
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modular synthesis of thirty lead-like scaffolds suitable for CNS drug discovery†
Joan
Mayol-Llinàs
 a,
William
Farnaby‡
b and
Adam
Nelson
*ac
a,
William
Farnaby‡
b and
Adam
Nelson
*ac
aSchool of Chemistry, University of Leeds, LS2 9JT, UK. E-mail: a.s.nelson@leeds.ac.uk
bTakeda Cambridge, Unit 418, Cambridge Science Park, Milton Road, Cambridge, CB4 0PA, UK
cAstbury Centre for Structural Molecular Biology, University of Leeds, LS2 9JT, UK
First published on 3rd November 2017
Abstract
A modular synthetic approach was developed that yielded thirty diverse lead-like scaffolds suitable for CNS drug discovery.
Controlling molecular properties is a challenge that is intrinsic to drug discovery.1 The challenge is intensified in central nervous system (CNS) programmes in order that CNS drugs are able to cross the blood–brain barrier.2 The properties of drugs differ from those of high-quality lead molecules since molecular weight, lipophilicity and complexity tend to increase during optimisation.3 Sourcing large numbers of lead-like screening compounds4 is, however, a significant challenge that is heightened by the limited scaffold diversity of historically explored chemical space.5 To address this challenge, the development of new synthetic methods to support discovery applications is being increasingly informed by prospective molecular property, diversity and novelty analyses.6,7 To facilitate the exploration of lead-like space for CNS drug discovery,8 we recently adapted a multi-parameter optimisation (MPO) system8b for scoring CNS drugs.9 We have now exploited our MPO scoring system to guide the development and exemplification of a unified synthetic approach to diverse sp3-rich10 lead-like scaffolds suitable for CNS drug discovery.11 In addition, the approach was designed to yield scaffolds adhering to accepted CNS design principles: small and rigid scaffolds8a that could yield screening compounds lacking functionality that could contribute to poor permeability and high efflux (e.g. carboxylate and sulfonyl groups and multiple amides, basic centres and hydrogen bond donors).8e These characteristics were largely captured in our filtering and scoring process.
Our synthetic approach (Scheme 1) exploits cyclisation precursors 1 with an embedded ring system (blue) and up to four functional group handles (green, light blue, beige and yellow blobs). It was envisaged that product scaffolds would be formed by reaction between pairs of functional groups, in some cases in conjunction with an external reactant (red). Crucially, the approach should result in significant product scaffold diversity including fused (light blue to yellow e.g.2; light blue to green e.g.3), bridged (green to beige e.g.4) and spirocyclic (green to yellow e.g.5 and 6) ring systems.
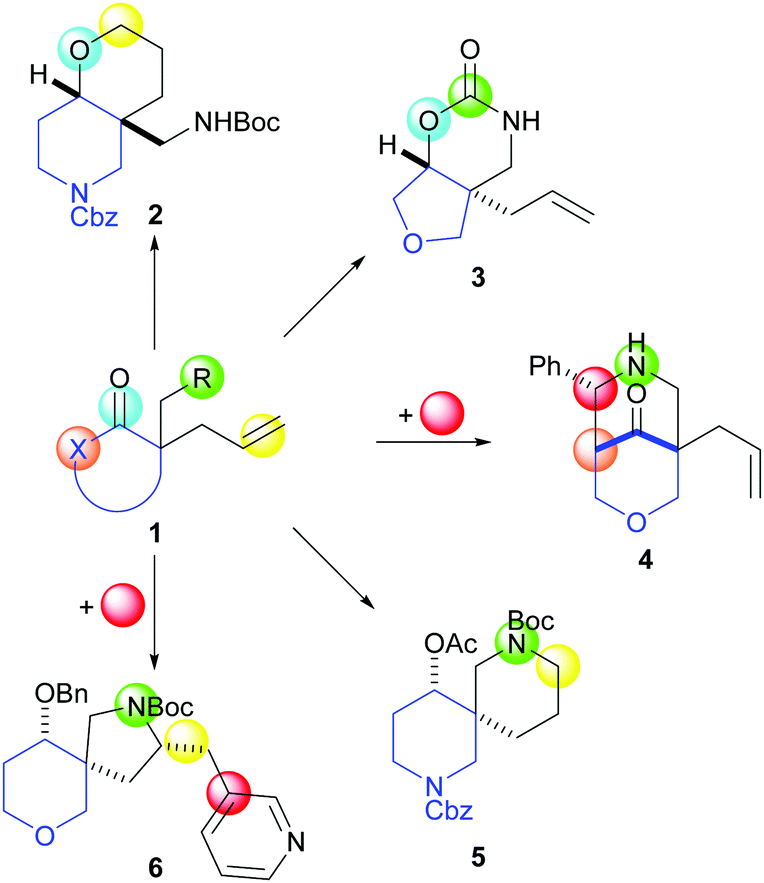 | ||
| Scheme 1 Envisaged approach for the synthesis of diverse lead-like scaffolds. | ||
A range of cyclisation precursors 1 was prepared (Scheme 2). For example, reaction of the lithium enolate of 7 with the carbonyl imidazole 8 (→ 9),12 and base-catalysed reaction with the α-NHBoc sulfone 10, gave the β-keto ester 11; Pd-catalysed decarboxylative allylation13 then gave the exemplar cyclisation precursor 1a (panel A).
