
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThiol–ene/oxidation tandem reaction under visible light photocatalysis: synthesis of alkyl sulfoxides†
Andrea
Guerrero-Corella
 a,
Ana
María Martinez-Gualda
a,
Fereshteh
Ahmadi
a,
Enrique
Ming
a,
Alberto
Fraile
*ab and
José
Alemán
*ab
a,
Ana
María Martinez-Gualda
a,
Fereshteh
Ahmadi
a,
Enrique
Ming
a,
Alberto
Fraile
*ab and
José
Alemán
*ab
aDepartamento de Química Orgánica (Módulo 1), Facultad de Ciencias, Universidad Autónoma de Madrid, 28049, Madrid, Spain. E-mail: jose.aleman@uam.es; Web: http://www.uam.es/jose.aleman
bInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, 28049, Madrid, Spain
First published on 4th September 2017
Abstract
The photocatalyzed synthesis of sulfoxides from alkenes and thiols has been carried out using Eosin Y. This is a metal-free method which uses a low catalyst loading, atmospheric oxygen as the oxidant, and visible light conditions (green light). A mechanism has been proposed that is consistent with the experimental results.
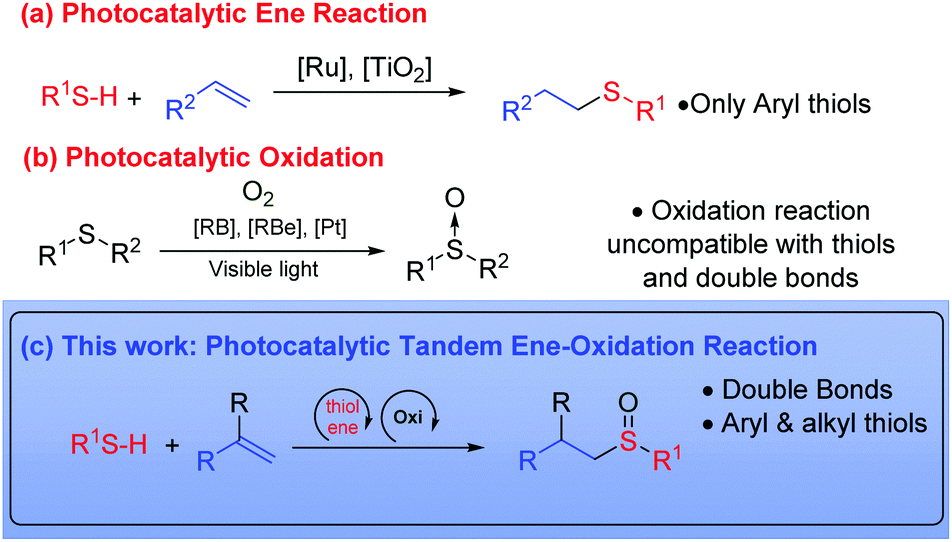
The radical addition of thiols to olefins was described by Posner in 1905,1 and has since emerged as one of the most direct methods for the construction of carbon–sulfur bonds. The thiol–ene reaction has attracted much scientific attention in recent years,2 particularly in the fields of polymers, materials and drug design. In the latter case, there are a wide variety of sulfur-containing pharmaceuticals and natural products e.g. zantac and romidepsin.3 In addition, this has increased its importance as ligands and chiral auxiliaries in synthetic chemistry.4 The thiol–ene coupling fulfills the requirements of the ‘click-chemistry’ concept5 due to the atom economy, efficiency and regioselectivity of the process. In this reaction, the anti-Markovnikov radical addition of the S–H bond occurs to an alkene (eqn (a), Scheme 1). This reaction can be initiated by thermal activation, using a radical initiator or by direct irradiation with UV light. However, this procedure often results in a low yield due to uncontrollable radical routes and the formation of different by-products. In the last years, visible light photoredox catalysis has emerged as a new powerful tool for the construction of organic molecules, using LEDs and domestic light bulbs which are cheaper and easier to handle than UV reactors.6 In 2012, Yoon's group demonstrated that thiol–ene reactions can occur by photo-redox using ruthenium catalysts with visible light.7 Two years later, Stephenson et al. proposed a mechanism in which a catalytic initiation and propagation of the reaction occurs in synergistic cycles.8 In this case bromotrichloromethane is reduced by initiation, generating the trichloromethyl radical, which promotes the addition and propagation reactions.8 More recently, semiconductor metal oxides (TiO2)9 and phenylglyoxylic acid10 have also been employed in the thiol–ene addition. All of these methods are exclusively used for the synthesis of thioethers by thiol–ene reaction.
