
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A highly active nickel electrocatalyst shows excellent selectivity for CO2 reduction in acidic media†
Gaia
Neri
 ,
Iain M.
Aldous
,
James J.
Walsh
,
Laurence J.
Hardwick
and
Alexander J.
Cowan
*
,
Iain M.
Aldous
,
James J.
Walsh
,
Laurence J.
Hardwick
and
Alexander J.
Cowan
*
Department of Chemistry, Stephenson Institute for Renewable Energy, The University of Liverpool, UK. E-mail: a.j.cowan@liverpool.ac.uk
First published on 24th November 2015
Abstract
The development of selective electrocatalysts for CO2 reduction in water offers a sustainable route to carbon based fuels and feedstocks. However, molecular catalysts are typically studied in non-aqueous solvents, in part to avoid competitive H2 evolution. [Ni(cyclam)]2+ (1) is one of the few known electrocatalysts that operate in water and 30 years after its report its activity remains a rarely surpassed benchmark. Here we report that [Ni(cyclam-CO2H)]2+ (cyclam-CO2H = 1,4,8,11-tetraazacyclotetradecane-6-carboxylic acid (2)) shows greatly enhanced activity versus1 for CO production. At pHs < pKa of the pendant carboxylic acid a large increase in catalytic activity occurs. Remarkably, despite the high proton concentration (pH 2), 2 maintains selectivity for CO2 reduction and is believed to be unique in operating selectively in such acidic aqueous solutions.
Introduction
The discovery of catalysts for the conversion of carbon dioxide (CO2) into fuels and feedstocks using renewable energy resources such as solar and wind generated electrical is amongst the most significant challenges in chemical research.1 Of particular interest is the reduction of CO2 to carbon monoxide (CO2 + 2e− + 2H+ → CO + H2O E0ap (VNHE) = −0.12 − 0.059 pH)2 as CO is a key industrial feedstock that can be used to generate a wide range of hydrocarbon products by Fischer–Tropsch chemistry. To enable practical utilisation, CO2 reduction electrocatalysts will need to be used in tandem with a sustainable oxidation reaction, such as water splitting (H2O → 2e− + 2H+ + 1/2O2, E0ap (VNHE) = 1.23–0.059 pH) making the development of low cost, selective CO2 reduction catalysts that operate in water at a range of pHs an imperative goal. However the majority of studies to date using molecular catalysts have been carried out in aprotic solvents such as dimethylformamide (DMF) and acetonitrile (CH3CN) with Brønsted acids added. Careful control of the acid