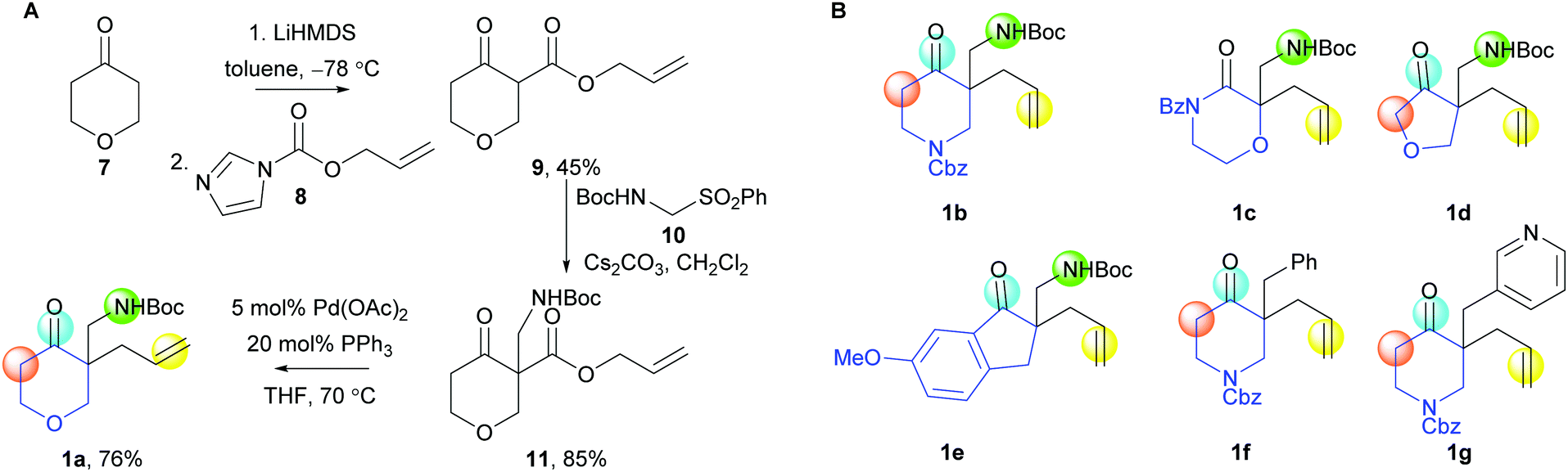 | ||
| Scheme 2 Synthesis of the cyclisation precursors 1. Panel A: Synthesis of 1a. Panel B: Other cyclisation precursors prepared (see ESI†). | ||
A toolkit of cyclisations was developed using the exemplar substrate 1a (Scheme 3). Initially, cyclisations to give fused scaffolds were investigated that involved the ketone (light blue) and either the alkene (yellow) or the Boc-protected amine (green) in the cyclisation precursor 1a. Hydroboration–oxidation of 1a gave the hemiacetal 12 which was reduced (Et3SiH, TFA) to give, after reprotection, 13 as a single diastereomer. Ozonolysis of 1a gave the hemiaminals 14 which, on treatment with benzylamine and NaBH(OAc)3, underwent double reductive amination to yield 15 as a 83![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :17 mixture of diastereomers. Alternatively, reduction of the ketone 1a with iBu2AlH gave the corresponding secondary alcohol 16a which iodocyclised to give 17 with high diastereoselectivity. Finally, treatment of the alcohol 16a with tBuOK gave the fused bicyclic carbamate 18.