 | ||
| Scheme 1 Previous reactions and tandem reaction of this work. | ||
On the other hand, the sulfinyl group can be found in several natural and pharmaceutical products such as esomeprazole, alliin and armodafinil. In addition, the sulfinyl group has been used as a chiral auxiliary11 or in many other reactions such as the Pummerer and Mislow–Evans rearrangements.12 The most traditional way for the synthesis of sulfoxides is the oxidation of sulfur derivatives.13 This oxidation is typically carried out using peroxides or peracids. However, the over-oxidation to sulfones and the safety aspects related to their use (m-CPBA, peracetic acid) are the main drawbacks associated with industrial processes. The oxidation of sulfides using atmospheric oxygen has proved to be a safer alternative (eqn (b), Scheme 1). This has been achieved using different photocatalysts.14 Rose Bengal,14a tetra-O-acetyl-riboflavin,14b or the platinum complex developed by our group,14c have showed to be good alternatives for this oxidation. Consequently, for the synthesis of alkylsulfoxide derivatives, a two-step process with two different catalytic systems is necessary. Therefore, two consecutive reactions are required as well as the isolation and the purification of the intermediates.15
Tandem reactions are considered to be a good alternative to economize the number of steps in the synthesis of products, by avoiding intermediate purification processes.16 Even though in the field of organocatalysis and metal-catalyzed reactions, tandem reactions have been widely developed, the number of publications relating to tandem processes in the photocatalytic field are scarce.17 Therefore, we considered whether it would be possible to carry out the tandem thiol–ene reaction and direct oxidation to sulfoxides, using a single photocatalyst and visible light (eqn (c), Scheme 1). However, in order to make both catalytic cycles (thiol–ene and oxidation) compatible, certain requirements must be met: (i) the formation of disulfides in the presence of oxygen should be avoided;18 (ii) the