concentration, coupled to the relatively high solubility of CO2 in these solvents minimises competitive H2 production (2H+ + 2e− → H2, E0ap (VNHE) = 0–0.059 pH). A further complication is that any CO2 electrolyser will require the cathode and anode to be separated by a membrane. To date the most effective membranes are proton exchange materials3 making the study of CO2 reduction in acidic conditions of particular interest. [Ni(cyclam)]2+ (1) is a low cost, highly selective CO2 reduction catalyst producing solely CO in water at pHs 7–4. Since the initial reports over 30 years ago,4–6 numerous attempts have been made to develop nickel cyclam catalysts with improved rate constants and onset potentials.7 However to the best of our knowledge only two reports observed an increase in the catalyst performance,8,9 with functionalisation of both the amines and carbon backbone typically causing losses in selectivity and excessive hydrogen production.The mechanism for the reduction of CO2 to CO by 1 has been extensively studied,5,10–13 and although the exact nature of the active species has yet to be unambiguously identified, several factors have been made clear. Firstly, [Ni(cyclam)]+ adsorbs on to some metal electrodes including Sn, Pb,14 and Hg,5 and adsorption onto the electrode is key for efficient CO2 reduction.111 has also been shown to act as a homogeneous CO2 reduction catalyst when used with a glassy carbon electrode (GCE)15 however the level of activity was significantly lower than can be achieved on Hg, which in part may be due to suppression of catalyst degradation pathways on Hg.16 Indeed Hg remains a common electrode for fundamental studies such as that presented here. At pH 5 adsorption initiates at potentials positive of the formal NiII/I couple in solution (−1.3 VNHE) and a monolayer is formed at ca. −1 VNHE. The adsorbed NiI complex is predicted to bind in an η1-C mode to CO2, prior to the transfer of a second electron to the catalyst centre.13 Computational studies13,17,18 indicate that the structure of the adsorbed complex is a trans-I conformation,19 with the axial amine hydrogens aiding CO2 binding. In solution 1 can adopt five different isomeric forms11 and for clarity structures of 1 and 2 are drawn in a planar geometry (Fig. 1). Numerous studies have proposed that the decline in activity upon cyclam modification is due to conformational changes limiting the availability of the N–H group suggesting their critical role in catalysis.5,13,17 Finally, 1 is most active at pH ∼4–5 indicating that dissolved CO2 and not HCO3− or CO32− is the preferred substrate. At pH values less than 4H2 evolution dominates and CO2 selectivity is lost.9
