
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA critical insight into the development pipeline of microfluidic immunoassay devices for the sensitive quantitation of protein biomarkers at the point of care
Ana I.
Barbosa
a and
Nuno M.
Reis
 *ab
*ab
aDepartment of Chemical Engineering, Loughborough University, Loughborough, Leicestershire LE11 3TU, UK
bDepartment of Chemical Engineering, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: n.m.reis@bath.ac.uk
First published on 31st January 2017
Abstract
The latest clinical procedures for the timely and cost-effective diagnosis of chronic and acute clinical conditions, such as cardiovascular diseases, cancer, chronic respiratory diseases, diabetes or sepsis (i.e. the biggest causes of death worldwide), involve the quantitation of specific protein biomarkers released into the blood stream or other physiological fluids (e.g. urine or saliva). The clinical thresholds are usually in the femtomolar to picolomar range, and consequently the measurement of these protein biomarkers heavily relies on highly sophisticated, bulky and automated equipment in centralised pathology laboratories. The first microfluidic devices capable of measuring protein biomarkers in miniaturised immunoassays were presented nearly two decades ago and promised to revolutionise point-of-care (POC) testing by offering unmatched sensitivity and automation in a compact POC format; however, the development and adoption of microfluidic protein biomarker tests has fallen behind expectations. This review presents a detailed critical overview into the pipeline of microfluidic devices developed in the period 2005–2016 capable of measuring protein biomarkers from the pM to fM range in formats compatible with POC testing, with a particular focus on the use of affordable microfluidic materials and compact low-cost signal interrogation. The integration of these two important features (essential unique selling points for the successful microfluidic diagnostic products) has been missed in previous review articles and explain the poor adoption of microfluidic technologies in this field. Most current miniaturised devices compromise either on the affordability, compactness and/or performance of the test, making current tests unsuitable for the POC measurement of protein biomarkers. Seven core technical areas, including (i) the selected strategy for antibody immobilisation, (ii) the surface area and surface-area-to-volume ratio, (iii) surface passivation, (iv) the biological matrix interference, (v) fluid control, (vi) the signal detection modes and (vii) the affordability of the manufacturing process and detection system, were identified as the key to the effective development of a sensitive and affordable microfluidic protein biomarker POC test.
 Ana I. Barbosa | Dr Ana I Barbosa obtained her PhD in Chemical Engineering from Loughborough University, UK, in 2016, where she worked on the development of a novel microfluidic platform, the Microcapillary Film (MCF), for the affordable sensitive quantitation of biomarkers. She currently works as a manufacturing scientist for Capillary Film Technology Ltd, a UK-based SME developing MCF technology for applications in life sciences and clinical diagnostics. Her research interest is the development of affordable miniaturised design approaches for the sensitive quantitation of protein biomarkers without sample preparation needs, combining aspects of analytical chemistry with microengineering and biology. |
 Nuno M. Reis | Dr Nuno M Reis is a Reader in Bioengineering and Biomedical Innovation at the University of Bath, UK, where his research group is particularly focused on the development of transformative microfluidic innovations based on melt-extruded Microcapillary Films (MCFs). He received both his undergraduate and PhD degrees in Biological Engineering from the University of Minho, Portugal. Previous to Bath, he held a lecturer position at Loughborough University, UK (2011–2016), and has 4.5 years’ post-doctoral experience at the University of Cambridge, UK (2006–2011). He is named co-inventor and co-author in several international patent applications and peer-reviewed publications specific to the application of MCFs to a range of bioanalytical applications. |
1. Introduction
Non-communicable diseases, including cardiovascular diseases, cancer and diabetes, are often associated with the ‘modern’ lifestyle in western countries and, therefore, are often considered a health challenge only in the developed countries.1,2 However, cardiovascular diseases and cancer are, since 2001, the top two causes of mortality in the developing world.3,4 From the 16 million deaths by cardiovascular diseases in 2001, 13 million occurred in the low-income and middle-income countries, compared with just 3 million in the high-income countries.5 This fact can be attributed to the rapid changes in lifestyle and an increase in the life expectancy of populations living in the developing regions of the planet, in addition to the chronic diseases linked to infectious conditions6,7 prevalent in these regions. The increased rate of incidence combined with the lack of proper diagnostics equipment and limited access to early treatment is dramatically shifting the focus of health challenges in the developing world from infectious diseases to non-communicable diseases.8Developing portable and affordable point-of-care (POC) tests capable of easily and accurately detecting non-communicable health conditions is now more urgent than ever before, and this should be regarded as a global challenge. Such tests could facilitate regular health check-ups, or offer a more cost-effective testing alternative to centralised pathology laboratory measurements by facilitating diagnosis in the comfort of the home, in community centres, or simply by enabling testing in surgeries and hospitals that lack the appropriate diagnostic equipment required for the effective diagnosis of non-communicable diseases. Early diagnosis enables early treatment, thus decreasing the number of deaths worldwide and the overall cost of patients treatment.7
The number of POC tests for non-communicable diseases currently available is very limited, which is certainly linked to the difficulty in developing robust tests capable of measuring very low concentrations of protein biomarkers in biological samples (e.g. whole blood, serum or urine) in a format that is compact, affordable and disposable. In the extreme scenario, POC tests are expected to meet the ASSURED policy published by WHO, which is still regarded as the international standard for developing POC tests.9 Consequently, the measurement of protein biomarkers is currently performed in centralised pathology laboratories using expensive and bulky equipment, in bioassay formats that take several hours to complete and that involve very complex fluid handling and pipetting.10,11
Microfluidic devices are a new and diverse technology, which uses fluids in micro environments in a controlled manner, and this distinguish them from conventional nitrocellulose lateral flow tests. They can be regarded as the ultimate technical solution for miniaturising protein biomarker immunoassays, by uniquely combining the advantages of simplified fluidics, a reduced amount of reagents and much shorter assay times.12 When translating an established commercial sensitive laboratory tests into a POC format, the ASSURED policy (affordable, sensitive, specific, user-friendly, rapid and robust, equipment-free and delivered) is regarded as the gold standard; however, so far there is no record of a microfluidic test that has been yet able to meet the expected ASSURED criteria for protein biomarkers measurement. For example, the lateral flow test currently available for measuring the cardiac biomarker Troponin I has a limit of detection around 0.5 ng m−1,13 which is around two orders of magnitude higher than the clinical threshold required for ruling out acute myocardial infarction.14 The lack of high-performance POC protein biomarker tests is linked to the reduced sensitivity of assays obtained in power-free POC tests, but also to the current prohibitive manufacturing costs of conventional microfluidic devices. The ASSURED criteria involve too many compromises that render the POC protein biomarker tests unsuitable for clinical use by underperforming compared to sophisticated centralised pathology testing. This review article provides an overview into an exciting pipeline of new microfluidic strategies for overcoming these challenges, and ultimately towards achieving miniaturised high-performance immunoassays effectively integrated in microfluidic formats.
2. Advantages and limitations of current microfluidics technology
Microfluidic devices can be broadly characterised by the precise control and manipulation of fluids at the submillimetre scale. Therefore, they usually involve small sample volumes (μl, nl, pl, fl), a small size (submillimetre channels or capillaries), reduced energy consumption and a controlled microenvironment.15 Current applications cover several scientific and commercial areas, including screening conditions for protein crystallisation,16 high-throughput screening in drug development,17 bioanalysis,18 single cell analysis19 and chemical synthesis,20,21 to name a few.Bioanalytical microfluidic systems, including the ones related to the quantitation of protein biomarkers for diagnostics, have rapidly developed over the past ten years, and now demonstrate the possibility to quantify low analyte concentrations in complex samples using small, miniaturised formats. Microfluidic devices appear to fulfil the technological gap between the simple-to-use ‘lateral flow’ POC tests and high-precision laboratory bioanalytical techniques. Better diagnostics are linearly correlated with an improved quality of life and a higher average life expectancy,22,23 and as part of this POC protein biomarkers quantitation is fundamental to the effective diagnosis of non-communicable diseases, which are the main causes of death worldwide. However, current microfluidics technology has several bottlenecks when it comes to the effective miniaturisation of POC protein biomarkers measurement. First, conventional microfluidic device fabrication methods are far from cost-effective. Second, the ASSURED criteria result in an inadequate limit of detection for protein biomarker analytes (<nM) in biological samples,24 requiring or lacking complex sample preparation and portable detection; however, it should not be compulsory that a modern high-performance near-the-patient test has to be portable or handheld although compactness would favour customer adoption. Biological samples, such as blood or faeces, are complex and their matrices interfere with the bioanalytical procedures, and therefore a better understanding of the interaction between the matrix components and the biosensing surface is required. Third, optical signal interrogation is commonly performed with a very sophisticated and bulky microscope located off the chip. The use of simpler and cheaper optical readout systems implies enhanced amplification and the use of multiple steps assays, resulting in a complex immunoassay procedure that is a challenge to miniaturise in POC tests.21 Therefore, finding new, cost-effective and simple approaches for optical signal detection or for understanding how simple established readout systems can provide sensitive interrogation is essential for the broader adoption and commercialisation of POC tests. An additional challenge often ignored in microfluidic bioanalytical systems is the fluid actuation and on-chip reagents storage.21
Despite the limitations highlighted, several microfluidic devices have been reported in the literature over the past 11 years with the capability of performing sensitive protein biomarker quantitation. The majority of these devices perform heterogeneous (solid phase) sandwich immunoassays. Table 1 summarises the pipeline of microfluidic devices reported in the literature for biomarker quantitation, specifying some of the key features related to their performance and methodology as reported by the authors. These microfluidic devices target protein biomarkers mostly related with the diagnosis of non-communicable diseases, being the most common cardiac biomarkers (e.g. troponin I (TnI), troponin T (TnT), creatine kinase (CK-MB), C-reactive protein (CRP) and myoglobin (Mb)),25,26 cancer biomarkers (e.g. prostate specific antigen, PSA; carcinoma embryonic antigen, CEA; α-fetaprotein, AFP and cancer antigen 125)27,28 and cytokines (e.g. TNF-α, IL-1, IL-4, IL-6 and IL-1) for sepsis29 and other inflammatory conditions.30
| Microfluidic system and publishing year | Protein biomarker (analyte) | Clinical threshold in blood samples (≥ng ml−1) | Manufact. process | Samp. vol. (μl) | LLoD (ng ml−1 or pM) | Surface passivation | Sample type | Immobilisation method/surface chemistry | Total assay time (min) | Fluid control | Detection mode | Readout system | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LLoD – lower limit of detection; TNF-α – tumour necrosis factor alpha; PSA – prostate specific antigen; TnI – troponin I; FABP – fatty-acid-binding proteins; IL-6 – interleukin 6; CRP – C-reactive protein; AFP – α-fetoprotein; IL-8 – interleukin-8; CEA – carcinoma embryonic antigen; TnT – troponin T; IL-4 – Interleukin-4; hCG – human chorionic gonadotropin;a this value corresponds to 6.7 mIU ml−1, based on 1 U equivalent to 1 μmol min−1, and the mass and molar concentrations herein mentioned for 1 min activity. RT – room temperature. | |||||||||||||
| PDMS microfluidic immunoassay mosaic (2005) | TNF-α | 0.014 (ref. 31) or 0.046 (ref. 32) | Reactive ion etching | 0.6 | ∼0.02 (0.38 pM) | 1% BSA in PBS for 10 s at room temperature (RT) | 1% BSA in buffer | Adsorption to PDMS | ∼12 | Capillary pump; continuous flow 30 nl min−1) | Fluorescence; fluorophore conjugation | Fluorescence scanner | 33 |
| Bio-barcode assay (2006) | PSA | 4 (ref. 34) | Multilayer soft lithography | 1 | 1.5 × 10−5 (5 × 10−4 pM) | 0.5% polyDuramide™ at RT | Goat serum | Covalent binding (gluteraldehyde-amine coupling on magnetic particle surface) | <60 | Pump; continuous flow (0.1 μl min−1) | Light scattered; silver-enhanced gold nanoparticles amplification | Verigene ID scanning system | 35 |
| Plasma panel capillary immunoassay (2007) | Myoglobin; CK-MB; TnI; FABP | 110 (ref. 36) | Glass capillaries manufacture | — | 1.2 (71 pM) | Casein for 1 h at RT | Diluted plasma (12.5%) | Covalent binding (glass pre-treated with (APDMES) (3-aminopropyltriethoxysilane and glutaraldehyde)) | <25 min | — | Chemiluminescence; enzymatic amplification | Photodiode detector | 40 |
| 70–110 (ref. 37) | 0.6 (7.14 pM) | ||||||||||||
| 0.006–0.05 (ref. 38) | 5.6 (233 pM) | ||||||||||||
| 4.3 (ref. 39) | 4 (267 pM) | ||||||||||||
| Dual network microfluidic chip (2008) | TNF-α | 0.014,31 0.046 (ref. 32) | Photolithography | 5–15 | 0.045 (0.9 pM) | 0.1% BSA in Tris for 4 h at 37 °C | Human serum | Covalent binding (Tosylactivated paramagnetic microbeads) | <60 | Pump; stop flow | Fluorescence; enzymatic amplification | Inverted fluorescence microscope | 41 |
| Digital microfluidic platform (2008) | Human insulin; IL-6 | 290–2900 (ref. 42) | Photolithography | <5 | — | Mouse IgG in HEPES buffer with BSA matrix; porcine, goat, bovine and mouse proteins suspended in surfactant matrix | Buffer | Adsorption to hydrophobised glass surfaces with Teflon AF | 7 | Magnetic bead manipulation; batch incubation | Chemiluminescence; enzymatic amplification | Photomultiplier tube | 42 |
| 0.001–0.1 (ref. 42 and 43) | |||||||||||||
| Optomagnetic immunoassay technology (2009) | TnI | 0.006–0.05 (ref. 38) | Injection moulding | 1 | 0.16 (3 pM) | 1% BSA and 10% sucrose in PBS for 1 h at RT | Non-diluted plasma | Adsorption to plastic surfaces | 5 | Magnetic particle control; stop flow | Label free; no amplification system | Total internal reflexion biosensor and a CCD camera | 44 |
| PDMS microfluidic assay capillary driven (2009) | CRP | 1000 (ref. 45)45 | Photolithography and photoplotted polymer masks | 5 | 1 (9 pM) | BSA for 15 min | Human Serum | Adsorption to Si wafers | 14 | Capillary Pump; continuous flow (82 nl min−1) | Fluorescence; fluorophore conjugation | Fluorescence microscope | 46 |
| BioCD protein array (2009) | PSA | 4 (ref. 34)34 | — | — | 4 (133 pM) | NaBH and 1% Casein | Diluted human serum (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) :4) |
Covalent binding (triethoxysilylbutyraldehyde (TESBA) cross-linking agent) | <120 | Pipetting; stop flow | Optical interferometry (label free) | BioCD scanning system | 47 |
| Immuno-pillar microfluidic assay (2010) | CRP | 1000 (ref. 45) | Injection moulding | 0.25 | 0.1 (0.9, 1.5, 3.3 pM) | 1% BSA in PBS for 45 min to 1 h at RT | Human serum | Adsorption to polystyrene beads | 12 | Pipetting; batch incubation | Fluorescence; fluorophore conjugation | Inverted fluorescence microscope | 49 |
| AFP | 10 (ref. 48) | ||||||||||||
| PSA | 4 (ref. 34) | ||||||||||||
| Microbead assay in a plastic chip (2010) | IL-8 | 1 (ref. 50) | Hot embossing | 3.3 | — | TBS starting block for 30 min at RT | — | Adsorption to magnetic particles | >65 | Pump; continuous flow (0.11 μl min−1) | Fluorescence detection; fluorophore conjugation | Epi-fluorescence upright microscope | 51 |
| Insulin | 290–2900 (ref. 42) | ||||||||||||
| Three dimensional helical glass tube with magnetic particles (2011) | CEA | 2.5 (ref. 52) | — | 30 | 4 × 10−3 (0.02 pM) | 1% BSA for 6 h at RT | Buffer | Covalent binding (paramagnetic spheres coated with epoxy group) | 8 | Pump; stop flow | Chemiluminescence; gold nanoparticles functionalised with DNAzyme | Spectofluormeter | 53 |
| Flow through detection cell with magnetic graphene nanosheets (2011) | CEA | 2.5 (ref. 52) | — | 200 | 1 × 10−3 (0.005 and 14.7 pM) | — | Buffer | Covalent binding (GOPS onto magnetic graphene nanosheets) | <30 | Pump; stop flow | Eletrochemical | Electrochemical analyser | 54 |
| AFP | 10 (ref. 48) | ||||||||||||
| Spiral flow-based separation microfluidic assay (2011) | TnT | 0.012 (ref. 55) | Rapid prototyping techniques | 1.5 | 10–100 (278–2780 pM) | Protein blocking solution for 2 min at RT | Whole blood (microfluidic device, including a flow-based separation channel) | Adsorption to cyclic olefin copolymer | 5 | Syringe with a pressure gauge; stop flow | Chemiluminsecence; enzymatic amplification | Photomultiplier tube and oscilloscope | 56 |
