, Y. Kinoshita and S. Kitagawa, J. Am. Chem. Soc., 2007, 129, 2607–2614 CrossRef CAS PubMed.
- Y. Horiuchi, T. Toyao, M. Fujiwaki, S. Dohshi, T.-H. Kim and M. Matsuoka, RSC Adv., 2015, 5, 24687–24690 RSC.
- M. J. Climent, A. Corma, S. Iborra and A. Velty, J. Mol. Catal. A: Chem., 2002, 182–183, 327–342 CrossRef CAS.
- M. Opanasenko, A. Dhakshinamoorthy, M. Shamzhy, P. Nachtigall, M. Horáček, H. Garcia and J. Čejka, Catal. Sci. Technol., 2013, 500–507 CAS.
- L. Mitchell, B. Gonzalez-Santiago, J. P. S. Mowat, M. E. Gunn, P. Williamson, N. Acerbi, M. L. Clarke and P. A. Wright, Catal. Sci. Technol., 2013, 3, 606–617 CAS.
- R. Ameloot, F. Vermoortele, J. Hofkens, F. C. De Schryver, D. E. De Vos and M. B. J. Roeffaers, Angew. Chem., Int. Ed., 2013, 52, 401–405 CrossRef CAS PubMed.
- T. Toyao, M. Fujiwaki, Y. Horiuchi and M. Matsuoka, RSC Adv., 2013, 3, 21582–21587 RSC.
- T. Kiyonaga, M. Higuchi, T. Kajiwara, Y. Takashima, J. Duan, K. Nagashima and S. Kitagawa, Chem. Commun., 2015, 51, 2728–2730 RSC.
- D. Loffreda, D. Simon and P. Sautet, J. Catal., 2003, 213, 211–225 CrossRef CAS.
- F. Calle-Vallejo, D. Loffreda, M. T. M. Koper and P. Sautet, Nat. Chem., 2015, 7, 403–410 CrossRef CAS PubMed.
- J.-M. Yan, Z.-L. Wang, L. Gu, S.-J. Li, H.-L. Wang, W.-T. Zheng and Q. Jiang, Adv. Energy Mater., 2015, 5, 1500107 CrossRef.
- F. Ke, L. Wang and J. Zhu, Nanoscale, 2015, 7, 1201–1208 RSC.
- M. Zhang, Y. Yang, C. Li, Q. Liu, C. T. Williams and C. Liang, Catal. Sci. Technol., 2014, 4, 329–332 CAS.
- L. Chen, H. Chen and Y. Li, Chem. Commun., 2014, 50, 14752–14755 RSC.
- L. Lin, T. Zhang, H. Liu, J. Qiu and X. Zhang, Nanoscale, 2015, 7, 7615–7623 RSC.
- W. Hou, N. Dehm and R. Scott, J. Catal., 2008, 253, 22–27 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: SEM, TEM, FTIR, XRD, XANES, FT-EXAFS, and TGA analyses on ceramics and MOFs, catalytic tests on deacetalization and Knoevenagel condensation reactions, and reusability test on hydrogenation reaction. See DOI: 10.1039/c7ce00390k |
| This journal is © The Royal Society of Chemistry 2017 |