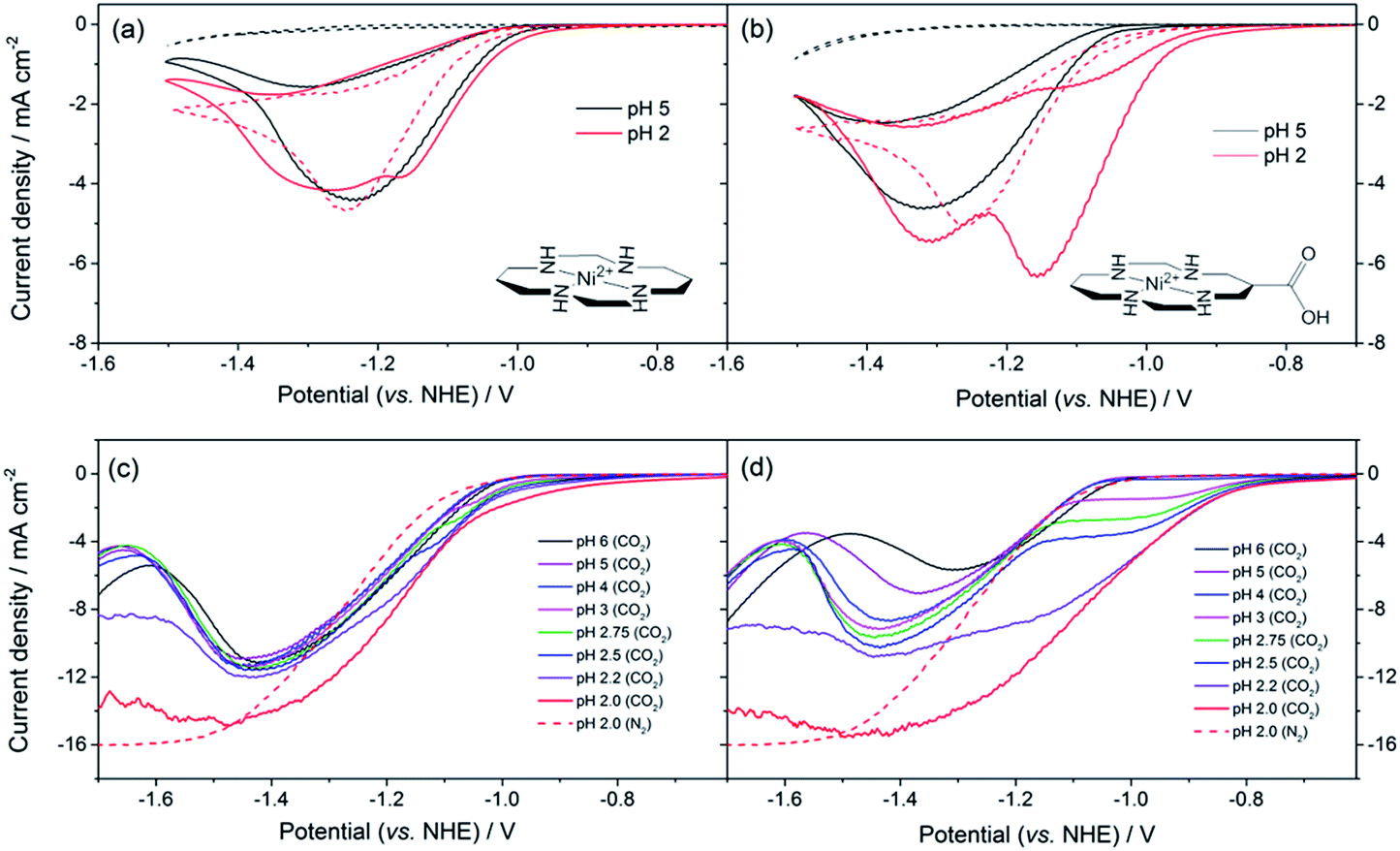 | ||
| Fig. 1 CVs of (a) 1 and (b) 2 (0.1 mM) under CO2 (solid lines) and Ar (dashed), at pH 5 (black) and 2 (red). Rotating disk electrode voltammetry of (c) 1 and (d) 2 (0.1 mM) under CO2 (solid lines) and Ar (dashed) recorded at 800 rpm at the pH indicated. All experiments are recorded using a Hg–Au amalgam electrode in 0.1 M NaClO4. | ||
Only a limited number of other classes of molecular CO2 reduction electrocatalysts for use in water are known.6,20–25 Of relevance are recent studies on organic catalysts including mercaptopteridine,26 an iridium pincer catalyst24 and very recently a water soluble iron porphyrin catalyst, labelled WSCAT,27 which preliminary data suggests is an extremely active catalyst at pH 6.7, although at lower pH values only H2 was produced. The limited pH range appears to be typical of CO2 reduction catalysts with most being studied between pH 6 and 7. In addition to WSCAT, Savéant et al.28,29 have also extensively studied other iron porphyrins for use in DMF. In an important breakthrough, a large increase in electrocatalytic activity for CO2 reduction to CO by an iron porphyrin modified with phenolic groups in DMF + 2.0 M H2O was reported.22 The acidic phenol groups on the catalyst framework acted as both a local proton source and to aid CO2 binding, greatly accelerating the proton coupled reduction of CO2, and similar approaches have now been employed by several groups studying a range of transition metal electrocatalysts for use in non-aqueous solvents.30–32 While these studies show that the addition of acidic groups can greatly accelerate the rate of reduction of CO2 in DMF, they have not been applied to catalysts that are active in water. Here we demonstrate that the modification of 1 with a carboxylic acid leads to a step change in catalytic activity in water with a five-fold increase in the observed rate constant (kobs), the turnover frequency per adsorbed catalyst, for 2 compared to 1, at −0.99 VNHE, near the foot of the catalytic wave. We also note an extremely high kobs = 3.4 (±1.0) × 103 s−1 at −1.25 VNHE. Perhaps most remarkable is that catalyst 2 operates in acidic conditions whilst maintaining selectivity towards CO2.