oxidation of the double bond with oxygen species should be eliminated (aldehyde formation)19 and (iii) the over-oxidation to sulfone must be controlled.13 In this work, we have developed the thiol–ene/oxidation tandem reaction, starting from alkenes. In addition, a plausible mechanism based on different mechanistic proofs is presented.
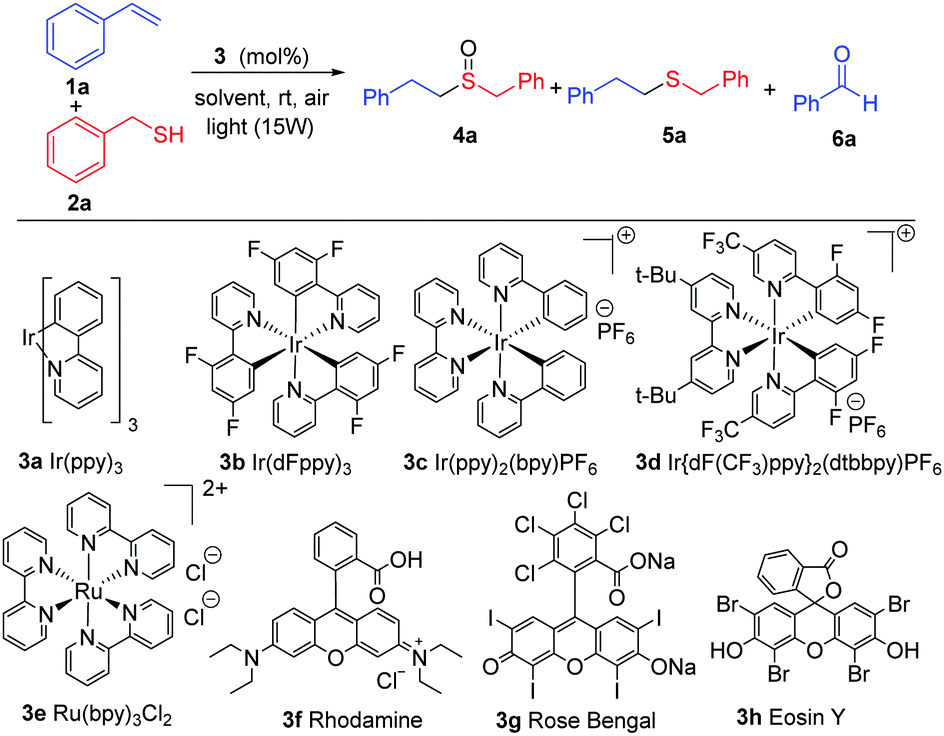
With the previous hypothesis, we started the screening reaction conditions by the addition of thiol 2a to styrene 1a in the presence of different photocatalysts using CH2Cl2 as solvent in an open-air vial (Table 1). Different light irradiation was chosen dependent on the absorption of the photocatalysts. Different iridium catalysts (3a–3d) enabled us to obtain as the major product the desired compound 4a, but they always resulted in a certain amount of the sulfur compound 5a and aldehyde 6a (entries 1–4). Other photocatalysts such as 3e, rhodamine 3f, led to a mixture of products, and Rose Bengal 3g did not evolve to the final oxidation after 36 hours. However, the inexpensive Eosin Y produced a full conversion in a very selective manner and yielded sulfoxide 4a (entry 8) exclusively. Other solvents or lower catalyst loading also led to the final product 4a, but with a lower selectivity (entries 10–13). The absence of Eosin Y did not afford any conversion (entry 14) in DCM or other solvents and blue light (see ESI† for details). With the best conditions ascertained (entry 8), we continued with different thiols (Scheme 2) and different double bonds (Schemes 3 and 4).
|
|
||||
|---|---|---|---|---|
| Entry | Photocatalyst | Light | Solvent | Ratio (4a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5a:6a)b :5a:6a)b |
| a Reactions were performed in 0.1 mmol scale of 1a in 0.360 mL of the indicated solvent after 36 h. b Determined by 1H-NMR. c Isolated yield after flash chromatography. | ||||
| 1 | 3a (2.5) | Blue | DCM | 59:32:8 |
| 2 | 3b (2.5) | Blue | DCM | 63:23:15 |
| 3 | 3c (2.5) | White | DCM | Decomposition |
| 4 | 3d (2.5) | White | DCM | 85:14:1 |