| Silicon photonic microring resonator (2011) | CEA | 2.5 (ref. 52) | Silicon-on-insulator | — | 25 (125 pM) | Starting block for 8 h at 4 °C | 100% FBS (fetal bovine serum) | Covalent (hydrazone-bond-formation chemistry) | 30 | Pump; continuous flow (10–30 μl min−1) | Label free (measure shifts in microring resonance) | Instrument that measures microring resonance | 57 |
| Silicon photonic microring resonator (2011) | CRP | 1000 (ref. 45) | Silicon-on-insulator | <10 | 0.02 (200 fM) | Starting block for 8 h at 4 °C | Diluted serum and plasma | Covalent (hydrazone-bond-formation chemistry) | ∼60 | Pump; continuous flow (10–30 μl min−1) | Resonance amplification through streptavidin-coated beads (∼10 μm diameter) | Instrument that measures microring resonance | 57 |
| Microfluidic nanoelectrode array (2011) | PSA | 4 (ref. 34) | UV lithography, electron-beam evaporation, and lift-off | 0.18 | 0.01 (0.33 pM) | — | Buffer | Covalent binding (self-assembled thiols monolayer to Au surface bound to a linker complex of metalised peptide nucleic acid conjugated with antibody) | ∼5 | Pump; stop flow | Electrochemical; enzymatic amplification: glucose oxidase PSA conjugated | Custom-built potentiostat, remote source meter, shielded probe station | 58 |
| Lab-on-paper (2011) | AFP; cancer antigen 125; CEA | 10 (ref. 48) | Paper manufacturing | 4 | 0.06 (0.9 pM) | 0.5% BSA + 0.5% casein for 15 min at RT | Buffer | Covalent binding (chitosan coating and glutaraldehyde cross-linking) | ∼6 | Passive flow; stop flow | Chemiluminescence; enzymatic amplification | Luminescence analyser | 60 |
| 17.5 × 109(35 U ml−1, ref. 59) | 6.6 × 107 (3.3 × 108 pM or 0.5–80.0 U ml−1) | ||||||||||||
| 2.5 (ref. 52) | 0.05 (0.25 pM) | ||||||||||||
| Microfluidic microtiter plate (2012) | PSA | 4 (ref. 34) | Injecting moulding | 5 | 0.016 (0.5 pM) | Optiblock flush at RT | Buffer | Adsorption to polystyrene | 120 | Gravity; stop flow | Chemifluorescence; enzymatic amplification | Fluorescence plate reader | 62 |
| IL-4 | 0.025 (ref. 61) | 2 × 10−4 (0.02 pM) | |||||||||||
| Multiplexed magnetic bead assay (2012) | IL-6 | 0.001–0.1 (ref. 42 and 43) | Soft lithography of PDMS | 5 | 0.01 (0.47 pM) to 1 (47.6 pM) | — | Buffer | Covalent binding (carboxyl terminated beads with sulfo-NHS and EDC chemistry) | ∼12 | Pump; continuous flow rate (1 μl min−1) | Fluorescence; fluorophore conjugation | Flow cytometer | 63 |
| TNF-α | 0.014,31 0.046 (ref. 32) | ||||||||||||
| Superparamagnetic beads (SPMBs) pattern-based immunoassay (2013) | CEA | 2.5 (ref. 52) | Soft lithography, electroplated nickel | ∼50 | 3.5 (17.5 pM) | 1% BSA for long periods of time at 4 °C | Serum | Covalent binding (iron oxide nanoparticles as the core with carboxyl groups on the surface) | 40 | Magnetic field manipulation; stop flow | Fluorescence; quantum dots | ICCD camera | 64 |
| AFP | 10 (ref. 48) | 3.9 (57.4 pM) | |||||||||||
| Immunoassay glass capillaries with ZnO nanorods (2013) | PSA | 4 (ref. 34) | Glass capillaries manufacture | — | 1 (33.3 pM) | 10 mg ml−1 BSA for 1 h at RT | Diluted human serum (10%) | Covalent binding (adding GPTS to ZnO nanorods) | 30 | Pump; continuous flow (50 μl min−1) | Fluorescence; fluorophore conjugation | Homemade fluorescence read out | 65 |
| AFP | 10 (ref. 48) | 5 (73.5 pM) | |||||||||||
| CEA | 2.5 (ref. 52) | 5 (25 pM) | |||||||||||
| Power-free chip enzyme immunoassay (2013) | PSA | 4 (ref. 34) | Laser cutting | 115 | 3.2 (107 pM) | 1% BSA in PBS for long periods at 4 °C | Non-diluted human serum | Covalent binding (APTMS functionalisation of magnetic particles) | 30 | Magnetic field manipulation; stop flow | Colorimetric; enzymatic amplification; | Cellphone camera | 66 |
| Silicon porous microarray (2013) | PSA | 4 (ref. 34) | Double-sided photolithography and chemical anisotropic wet-etching using KOH | — | 1.7 (56.7 pM) | 5% non-fat powered milk | Whole blood (integrated acousto-phoresis separation plasma) | Adsorption to porous silicon chips | 15 | Pump; continuous flow (50 μl min−1) | Fluorescence; fluorophore conjugation | Confocal microscope | 67 |
| Gold/Graphene origami – immunosensor (2013) | CEA | 2.5 (ref. 52) | Paper manufacturing | 2 | 8 × 10−4 (0.004 pM) | 0.5% BSA + 0.5% casein for 1 h at RT | Human serum | Adsorption to gold/graphene | ∼60 | Passive flow; stop flow | Electrochemical | Photomultiplier tube | 68 |
| Autonomus capillary system (2014) | TnI | 0.006–0.05 (ref. 38) | Laser etching | 15 | 0.024 (1 pM) | 1 mg ml−1 BSA for 2 h at RT | Buffer | Covalent binding (PMMA with APTES and cross-linked glutaraldehyde) | 7 to 9 | Capillary pump; continuous flow assay (0.32 nl min−1) | Fluorescence labelling | House built fluorescence reader | 69 |
| Microcapillary film (MCF) (2014) | PSA | 4 (ref. 34) | Melt-extrusion | 150 | 0.04 to 0.9 (1.54 to 35 pM) | 3% BSA for 2 h at RT | Whole blood, serum or buffer | Adsorption to FEP-Teflon | 15 to 50 min | Manual syringe control (multiple syringe device) | Colorimetric and fluorescence; enzymatic amplification | Flatbed scanner/smartphone | 72–74 |
| IL-1β | 0.005 (ref. 70) | 0.007 (0.426 pM) | Superblocking for 2 h at RT | ||||||||||
| TNF-α | 0.014 (ref. 31)31 | 0.007 (0.114 pM) | |||||||||||
| IL-6 | 0.001–0.1 (ref. 42 and 43) | 0.015 (0.713 pM) | |||||||||||
| IL-12 | 0.5 (ref. 71) | 0.002 (0.035 pM) | |||||||||||
| Microfluidic multilayer array (2014) | PSA | 4 (ref. 34) | Soft lithography | 5 nL | 0.030 | 1% Casein in PBS | Human serum | Covalent to coated glass slides with epoxysilane | 14 | Pipetting; stop flow | Fluorescence; fluorophore conjugation | Fluorescence microarray scanner | 75 |
| TNF-α | 0.014 (ref. 31) | 0.052 | |||||||||||
| IL-1β | 0.005 (ref. 70)70 | 0.017 | |||||||||||
| IL-6 | 0.001–0.1 (ref. 42 and 43) | 0.021 (1 pM) | |||||||||||
| 3D paper immunoassay (2014) | hCG | 2.4 × 105 (10 mIU ml−1 to 100 mIU ml−1)76 | — | 20 | 2.4 × 105 (6.7 × 106 pM)a | 0.1% Tween20, 5% sucrose, 1% casein, 0.1% proclin in BBS | Urine | Adsorption (hydrophilic nylon membrane) | 10 | Passive flow; stop flow | Colloidal gold nanoparticles | Flatbed scanner | 77 |
| Microfluidic microarray immunoassays (2014) | IL-6 | 0.001–0.1 (ref. 42 and 43) | Multilayer soft-lithography | 5 | 0.084 (4 pM) | — | Buffer | Covalent (glass slides with epoxy silane) | <3 h | Pipetting; stop flow | Fluorescence; fluorophore conjugation | Fluorescence microarray scanner | 78 |
| IL-1β | 0.005 (ref. 70) | 0.07 (4 pM) | |||||||||||
| TNF-α | 0.014 (ref. 31) | 1.6 (30 pM) | |||||||||||
| PSA | 4 (ref. 34) | 0.45 (15 pM) | |||||||||||
| Microtiter graphene based immunoassay (2014) | CRP | 1000 (ref. 45) | Injection moulding | — | 0.07 (0.6 pM) | 5% BSA for 30 min at 37 °C | Diluted whole blood and plasma | Covalent binding (graphene nanoplatelets and APTES to polystyrene surface) | <30 | Pipetting; batch incubation | Colorimetric; enzymatic amplification | Smartphone | 79 |
| Lab-on-a-disc with TiO2 fibrous mat (2015) | CRP | 1000 (ref. 45) | CNC | 10 | 8 × 10−4 (∼6 fM) | 1% BSA in PBS for 1 h at 37 °C | Whole blood (blood cell separation on the disc) | Covalent binding (PDMS coated with silicon and nanofibres of TiO2 treated with GPDES) | 30 | Rotation actuation; stop flow | Chemiluminescence; enzymatic amplification | Homebuilt with cooled PMT module and CCD camera | 80 |
| TnI | 0.006–0.05 (ref. 38) | Micromachining | 0.037 (1.5 pM) | ||||||||||
| Surface plasmon resonance-based immunoassay (2015) | CRP | 1000 (ref. 45) | — | 50 | 1.2 (11 pM) | 1% BSA for 30 min at RT | Diluted (1:1000) whole blood, serum and plasma |
Affinity binding (protein A/G covalently bound to the surface) | 3 | Pump; continuous flow (10 μl min−1) | Label free (surface plasmon resonance) | BIA core surface plasmon resonance | 81 |
A sandwich immunoassay performed in a microfluidic device involves a complex sequence of biochemical reactions and physical interactions with the surface of the miniaturised system. The development of microfluidic devices for sensitive protein quantitation demands an understanding of each immunoassay reaction independently of the end result.
This is particularly significant when the sensitive quantitation is bound to the affordability of the device, therefore requiring the use of cheap optoelectronic components. This review critically discusses the latest technical development in seven key areas that are believed to be fundamental for the effective development of sensitive and affordable microfluidic protein biomarker POC tests, namely: (i) the selected strategy for antibody immobilisation, (ii) the surface area and surface-area-to-volume ratio, (iii) the effect of biological matrix interference, (iv) the significance of fluid control, (v) the signal detection modes, (vi) the manufacturing process and (vii) surface passivation.
3. Clarifying the concepts of sensitivity and the lower limit of detection
In this review, the terms sensitivity and the lower limit of detection (LLoD) are highly used throughout, it is therefore important to understand their meaning, relationship and analytical significance. Analytical sensitivity is a term that indicates the capacity of the method to differentiate between two very close concentrations of protein biomarkers, which is usually given by the slope of the response curve. This means sensitivity does not consider the value of the noise (i.e. background). On the other hand, the LLoD describes the minimum protein concentration that a device/test can quantify with a specified precision and reproducibility. The smaller the LLoD value, the better the performance of the immunoassay. Most frequently, the LLoD is defined in terms of the concentration producing a signal equivalent to three times the standard deviation of a series of blank (baseline) measurements. This definition is equivalent to a 99% confidence level.82 The detection limit is intrinsically a function of both the signal strength and signal stability, because of this the term is related to the signal-to-noise ratio, or SNR in short. Thus, a signal-to-noise ratio of 3 is considered equivalent to blank + (3*σ).83 Although in this review the terms LLoD and sensitivity are somehow used interchangeably, it is important to understand that assay performance should consider simultaneously the lowest concentration that significantly differs from the blank (LLoD) and the response curve slope that differentiates between two very close protein concentrations points in the response or calibration curve. It is also important to note that for the same biomarker, the assay detection limit varies for different specimen types, such as buffer versus serum; therefore, Table 1 presents a column that specifies which sample type was used for each microfluidic assay.4. Strategies for antibody immobilisation
A universal feature in heterogeneous immunoassays is the presence of a solid phase that enables the capture of the targeted biomarker protein and the separation of bound and free reagents and the analyte. The first stage in every sandwich immunoassay is surface preparation, which includes the immobilisation of the capture antibody (CapAb) or antigen and effective blocking of the remaining binding sites (surface passivation), which can also include protein immobilisation (e.g. BSA, casein, gelatin). Immobilised antibodies must have the complementary-determining regions (CDRs) available for the targeted analyte/antigen to bind, which means the immobilisation technique has to provide proper antibody orientation. The strength of the binding between antibody–antigen, also called the affinity, will differ depending on the antibody immobilisation process and the surface where it is immobilised, since denaturation and conformational changes in antibodies can alter the structure of their CDRs.84 Hence in the development of solid-phase immunoassays, antibody–antigen avidity (the sum of multiple antibody–antigen non-covalent interactions)85 becomes an important parameter to consider. Several antibody immobilisation procedures have been implemented and the choice depends on the microfluidic surface characteristics and on the long-term interactions between the antibodies and the surface.4.1 Passive adsorption to surfaces
Passive adsorption to surfaces is the simplest method for antibody immobilisation in microfluidic devices. This requires placing the antibodies in direct contact with the surface. This interaction will determine the amount of antibody adsorbed, known as the surface capacity, and the orientation of the antibody. The reported drawbacks of physisorption or passive/physical adsorption mechanisms are random orientation and a weak attachment to certain surfaces, since proteins may be removed by some buffers or surfactants when performing the assays.86 An ideal antibody adsorption surface should have a high affinity to the antibody constant fragment (Fc), so that the variable region (Fab) remains available for binding. The binding between the antibody and surface has to be strong enough to avoid removal of immunoreagents from the surface, but cannot be too strong to denature completely the antibody or change conformation of the Fab region and the antibodies affinity. In addition, a high antibody–antigen affinity compared to the surface–antibody or surface–antigen affinity is required in order to maximise sensitivity of the assay. Therefore, is clear that the individual interaction between the CapAb and the surface will affect the assay performance. However, it is important to understand that the CapAb ‘binding activity’ is highly dependent on the relative amount of antibodies on a surface, i.e. surface packing. Several studies describing antibody adsorption to hydrophobic (e.g. plastics) and hydrophilic surfaces (e.g. silicon) concluded that around half a monolayer coverage yields the optimum condition for conservation of the CapAb binding activity, with this characterised by antibodies being vertically orientated to favour a more effective capture and binding of antigens. At lower concentrations, CapAb molecules have been found to adsorb with a flat-on orientation, yielding a strong ‘binding’ with the surface, thereby complicating the chemical binding to antigen molecules. On the other hand, a full monolayer coverage is characterised by a reduced level in probe binding per antibody and an unstable adsorption of CapAb molecules on the surface, this is due to the increase in both the packing density and aggregation, which causes steric hindrance to CapAb–antigen binding. This has been shown to lead to a sharp reduction in hCG binding capacity.87,88 Microfluidic immunoassays have been developed from different surfaces for antibody adsorption, such as plastic, glass, silicon and PDMS. Plastics are usually preferred as surfaces for passive antibody adsorption, due to their hydrophobic nature, as antibodies adsorb mainly by attaching their non-polar domains (CH3 and CH2) to the surface and establishing strong intermolecular binding. The most common example of antibody adsorption in immunoassays is the immobilisation of antibodies onto a polystyrene microtiter plate (MTP), which is the gold standard for laboratory immunoassays for protein biomarker quantitation. Other plastics, such as polypropylene and polyvinyl chloride (PVC), are also common in the diagnostics industry.89 Antibody adsorption onto plastics has been successfully used in microfluidic tests, with fluorinated ethylene propylene (FEP-Teflon®) microcapillaries,72 polystyrene channels,62 glass surfaces hydrophobised with Teflon® AF42 and cyclic olefin copolymer56 being some examples in a large spectrum of plastic adsorption microfluidic surfaces used for protein biomarkers quantitation, with some authors reporting fM detection levels.74Protein adsorption onto glass appears to occur mainly due to electrostatic interactions, which does not favour quantitative immunoassays. Antibodies tend to form multilayers in which adsorbed molecules become polar binding towards other antibodies, which is undesirable in quantitative immunoassays. Consequently, microfluidic devices intended for sensitive protein quantitation fabricated from glass usually use covalent immobilisation procedures.
Silicon is another popular material used for antibody adsorption on microfluidic devices, but presents the major drawback of antibodies adsorbing less to silicon surfaces due to reversible binding,90 with covalent immobilisation preferred. However, some microfluidic devices have been able to quantify CRP with a lower limit of detection, with a LLoD of 1 ng ml−1 or 9 pM, and PSA, with a LLoD of 1.7 ng ml−1 or 56.7 pM, using antibodies adsorbed onto silicon wafers.46,67
PDMS is the preferred material for microfluidic researchers due to simple manufacture prototyping. Although hydrophobic, this polymer presents problems related to non-specific adsorption, which is undesirable in POC tests.91 Nevertheless, the sensitive quantitation of TNF-α with a LLoD of 0.02 ng ml−1 (0.38 pM) has been reported based on antibody adsorption to PDMS surfaces.33 The covalent attachment of antibodies combined with previous surface modification appears to be the most common approach used for immunoassays in PDMS devices. Detection of the pregnancy hormone hCG was reported using a hydrophilic nylon membrane based on antibody adsorption with a detection limit of 6.7 × 106 pM (i.e. 6.7 mIU ml−1). Note, however, that the detection limit of the pregnancy biomarker hCG, even at the early stages of pregnancy, is several orders of magnitude higher than the LLoD required for cancer and cardiac protein biomarkers.77
Gold surfaces have been used in microfluidic assays for antibody adsorption, achieving a LLoD of 8 × 10−4 ng ml−1 (0.004 pM) for CEA biomarker based on a gold and graphene origami-immunosensor.92
Different surface chemistries promote different types of intermolecular binding, which interfere with the signal-to-noise ratio, an important feature in quantitative immunoassays. Consideration of the surface properties and chemistries is therefore paramount to achieving high sensitivity and lower LLoDs in microfluidic devices relying on physisorption of CapAb or antigens.