Results
The synthesis of 2, a derivative of 1 with a carboxylate group on the carbon backbone (Fig. 1) has been reported elsewhere, where we examined the immobilisation of 2 on metal oxide surfaces for the development of a photocatalytic system.33 Cyclic voltammograms (CVs) of 1 and 2 on a Hg/Au electrode at pH 5, higher than the pKa of the carboxylic acid group of 2 are in line with past reports, Fig. 1a and b. A NiII/I couple is present under argon at −1.30 VNHE (1) and −1.33 VNHE (2), (see Fig. S1† for an expansion). Under CO2 a large current enhancement, of similar magnitude for both 2 and 1 indicates that catalytic CO2 reduction is occuring.4,33 At pH 2 under argon the NiII/I couples of both 1 and 2 are no longer visible by CV, and a catalytic curve due to proton reduction at potentials negative of −1.1 VNHE is observed, Fig. 1a and b. The addition of CO2 to 1 at pH 2 leads to only a slight increase in current density, indicating that some CO2 reduction may occur at this pH (Fig. 1a), although we show below that H2 evolution dominates. In contrast, the current density of 2 under CO2 is notably increased and shifted anodic of the current response under argon (Fig. 1b, pH 2) indicating that 2 is an extremely active catalyst for CO2 reduction even at very low pHs.Bulk electrolysis experiments (Fig. S7†) confirm that 2 remains selective towards CO production at pH 2 and that its activity exceeds that of the parent catalyst 1. 5.2 ± 0.3 C of charge is passed within 1 hour during the electrolysis of an unstirred solution (3 μM) of 2 at −0.99 VNHE with a very good selectivity towards CO production >4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CO:H2 (faradaic efficiency (FE), total = 81%, H2 = 15 ± 5%, CO = 66 ± 9%, with errors being the result of 3 experiments). This corresponds to an average bulk turnover number of 591 for CO in 1 hour. In contrast 1 passes only 2.0 ± 0.2C in 1 hour with a lower selectivity 0.2:1 CO:H2 (FE, total = 86%, H2 = 73 ± 16%, CO = 13 ± 10%), and a bulk turnover number of ca. 45 for CO production. No liquid phase products were detected by NMR. [Ni(cyclam)]2+ and its derivatives are known to form inactive species in the presence of CO,34 however activity can be maintained through constant CO2 purging and experiments with 2 over a 7.5 hour period show activity being maintained, Fig. S8.†
:1 CO:H2 (faradaic efficiency (FE), total = 81%, H2 = 15 ± 5%, CO = 66 ± 9%, with errors being the result of 3 experiments). This corresponds to an average bulk turnover number of 591 for CO in 1 hour. In contrast 1 passes only 2.0 ± 0.2C in 1 hour with a lower selectivity 0.2:1 CO:H2 (FE, total = 86%, H2 = 73 ± 16%, CO = 13 ± 10%), and a bulk turnover number of ca. 45 for CO production. No liquid phase products were detected by NMR. [Ni(cyclam)]2+ and its derivatives are known to form inactive species in the presence of CO,34 however activity can be maintained through constant CO2 purging and experiments with 2 over a 7.5 hour period show activity being maintained, Fig. S8.†
To understand the factors behind the enhanced activity of 2 at low pH we have examined the electrochemical response of 1 and 2 over a wide pH (6–2) range using rotating disc electrode (RDE) voltammetry, (Fig. 1c and d), differential pulse voltammetry (DPV, Fig. S2 and S3†) and CV measurements (Fig. S4–6†). RDE measurements are employed to study the catalysis under CO2 as they minimise the effects of substrate diffusion and product inhibition, simplifying the analysis of the electrochemical response. Between pHs 6 and 4 RDE measurements of 2 under CO2 show only a slight increase in plateau current density, Fig. 1d. Between pH 3 and 2 a dramatic change is noted with a new reductive feature (ca. −0.95 V) growing in under CO2 as the pH decreases, which is shown above to be due to catalytic CO2 reduction. This leads to a large decrease in the potential necessary for catalysis between pH 5 and 2 of ca. 240 mV versus the normal hydrogen electrode. In the RDE measurements we define the potential necessary for catalysis as being when the current density exceeds 2 mA cm−2.35 In contrast with 1 we only measure a very small shift (ca. 50 mV) in the potential necessary for catalysis between pH 6 and 2, which will be at least in part due to the increased level of H2 production at low pHs. By pH 2 there is minimal separation of the RDE curves of 1 in the presence and absence of CO2, Fig. 1c. This step change in behaviour of 2 but not 1 is indicative of a change in catalytic mechanism for 2 between pH 3 and 2. Furthermore whilst the variation in overpotential for CO2 reduction brought about by a change in pH (0.18 V) is equivalent for both catalysts only 2 shows a significant change in potential necessary for catalysis. The lack of a pH dependence for 1 is further explored in the ESI (Fig. S2 and S3†) where we demonstrate that the NiII/I couple under argon is independent of pH.