| 5 | 3e (2.5) | Blue | DCM | 45:45:10 |
| 6 | 3f (2.5) | Blue | DCM | 45:40:15 |
| 7 | 3g (2.5) | Green | DCM | 16:83:1 |
| 8 | 3h (2.5) | Green | DCM |
99:0:1 (81%)c |
| 9 | 3h (2.5) | Green | DCE | 98:0:2 |
| 10 | 3h (2.5) | Green | Toluene | 88:0:12 |
| 11 | 3h (2.5) | Green | MECN | 84:0:16 |
| 12 | 3h (2.5) | Green | DMF | Complex mixture |
| 13 | 3h (1.0) | Green | DCM | 30:69:1 |
| 14 | No catalyst | Green | DCM | No reaction |
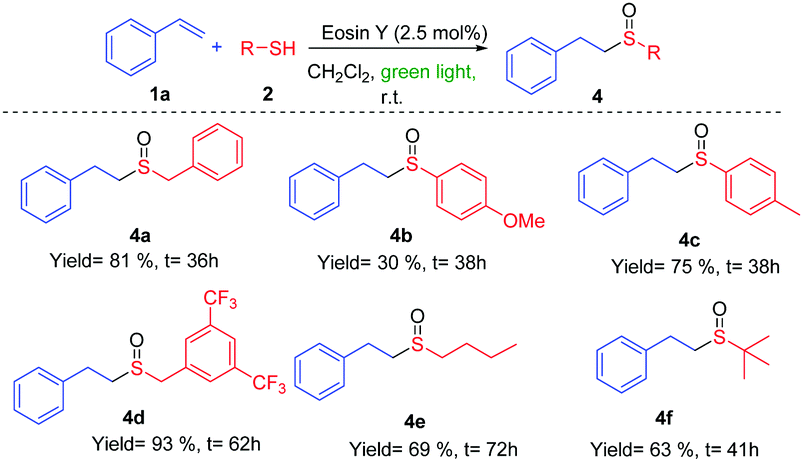 | ||
| Scheme 2 Reactions of styrene 1a with different thiols 2. | ||
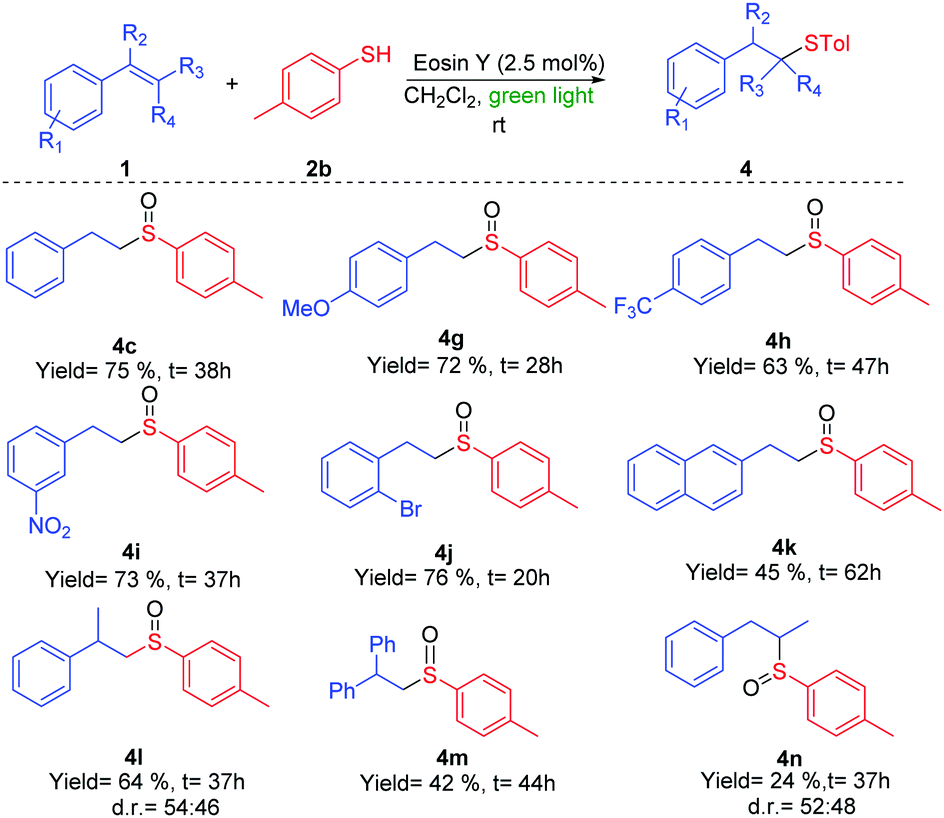 | ||
| Scheme 3 Reactions of different double bonds 1 with thiol 2b. | ||
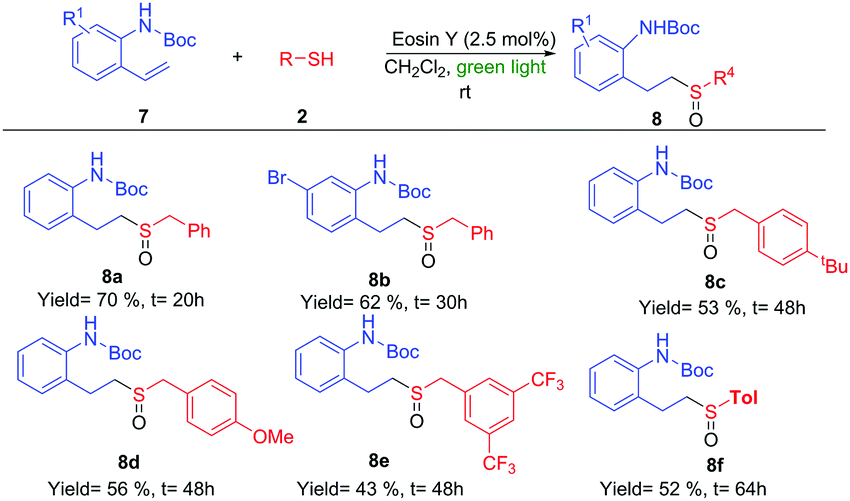 | ||
| Scheme 4 Sulfoxidation of vinyl-amino derivatives 7. | ||
The reaction of phenyl derivatives, containing electron-donating groups, allowed the final products 4b and 4c with lower yield than the benzyl group (4a) due to the formation of different byproducts, whereas the electron-withdrawing groups (EWGs) achieved a better yield (4d) (Scheme 2). The alkyl thiols were also compatible with the tandem reaction (4e), even in the case of the bulkiest of them (4f). Different EWGs and electron-donating groups (EDGs) at the double bond were tolerated under these reaction conditions (Scheme 3). The use of a methoxy group or CF3 at the phenyl derivative produced 4g and 4h in good yields. Interestingly, the presence of those groups sensitive to radical conditions, such as the nitro group (1i) or the bromine atom (1j), allowed the synthesis of sulfoxides 4i and 4j in good yields. However, the presence of a bulkier substituent such as the naphthyl group (1k) gave a moderate yield. The reaction also worked with trisubstituted double bonds from moderate to good yields with α-methyl (4l), α-phenyl (4m) and β-methyl (4n) substituents. The reaction with oct-1-ene did not work.