4.2 Covalent binding
The selection of strategy for the covalent immobilisation of antibodies onto microfluidic surfaces depends on the type of surface being used, since it requires an intermediate linker to bind the antibody molecules. Consequently, a wide variety of methods have been utilised and reported in the literature, and these have been extensively reviewed by Kim et al.93 Covalent immobilisation is usually regarded as more stable and offering a higher surface coverage, two important features for sensitive immunoassays. Nevertheless, covalent antibody immobilisation strategies usually involve complex chemistries, which increases the manufacturing complexity of the microfluidic tests, and furthermore there is usually no guarantee that the covalently immobilised antibody molecules will be correctly orientated or will remain active after linkage to the active antibody sites.94,95The majority of recently reported microfluidic devices use surface silanisation for antibody immobilisation. Silanisation involves covering a surface with self-assembly organofunctional alkoxysilane molecules.96 Mineral components, such as mica, glass and metal oxide surfaces, can all be silanised, because they contain hydroxyl groups (–OH), which attack and displace the alkoxy groups on the silane, thus forming a covalent –Si–O–Si– bond. Typical organofunctional alkoxysilanes include APTES ((3-aminopropyl)-triethoxysilane), APDMES ((3-aminopropyl)-dimethyl-ethoxysilane), APTMS ((3-aminopropyl)-trimethoxysilane), GPMES ((3-glycidoxypropyl)-dimethyl-ethoxysilane) and MPTMS ((3-mercaptopropyl)-trimethoxysilane).96
Proteins have a number of potential immobilising sites, namely: (i) the α-amino groups of the chain and the ε-amino groups of lysine and arginine, (ii) the α-carboxyl groups of the chain end and the β- and γ-carboxyl groups of aspartic and glutamic acids, (iii) the phenol ring of tyrosine, (iv) the thiol group of cysteine, (v) the hydroxyl groups of serine and threonine, (vi) the imidazole group of histidine and (vii) the indole group of tryptophan. Further details about these functional chemical groups are summarised in Table 2.97
| Side groups | Amino acids | Surfaces |
|---|---|---|
| —NH2 | Lys, hydroxyl-Lys | Carboxylic acid active ester (NHS), epoxy, aldehyde |
| —SH | Cys | Maleimide, pyridyil disulphide, vinyl sulfone |
| —COOH | Asp, Glu | Amine |
| —OH | Ser, Thr | Epoxy |
Antibodies can directly bind to a silanised surface, which has organofunctional alkoxy silanes, amine groups and epoxy groups. This procedure is common with microfluidic surfaces that undergo modification for further antibody immobilisation. In respect to sensitive biomarker quantitation, different approaches have been reported in the literature, including TiO2 nanofibres treated with GPDES (3-glycidoxypropyl) methyldiethoxysilane (Fig. 1A);80 inner glass capillary surfaces with ZnO nanorods modified with (3-glycidoxypropyl) trimethoxy silane (GPTS);65 graphene nanosheets treated with 3-glycidyloxypropyl trimethoxysilane (GOPS);54 glass slides silanised with epoxysilane surface75,78 and functionalised graphene nanoplatelets with APTES (3-aminopropyl)-triethoxysilane.79
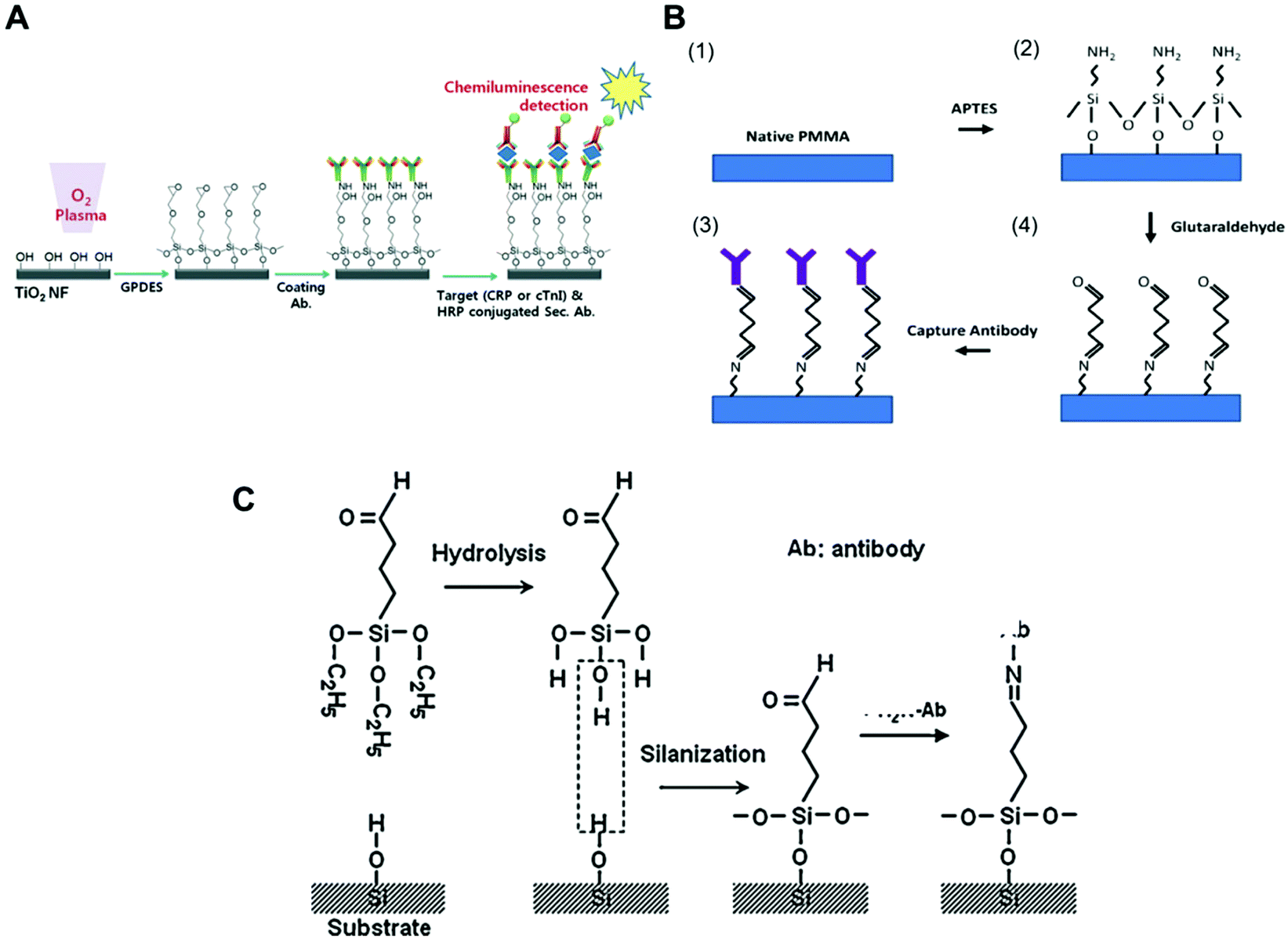 | ||
| Fig. 1 Examples of surface chemistries and the strategies exploited for the covalent immobilisation of antibodies in microfluidic devices used for protein biomarkers quantitation. (A) Schematic of antibody immobilisation and the immunoassay on TiO2 nanofibres (NFs), starting with plasma activation of the surface and the silanisation process using GPDES ((3-glycidoxypropyl) methyldiethoxysilane).80 (B) Silanization on PMMA (poly(methyl methacrylate)) using APTES ((3-aminopropyl)-triethoxysilane) followed by glutaraldehyde.69 (C) Aldehyde modification of a SiO2 surface and antibody immobilisation, using triethoxysilylbutyraldehyde (TESBA).47 Figures reprinted from ref. 80 with permission from the Royal Society of Chemistry; ref. 47 and 69 with permission from Elsevier. | ||
Silanisation and other surface modification chemistries also use aldehydes as cross-linkers for protein immobilisation. Some studies showed that amine derivatization followed by glutaraldehyde (GA) cross-linking yielded supports with greater amounts of immobilised enzymes and higher activity.98 Aldehyde is a reactive compound that forms a labile Schiff base with the amine and can be further reduced to form a stable secondary amine bond using NaCNBH3 or NaBH4. GA is a bis-aldehyde compound that has two reactive ends, and therefore can cross-link two amine functional groups, which can be two proteins or a protein and a surface polymer with amine groups, such as the organofunctional alkoxy silanes.93 Consequently, GA has been used as cross-linker for antibody immobilisation in microfluidic chips with APTES ((3-aminopropyl) triethoxysilane) for antibody covalent immobilisation to PMMA (poly(methylmethacrylate)), (Fig. 1B),69 but also with glass surfaces,40 magnetic particles35 and for the aldehyde surface modification of silica (Fig. 1C).47
4.3 Hybrid immobilisation strategies
The combination of covalent binding with passive adsorption and affinity binding has also been explored for immobilising the CapAb. Theoretically, this approach allows the best control of both the antibody orientation and activity, since a specific and known affinity binding is promoted. The two most common techniques for antibody immobilisation by affinity binding are by using avidin–streptavidin and protein A/G. The first uses the strongest non-covalent bond in nature (Kd = 1015 M−1). This high affinity ensures that once the complex is formed, it is not disturbed by extreme changes in pH and temperature, or by the use of organic solvents, denaturants, detergents and even proteolytic enzymes;99,100 therefore, it is stable during biochemical assays. The second common technique is protein A/G immobilisation, which relies on the specific interaction with the Fc constant region of IgG molecules;86 however, the affinity constant of this interaction is lower (approximately 1.13 × 108 and 2.90 × 107 M−1 for protein G and protein A, respectively) than the biotin–avidin binding, making the complex more vulnerable to surface washing. Recently, the conjugation of antibodies with DNA,101 synthetic peptides102 or oligonucleotides103 has also been applied in antibody immobilisation. All these techniques apparently lead to a higher affinity of the immobilised antibody, due to the proper orientation and good antibody density control. Nevertheless, only a few examples can be found of microfluidic assays that use this immobilisation strategy. This might be due to the complex immobilisation chemistry involved, since a combination of immobilising techniques must be considered. For example, achieving the proper orientation and activity of immobilised protein A is challenging by itself and affects the antibody immobilisation step.104 A surface plasmon resonance immunoassay detected PSA after covalently immobilising protein A/G to a glass surface to promote the affinity binding of antibodies. A microfluidic nanoelectrode array was also able to quantify PSA using antibodies conjugated with a linker complex of a metalised peptide nucleic acid that was covalently attached to a self-assembled thiols monolayer.585. Relevance of the surface area and surface-area-to-volume ratio
Analysis of the antibody–antigen equilibrium shows that a higher concentration of immobilised CapAb in a reaction medium will push the equilibrium towards the formation of an antibody–antigen complex. In high-sensitivity systems, the concentration of antigen available is very low, usually in the order of pM to fM. A good control on the total amount, density and activity of CapAb is therefore essential for achieving the required LLoD. In practice, the total amount of antibody that can be immobilised on a microfluidic channel well is limited and is smaller than a monolayer in order to avoid steric hindrance.88 This is ultimately linked to the surface chemistry selected for immobilising the antibody but also to the limited total surface area available. Since antibody immobilisation is a reaction process between a ‘clean’ high-affinity surface and an antibody solution, the surface-area-to-volume ratio of the microchannel or microcapillary is paramount to the overall performance of the microfluidic test. This depends on the antibody surface affinity, which in turn relies on the antibody immobilisation technique used. There are several examples of microfluidic platforms that have achieved high sensitivity by enhancing the surface area available. For example, a ‘lab-on-a-disc’ device used an antibody immobilised on coated polystyrene beads to yield LLoDs of 0.27 ng ml−1 (11.3 pM) for TnI, 0.27 ng ml−1 (1.45 pM) for CRP and 0.32 ng ml−1 (37.7 pM) for NTproBNP from whole blood samples.105 Using a similar device but with electrospun TiO2 nanofibres printed onto the surface of the chambers, the LLoD achieved for TnI in whole blood was 0.037 ng ml−1 (1.5 pM) and for CRP in serum was 8 × 10−4 ng ml−1 (0.007 pM) (Fig. 2A).80 This represented a 7-fold reduction in the LLoD for TnI and about a 300-fold reduction in the LLoD for CRP, by simply increasing the overall surface available for immobilising the CapAb. | ||
| Fig. 2 Strategies used for enhancing the surface area in microfluidic devices for antibody immobilisation. (A) TiO2 nanofibres used in a ‘lab-on-a-disc’ for CRP and TnI detection. SEM images of the TiO2 nanofibres (NFs): (i) top and (iii) side views of the low-density TiO2 NFs remaining on the donor Si substrate and (ii) top and (iv) side views of a high-density TiO2 NF mat transferred to the target Si substrate; insets 1 and 2 are the photographs of the TiO2 NFs (2 cm × 2 cm).80 (B) SEM images of ZnO nanorods grown on the inner surface of a glass capillary. (i) to (iii) Top-view; (iv) cross-sectional view; the inset of (i) shows the optical images of a capillary after (left) and before (right) the nanorod growth.65 (C) SEM images of the porous silicon network. (i) Cross-sections and (ii) top views of the rigid sponge-like porous silicon network structure.107 (D) Electron micrograph of a hot embossed microwell containing a microbead. The scale bar of the image is 4 μm, with a ×30000 magnification.51 Figures adapted from ref. 65 and 80 with permission from Royal Society of Chemistry;51 with permission from Institute of Physics; ref. 107 with permission from the American Chemical Society. | ||
A glass capillary device was able to quantify PSA, AFP and CEA in serum with a LLoD between 1 and 5 ng ml−1 (33.3 pM for PSA, 74 pM for AFP and 25 pM for CEA) based on ZnO nanorods deposited within the glass capillaries (Fig. 2B).65 A porous silicon array was able to increase the LLoD for PSA from 1.7 ng ml−1 (56.6 pM)67 to 800 fg ml−1 (0.027 pM) just by increasing the concentration of CapAb for passive adsorption.106 This reduction of more than 2000-fold in the LLoD for PSA was only possible due to the larger surface area of the porous substrate produced by the electrochemical dissolution of monocrystalline silicon (Fig. 2C).107 A popular approach used for enhancing the surface area is to immobilise the antibodies onto small beads (Fig. 2D). The CapAb–antigen complex immobilised onto the surface of the beads can then be detected with a second labelled antibody than binds specifically to the CapAb–antigen complex;35,41,49,51,53,63,66,108 alternatively, a secondary antibody immobilised onto the inner surface of the channels captures the complex bead-antibody–antigen.44 The beads can be magnetic, which facilitates the fluid actuation, mixing and separation of the bound and unbound antigen (washing). The use of magnetic beads in microfluidics has been fully reviewed by Tekin et al.109 Other authors have reviewed the use of beads in microfluidic immunoassays more broadly.109
6. Importance of non-specific binding and surface passivation
In a solid-phase immunoassay, non-specific binding of an antigen or detection antibody to the surface usually occurs, increasing the LLoD and reducing the signal-to-noise ratio. For optimised performance of the assay, two steps are essential: surface washing and blocking. The washing consists of removing molecules that are unbound or that present a lower affinity to the surface, whereas blocking consists of occupying any remaining binding sites on the surface with an inert agent to reduce the non-specific binding. It was discussed in section 4.1 that a half monolayer appears to favour CapAb–antigen binding, consequently many binding sites can remain available on the surface at the end of the first stage of CapAb immobilisation, which is especially true when the immobilisation of CapAb relies on physical adsorption.Although surface washing and blocking are routinely used in high-performance immunoassays, the specific methodologies and reagents used are often the result of an extensive empiric optimisation that provides the best signal-to-noise ratio and most robust performance for a given immunoassay. Nevertheless, the dependence on several physical and chemical variables, such as surface chemistry, antibodies purity and antibody affinity, is easily understandable. By analysing Table 1, it is clear that bovine serum albumin (BSA) is the most popular surface passivation agent used in microfluidic immunoassays when it comes to the quantitation of protein biomarkers. The composition of BSA blocking solution used varies from 0.1 to 3% w/v, with incubation times that can go from seconds to several hours.33,41,46,49,53,64,65,69,72–74,79–81 This suggests that BSA has a broad capacity of surface passivation, which is independent of the surface chemistry and assay reagents.110 CRP and CEA were quantified with LLoDs of 8 × 10−4 and 4 × 10−3 ng ml−1 in different microfluidic surfaces, such as TiO2 fibres and glass.53,80 BSA is also used in mixtures with other molecules, such as casein60,68 and sucrose. A paper microfluidic device reported LLoDs of 8 × 10−4 ng ml−1 for CEA using 0.5% BSA and 0.5% casein for surface passivation.68 A signal-to-noise ratio of 2300 was reported for 500 pM of TnI in an assay with 1% BSA and 10% sucrose in PBS for 1 h, with a LLoD of 0.16 ng ml−1 (3 pM).44 Casein was also used on its own for the surface passivation of treated glass slides, achieving LLoDs of 0.017 and 0.02 ng ml−1 for IL-1β and IL-6, respectively.40,47,75 Non-fat powered milk was used for PSA quantitation in an assay that achieved a LLoD of 1.7 ng ml−1 in silicon surfaces.67
The wide spectrum of traditional protein blockers used in microfluidic immunoassays has resulted in some impressive low LLoDs values, as can be seen in Table 1; however, the availability of modern microfluidic substrates has triggered the development of novel polymer matrices for surface passivation methods that are more effective and universal compared to protein blockers. For example, PDMS-based devices suffer low wettability and biofouling problems from non-specific protein/hydrophobic analyte adsorption.111 To overcome this issue a bio-barcode assay, which claimed attomolar sensitivity for PSA quantitation, achieved a LLoD of 1.5 × 10−5 ng ml−1 using polyDuramide™ for surface passivation. The polyDuramide™ polymer matrix adsorbs onto the glass and PDMS through hydrogen binding, reducing the non-specific signal and increasing the signal-to-noise ratio of the assay by at least 8-fold.35 Although anti-fouling coatings are still not widely used in microfluidic immunoassays, they could become key to the development of highly sensitive immunoassays to help achieve a very low LLoD. In general, the basic purpose of anti-fouling coatings is to minimise the intermolecular forces and interactions between ‘contaminating’ matter in the sample matrix and the surface of the microfluidic substrate, such that adhered molecules can be easily detached and released under low shear rates. Consequently, polymers with anti-fouling properties should be hydrophilic and electrically neutral, and should have hydrogen bond acceptors but no hydrogen bond donors. Materials/polymers, such as poly(ethylene oxide), PEG and polyzwitterion, polyhydroxy, have been used for anti-fouling coatings in PDMS devices, and this has been extensively reviewed by Zhang and Chiao (2015) and elsewhere.112,113 The preparation of superhydrophobic surface coatings with micro- and nanoscale feature dimensions has also been described with an aim to reduce the amount of surface contamination as well as to induce self-cleaning under flow conditions.114
7. Sample preparation
Most protein biomarkers produced in the human body are released into the blood stream, urine or other biological fluids. Nevertheless, the protein biomarkers currently used in healthcare to diagnose non-communicable diseases mostly target the blood stream, and for this reason, this review focuses only on preparations of blood samples. There are some recent efforts made towards the discovery and development of new protein biomarker immunoassays for non-communicable diseases that can be measured in urine and saliva, since these samples are less invasive and easier to collect compared to intravenous blood, which is also therefore ideal for POC testing.115,116 Independently of the biological sample used, the detection and quantitation of biomaker molecules involves a specific antibody–antigen reaction in a multi-component complex medium. Consequently, sample preparation is a major step in any high-sensitivity immunoassay, since the matrix components usually interfere with the assay performance.The use of biological samples is fundamental for the validation of an assay's performance; however, most of the reported microfluidic immunoassay devices have not been tested with real human samples, and the available data are mostly limited to a buffer spiked with recombinant or purified protein biomarker molecules.33,42,53,54,60,62,63,69,78 Some studies used other types of biological matrices as analyte diluents, in an attempt to mimic human biological matrices, such as undiluted goat serum,35 or fetal bovine serum,56 while other studies have relied on diluted human whole blood,81 plasma40,81 or serum.47,57,65,81 Although there are some examples of microfluidic devices that were able to quantify protein biomarkers in undiluted human plasma44 or human serum,41,46,49,66,68,75,108 only a very few studies reported the quantitation of protein biomarkers in microfluidic devices using undiluted whole blood samples. This includes the work with a novel fluoropolymer microfluidic material called a Microcapillary Film (MCF) for the quantitation of PSA in whole blood samples without sample treatment (Fig. 3) based on a heterogeneous sandwich immunoassay.72,73 From the perspective of the commercialisation of microfluidic diagnostics tests, this new alternative of using no sample preparation is by far the most appealing and promising,117 as miniaturisation of the sample preparation steps remains by far one of the biggest challenges within the microfluidics community.