In order to assess if the change in current density under CO2 with pH is due to the protonation of the carboxylic acid of 2 we have measured the pKa of this group by Fourier-Transform Infrared (FTIR) spectroscopy in solution (Fig. S9†). The spectra were recorded in a 0.1 mm path length CaF2 IR cell. The initially synthesised catalyst is prepared in basic conditions and the carboxylate has νas(CO2−) at 1575 cm−1 and νs(CO2−) modes at 1375 cm−1 in line with literature reports for similar complexes.36 Titration of a 0.1 M solution of 2 in D2O (initial pD = 9.61) with DCl showed the clear emergence of the carboxylic acid form of 2 with νas(CO) at 1706 cm−1 in D2O, with pKa ∼ 2.6. Deuterated solvents are required to avoid the δ(HOH) mode of H2O masking the spectral window of interest. There is an excellent correlation between the relative concentration of the protonated carboxylic acid in solution and the current density for 2 measured under CO2 using RDE at −0.99 VNHE (Fig. 2b) and −1.1 VNHE (Fig. S10†). This clearly shows that the enhancement in catalytic activity towards CO2 reduction is due to the availability of the protonated carboxylic acid group of 2 at low pH values. In contrast a similar pH titration of catalyst 1 shows no clear changes in the spectral and pH region studied.
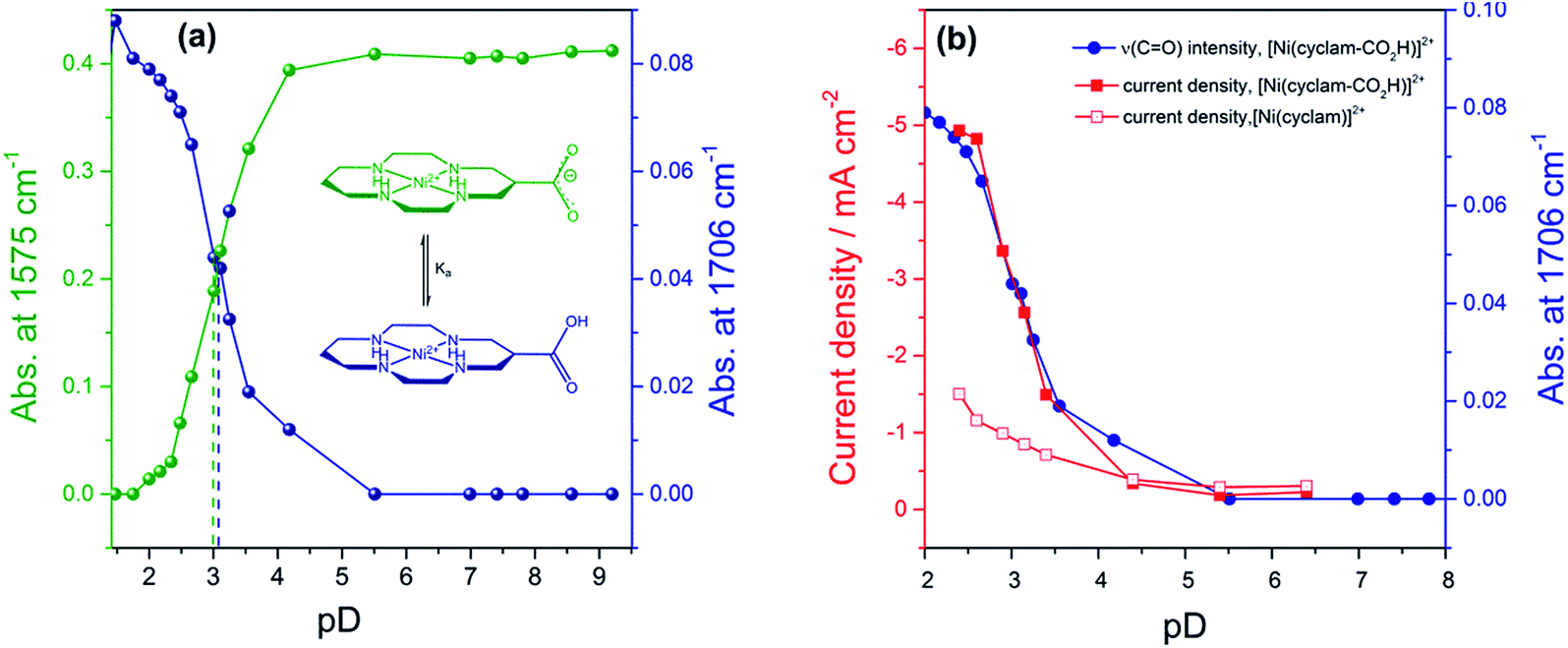 | ||
| Fig. 2 (a) pD titration curves obtained by plotting the IR intensity of the peaks of the carboxylic acid (1706 cm−1, blue) and carboxylate (1575 cm−1, green). (b) Relationship between CO2 reduction current measured at −0.99 VNHE of 2 (filled squares) by rotating disk electrode voltammetry (800 rpm, 100 mV s−1), relative concentration of carboxylic acid (given by the νas(CO) at 1706 cm−1, blue circles) and pD. The current density of 1 under CO2 with pH is also shown (open squares). pD = pH + 0.4. | ||