:17 mixture of diastereomers. Alternatively, reduction of the ketone 1a with iBu2AlH gave the corresponding secondary alcohol 16a which iodocyclised to give 17 with high diastereoselectivity. Finally, treatment of the alcohol 16a with tBuOK gave the fused bicyclic carbamate 18.
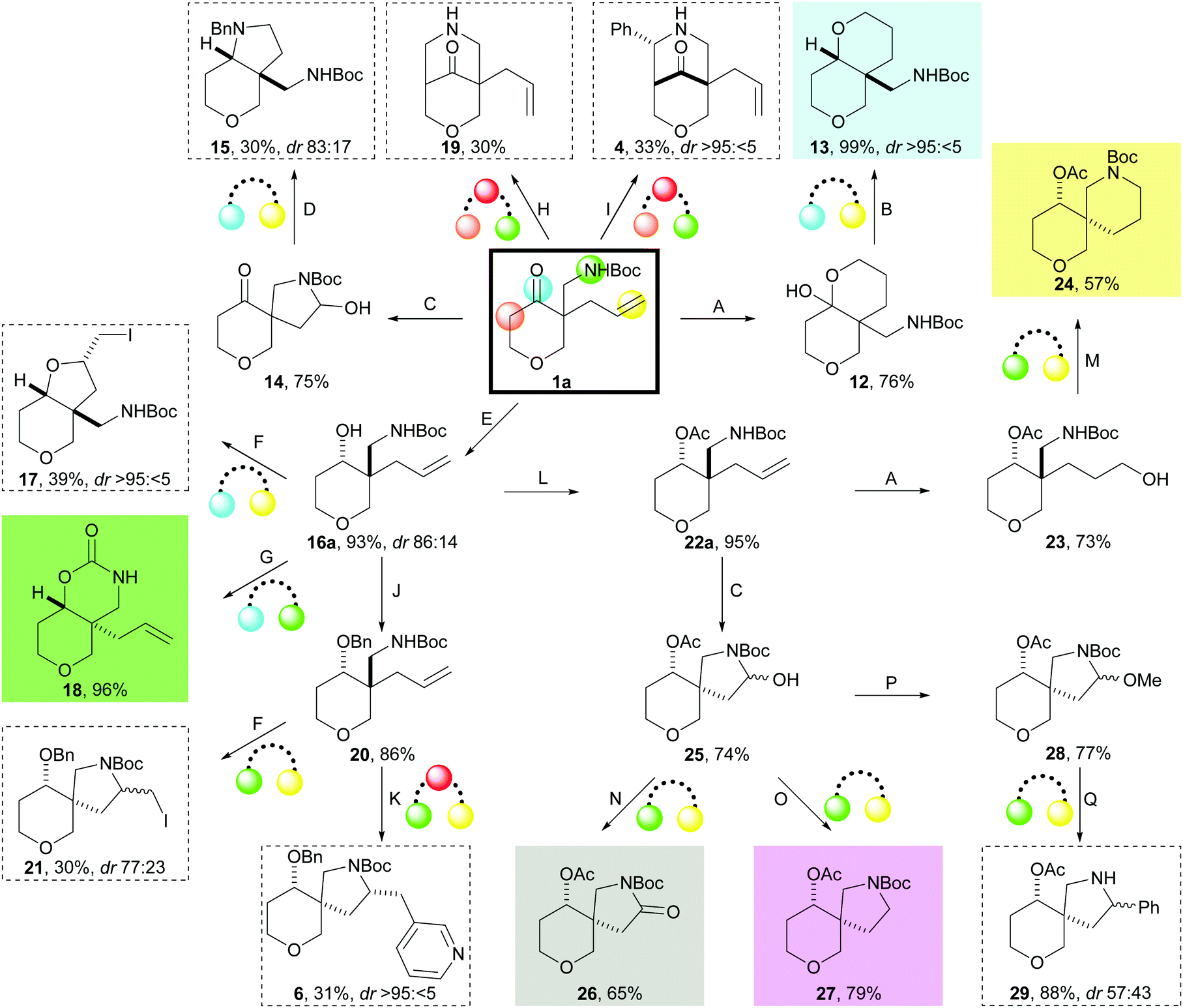 | ||
| Scheme 3 Identification of cyclisations for the synthesis of lead-like scaffolds using the precursor 1a (bold box). Scaffolds prepared using prioritised (coloured background; see Scheme 4) and other (dashed boxes) methods are shown. The functional groups that react to form rings are denoted by colour. Methods: A: disiamylborane, THF, 0 °C then NaBO3·4H2O, THF–H2O; B: Et3SiH, TFA, CH2Cl2 then Boc2O, Et3N, CH2Cl2; C: O3, CH2Cl2, −78 °C then Me2S, CH2Cl2; D: BnNH2, NaBH(OAc)3, CH2Cl2; E: iBu2AlH, CH2Cl2, −78 °C; F: I2, NaHCO3, MeCN; G: tBuOK, THF, 0 °C; H: paraformaldehyde, TFA, MeOH, 65 °C; I: PhCHO, TFA, MeOH, 65 °C; J: NaH, BnBr, cat. Bu4NI, THF, 0 °C; K: 3-bromopyridine, 5 mol% Pd(OAc)2, 10 mol% DPE-Phos, Cs2CO3, dioxane, 105 °C; L: Ac2O, pyridine; M: MsCl, Et3N, CH2Cl2 then TFA, CH2Cl2 then Et3N, CH2Cl2 then Boc2O, Et3N, CH2Cl2; N: PDC, celite, CH2Cl2; O: NaBH(OAc)3, AcOH; P: cat. TsOH, MeOH; Q: PhMgBr, CuBr·Me2S, Et2O·BF3, Et2O, 0 °C. | ||
Cyclisations between the distal α carbon (beige) and the Boc-protected amine (green) yielded bridged scaffolds. Thus, intramolecular Mannich reaction14 with either formaldehyde (→ 19) or benzaldehyde (→ 4) gave the bridged scaffolds.