 | ||
| Fig. 3 Microcapillary film (MCF) platform for a sandwich immunoassay using whole blood samples without sample treatment. (A) Microphotograph of 10-bore MCF containing 200 μm internal diameter microcapillaries. (B) ‘Lab-on-a-stick’ showing the loading of a blood sample by capillary action based on a fluoropolymer MCF coated with hydrophilic polyvinyl alcohol. | ||
Several studies have reported biomarker quantitation in microfluidic devices using pre-treated whole blood samples, with sample preparation structures embedded in the chip. For example, a lab-on-a-disc was capable of quantifying CRP and TnI from whole blood samples by separating the red blood cells through centrifugation,80 while a silicon porous microarray was integrated with an acoustophoresis system for plasma separation from whole blood samples (Fig. 4A)67 and other microfluidic devices have incorporated a flow-based blood separation channel for whole blood protein quantitation (Fig. 4B).56
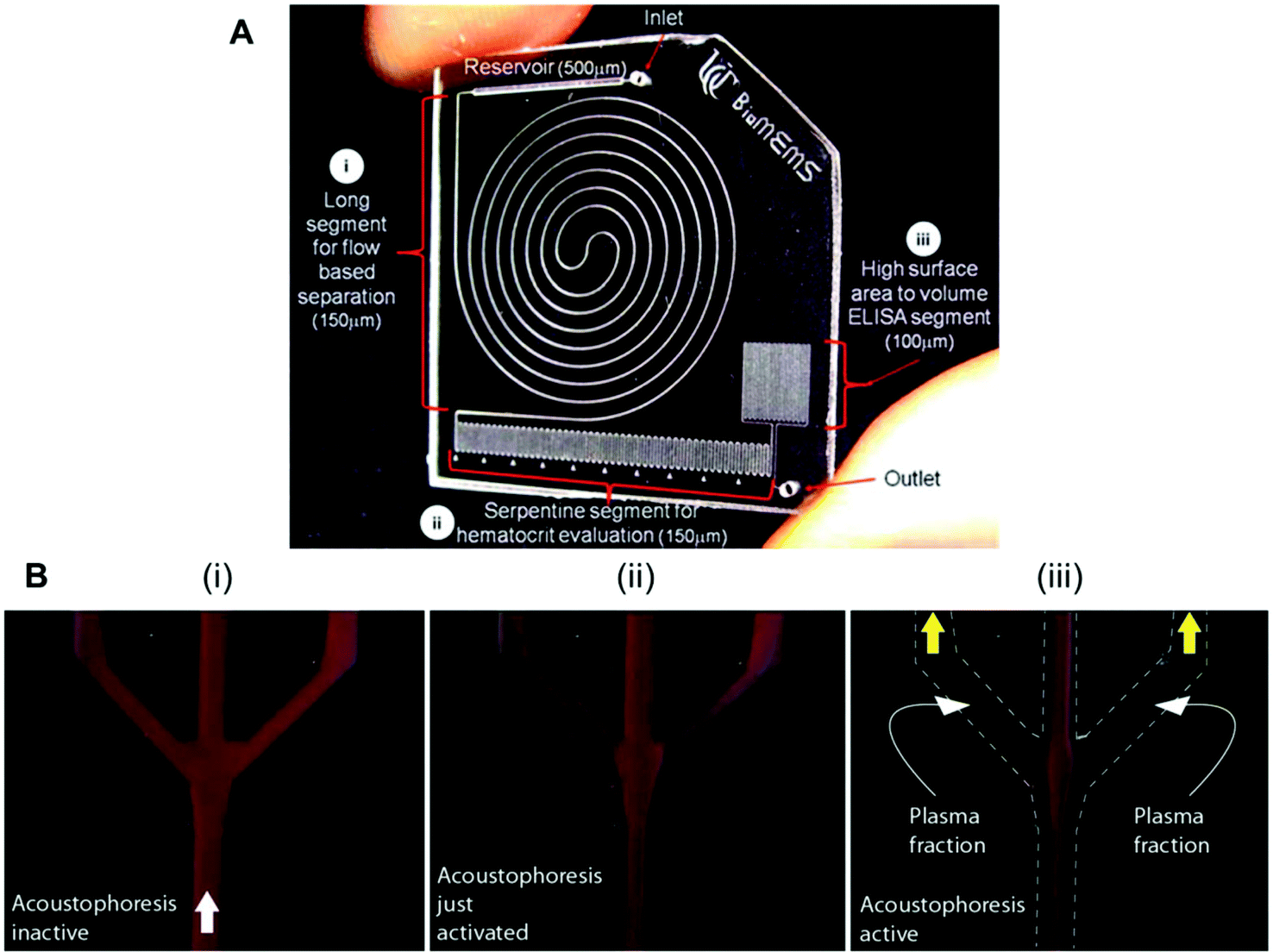 | ||
| Fig. 4 Examples of microfluidic approaches for whole blood sample treatment. (A) Integrated blood analysis chip design fabricated in COC (cyclic olefin copolymer): (i) a blood sample is injected into a long spiral flow-based separation channel; (ii) haematocrit is evaluated based on the number of serpentine switchbacks that are filled with packed erythrocytes; (iii) the blood sample is then flowed into a high surface-area-to-volume ratio ELISA protein quantitation segment where a biomarker of interest is evaluated.56 (B) Sequence showing the starting phase of plasma production (i) with inactive ultrasound, (ii) starting acoustophoresis, and (iii) continuous phase of plasma production, with the final fractions of red blood cells removed via the central outlet.67 Figures adapted from ref. 56 and 67 with permission from the Royal Society of Chemistry. | ||
8. Fluid handling control
The standard fluid handling procedure in sandwich heterogeneous immunoassays involves several steps, with multiple sequential reagents addition and intensive washings between reagents incubation. This procedure allows a higher amount of antigen to be bound, due to the extended sample incubation time. The washings are important to reduce the non-specific signal resulting from the antigen detection antibody (DetAb) or enzyme molecules that eventually bind non-specifically to the solid surface. Consequently, high-performance sandwich assays require a multistep procedure to achieve the required sensitivity.24 There are some successfully reported attempts that have used microfluidic technologies for the automated sequential delivery of reagents that have converted the overall procedure into a single step for the operator.46,69 For example, Gervais et al. were able to quantify CRP protein with a LLoD of 10 ng ml−1 (90 pM) in 3 min, and <1 ng ml−1 (9 pM) in 14 min, in a PDMS chip involving integrated reagents with the flow controlled by an embedded capillary pump.46 This was possible due to the extremely low flow rates (30 nl min−1) promoted by the capillary pump. Overall, both fluid handling and actuation are paramount factors for achieving sensitive protein quantitation in microfluidic protein biomarker immunoassays.8.1 Pressure-driven systems
Pressure-driven flow systems presuppose the use of an external fluid control actuation. The most common external fluid-control devices in microfluidic systems are flow pumps, typically syringe pumps. Pumps can deliver flow rates over several orders of magnitude, from pl min−1 to several ml min−1, as well as allowing stop and continuous flow procedures, which are important for precise reagent loading and incubation in sandwich immunoassays. These are also easily connected to microfluidic microchannels or microcapillaries, with the flow being typically laminar in the biosensing region due to the small dimensions of the devices. However, pumps are normally expensive and require a continuous power supply, thus compromising the portability of the system. Most microfluidic devices reported in the literature use external pumps for fluid control in order to achieve sensitive protein biomarkers quantitation in sandwich immunoassays. The bulky aspects of syringe pumps is naturally a challenge for the successful miniaturisation of POC tests.Several microfluidic devices use an immunoassay procedure based on a continuous flow of reagents at variable flow rates,35,51,56,63,65,81 but some studies used stopped flow during the incubation of reagents.41,53,54,58,67 The choice of flow mode appears to be more related to the personal preference of the authors, as currently these is a lack of literature on the effect of flow on the immunoassays performance. The dimensions of the microfluidic devices used are also very variable; however, all the studies herein reported used microchannels or microcapillaries as reaction chambers. The MCF technology allows interfacing of the microfluidic strips directly with the reagent wells, and uses a pressure-driven system that relies on disposable and low-cost fluid-control devices, named a Multiple Syringe Aspirator (MSA), capable of loading solutions into 80 capillaries simultaneously using an array of 1 ml plastic syringes through a simple rotation of a central knob (Fig. 5A).72
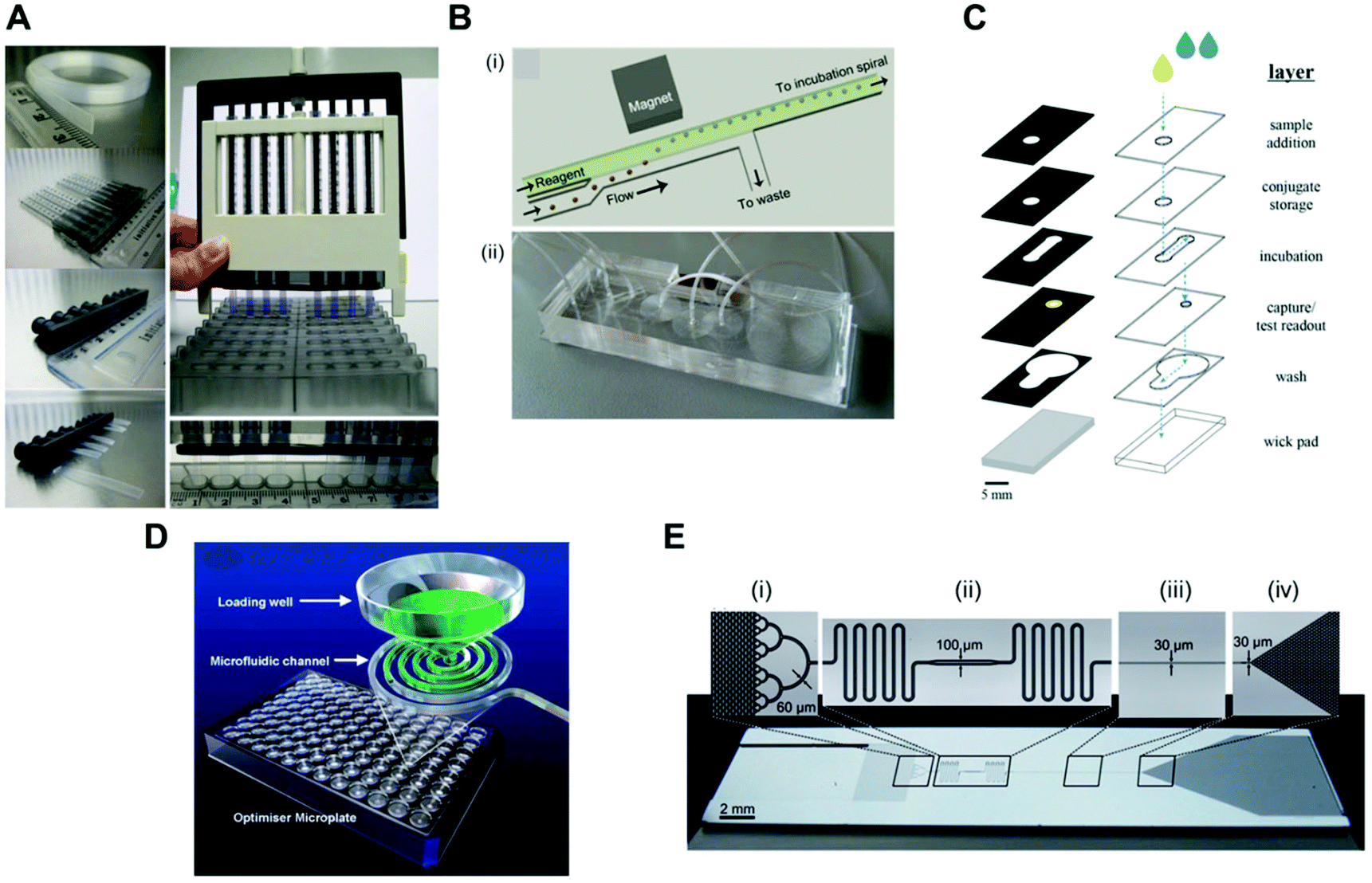 | ||
| Fig. 5 Fluid-control approaches implemented in microfluidic devices for protein biomarker quantitation. (A) Multiple syringe aspirator (MSA) used in microengineering fluoropolymer microcapillary film (MCF) strips and disposable 1 ml syringes for generating pressure-driven flow through 80 parallel microcapillaries.73 (B) Magnetic automated bead transfer device: (i) the magnet pulls the beads from the carrier stream to the reagent stream, whereas the current stream is diverted to waste; (ii) an assembled three-layer PDMS microdevice.63 (C) Fluid handling through a 3D microfluidic paper device with hydrophobic patterned barriers (black areas).77 (D) Microfluidic microtiter plate (optimiser microplate) with gravity controlling the fluid flow.62 (E) Fluidic control in a microchannel using capillary pumps with an average flow rate of 82 nl min−1: (i) sample collector ending with hierarchical delay valves; (ii) flow resistors and central deposition zone for the detection antibodies; (iii) reaction chamber and (iv) capillary pump.46 Figures adapted from ref. 46, 62, 73 and 77 with permission from the Royal Society of Chemistry;63 with permission from Springer. | ||
8.2 Centrifugal forces
Many microfluidic devices use centrifugal forces for fluid flow control in protein biomarker immunoassays. For example, a lab-on-a-disc capable of measuring CRP and TnI, with a LLoD of 0.27 ng ml−1 (11.3 pM) and BNP with a LLoD of 0.32 ng ml−1 (37.6 pM), moved reagents from one chamber to the other based on a rotating disc and a specific valve actuation.80,105 Honda et al. also described a disc-based microfluidic platform capable of quantifying AFP, IL-6 and CEA, with detection limits of 0.01, 0.026 and 0.26 ng ml−1 (0.14, 1.24, 1.3 pM), respectively, using centrifugal forces. Other immunoassay devices used centrifugal force for reagents actuation, as critically discussed by Gorkin et al.1188.3 Magnetic forces
The use of a magnetic field is another form of fluid actuation successfully implemented in microfluidic devices, and allows performing heterogeneous immunoassays involving multiple steps. This has been successfully used for the quantitation of protein biomarkers.119 Here, CapAb is usually immobilised onto the magnetic beads surface and then moved through a sequence of chambers containing a series of reagents (Fig. 5B). LLoDs of 3.5 ng ml−1 for CEA, 3.9 ng ml−1 for AFP and 3.2 ng ml−1 for PSA have been reported in human serum samples. These are clinically relevant for AFP and PSA biomarker detection; however, they are not sufficient to meet not sufficient to meet clinical requirement for CEA biomarker, which presents a clinical threshold above 2.5 ng ml−1.42,108,120 Alternatively, the magnetic beads containing the antigen already captured may bind to a coated surface, achieving a LLoD of 0.16 ng ml−1 for TnI in undiluted plasma.44 The ability to move the ‘reaction’ surface instead of the liquid reagents allows achieving a high degree of simplification of the immunoassay procedure.8.4 Passive-flow systems
In passive-flow systems, the fluid actuation is not promoted by an external mechanical or magnetic force, but rather by an intrinsic device characteristic, such as the surface properties. For example, lateral flow ‘dipstick’ technology relies on hydrophilic strip properties and the geometry of nitrocellulose membranes. In analogy, some microfluidic systems were developed operating on the same principle of passive flow, with flow rates being determined by the intrinsic design of the device. The typical driving forces for propelling liquids in passive microfluidics are, for example, chemical gradients on surfaces, osmotic pressure, degassed PDMS,121 permeation in PDMS,122 gravity and capillary forces.123 The drawback of this fluid strategy is generally the difficulty in controlling the flow rate, sample volumes and incubation times in these systems, and for that reason, a number of microfluidic strategies were developed for enhancing the passive-flow control in immunoassay devices. For example, wax patterning as hydrophobic barriers onto hydrophilic paper creates microchannels and reaction chambers, which allowed a ‘lab-on-a-paper’ device to quantify α-AFP, cancer antigen 125 and CEA with LLoDs of 0.06 ng ml−1 (0.9 pM), 6.6 × 107 ng ml−1 (0.33 U ml−1) and 0.05 ng ml−1 (0.25 pM), respectively.60 Also, an origami gold/graphene paper immunosensor was able to quantify CEA with a LLoD of 8 × 10−4 ng ml−1 (0.004 pM).68 The pregnancy hormone hCG was also quantitated, with a LLoD of 6.7 × 106 pM (6.7 mIU ml−1) in a paper microfluidic device (Fig. 5C).77 Paper microfluidic devices achieved good LLoDs for protein quantitation based on printed channels and architectured 3D paper structures for controlling antibody immobilisation and reagent incubation times. These features are not found in lateral flow devices though, which probably explains the lack of sensitivity of conventional ‘dipstick’ tests. The use of paper microfluidics in diagnostics was fully reviewed by Yetisen et al.124In addition to capillary forces (which are linked to wettability of the microfluidic device), gravity can also be effective in generating continuous fluid movement along a microfluidic surface. A novel microfluidic microtiter plate was able to quantity PSA and IL-4 with LLoDs of 0.016 ng ml−1 (0.53 pM) and 2 × 10−4 ng ml−1 (13.3 pM), respectively, only based on gravity (Fig. 5D).62
A more sophisticated yet challenging approach in respect to microfabrication was proposed by Zimmermann et al.123 and involved a series of autonomous capillary systems with liquids displaced by capillarity to enable accurate volumes of liquids and precise flow rates to be achieved. The capillary pumps comprised microstructures of various shapes with dimensions ranging from 15 to 250 mm, positioned in the capillary pumps to encode a desired capillary pressure and to provide a flow rate between 12 and 222 nl min−1.123 Capillary pumps integrated in microfluidic devices have been used to quantify TnI with a LLoD of 0.024 ng ml−1 (1 pM),69 TNF-α with a LLoD of 0.02 ng ml−1 (0.38 pM)33 and CRP with a LLoD of 1 ng ml−1 (9 pM).46 CRP was quantified using a one-step sandwich assay, using reagents integrated in the microfluidic device and an immunoassay triggered upon the addition of a sample (Fig. 5E).