It has been shown that for 1 the active CO2 reduction catalyst is adsorbed onto Hg electrodes.11 It is therefore important to ascertain if the active form of 2 is also an adsorbed species. The current density under CO2 of 2 on a GCE is found to be significantly lower than that measured on a HMDE (Fig. S11 and S12†) suggesting that the active catalyst is indeed surface adsorbed 2. The surface concentration of 2 on the HMDE electrode has been measured using double-potential-step chronocoulometry11 (Tables S1–S3†) and is found to be 2.0 (±0.2) × 10−10 mol cm−2 at −0.99 VNHE at pH 5, similar to that previously reported for 1, 1.6 (±0.2) × 10−10 mol cm−2.34 At pH 2 the surface concentration of both 1 and 2 are changed by a minimal amount (2.2 (±0.2) × 10−10 mol cm−2 and 1.6 (±0.2) × 10−10 mol cm−2 respectively at −0.99 VNHE), indicating that the large increase in activity of 2 cannot be attributed to a change in the surface coverage of the catalyst with pH.
The kinetic behaviour of 1 and 2 at pH 2 can be obtained from the RDE measurements carried out at different rotation rates (Fig. S15†). We calculate the kinetic activity of the catalyst from voltammetric sweep measurements as it has recently been highlighted that turnover frequencies obtained from long-term bulk electrolysis measurements at high current densities can be complicated by a range of factors including (i) substrate diffusion, (ii) product inhibition and (iii) catalyst deactivation.37
Using the limiting current obtained from the intercepts of Koutecký–Levich plots (Fig. S14 and S15†) we can obtain an apparent first order rate constant (kobs, s−1), i.e. the turnover frequency (TOF) per adsorbed catalyst using eqn (1).38
| icat = nFΓAkobs | (1) |
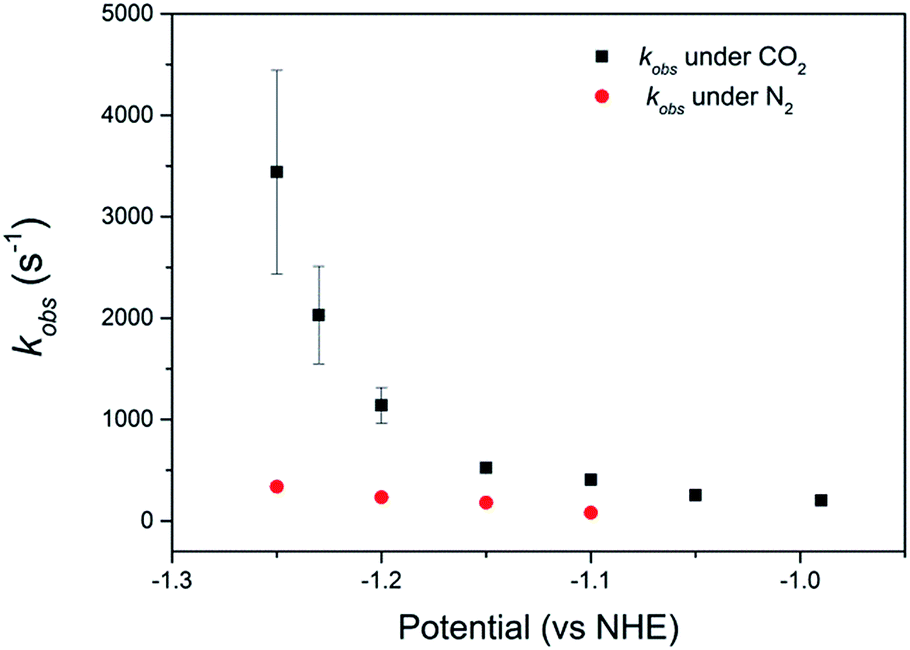 | ||
| Fig. 3 Plot of kobs of catalyst 2, calculated from the intercepts of Koutecký–Levich plots, vs. potential at pH 2. Values are obtained averaged from 3 independent measurements with the error bars calculated from the uncertainties in the intercepts of the Koutecký–Levich plots. | ||
Discussion