The importance of the nitrogen atom in the pharmaceutical industry is reflected by the large number of products containing nitrogen with biological activity.20 Therefore, the inclusion of a nitrogen atom in these styrene derivatives would increase the broad spectrum of this methodology. We started using ortho-vinylamino substituted derivatives 7. After some trials with different protecting groups (see ESI†), we found that the use of the Boc group was the most appropriate. The use of 7a in optimal reaction conditions resulted in the synthesis of the amino-sulfoxide derivative 8a in a good yield after 20 hours (Scheme 4). The bromo substituent (8b) and other benzyl derivatives (8c, 8d and 8e) were also tolerated but with a slightly lower isolated yields. The use of an aromatic group (Tol, 7f) worked with a 52% yield. As example of the potential of this methodology, we carried out one transformation to yield seven-membered rings (Scheme 5). Compound 8a was placed under Pummerer reaction conditions, in which the more acidic benzylic position was prone to be functionalized, giving the cyclized product (9) with 71% yield.
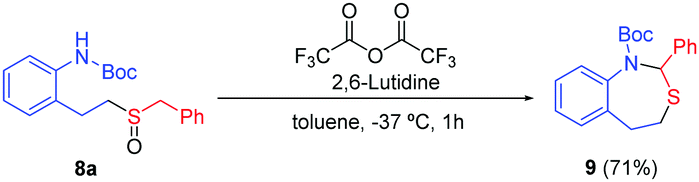 | ||
| Scheme 5 Pummerer reaction over the sulfoxide 8a. | ||
In order to propose a plausible mechanism for the reaction, different studies were carried out. Firstly, we performed two independent reactions to ensure that the two individual steps of the tandem reaction (thiol–ene reaction and oxidation) were taking place. In the absence of oxygen the thiol–ene reaction was stopped and no oxidized product 4j was observed, giving the sulfur product 11j in good yield (eqn (a), Scheme 6). In addition, when product 11j was placed under the same reaction conditions in the presence of oxygen, the oxidation gave the sulfoxide 4j in good yield. Then, Stern–Volmer fluorescence quenching studies with thiol 2b and the alkene 1j were carried out (see ESI†).21 This experiment makes possible to determine if a reaction between the excited state of the EY* and one or more substrates occur. The fluorescence of EY* was quenched in the presence of the thiol (see ESI†). Therefore, the photooxidation of the thiol by EY is thermodynamically favorable and it should be the first step in our reaction.