9. Detection modes, signal amplification and readout systems
An essential feature in quantitative immunoassays is the detection of the antibody–antigen complex. Miniaturised systems, due to the small volumes (pl to μl) used, are able to detect simultaneously a wide a range of molecules, a technique called multiplexing. Most heterogeneous immunoassays reported have implemented optical detection based on labels, which are molecules that can produce a detectable signal. Depending on the nature of the signal, immunoassays can be segmented as colorimetric, fluorescent or chemiluminescent. Other immunoassay detection modes are based on electrochemical signal changes or refractive index changes, with techniques called label-free techniques, since they do not rely on labels.Colorimetric assays measure the antibody–antigen complex through the colour intensity of a solution or particles. Colorimetric detection is inherently less sensitive than fluorescence and chemiluminescence, since in order to measure low concentrations of a chromogen, small differences in intensity must be measured at a high light intensity, which limits the LLoD. Also, the relationship between the optical absorbance and intensity of transmitted light is logarithmic. Therefore, at high chromogen concentrations, large differences in optical absorbance can still lead to small differences in the intensity of transmitted light, which usually corresponds to a narrow dynamic range for immunoassays.125 Nevertheless, chromogenic substrates offer speed, simplicity, a well-established assay chemistry, high quality reagents and the widespread availability of cost-effective readers. For this reason, several studies have presented new ways to increase the performance of colorimetric microfluidic detection, for example through enzymatic amplification systems126,127 with a detectable chromogen in solution or through the use other amplification systems, such as gold nanoparticles silver enhancement,128–130 with the colour intensity given by small particles.
Enzyme amplification depends on the biocatalytic capability of these molecules, as a single enzyme molecule can produce up to 107 molecules of substrate per minute, increasing the strength of the signal and therefore the sensitivity a million fold, when compared to a label that produces just a single event.131 Independently of the selected colorimetric or fluorescence mode, enzymatic amplification is one of the most powerful aspects of an immunoassay in a microfluidic device for measuring protein biomarkers, as concentration can rapidly increase in very small volumes and without relying on mixing or long diffusion distances.
Silver enhancement is an amplification technique that makes use of larger gold nanoparticles, which in theory are easier to detect at low concentrations. This technique depends on silver ions adhering to the surface of the gold nanoparticles. Gold has the capacity to catalyse the silver ions, reducing these to silver atoms, promoted by electrons released from the reducing molecules in solution around the gold nanoparticles. Silver atoms have the same catalytic capability as gold nanoparticles, and therefore successive layers of silver atoms are deposited, thus increasing the particle size.129
There are no reports in literature of colorimetric microfluidic immunoassays applied to sensitive protein biomarkers quantitation without amplification, which is to some extent no surprise. A microfluidic paper device was able to quantify hCG, the pregnancy hormone, using only colloidal gold nanoparticles and a flatbed scanner as a readout system; however, pregnancy tests LLoDs are much higher than cancer and cardiovascular diseases LLoD tests.77 For example, PSA was quantified on microfluidic platforms using colorimetric enzymatic amplification and smartphones with LLoDs of 3.2 ng ml−1 (107 pM) for a PDMS device66 and 0.9 ng ml−1 for a MCF platform.73 CRP was quantified with a LLoD of 0.07 ng ml−1 (0.6 pM), also based on colorimetric enzymatic amplification and a smartphone camera.79 The bio-barcode was able to quantify PSA using silver-enhanced gold nanoparticles, with a LLoD of 1.5 × 10−5 ng ml−1 (5 × 10−4 pM).35
Although colorimetric detection has been used successfully for protein biomarker quantitation in microfluidic devices, fluorescence is by far the most common detection mode used for sensitive microfluidic immunoassays, as can also be seen in Table 1. This is probably due to the fact that fluorescence detection systems are intrinsically more sensitive, as they are measured relative to the absence of light. Also, fluorescent signals respond linearly to excitation light intensity, up to the limit of quenching and photo-bleaching.125 Fluorescence occurs due to certain molecules, called fluorophores, that emit light at a certain wavelength. For the emission to occur, fluorophores need to absorb light at a different wavelength that will excite electrons forcing them to move to a superior energetic level. The excitation and emission wavelength depends on the fluorescent molecule. Several microfluidic devices were able to detect protein biomarkers without the need for further amplification, using fluorophores as assay labels. Although conjugating antibodies directly to fluorophores offers the possibility of simplifying the assay procedure, these immunoassays use expensive and bulky readout equipment. For instance, an immuno-pillar platform was able to quantify CRP, α-AFP and PSA with a LLoD of 0.1 ng ml−1, using fluorophores (FITC, Alexa fluor 555, and Dylight 649) directly conjugated to the DetAb and an inverted fluorescence microscope.49 The CRP detection, with a LLoD of 1 ng ml−1 was performed by a microfluidic assay using Alexa Fluor 647 and a fluorescence microscope.46 Interleukin-8 and insulin were quantified in a microfluidic immunoassay using Alexa fluor 488 and an epifluorescence upright microscope.51 IL-6 and TNF-α were quantitated with a LLoD between 0.01 ng ml−1 and 1 ng ml−1 using phycoerythrin and a Bio-Plex 200 array reader as a readout system.63 PSA was quantified with a 1.7 ng ml−1 LLoD, in a porous silicon substrate, using FITC and a confocal microscope as a readout system.67
Fluorescent scanners were also successfully used in protein biomarkers quantitation with fluorescent signal detection without further signal amplification. For example, TNF-α was detected with a LLoD of 0.02 ng ml−1 in a mosaic microfluidic platform using detection antibodies directly conjugated to the fluorophores Cy5 and Alexafluor 647.33 PSA, TNF-α, IL-1β and IL-6 were quantitated with a LLoD of 1 pg ml−1 using the neutravidin-conjugated fluorophores Dylight 488, 550 and 650.75 Also, IL-6, IL-1β, TNF-α and PSA were quantified with LLoDs between 4 and 30 pM with the fluorophores Alexa fluor 647, phycoerythrin and Alexa fluor 546, directly conjugated to DetAb.78 As expensive and bulky equipment is incompatible with the product specifications of microfluidic POC diagnostic tests, several studies used portable, low-cost and sensitive fluorescent readout systems, capable of reading fluorescent signals. For example, TnI was quantified with a LLoD of 0.024 ng ml−1 using detection antibodies conjugated with FITC (fluorescein isothiocyanate) with a homebuilt readout system, with dimensions of 10 × 7 × 7 cm3, an LED (Nichia ultrabright blue LED) for fluorescence excitation, an excitation and emission filter, a 10× objective and a detector (H9858 photosensor module) (Fig. 6A).69 By using a smartphone, a portable black UV light and a dichroic filter for illumination system, PSA was quantified with a LLoD of 0.04 ng ml−1 (Fig. 6B).65
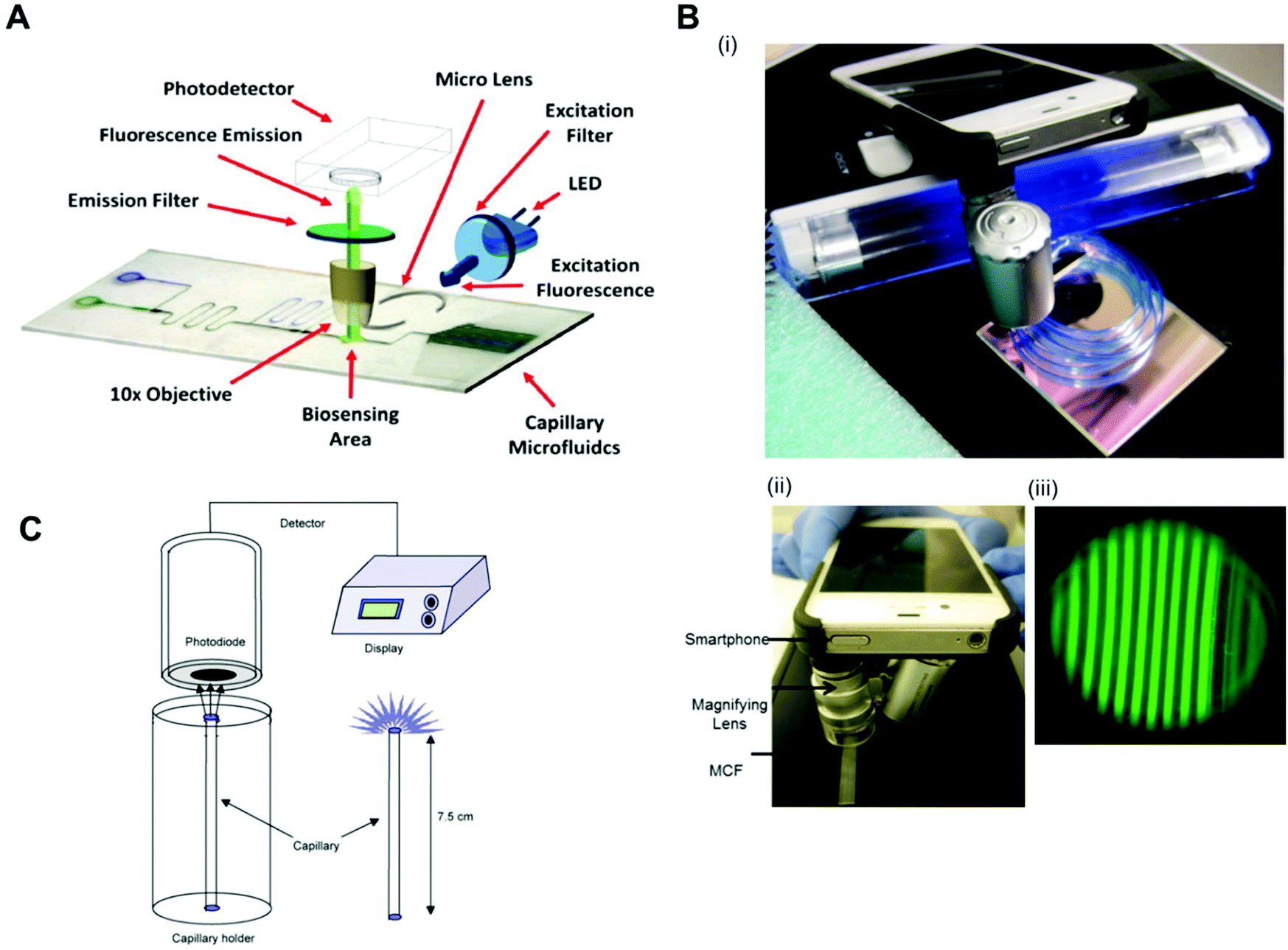 | ||
| Fig. 6 Detection modes and readout systems used in microfluidic devices for protein biomarker quantitation. (A) Configuration of a fluoroimmunosensing device for an autonomous capillary microfluidic signal detection system.69 (B) Smartphone fluorescence detection system in a microcapillary film: (i) MCF phone components; (ii) MCF phone detection and (iii) smartphone fluorescence image of microcapillaries.73 (C) The set-up for the measurement of chemiluminescence using a photodiodetector and the special stand for the vertical positioning of the capillaries.40 Figures adapted from ref. 40, 69 and 73 with permission from Elsevier. | ||
Fluorescence was also detected with quantum dots nanocrystals, with the quantum mechanical properties and excitation confined to the nanocrystal. For example, CEA and α-AFP were quantified with LLoDs of 3.5 (17.5 pM) and 3.9 (57.3 pM) ng ml−1, using streptavidin conjugated to quantum dots and an ICCD camera.108
Chemiluminescence is caused by a molecular reaction of two (or more) ground-state molecules producing a final molecule in an excited state. The energy in the reactants is transferred to the products, which are also excited while they are being formed. Contrary to fluorescence, in chemiluminescence there is no need for an excitation light source, which simplifies the optics, which therefore makes it highly desirable for POC. On the other hand, the signal has to be measured in the absolute dark, similar to fluorescent measurements, with a deep cooled camera. In general, chemiluminescence allows an improvement in terms of higher sensitivity and lower LLoDs, but the design of robust portable chemiluminescence detectors is naturally challenging.