Finally, cyclisations between the Boc-protected amine (green) and the alkene (yellow) in 1a gave spirocyclic scaffolds. Iodocyclisation of benzyl-protected 20 gave 21 (dias: 77:23), whilst Pd-catalysed aminoarylation15 with 3-bromopyridine gave the spirocycle 6. Alternatively, hydroboration–oxidation of acetyl-protected 22a, followed by sulfonylation and cyclisation, gave, after reprotection, the spirocycle 24 in 57% overall yield. Finally, ozonolysis of 22a gave a mixture of hemiaminals 25 that could be converted into three different spirocyclic scaffolds: oxidation with PDC gave the γ-lactam 26; reduction with NaBH(OAc)3 in AcOH gave 27; and conversion into the aminals 28 and treatment with PhMgBr and CuBr·Me2S gave 29.
In total, twelve scaffolds16 were prepared from the precursor 1a using the toolkit of cyclisation reactions. High scaffold diversity was possible, with product scaffolds based on fused, spiro and bridged bicyclic ring systems.
The toolkit of cyclisations was exploited in the synthesis of additional scaffolds by changing the precursor 1 (Scheme 4). Product scaffolds were selected on the basis of the potential of their derivatives to serve as high-quality leads for CNS drug discovery.9 Thus, potential scaffolds were decorated once or twice with exemplar capping groups, and the CNS lead-likeness of the resulting virtual compounds scored (see ref. 9 and ESI†). Thus, desirability scores (0.05–1) were determined for six properties (clogP; clogD at pH 7.4; pKa; number of H bond donors; molecular weight; and topological polar surface area), and summed to give a CNS Lead MPO score (0.3–6) (Fig. 1). Lead-like scaffolds for CNS drug discovery would have many derivatives with high (>∼4) CNS Lead MPO scores. In addition, the five cyclisations that had proceeded in high yield and diastereoselectivity with 1a were prioritised (see Scheme 3).