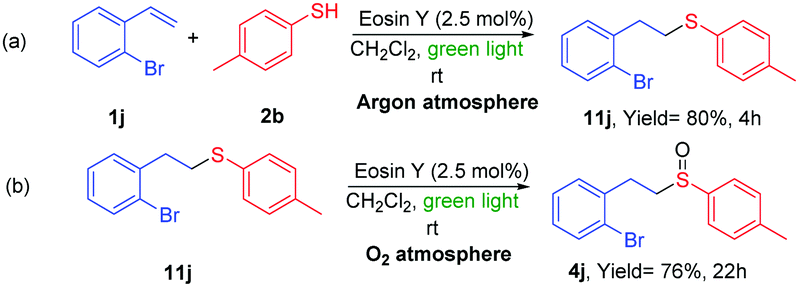 | ||
| Scheme 6 Reactions of ene-addition and oxidation of compounds 1j and 11j. | ||
Once the first stage of the reaction had been determined, the next study consisted of measuring the quantum yield in the addition of thiols to the double bond.21,22 This indicates the relationship between the absorbed photons and the emitted photons. If the result is larger than one this means that a chain propagation mechanism is taking place because each mole of photons absorbed gives rise to more than one mole of product. On the other hand, if the result is less than one, this means that it is a light dependent process, although a chain propagation process cannot be eliminated. The quantum yield measured for this thiol–ene reaction was 5.8, pointing to a chain propagation mechanism.22
For the second reaction, the oxidation step, two mechanisms are proposed for the oxidation of the sulfur atom. The first consists of a process of energy transfer to form singlet oxygen, and the second involves the transfer of an electron to form the superoxide radical anion.23 The discrimination between these two pathways can be demonstrated by indirect studies to determine which is the predominant one (see Table SI-2 in ESI†). The addition of DABCO, which acts as quencher of 1O2,23,24 almost completely inhibits the oxidation reaction (conversion = 14%). Interestingly, benzoquinone, which acts as scavenger of the superoxide radical anion,23 did not significantly change the final conversion (87%), indicating that the formation of this radical derivative is not taking place. The iodine test25 did not produce the blue dark colour indicative of the formation of peroxide radical species. It is well known that oxidations via singlet oxygen can be accelerated using deuterated solvents,26 and this was observed when the reaction was performed in MeOH (conversion = 60%, 3 h) and in CD3OD (conversion = 95%, 3 h, see the kinetic profile in ESI†). The quantum yield was also calculated for this process. We found a Φ = 0.003, showing that it is a light dependent process (see ESI,†).27 To confirm this hypothesis the reaction was carried out both in the presence, and the absence of light (see ESI†). The results indicated that the product formation only occurred during the periods of visible light irradiation, therefore the process is light dependent. These mechanistic proofs point to the following proposal (Fig. 1). Firstly, the EY is excited when irradiated with green light from the ground state to the triplet excited state (E = 1.91 eV).6c,28 The photoexcited catalyst (EY*, E1/2 = +0.83 V vs. SCE (saturated calomel electrode)6c,28) oxidises the thiol I, which acts as a donor via a hydrogen atom transfer process (HAT)29 (E1/2 = +0.45 V vs. SCE),30 generating a thiyl radical II. Then, alkene III reacts with the thiyl radical to form IV. This latter radical (IV) can be reduced by the oxidized EY to yield the final thioether V, recovering the initial photocatalyst (EY). A mechanism of chain propagation is also possible according to the quantum yield (Φ = 5.8) which was measured for this reaction (eqn (a), Scheme 6). Therefore, in this propagation mechanism, the radical IV can be reduced via a HAT process29 by an electron and a proton from the thiol I to form V (right, Fig. 1). In fact, we have used a deuterated thiol (TolSD) which has been incorporated to the final product D-V (R = o-Br–C6H4) in an anti-Markovnikov manner (Fig. 1 and ESI†). In the second oxidation cycle the Eosin Y is excited to the triplet state, and by means of an energy transfer mechanism, results in the formation of singlet oxygen31 which can react with the thioether V to give the final sulfoxide 4. This mechanism is possible because the activation energy of singlet oxygen is 0.95 eV,31a,32 which indicates that the energy transfer of the excited EY (1.91 eV6c,28) is thermodynamically flexible.