Chemiluminescence requires enzymatic signal amplification (more commonly, HRP) and a chemiluminescent substrate (the most common is Luminol), which adds one more step to the microfluidic immunoassay compared to traditional fluorescence. Several microfluidic devices use chemiluminescence for sensitive protein quantitation. For example, CRP and TnI were quantified using HRP with LLoDs of 8 × 10−4 and 0.037 ng ml−1, respectively, measuring the chemiluminescent signal with a homebuilt system, comprising a cooled PMT module and a CCD camera.80 IL-4 and PSA (LLoDs of 2 × 10−4 ng ml−1 and 0.016 ng ml−1, respectively) were also quantitated based on chemiluminescence, HRP and a microplate fluorescent reader.62 Insulin and IL-6 were also quantified by chemiluminescence, using biotinylated AP bound to streptavidin magnetic beads and a photomultiplier tube.42 CEA was quantified with a LLoD of 0.041 ng ml−1 with gold nanoparticles functionalised with DNAzyme.53 Troponin T was quantified with a LLoD in the range of 10 to 100 ng ml−1 with HRP, using a photomultiplier and an oscilloscope.56 Myoglobin, CK-MB, TnI and FABP were quantified with LLoDs of 1.2, 0.6, 5.6 and 4 ng ml−1 respectively, based on chemiluminescence, with HRP and a photodiode detector (Fig. 6C).40 AFP, cancer antigen 125 and CEA were quantified with LLoDs of 0.06 ng ml−1, 6.6 × 107 ng ml−1 and 0.05 ng ml−1, respectively, using chemiluminescence with HRP and a luminescence analyser.60
Other detection modes used for microfluidic protein quantitation involve non-optical detection modes, such as electrochemical detection, which is important for opaque substrates and dense optical matrices.132 These have reported PSA quantitation of 0.01 ng ml−1 using glucose oxidase PSA conjugated in a competitive assay and a custom built-in potentiostat as the readout system.58 CEA and AFP were quantitated with LLoDs of 1 × 10−3 ng ml−1 using electrochemical detection and an electrochemical analyser.54
Label-free techniques based on refractive index changes of magnetic beads attachment to a surface were able to quantify TnI with a LLoD of 0.024 ng ml−1, using a total internal reflexion biosensor and a CCD camera.44 CRP was quantified with a LLoD of 1.2 ng ml−1, using Biacore surface plasmon resonance.81 Label-free techniques involve fewer steps and therefore are usually faster to perform; however, they are not always as sensitive as the labelled techniques, and very often they require very expensive equipment. For example, a label-free technique based on measuring the shifts in microring resonance was able to increase the sensitivity from μg ml−1 to pg ml−1 by amplifying the signal with streptavidin-coated microbeads (Fig. 7).57 On the other hand, some other technologies, such as nanowire biosensors, present great potential for the quantitation of protein biomarkers at POC settings, as these can be cost-effective133 and allow sensitive detection without labels.134,135 It has been shown that silicon nanowires with a primary antibody covalently bound to their surface enable the detection of biomarkers by registering a change in the conductance, which is proportional to the amount of antigen bound.134 Although nanowire sensors are in the early stage of development, PSA and CEA were quantified with a LLoD of 9 × 10−4 ng ml−1 in a multiplex assay using a nanowire sensor with human serum samples.135
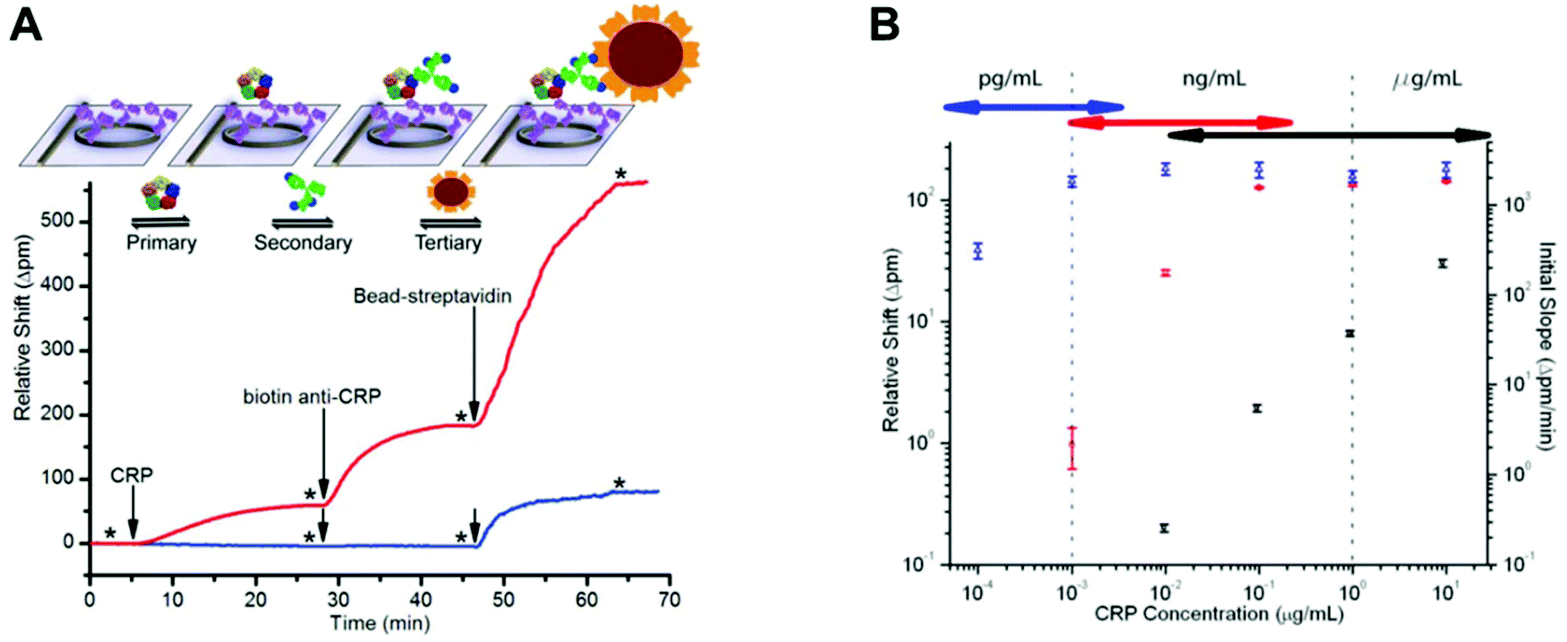 | ||
| Fig. 7 Signal amplification impact on CRP assay sensitivity and dynamic range using a microring resonator. (A) Schematic and real-time data plot showing the sequential addition of CRP, the biotinylated secondary antibody and streptavidin-functionalised beads on the microring resonators. The red trace is 1 μg ml−1 of CRP, while the blue trace is 0.01 μg ml−1 of CRP. (B) A log–log calibration plot showing the response of the microring resonators to varying concentrations of CRP using the three-step assay. Black squares indicate the initial slope of the primary binding (right axis), while the red circles indicate the secondary antibody shift and the blue triangles indicate the bead shift (left axis).57 Reproduced from ref. 57 with permission from the Royal Society of Chemistry. | ||
Over 90% of the microfluidic immunoassays summarised in Table 1 used complex, non-portable and expensive readout systems to quantify protein biomarkers, with only a few studies using microfluidic devices with a readout system comprising low-cost optoelectronic components, such as a flatbed scanner77 or a smartphone.66,79 Although the use of equipment for the quantitation of immunoassays is not compatible with the ASSURED criteria, WHO's policy is perhaps disconnected from the current reality in rapid technological progress. The cost of optoelectronic components has dropped massively over the last decade136 and are now found in most portable gadgets and home smart equipment. It is now possible to use optical readout systems that are very low-cost or even disposable; one example, is the latest ‘digital’ semi-quantitative pregnancy test from ClearBlue.137
10. Importance of affordable microfluidic manufacturing and compact low-cost detection
Sensitive biomarker quantitation in microfluidic systems is only possible by manipulating a complex interaction of effects associated with the miniaturisation of the sandwich immunoassays and technological instrumentation available. These two aspects have to be integrated in such a way that sensitive detection is enabled with cost-effective technologies, otherwise it loses its POC applicability. The starting point of this integration is the manufacturing process selected for microfluidic devices, which needs first of all to allow mass production, in order to make it cost effective. Another aspect to consider is the use of a compact low-cost detection mode without compromising sensitivity, which is important for the adoption of technology for POC testing. This second aspect is also intrinsically linked to the manufacturing of microfluidic devices, as non-opaque microfludic materials cannot be used for optical detection, while auto-fluorescent materials will not achieve the sensitivity required for the diagnostic of non-communicable diseases based on fluorescence detection. In other words, the optimisation of a microfluidic immunoassay ultimately depends on the manufacturing technique selected and available, which sets the geometry and materials used, and ultimately the economics, which in practice dictates the adoption of the microfluidic POC test.According to Becker,138 the limited success of microfluidic devices being commercialised is associated with underestimating the challenges of microfluidics manufacturing processes, which are usually overlooked by the designers and people working on the application areas. Becker138 claims that there are no technical barriers to build microfluidic devices; however, to be able to compete with conventional solutions, a thoughtful study of the design and manufacturing planning must be performed. For example, the number of produced units will influence the cost of the microfluidic device; therefore for low to medium volumes of manufacturing processes, lower initial investments are preferred, such as elastomer casting of soft polymers, including PDMS, and hot embossing. These are the most popular manufacturing techniques used within the academic environment. If a large volume of products is desired, for example, in the field of POC diagnostics, injection moulding is more suited, although it requires a high initial investment, but this is compensate for at high product volumes with the low cost of the raw materials.138 The manufacturing techniques and materials used for the fabrication of microfluidic devices were critically reviewed by Waldbaur et al.139
An analysis of the manufacturing processes used in microfluidics shows that most protein quantitation devices are fabricated for small-scale production. Therefore, soft litography and fast prototyping techniques are the most popular manufacturing processes used.33,35,42,46,56,58,63–67,69,75,78,80 This is certainly one of the reasons why microfluidics are still not widely commercialised, as those techniques lack scalability, yet alternative technologies are expensive with a complex manufacturing process involving many steps. Nevertheless, some microfluidic devices already use a scalable manufacturing process adequate for the mass production of POC diagnostic devices, such as injection moulding.44,49,62,79 Several studies developed the sensitive quantitation of protein biomarkers in paper, due to the low cost of paper manufacturing.60,68,79 A further innovative approach is the use of mass-manufactured melt-extruded film for the quantitation of protein biomarkers;72,73 whereby melt-extrusion is perhaps the most cost-effective method for embedding microengineering features in thermoplastics, and this technique allows fabricating several kilometres of material per day with a single extrusion line, sufficient to produce up to 1 million test strips.
11. Conclusions and future outlook
Microfluidic protein quantitation is a promising area for the POC diagnosis of non-communicable diseases and severe infectious, such as sepsis. Diagnosing these health conditions requires the accurate quantitation of very small amounts of biomarkers present in biological samples. The main challenge for the successful commercialisation of POC microfluidic diagnostic tools is to combine a good sensitivity of the test with low-cost manufacturing and affordable and compact signal detection. Understanding the intrinsic science of immunoassays and miniaturisation is essential to overcome current barriers.The effective miniaturisation of immunoassays requires a deep understanding of antibody immobilisation, biological matrix interference, fluid control, surface passivation and signal detection modes. These are fundamental aspects of microfluidic immunoassays, and the interaction between all these aspects should not be disregarded when it comes to achieving the highly sensitive quantitation of protein biomarkers. Microfluidic devices to date have used several methods for antibody immobilisation, including passive adsorption, which is common with plastic surfaces, covalent binding, where silanisation seems to be the base of most of covalent binding techniques, and a combination of the two techniques together with some antibody-orientated techniques, which are still not widely used. The covalent binding of an antibody is the most popular method used for antibody immobilisation onto the surfaces of microfluidic devices. Most devices reported in the literature have reported data for the detection in buffers or non-biological matrices that mimic biological samples in immunoassays; however, there is some success in embedding structures within the microfluidic device for plasma separation from whole blood samples. Sample preparation remains one of the main challenges for immunoassay miniaturisation; however, there is now a precedent in carrying out sensitive protein biomarkers detection without sample preparation that used fluoropolymer microfluidic strips. Although relying on empirical rules, effective surface passivation and washings are essential to yield very low LLoD values.
Fluid control remains mostly performed by pumps, which are instruments external to the chip, with reagents loaded through pressure-driven systems capable of stopping the flow during the incubation of reagents or performing multiple assay steps.
The most common detection mode utilised is optical fluorescence, which uses complex and expensive readout systems, such as microscopes, flow cytometers or fluorescent scanners. Signal amplification is often used in microfluidic protein biomarker quantitation and is usually related to the detection mode and readout system. With the rapid decrease in the cost of optoelectronic components, now is the ideal moment to implement more effective and affordable optical interrogation strategies in microfluidic tests that do not rely on expensive, bulky and ultra-sensitive detection equipment.
The seven core technical aspects discussed in this review (antibody immobilisation, surface area, surface passivation, sample preparation, fluid control, signal detection and affordability of both manufacturing and the detection system) should be considered much earlier during the development of novel microfluidic devices for protein biomarker measurement through applying a highly integrated approach. The unmet medical need or specific application should feed the technical specifications, and not the other way round. Sensitivity can potentially be achieved in many different ways; however, scalability and effective product adoption will require several technical compromises that clearly are not yet being met by the majority of microfluidic tests under development. Overall, microfluidics research appears to be still at the very early stage of demonstration; there is some success in demonstrating the possible of quantifying proteins ‘on a chip’ in academic environments; however, most technological solutions currently being explored remain somehow disconnected from the industrial and societal realities. This is well illustrated by the large amount of microfluidic devices that rely on expensive and bulky external pumps and expensive and non-portable readout systems for the immunoassay quantitation of protein biomarkers. In addition, most microfluidic devices are manufactured by prototyping techniques, instead of easily scalable manufacturing processes.
The future of microfluidic protein biomarker quantitation should involve the development of manufacturing techniques that use low-cost raw materials and designs that are more easily scalable. Also, simplifying the immunoassay procedure without compromising the sensitivity opens up the possibility of eliminating external powered instruments, such as pumps and microscopes. This has to be achieved by the proper integration of all the microfluidic immunoassays aspects, so that the end product is a commercially viable POC device. For example, adding a signal amplification step to a POC test might eliminate the need to use an expensive readout system, while eliminating sample preparation has the potential to reduce the complexity and cost of the microfluidic diagnostic tests.
A better understanding of miniaturised immunoassays is essential for designing and planning the future manufacturing of microfluidic devices for sensitive POC diagnostics to enable them to contribute to earlier diagnostics and to support a reduction in the number of deaths from chronic diseases, such as cancer and cardiovascular diseases, around the world.
Notes and references
- G. Egger, In Search of a Germ Theory Equivalent for Chronic Disease, Prev. Chronic Dis., 2012, 9, 110301 Search PubMed.
- G. Egger and J. Dixon, Beyond Obesity and Lifestyle: A Review of 21st Century Chronic Disease Determinants, BioMed Res. Int., 2014, 2014, 731685 CrossRef PubMed.
- D. Yach, C. Hawkes, C. L. Gould and K. J. Hofman, et al., The Global Burden of Chronic Diseases: Overcoming impediments to prevention and control, J. Am. Med. Assoc., 2004, 291(21), 2616–2622 CrossRef CAS PubMed.
- R. Beaglehole and D. Yach, Globalisation and the Prevention and Control of Non-Communicable Disease: The Neglected Chronic Diseases of Adults, Lancet, 2003, 362, 903–908 CrossRef CAS.
- A. D. Lopez, C. D. Mathers, M. Ezzati, D. T. Jamison and C. J. Murray, Global Burden of Disease and Risk Factors, World Bank, 2006 Search PubMed.
- P. Kanavos, The Rising Burden of Cancer in the Developing World, Ann. Oncol., 2006, 17(Suppl 8), viii15–viii23 Search PubMed.
- D. S. Celermajer, C. K. Chow, E. Marijon, N. M. Anstey and K. S. Woo, Cardiovascular Disease in the Developing World: Prevalences, Patterns, and the Potential of Early Disease Detection, J. Am. Coll. Cardiol., 2012, 60, 1207–1216 CrossRef PubMed.
- C. D. Mathers and D. Loncar, Projections of Global Mortality and Burden of Disease from 2002 to 2030, PLoS Med., 2006, 3, e442 Search PubMed.
- H. Kettler, K. White and S. Hawkes, Mapping the landscape of diagnostics for sexually transmitted infections, WHO/TDR, Geneva, 2004 Search PubMed.
- D. A. Giljohann and C. A. Mirkin, Drivers of Biodiagnostic Development, Nature, 2009, 462, 461–464 CrossRef CAS PubMed.
- P. Yager, T. Edwards, E. Fu, K. Helton, K. Nelson, M. R. Tam and B. H. Weigl, Microfluidic Diagnostic Technologies for Global Public Health, Nature, 2006, 442, 412–418 CrossRef CAS PubMed.
- C.-C. Lin, J.-H. Wang, H.-W. Wu and G.-B. Lee, Microfluidic Immunoassays, J. Assoc. Lab. Autom., 2010, 15, 253–274 CrossRef CAS.
- J. Zhu, N. Zou, D. Zhu, J. Wang, Q. Jin, J. Zhao and H. Mao, Simultaneous Detection of High-Sensitivity Cardiac Troponin I and Myoglobin by Modified Sandwich Lateral Flow Immunoassay: Proof of Principle, Clin. Chem., 2011, 57, 1732–1738 CAS.