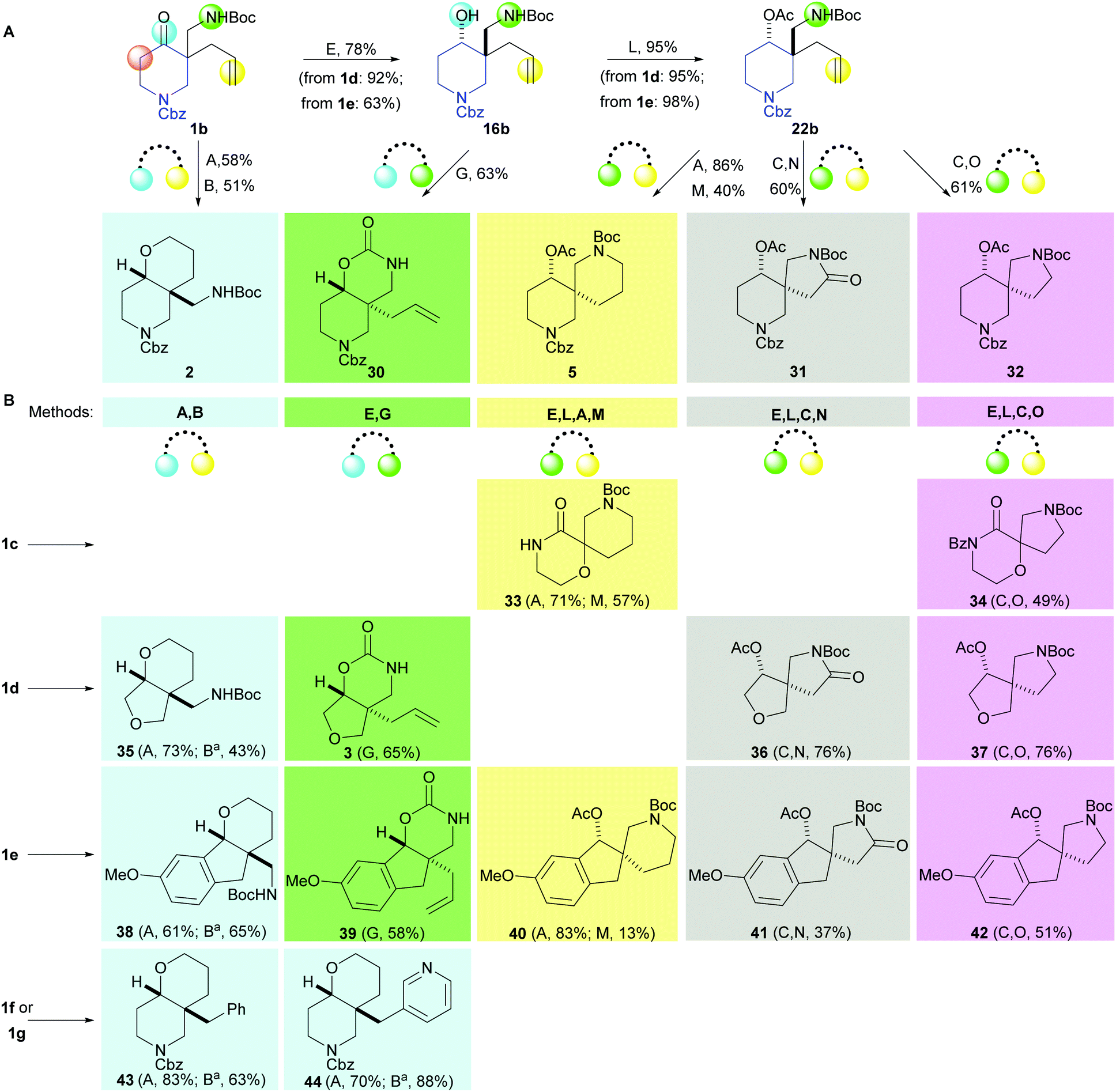 | ||
| Scheme 4 Eighteen additional scaffolds prepared using the prioritised cyclisation toolkit. Standard methods: A: disiamylborane, THF, 0 °C then NaBO3·4H2O, THF–H2O; B: Et3SiH, TFA, CH2Cl2 then Boc2O, Et3N, CH2Cl2; C: O3, CH2Cl2, −78 °C then Me2S, CH2Cl2; E: iBu2AlH, CH2Cl2, −78 °C; G: tBuOK, THF, 0 °C; L: Ac2O, pyridine; M: MsCl, Et3N, CH2Cl2 then TFA, CH2Cl2 then Et3N, CH2Cl2 then Boc2O, Et3N, CH2Cl2; N: PDC, celite, CH2Cl2; O: NaBH(OAc)3, AcOH. See ESI† for diastereoselectivities. aSee ESI† for details of method variations. | ||
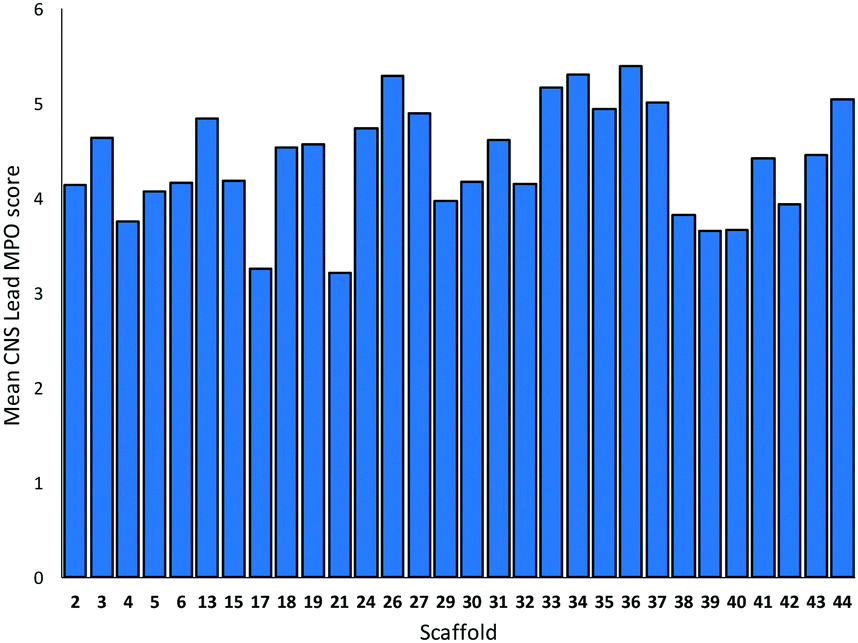 | ||
| Fig. 1 Mean CNS lead MPO scores of virtual compounds generated from the thirty prepared scaffolds and one or two decorations with exemplar medicinal chemistry capping groups (see ref. 9 and ESI† for details). Individual compounds received an MPO score for CNS lead-likeness on a 0.3–6 scale (see text). | ||
Remarkably, our synthetic approach was highly modular. By varying both the precursor (1a–g) and the cyclisation, it was possible to vary independently the rings in product scaffolds.17 The approach was also highly efficient, delivering 30 scaffolds in a total of 51 steps from the precursors 1a–g.18 We note that, in addition to specific application in CNS drug discovery, simple derivatives of scaffolds would be highly distinctive fragments with suitable properties for fragment-based discovery.19