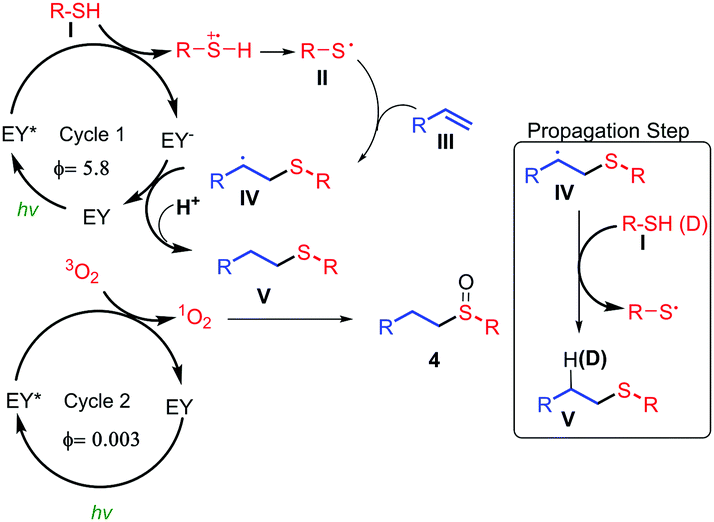 | ||
| Fig. 1 Plausible mechanism based on different mechanism observations. | ||
In conclusion, the first photo-catalyzed synthesis of sulfoxides from alkenes and thiols was carried out using Eosin Y, which has been shown to be the best catalyst.
Spanish Government (CTQ2015-64561-R) and the ERC Council (ERC-CG, contract number: 647550) are acknowledged.
Conflicts of interest
There are no conflicts to declare.Notes and references
- T. Posner, Ber. Dtsch. Chem. Ges., 1905, 38, 646 CrossRef.
- (a) J. Xu and C. Boyer, Macromolecules, 2015, 48, 520 CrossRef CAS; (b) A. B. Lowe, Polym. Chem., 2014, 5, 4820 RSC; (c) A. B. Lowe, Polymer, 2014, 55, 5517 CrossRef CAS.
- M. Feng, B. Tang, S. H. Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200 CrossRef CAS PubMed.
- H. Pellissier, Chiral Sulfur Ligands, The Royal Society of Chemistry, 2009 Search PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS PubMed.
- For some selected reviews in photocatalysis, see: (a) C. Prier, D. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (b) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC; (c) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed.
- E. L. Tyson, M. S. Ament and T. P. Yoon, J. Org. Chem., 2013, 78, 2046 CrossRef CAS PubMed.
- M. H. Keylor, J. E. Park, C.-J. Wallentin and C. R. J. Stephenson, Tetrahedron, 2014, 70, 4264 CrossRef CAS.
- V. T. Bhat, P. A. Duspara, S. Seo, N. S. B. Abu Bakar and M. F. Greaney, Chem. Commun., 2015, 51, 4383 RSC.
- D. Limnios and C. G. Kokotos, Adv. Synth. Catal., 2017, 359, 323 CrossRef CAS.
- B. M. Trost and M. Rao, Angew. Chem., Int. Ed., 2015, 54, 5026 CrossRef CAS PubMed.
- For Pummerer reaction, see: S. K. Bur and A. Padwa, Chem. Rev., 2004, 104, 2401 CrossRef CAS PubMed.
- (a) C. M. Rayner, Contemp. Org. Synth., 1995, 2, 409 RSC; (b) M. Madesclaire, Tetrahedron, 1986, 42, 5459 CrossRef CAS.
- (a) X. Gu, X. Li, Y. Chai, Q. Yang, P. Li and Y. Yao, Green Chem., 2013, 15, 357 RSC; (b) J. Dadová, E. Svobodová, M. Sikorski, B. König and R. Cibulka, ChemCatChem, 2012, 4, 620 CrossRef; (c) A. Casado-Sanchez, R. Gomez-Ballesteros, F. Tato, F. J. Soriano, G. Pascual-Coca, S. Cabrera and J. Aleman, Chem. Commun., 2016, 52, 9137 RSC.