- L. Cullen, W. Parsonage, M. Than, S. Goodacre, L. Cullen, J. Greenslade, K. Merollini, R. Body and S. Carley, et al., Myocardial Infarction: Rapid Ruling out in the Emergency Room, Lancet, 2015, 386, 2449–2450 CrossRef.
- E. K. Sackmann, A. L. Fulton and D. J. Beebe, The Present and Future Role of Microfluidics in Biomedical Research, Nature, 2014, 507, 181–189 CrossRef CAS PubMed.
- C. Longuet, A. Yamada, Y. Chen, D. Baigl and J. Fattaccioli, Spatially-Controlled Protein Crystallization in Microfluidic Chambers, J. Cryst. Growth, 2014, 386, 179–182 CrossRef CAS.
- D. An, K. Kim and J. Kim, Microfluidic System Based High Throughput Drug Screening System for Curcumin/TRAIL Combinational Chemotherapy in Human Prostate Cancer PC3 Cells, Biomol. Ther., 2014, 22, 355–362 CrossRef CAS PubMed.
- M. K. Araz, A. M. Tentori and A. E. Herr, Microfluidic Multiplexing in Bioanalyses, J. Lab. Autom., 2013, 18, 350–366 CrossRef CAS PubMed.
- M. Werner, R. Palankar, L. Arm, R. Hovius and H. Vogel, Microfluidic Single-Cell Analysis with Affinity Beads, Small, 2015, 11, 2607–2613 CrossRef CAS PubMed.
- K. S. Elvira, X. Casadevall i Solvas, R. C. R. Wootton and A. J. deMello, The Past, Present and Potential for Microfluidic Reactor Technology in Chemical Synthesis, Nat. Chem., 2013, 5, 905–915 CrossRef CAS PubMed.
- G. M. Whitesides, The Origins and the Future of Microfluidics, Nature, 2006, 442, 368–373 CrossRef CAS PubMed.
- M. May, M. Gompels, V. Delpech, K. Porter, F. Post, M. Johnson, D. Dunn, A. Palfreeman, R. Gilson and B. Gazzard, et al., Impact of Late Diagnosis and Treatment on Life Expectancy in People with HIV-1: UK Collaborative HIV Cohort (UK CHIC) Study, Br. Med. J., 2011, 343, d6016 CrossRef PubMed.
- A. K. Arrington, L. Goldstein, L. Kruper, C. Vito, J. Yim and S. L. Chen, Life Expectancy after Curative-Intent Treatment of Breast Cancer: Impact on Long-Term Follow-up Care, Am. Surg., 2014, 80, 604–609 Search PubMed.
- C. D. Chin, V. Linder and S. K. Sia, Commercialization of Microfluidic Point-of-Care Diagnostic Devices, Lab Chip, 2012, 2118–2134 RSC.
- A. S. Jaffe, L. Babuin and F. S. Apple, Biomarkers in Acute Cardiac Disease: The Present and the Future, J. Am. Coll. Cardiol., 2006, 48, 1–11 CrossRef CAS PubMed.
- H. C. Beck, M. Overgaard and L. Melholt Rasmussen, Plasma Proteomics to Identify Biomarkers – Application to Cardiovascular Diseases, Transl. Proteomics, 2015, 7, 40–48 CrossRef CAS.
- A. K. Füzéry, J. Levin, M. M. Chan and D. W. Chan, Translation of Proteomic Biomarkers into FDA Approved Cancer Diagnostics: Issues and Challenges, Clin. Proteomics, 2013, 10, 13 CrossRef PubMed.
- N. L. Henry and D. F. Hayes, Cancer Biomarkers, Mol. Oncol., 2012, 6, 140–146 CrossRef CAS PubMed.
- F. A. Bozza, J. I. Salluh, A. M. Japiassu, M. Soares, E. F. Assis, R. N. Gomes, M. T. Bozza, H. C. Castro-Faria-Neto and P. T. Bozza, Cytokine Profiles as Markers of Disease Severity in Sepsis: A Multiplex Analysis, Crit. Care, 2007, 11, R49 CrossRef PubMed.
- C. L. Mack, Serum Cytokines as Biomarkers of Disease and Clues to Pathogenesis, Hepatology, 2007, 46, 6–8 CrossRef CAS PubMed.
- A. Ferrajoli, M. J. Keating, T. Manshouri, F. J. Giles, A. Dey, Z. Estrov, C. A. Koller, R. Kurzrock, D. A. Thomas and S. Faderl, et al., The Clinical Significance of Tumor Necrosis Factor-Alpha Plasma Level in Patients Having Chronic Lymphocytic Leukemia, Blood, 2002, 100, 1215–1219 CAS.
- M. Li, H. Wang, X. Wang, J. Huang, J. Wang, X. Xi, M. Marel, M. Zrustova, B. Stasny and R. Light, et al. Diagnostic Accuracy of Tumor Necrosis Factor-Alpha, Interferon-Gamma, Interlukine-10 and Adenosine Deaminase 2 in Differential Diagnosis between Tuberculous Pleural Effusion and Malignant Pleural Effusion, J. Cardiothorac. Surg., 2014, 9, 118 CrossRef PubMed.
- S. Cesaro-Tadic, G. Dernick, D. Juncker, G. Buurman, H. Kropshofer, B. Michel, C. Fattinger and E. Delamarche, High-Sensitivity Miniaturized Immunoassays for Tumor Necrosis Factor Alpha Using Microfluidic Systems, Lab Chip, 2004, 4, 563–569 RSC.
- I. M. Thompson, D. K. Pauler, P. J. Goodman, C. M. Tangen, M. S. Lucia, H. L. Parnes, L. M. Minasian, L. G. Ford, S. M. Lippman and E. D. Crawford, et al., Prevalence of Prostate Cancer among Men with a Prostate-Specific Antigen Level ≤4.0 Ng per Milliliter, N. Engl. J. Med., 2004, 350, 2239–2246 CrossRef CAS PubMed.
- E. D. Goluch, J.-M. Nam, D. G. Georganopoulou, T. N. Chiesl, K. A. Shaikh, K. S. Ryu, A. E. Barron, C. A. Mirkin and C. Liu, A Bio-Barcode Assay for on-Chip Attomolar-Sensitivity Protein Detection, Lab Chip, 2006, 6, 1293–1299 RSC.
- J. A. de Lemos, D. A. Morrow, C. M. Gibson, S. A. Murphy, M. S. Sabatine, N. Rifai, C. H. McCabe, E. M. Antman, C. P. Cannon and E. Braunwald, The Prognostic Value of Serum Myoglobin in Patients with Non-ST-Segment Elevation Acute Coronary Syndromes. Results from the TIMI 11B and TACTICS-TIMI 18 Studies, J. Am. Coll. Cardiol., 2002, 40, 238–244 CrossRef CAS PubMed.
- V. G. Davila-Roman, Cardiac Biomarkers: Expert Advice for Clinicians, Circulation, 2012, 126, e264–e265 CrossRef.
- V. S. Mahajan and P. Jarolim, How to Interpret Elevated Cardiac Troponin Levels, Circulation, 2011, 124, 2350–2354 CrossRef PubMed.
- J. M. Young, J. W. Pickering, P. M. George, S. J. Aldous, J. Wallace, C. M. Frampton, R. W. Troughton, M. A. Richards, J. H. Greenslade and L. Cullen, et al., Heart Fatty Acid Binding Protein and Cardiac Troponin: Development of an Optimal Rule-out Strategy for Acute Myocardial Infarction, BMC Emerg. Med., 2016, 16, 34 CrossRef PubMed.
- F. Torabi, H. R. Mobini Far, B. Danielsson and M. Khayyami, Development of a Plasma Panel Test for Detection of Human Myocardial Proteins by Capillary Immunoassay, Biosens. Bioelectron., 2007, 22, 1218–1223 CrossRef CAS PubMed.
- M. Herrmann, T. Veres and M. Tabrizian, Quantification of Low-Picomolar Concentrations of TNF-Alpha in Serum Using the Dual-Network Microfluidic ELISA Platform, Anal. Chem., 2008, 80, 5160–5167 CrossRef CAS PubMed.
- R. S. Sista, A. E. Eckhardt, V. Srinivasan, M. G. Pollack, S. Palanki and V. K. Pamula, Heterogeneous Immunoassays Using Magnetic Beads on a Digital Microfluidic Platform, Lab Chip, 2008, 8, 2188–2196 RSC.
- T. Ashizawa, R. Okada, Y. Suzuki, M. Takagi, T. Yamazaki, T. Sumi, T. Aoki, S. Ohnuma and T. Aoki, Clinical Significance of Interleukin-6 (IL-6) in the Spread of Gastric Cancer: Role of IL-6 as a Prognostic Factor, Gastric Cancer, 2005, 8, 124–131 CrossRef CAS PubMed.
- D. M. Bruls, T. H. Evers, J. A. H. Kahlman, P. J. W. van Lankvelt, M. Ovsyanko, E. G. M. Pelssers, J. J. H. B. Schleipen, F. K. de Theije, C. A. Verschuren and T. van der Wijk, et al., Rapid Integrated Biosensor for Multiplexed Immunoassays Based on Actuated Magnetic Nanoparticles, Lab Chip, 2009, 9, 3504–3510 RSC.
- J. Salazar, M. S. Martínez, M. Chávez, A. Toledo, R. Añez, Y. Torres, V. Apruzzese, C. Silva, J. Rojas and V. Bermúdez, et al., C-Reactive Protein: Clinical and Epidemiological Perspectives, Cardiol. Res. Pract., 2014, 2014, 605810 Search PubMed.
- L. Gervais and E. Delamarche, Toward One-Step Point-of-Care Immunodiagnostics Using Capillary-Driven Microfluidics and PDMS Substrates, Lab Chip, 2009, 9, 3330–3337 RSC.
- X. Wang, M. Zhao, D. D. Nolte and T. L. Ratliff, Prostate Specific Antigen Detection in Patient Sera by Fluorescence-Free BioCD Protein Array, Biosens. Bioelectron., 2011, 26, 1871–1875 CrossRef CAS PubMed.
- M. Biselli, F. Conti, A. Gramenzi, M. Frigerio, A. Cucchetti, G. Fatti, M. D'Angelo, M. Dall'Agata, E. G. Giannini and F. Farinati, et al., A New Approach to the Use of α-Fetoprotein as Surveillance Test for Hepatocellular Carcinoma in Patients with Cirrhosis, Br. J. Cancer, 2015, 112, 69–76 CrossRef CAS PubMed.
- M. Ikami, A. Kawakami, M. Kakuta, Y. Okamoto, N. Kaji, M. Tokeshi and Y. Baba, Immuno-Pillar Chip: A New Platform for Rapid and Easy-to-Use Immunoassay, Lab Chip, 2010, 10, 3335–3340 RSC.
- A. R. Franz, S. Sieber, F. Pohlandt, M. Kron and G. Steinbach, Whole Blood Interleukin 8 and Plasma Interleukin 8 Levels in Newborn Infants with Suspected Bacterial Infection, Acta Paediatr., 2004, 93, 648–653 CrossRef CAS PubMed.
- J. A. Thompson, X. Du, J. M. Grogan, M. G. Schrlau and H. H. Bau, Polymeric Microbead Arrays for Microfluidic Applications, J. Micromech. Microeng., 2010, 20, 115017 CrossRef.
- D. S. Thomas, E.-O. Fourkala, S. Apostolidou, R. Gunu, A. Ryan, I. Jacobs, U. Menon, W. Alderton, A. Gentry-Maharaj and J. F. Timms, Evaluation of Serum CEA, CYFRA21-1 and CA125 for the Early Detection of Colorectal Cancer Using Longitudinal Preclinical Samples, Br. J. Cancer, 2015, 113, 268–274 CrossRef CAS PubMed.
- C. Wang, J. Wu, C. Zong, H. Ju and F. Yan, Highly Sensitive Rapid Chemiluminescent Immunoassay Using the DNAzyme Label for Signal Amplification, Analyst, 2011, 136, 4295–4300 RSC.
- J. Tang, D. Tang, R. Niessner, G. Chen and D. Knopp, Magneto-Controlled Graphene Immunosensing Platform for Simultaneous Multiplexed Electrochemical Immunoassay Using Distinguishable Signal Tags, Anal. Chem., 2011, 83, 5407–5414 CrossRef CAS PubMed.
- E. Christenson and R. H. Christenson, The Role of Cardiac Biomarkers in the Diagnosis and Management of Patients Presenting with Suspected Acute Coronary Syndrome, Ann. Lab. Med., 2013, 33, 309–318 CrossRef CAS PubMed.
- A. W. Browne, L. Ramasamy, T. P. Cripe and C. H. Ahn, A Lab-on-a-Chip for Rapid Blood Separation and Quantification of Hematocrit and Serum Analytes, Lab Chip, 2011, 11, 2440–2446 RSC.
- M. S. Luchansky, A. L. Washburn, M. S. McClellan and R. C. Bailey, Sensitive on-Chip Detection of a Protein Biomarker in Human Serum and Plasma over an Extended Dynamic Range Using Silicon Photonic Microring Resonators and Sub-Micron Beads, Lab Chip, 2011, 11, 2042–2044 RSC.
- N. Triroj, P. Jaroenapibal, H. Shi, J. I. Yeh and R. Beresford, Microfluidic Chip-Based Nanoelectrode Array as Miniaturized Biochemical Sensing Platform for Prostate-Specific Antigen Detection, Biosens. Bioelectron., 2011, 26, 2927–2933 CrossRef CAS PubMed.
- C. Peters-Engl, M. Medl, E. Ogris and S. Leodolter, Tumor-Associated Trypsin Inhibitor (TATI) and Cancer Antigen 125 (CA125) in Patients with Epithelial Ovarian Cancer, Anticancer Res., 1995, 15, 2727–2730 CAS.
- S. Wang, L. Ge, X. Song, J. Yu, S. Ge, J. Huang and F. Zeng, Paper-Based Chemiluminescence ELISA: Lab-on-Paper Based on Chitosan Modified Paper Device and Wax-Screen-Printing, Biosens. Bioelectron., 2012, 31, 212–218 CrossRef CAS PubMed.
- D. W. Jekarl, J. Y. Kim, S. Lee, M. Kim, Y. Kim, K. Han, S. H. Woo and W. J. Lee, Diagnosis and Evaluation of Severity of Sepsis via the Use of Biomarkers and Profiles of 13 Cytokines: A Multiplex Analysis, Clin. Chem. Lab. Med., 2015, 53, 575–581 CrossRef CAS PubMed.
- J. Kai, A. Puntambekar, N. Santiago, S. H. Lee, D. W. Sehy, V. Moore, J. Han and C. H. Ahn, A Novel Microfluidic Microplate as the next Generation Assay Platform for Enzyme Linked Immunoassays (ELISA), Lab Chip, 2012, 12, 4257–4262 RSC.
- L. A. Sasso, I. H. Johnston, M. Zheng, R. K. Gupte, A. Ündar and J. D. Zahn, Automated Microfluidic Processing Platform for Multiplexed Magnetic Bead Immunoassays, Microfluid. Nanofluid., 2012, 13, 603–612 CrossRef CAS PubMed.
- X. Yu, H.-S. Xia, Z.-D. Sun, Y. Lin, K. Wang, J. Yu, H. Tang, D.-W. Pang and Z.-L. Zhang, On-Chip Dual Detection of Cancer Biomarkers Directly in Serum Based on Self-Assembled Magnetic Bead Patterns and Quantum Dots, Biosens. Bioelectron., 2013, 41, 129–136 CrossRef CAS PubMed.
- W. Hu, Z. Lu, Y. Liu, T. Chen, X. Zhou and C. M. Li, A Portable Flow-through Fluorescent Immunoassay Lab-on-a-Chip Device Using ZnO Nanorod-Decorated Glass Capillaries, Lab Chip, 2013, 13, 1797–1802 RSC.
- H. Adel Ahmed and H. M. E. Azzazy, Power-Free Chip Enzyme Immunoassay for Detection of Prostate Specific Antigen (PSA) in Serum, Biosens. Bioelectron., 2013, 49, 478–484 CrossRef CAS PubMed.
- A. A. Tajudin, K. Petersson, A. Lenshof, A.-M. Swärd-Nilsson, L. Aberg, G. Marko-Varga, J. Malm, H. Lilja and T. Laurell, Integrated Acoustic Immunoaffinity-Capture (IAI) Platform for Detection of PSA from Whole Blood Samples, Lab Chip, 2013, 13, 1790–1796 RSC.
- J. Yan, M. Yan, L. Ge, S. Ge and J. Yu, An Origami Electrochemiluminescence Immunosensor Based on Gold/graphene for Specific, Sensitive Point-of-Care Testing of Carcinoembryonic Antigen, Sens. Actuators, B, 2014, 193, 247–254 CrossRef CAS.
- M. I. Mohammed and M. P. Y. Desmulliez, Autonomous Capillary Microfluidic System with Embedded Optics for Improved Troponin I Cardiac Biomarker Detection, Biosens. Bioelectron., 2014, 61, 478–484 CrossRef CAS PubMed.
- T. Naito, M. Ohtsuka, H. Ishikawa, H. Satoh and S. Hasegawa, Clinical Significance of Cytokine Measurement in Pleural Effusion, Kekkaku, 1997, 72, 565–572 CAS.