In conclusion, a highly modular and efficient approach for the synthesis of structurally diverse molecular scaffolds was developed. Crucially, it was demonstrated that independent variation of the rings within the scaffolds was possible, enabling the synthesis of thirty diverse and lead-like molecular scaffolds suitable for CNS drug discovery programmes. We thank Takeda, the University of Leeds and EPSRC (EP/N025652/1) for funding, and Dr Andrew Ayscough for discussions.
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. A. Lipinski, Drug Discovery Today, 2004, 1, 337 CrossRef CAS PubMed.
- (a) Z. Rankovic, J. Med. Chem., 2015, 58, 2584 CrossRef CAS PubMed; (b) Z. Rankovic, J. Med. Chem., 2017, 60, 5943 CrossRef CAS PubMed.
- S. J. Teague, A. M. Davis, P. D. Leeson and T. Oprea, Angew. Chem., Int. Ed., 1999, 38, 3743 CrossRef CAS PubMed.
- A. Nadin, C. Hattotuwagama and I. Churcher, Angew. Chem., Int. Ed., 2012, 51, 1114 CrossRef CAS PubMed.
- (a) A. H. Lipkus, Q. Yuan, K. A. Lucas, S. A. Funk, W. F. Bartelt III, R. J. Schenck and A. J. Trippe, J. Org. Chem., 2008, 73, 4443 CrossRef CAS PubMed; (b) M. Garcia-Castro, S. Zimmermann, M. G. Sankar and K. Kumar, Angew. Chem., Int. Ed., 2016, 55, 7586 CrossRef CAS PubMed; (c) S. R. Langdon, N. Brown and J. Blagg, J. Chem. Inf. Model., 2011, 51, 2174 CrossRef CAS PubMed.
- (a) D. Foley, A. Nelson and S. Marsden, Angew. Chem., Int. Ed., 2016, 55, 13650 CrossRef CAS PubMed; (b) I. Colomer, C. J. Empson, P. Craven, Z. Owen, R. G. Doveston, I. Churcher, S. P. Marsden and A. Nelson, Chem. Commun., 2016, 52, 7209 RSC.