- The synthesis of β-ketosulfoxides, starting from double bond and thiols, was recently reported. However, our and other external group, were not able to replicate these results, obtaining always starting material and small amount of sulfoxides 4. (a) See: T. Keshari, V. K. Yadav, V. Srivastava and L. D. S. Yadav, Green Chem., 2014, 16, 3986 RSC . For a recent related work, see: ; (b) H. Cui, W. Wei, D. Yang, Y. Zhang, H. Zhao, L. Wang and H. Wang, Green Chem., 2017, 19, 3520 RSC.
- (a) W. Zhao and F.-E. Chen, Curr. Org. Synth., 2012, 9, 873 CrossRef CAS; (b) B. Ganem, Acc. Chem. Res., 2009, 42, 463 CrossRef CAS PubMed; (c) D. J. Ramon and M. Yus, Angew. Chem., Int. Ed., 2005, 44, 1602 CrossRef CAS PubMed.
- See e.g. (a) J.-R. Chen, D.-M. Yan, Q. Wei and W.-J. Xiao, ChemPhotoChem, 2017, 1, 148 CrossRef; (b) B. Hu, W. Dong, Z. Feng, X. Gao, H. Gao, X. Xie and Z. Zhang, Asian J. Org. Chem., 2016, 5, 1467 CrossRef CAS; (c) S. Bloom, D. D. Bume, C. R. Pitts and T. Lectka, Chem. – Eur. J., 2015, 21, 8060 CrossRef CAS PubMed; (d) S. Jana, A. Verma, R. Kadu and S. Kumar, Chem. Sci., 2017, 8, 6633 RSC; (e) S. K. Pagire and O. Reiser, Green Chem., 2017, 19, 1721 RSC; (f) D. Alpers, M. Gallhof, C. B. W. Starka and M. Brasholz, Chem. Commun., 2016, 52, 1025 RSC.
- A. Talla, B. Driessen, N. J. W. Straathof, L.-G. Milroy, L. Brunsveld, V. Hessel and T. Noël, Adv. Synth. Catal., 2015, 357, 2180 CrossRef CAS.
- Y. Deng, X.-J. Wei, H. Wang, Y. Sun, T. Noël and X. Wang, Angew. Chem., Int. Ed., 2017, 56, 832 CrossRef CAS PubMed.
- A. S. Franklin and L. E. Overman, Chem. Rev., 1996, 96, 505 CrossRef CAS PubMed.
- D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803 RSC.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426 RSC.
- S. M. Bonesi, I. Manet, M. Freccero, M. Fagnoni and A. Albini, Chem. – Eur. J., 2006, 12, 4844 CrossRef CAS PubMed.
- C. Ouannes and T. Wilson, J. Am. Chem. Soc., 1968, 90, 6527 CrossRef CAS.
- G. K. Fekarurhobo, S. S. Angaye and F. G. Obomanu, J. Emerging Trends Eng. Appl. Sci., 2013, 4, 394 CAS.
- R. Lechner, S. Kummel and B. König, Photochem. Photobiol. Sci., 2010, 9, 1367 CAS.
- The overall quantum yield was 0.006 (see ESI,†).
- D. P. Hari and B. König, Chem. Commun., 2014, 50, 6688 RSC.
- L. Capaldo and D. Ravelli, Eur. J. Org. Chem., 2017, 2056 CrossRef CAS.
- F. G. Bordwell, X.-M. Zhang, A. V. Satish and J.-P. Cheng, J. Am. Chem. Soc., 1994, 116, 6605 CrossRef CAS.
- (a) M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351 CrossRef CAS; (b) J. Zhang, L. Wang, Q. Liu, Z. Yang and Y. Huang, Chem. Commun., 2013, 49, 11662 RSC.
- E. Baciocchi, T. Del Giacco, F. Elisei, M. F. Gerini, M. Guerra, A. Lapi and P. Liberali, J. Am. Chem. Soc., 2003, 125, 16444 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cc05672a |
| This journal is © The Royal Society of Chemistry 2017 |