- L. Valdés, E. S. José, J. M. A. Dobaño, A. Golpe, J. M. Valle, P. Penela and F. J. G. Barcala, Diagnostic Value of Interleukin-12 p40 in Tuberculous Pleural Effusions, Eur. Respir. J., 2009, 33, 816–820 CrossRef PubMed.
- A. I. Barbosa, A. P. Castanheira, A. D. Edwards and N. M. Reis, A Lab-in-a-Briefcase for Rapid Prostate Specific Antigen (PSA) Screening from Whole Blood, Lab Chip, 2014, 2918–2928 RSC.
- A. I. Barbosa, P. Gehlot, K. Sidapra, A. D. Edwards and N. M. Reis, Portable Smartphone Quantitation of Prostate Specific Antigen (PSA) in a Fluoropolymer Microfluidic Device, Biosens. Bioelectron., 2015, 70, 5–14 CrossRef CAS PubMed.
- A. P. Castanheira, A. I. Barbosa, A. D. Edwards and N. M. Reis, Multiplexed Femtomolar Quantitation of Human Cytokines in a Fluoropolymer Microcapillary Film, Analyst, 2015, 140, 5609–5618 RSC.
- J. L. Garcia-Cordero and S. J. Maerkl, A 1024-Sample Serum Analyzer Chip for Cancer Diagnostics, Lab Chip, 2014, 14, 2642–2650 RSC.
- G. S. de Moraes, R. do Amaral Cristovam and R. F. Savaris, Comparative Analysis of the Accuracy of Urinary hCG Tests in Vitro, Rev. Assoc. Med. Bras., 2011, 57, 516–522 CrossRef.
- J. E. Schonhorn, S. C. Fernandes, A. Rajaratnam, R. N. Deraney, J. P. Rolland and C. R. Mace, A Device Architecture for Three-Dimensional, Patterned Paper Immunoassays, Lab Chip, 2014, 14, 4653–4658 RSC.
- F. Volpetti, J. Garcia-Cordero and S. J. Maerkl, A Microfluidic Platform for High-Throughput Multiplexed Protein Quantitation, PLoS One, 2015, 10, e0117744 Search PubMed.
- S. K. Vashist, E. Marion Schneider, R. Zengerle, F. von Stetten and J. H. T. Luong, Graphene-Based Rapid and Highly-Sensitive Immunoassay for C-Reactive Protein Using a Smartphone-Based Colorimetric Reader, Biosens. Bioelectron., 2015, 66, 169–176 CrossRef CAS PubMed.
- W. S. Lee, V. Sunkara, J.-R. Han, Y.-S. Park and Y.-K. Cho, Electrospun TiO2 Nanofiber Integrated Lab-on-a-Disc for Ultrasensitive Protein Detection from Whole Blood, Lab Chip, 2015, 15, 478–485 RSC.
- S. K. Vashist, E. M. Schneider and J. H. T. Luong, Surface Plasmon Resonance-Based Immunoassay for Human C-Reactive Protein, Analyst, 2015, 140, 4445–4452 RSC.
- D. A. Armbruster and T. Pry, Limit of Blank, Limit of Detection and Limit of Quantitation, Clin. Biochem. Rev., 2008, 29(Suppl 1), S49–S52 Search PubMed.
- A. Shrivastava and V. B. Gupta, Methods for the Determination of Limit of Detection and Limit of Quantitation of the Analytical Methods, Chron. Young Sci., 2011, 2, 21–25 CrossRef.
- A. W. P. Vermeer, M. G. E. G. Bremer and W. Norde, Structural Changes of IgG Induced by Heat Treatment and by Adsorption onto a Hydrophobic Teflon Surface Studied by Circular Dichroism Spectroscopy, Biochim. Biophys. Acta, Gen. Subj., 1998, 1425, 1–12 CrossRef CAS.
- R. G. Feldman, M. E. Hamel, M. A. Breukels, N. F. Concepcion and B. F. Anthony, Solid-Phase Antigen Density and Avidity of Antibodies Detected in Anti-Group B Streptococcal Type III IgG Enzyme Immunoassays, J. Immunol. Methods, 1994, 170, 37–45 CrossRef CAS PubMed.
- F. Rusmini, Z. Zhong and J. Feijen, Protein Immobilization Strategies for Protein Biochips, Biomacromolecules, 2007, 8, 1775–1789 CrossRef CAS PubMed.
- M. E. Wiseman and C. W. Frank, Antibody Adsorption and Orientation on Hydrophobic Surfaces, Langmuir, 2012, 28, 1765–1774 CrossRef CAS PubMed.
- X. Wang, Y. Wang, H. Xu, H. Shan and J. R. Lu, Dynamic Adsorption of Monoclonal Antibody Layers on Hydrophilic Silica Surface: A Combined Study by Spectroscopic Ellipsometry and AFM, J. Colloid Interface Sci., 2008, 323, 18–25 CrossRef CAS PubMed.
- S. S. Deshpande, Enzyme Immunoassays: From Concept to Product Development, Springer Science & Business Media, 1996 Search PubMed.
- A. Sellborn, M. Andersson, J. Hedlund, J. Andersson, M. Berglin and H. Elwing, Immune Complement Activation on Polystyrene and Silicon Dioxide Surfaces. Impact of Reversible IgG Adsorption, Mol. Immunol., 2005, 42, 569–574 CrossRef CAS PubMed.
- E. Eteshola and D. Leckband, Development and Characterization of an ELISA Assay in PDMS Microfluidic Channels, Sens. Actuators, B, 2001, 72, 129–133 CrossRef CAS.
- S. K. Vashist, C. K. Dixit, B. D. MacCraith and R. O'Kennedy, Effect of Antibody Immobilization Strategies on the Analytical Performance of a Surface Plasmon Resonance-Based Immunoassay, Analyst, 2011, 136, 4431–4436 RSC.
- D. Kim and A. E. Herr, Protein Immobilization Techniques for Microfluidic Assays, Biomicrofluidics, 2013, 7, 41501 CrossRef PubMed.
- R. A. Vijayendran and D. E. Leckband, A Quantitative Assessment of Heterogeneity for Surface-Immobilized Proteins, Anal. Chem., 2001, 73, 471–480 CrossRef CAS PubMed.
- A. K. Trilling, J. Beekwilder and H. Zuilhof, Antibody Orientation on Biosensor Surfaces: A Minireview, Analyst, 2013, 138, 1619 RSC.
- N. R. Glass, R. Tjeung, P. Chan, L. Y. Yeo and J. R. Friend, Organosilane Deposition for Microfluidic Applications, Biomicrofluidics, 2011, 5, 36501–365017 CrossRef PubMed.
- J. Krenková and F. Foret, Immobilized Microfluidic Enzymatic Reactors, Electrophoresis, 2004, 25, 3550–3563 CrossRef PubMed.
- A. K. Singh, A. W. Flounders, J. V. Volponi, C. S. Ashley, K. Wally and J. S. Schoeniger, Development of Sensors for Direct Detection of Organophosphates. Part I: Immobilization, Characterization and Stabilization of Acetylcholinesterase and Organophosphate Hydrolase on Silica Supports, Biosens. Bioelectron., 1999, 14, 703–713 CrossRef CAS PubMed.
- C. Rösli, J.-N. Rybak, D. Neri and G. Elia, Quantitative Recovery of Biotinylated Proteins from Streptavidin-Based Affinity Chromatography Resins, in Avidin-Biotin Interactions, Humana Press, Totowa, NJ, 2008, pp. 89–100 Search PubMed.
- E. P. Diamandis and T. K. Christopoulos, The Biotin-(Strept)avidin System: Principles and Applications in Biotechnology, Clin. Chem., 1991, 37, 625–636 CAS.
- Y. Jung, J. M. Lee, H. Jung and B. H. Chung, Self-Directed and Self-Oriented Immobilization of Antibody by Protein G-DNA Conjugate, Anal. Chem., 2007, 79, 6534–6541 CrossRef CAS PubMed.
- Y. Jung, H. J. Kang, J. M. Lee, S. O. Jung, W. S. Yun, S. J. Chung and B. H. Chung, Controlled Antibody Immobilization onto Immunoanalytical Platforms by Synthetic Peptide, Anal. Biochem., 2008, 374, 99–105 CrossRef CAS PubMed.
- I. A. Kozlov, P. C. Melnyk, K. E. Stromsborg, M. S. Chee, D. L. Barker and C. Zhao, Efficient Strategies for the Conjugation of Oligonucleotides to Antibodies Enabling Highly Sensitive Protein Detection, Biopolymers, 2004, 73, 621–630 CrossRef CAS PubMed.
- A. Hirlekar Schmid, S. E. Stanca, M. S. Thakur, K. R. Thampi and C. Raman Suri, Site-Directed Antibody Immobilization on Gold Substrate for Surface Plasmon Resonance Sensors, Sens. Actuators, B, 2006, 113, 297–303 CrossRef CAS.
- J. Park, V. Sunkara, T.-H. Kim, H. Hwang and Y.-K. Cho, Lab-on-a-Disc for Fully Integrated Multiplex Immunoassays, Anal. Chem., 2012, 84, 2133–2140 CrossRef CAS PubMed.
- S. Lee, S. Kim, J. Malm, O. C. Jeong, H. Lilja and T. Laurell, Improved Porous Silicon Microarray Based Prostate Specific Antigen Immunoassay by Optimized Surface Density of the Capture Antibody, Anal. Chim. Acta, 2013, 796, 108–114 CrossRef CAS PubMed.
- A. Ressine, S. Ekström, G. Marko-Varga and T. Laurell, Macro-/nanoporous Silicon as a Support for High-Performance Protein Microarrays, Anal. Chem., 2003, 75, 6968–6974 CrossRef CAS PubMed.
- X. Yu, H.-S. Xia, Z.-D. Sun, Y. Lin, K. Wang, J. Yu, H. Tang, D.-W. Pang and Z.-L. Zhang, On-Chip Dual Detection of Cancer Biomarkers Directly in Serum Based on Self-Assembled Magnetic Bead Patterns and Quantum Dots, Biosens. Bioelectron., 2013, 41, 129–136 CrossRef CAS PubMed.
- H. C. Tekin and M. A. M. Gijs, Ultrasensitive protein detection: a case for microfluidic magnetic bead-based assays, Lab Chip, 2013, 13, 4711–4739 Search PubMed.
- B. Sweryda-Krawiec, H. Devaraj, G. Jacob and J. J. Hickman, A New Interpretation of Serum Albumin Surface Passivation, 2004 Search PubMed.
- I. Wong and C.-M. Ho, Surface Molecular Property Modifications for Poly(dimethylsiloxane) (PDMS) Based Microfluidic Devices, Microfluid. Nanofluid., 2009, 7, 291–306 CrossRef CAS PubMed.
- H. Zhang and M. Chiao, Anti-Fouling Coatings of Poly(dimethylsiloxane) Devices for Biological and Biomedical Applications, J. Med. Biol. Eng., 2015, 35, 143–155 CrossRef PubMed.
- I. Banerjee, R. C. Pangule and R. S. Kane, Antifouling Coatings: Recent Developments in the Design of Surfaces That Prevent Fouling by Proteins, Bacteria, and Marine Organisms, Adv. Mater., 2011, 23, 690–718 CrossRef CAS PubMed.
- N. J. Shirtcliffe and P. Roach, Superhydrophobicity for Antifouling Microfluidic Surfaces, in Methods in molecular biology, Clifton, N.J., 2013, vol. 949, pp. 269–281 Search PubMed.
- N. J. Bonne and D. T. Wong, Salivary Biomarker Development Using Genomic, Proteomic and Metabolomic Approaches, Genome Med., 2012, 4, 82 CrossRef PubMed.
- A. Courbet, E. Renard and F. Molina, Bringing next-Generation Diagnostics to the Clinic through Synthetic Biology, EMBO Mol. Med., 2016, 8, 987–991 CrossRef CAS PubMed.
- W. S. Mielczarek, E. A. Obaje, T. T. Bachmann and M. Kersaudy-Kerhoas, Microfluidic Blood Plasma Separation for Medical Diagnostics: Is It Worth It?, Lab Chip, 2016, 16, 3441–3448 RSC.
- R. Gorkin, J. Park, J. Siegrist, M. Amasia, B. S. Lee, J.-M. Park, J. Kim, H. Kim, M. Madou and Y.-K. Cho, Centrifugal Microfluidics for Biomedical Applications, Lab Chip, 2010, 10, 1758–1773 RSC.
- V. Mani, B. V. Chikkaveeraiah and J. F. Rusling, Magnetic Particles in Ultrasensitive Biomarker Protein Measurements for Cancer Detection and Monitoring, Expert Opin. Med. Diagn., 2011, 5, 381–391 CrossRef CAS PubMed.
- H. Adel Ahmed and H. M. E. Azzazy, Power-Free Chip Enzyme Immunoassay for Detection of Prostate Specific Antigen (PSA) in Serum, Biosens. Bioelectron., 2013, 49, 478–484 CrossRef CAS PubMed.
- K. Hosokawa, K. Sato, N. Ichikawa and M. Maeda, Power-Free Poly(dimethylsiloxane) Microfluidic Devices for Gold Nanoparticle-Based DNA Analysis, Lab Chip, 2004, 4, 181–185 RSC.
- G. C. Randall and P. S. Doyle, Permeation-Driven Flow in Poly(dimethylsiloxane) Microfluidic Devices, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10813–10818 CrossRef CAS PubMed.
- M. Zimmermann, H. Schmid, P. Hunziker and E. Delamarche, Capillary Pumps for Autonomous Capillary Systems, Lab Chip, 2007, 7, 119–125 RSC.
- A. K. Yetisen, M. S. Akram and C. R. Lowe, Paper-Based Microfluidic Point-of-Care Diagnostic Devices, Lab Chip, 2013, 13, 2210–2251 RSC.
- J. P. Gosling, Enzyme Immunoassay, in Immunoassay, Elsevier, 1996, pp. 287–308 Search PubMed.
- Z. Gao, L. Hou, M. Xu and D. Tang, Enhanced Colorimetric Immunoassay Accompanying with Enzyme Cascade Amplification Strategy for Ultrasensitive Detection of Low-Abundance Protein, Sci. Rep., 2014, 4, 3966 Search PubMed.
- J. L. Brooks, B. Mirhabibollahi and R. G. Kroll, Increased Sensitivity of an Enzyme-Amplified Colorimetric Immunoassay for Protein A-Bearing Staphylococcus Aureus in Foods, J. Immunol. Methods, 1991, 140, 79–84 CrossRef CAS PubMed.
- Y. Liu, D. Zhang, E. C. Alocilja and S. Chakrabartty, Biomolecules Detection Using a Silver-Enhanced Gold Nanoparticle-Based Biochip, Nanoscale Res. Lett., 2010, 5, 533–538 CrossRef CAS PubMed.
- R. Liu, Y. Zhang, S. Zhang, W. Qiu and Y. Gao, Silver Enhancement of Gold Nanoparticles for Biosensing: From Qualitative to Quantitative, Appl. Spectrosc. Rev., 2013, 121–138 CAS.
- C.-H. Yeh, C.-Y. Hung, T. C. Chang, H.-P. Lin and Y.-C. Lin, An Immunoassay Using Antibody-Gold Nanoparticle Conjugate, Silver Enhancement and Flatbed Scanner, Microfluid. Nanofluid., 2008, 6, 85–91 CrossRef.
- I. Weeks and L. J. Kricka, The Immunoassay Handbook, in The Immunoassay Handbook, Elsevier, 2013, pp. 267–285 Search PubMed.
- A. Warsinke, A. Benkert and F. W. Scheller, Electrochemical Immunoassays, Fresenius J. Anal. Chem., 2000, 366(6–7), 622–634 CrossRef CAS PubMed.
- K. Sun, I. Zeimpekis, C. Hu, N. M. J. Ditshego, O. Thomas, M. R. R. de Planque, H. M. H. Chong, H. Morgan and P. Ashburn, Low-Cost Top-down Zinc Oxide Nanowire Sensors through a Highly Transferable Ion Beam Etching for Healthcare Applications, Microelectron. Eng., 2016, 153, 96–100 CrossRef CAS.
- F. Patolsky, G. Zheng and C. M. Lieber, Fabrication of Silicon Nanowire Devices for Ultrasensitive, Label-Free, Real-Time Detection of Biological and Chemical Species, Nat. Protoc., 2006, 1, 1711–1724 CrossRef CAS PubMed.
- C. Rivet, H. Lee, A. Hirsch, S. Hamilton and H. Lu, Microfluidics for Medical Diagnostics and Biosensors, Chem. Eng. Sci., 2011, 66, 1490–1507 CrossRef CAS.
- S. Bernabé, in Advanced Packaging Concepts for Low-Cost Optoelectronic Devices, ed. P.-Y. Fonjallaz and T. P. Pearsall, 2006, vol. 6350, p. 635008 Search PubMed.
- C. Gnoth and S. Johnson, Strips of Hope: Accuracy of Home Pregnancy Tests and New Developments, Geburtshilfe Frauenheilkd., 2014, 74, 661–669 CrossRef CAS PubMed.
- H. Becker, It's the Economy, Lab Chip, 2009, 9, 2759–2762 RSC.
- A. Waldbaur, H. Rapp, K. Länge and B. E. Rapp, Let There Be Chip—towards Rapid Prototyping of Microfluidic Devices: One-Step Manufacturing Processes, Anal. Methods, 2011, 3, 2681 RSC.
| This journal is © The Royal Society of Chemistry 2017 |