- See: (a) A. V. Borisov, V. V. Voloshchuk, M. A. Nechayev and O. O. Grygorenko, Synthesis, 2013, 2413 CAS; (b) R. G. Doveston, P. Tosatti, M. Dow, D. J. Foley, H. Y. Li, A. J. Campbell, D. House, I. Churcher, S. P. Marsden and A. Nelson, Org. Biomol. Chem., 2015, 13, 859 RSC; (c) M. Lüthy, M. C. Wheldon, C. Haji-Cheteh, M. Atobe, P. S. Bond, P. O’Brien, R. E. Hubbard and I. J. S. Fairlamb, Bioorg. Med. Chem., 2015, 23, 2680 CrossRef PubMed.
- (a) A. K. Ghose, T. Herbertz, R. L. Hudkins, B. D. Dorsey and J. P. Mallamo, ACS Chem. Neurosci., 2012, 3, 50 CrossRef CAS PubMed; (b) T. T. Wager, X. Hou, P. R. Verhoest and A. Villalobos, ACS Chem. Neurosci., 2010, 1, 435 CrossRef CAS PubMed; (c) H. Gunaydin, ACS Med. Chem. Lett., 2016, 7, 89 CrossRef CAS PubMed; (d) T. T. Wager, X. Hou, P. R. Verhoest and A. Villalobos, ACS Chem. Neurosci., 2016, 7, 767 CrossRef CAS PubMed; (e) S. A. Hitchcock, J. Med. Chem., 2012, 55, 4877 CrossRef CAS PubMed.
- J. Mayol-Llinàs, A. Nelson, W. Farnaby and A. Ayscough, Drug Discovery Today, 2017, 22, 965 CrossRef PubMed.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752 CrossRef CAS PubMed.
- For an approach to the synthesis of scaffolds for CNS drug discovery, see: J. T. Lowe, M. D. Lee IV, L. B. Akella, E. Davoine, E. J. Donckele, L. Durak, J. R. Duvall, B. Gerard, E. B. Holson, A. Joliton, S. Kesavan, B. C. Lemercier, H. Liu, J. Marie, C. A. Mulrooney, G. Muncipinto, M. Welzel-O’Shea, L. M. Panko, A. Rowley, B. Suh, M. Thomas, F. F. Wagner, J. Wei, M. A. Foley and L. A. Marcaurelle, J. Org. Chem., 2012, 77, 7187 CrossRef CAS PubMed.
- B. M. Trost and J. Xu, J. Org. Chem., 2007, 72, 9372 CrossRef CAS PubMed.
- Y. Numajiri, B. P. Pritchett, K. Chiyoda and B. M. Stoltz, J. Am. Chem. Soc., 2015, 137, 1040 CrossRef CAS PubMed.
- (a) D. Stead, P. O’Brien and A. J. Sanderson, Org. Lett., 2005, 7, 4459 CrossRef CAS PubMed; (b) H. Brice, D. M. Gill, L. Goldie, P. S. Keegan, W. J. Kerr and P. H. Svensson, Chem. Commun., 2012, 48, 4836 RSC.
- M. B. Bertrand, M. L. Leathen and J. P. Wolfe, Org. Lett., 2007, 9, 457 CrossRef CAS PubMed.
- Capturing graph-node-bond frameworks and multiple bonds to α atoms.
- Some cyclisations were not possible with specific precursors (e.g. no ketone in 1c; no NHBoc in 1f or 1g) or were deprioritised due to poor leadlikeness (e.g. two lactams with 1c and methods C,N).
- Counted steps resulted in isolated and characterised compounds.
- (a) C. W. Murray and D. C. Rees, Angew. Chem., Int. Ed., 2016, 55, 488 CrossRef CAS PubMed; (b) A. D. Morley, A. Pugliese, K. Birchall, J. Bower, P. Brennan, N. Brown, T. Chapman, M. Drysdale, I. H. Gilbert, S. Hoedler, A. Jordon, S. V. Ley, A. Merritt, D. Miller, M. E. Swarbrick and P. G. Wyatt, Drug Discovery Today, 2013, 18, 1221 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1566497–1566499. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc06078e |
| ‡ Current address: Division of Biological Chemistry and Drug Discovery, School of Life Sciences, University of Dundee, Dow Street, Dundee, DD1 5EH, UK. |
| This journal is © The Royal Society of Chemistry 2017 |