Comparison of the catalyst performance with existing benchmarks is ideally carried out by comparison of the overpotential dependence of the catalytic rate constant.1 Although these data are becoming increasingly reported for catalysts in aprotic solvents, we are unaware of its availability for the few CO2 reduction catalysts that operate in water.27 The value of kobs = 3.4 (±1.0) × 103 s−1 for 2 at pH 2 under CO2, measured at a single potential (−1.25 VNHE) exceeds the reported TOF of the majority of known water soluble CO2 reduction catalysts,9,24,34 including 1 (6.3 × 101 s−1).34 To the best of our knowledge there has only been one reported water soluble catalyst that operates at a greater rate, the recently reported iron porphyrin catalyst WSCAT.27 We also note that the measured rate constant for 2 under CO2 also exceeds that of many of the most commonly studied CO2 reduction catalysts operating in aprotic solvents,1 which is perhaps surprising given the significantly lower dissolved CO2 concentration in water (0.28 M (CH3CN), 34 mM (H2O)).However the most significant feature of 2 is its selectivity towards CO2 even under acidic conditions. All previously reported derivatives of 1 have shown predominantly hydrogen production outside of a small pH window7,11 and we note that the majority of CO2 reduction catalysts are reported at pHs close to neutral (5–7),24,25,27 making the ability of 2 to operate at pHs as low as 2 unusual. The correlation between the current density under CO2 and the protonation state of the carboxylic acid group of 2 suggests that the protonation state of the catalyst is an important factor in the enhanced TOF, and hence selectivity towards CO2 of 2 in acidic solutions (Fig. 2b). It may be envisaged that protonation of the carboxylic acid group leads to 2 being more readily reduced to form the active NiI catalyst, however DPV studies indicate the NiII/I couple to be pH independent under argon, Fig. S2 and 3.† Alternatively previous studies have shown that the presence of a local proton source can accelerate CO2 reduction and it is viable that the acid group may also aid catalysis here.28,30–32 In the homogenous reduction of CO2 by 1 in acetonitrile a proton concentration dependent peak in the CV, similar to the feature observed by RDE (Fig. 1d) here at ca. −0.95 VNHE was reported.16 This peak was assigned to the reduction of a protonated CO2 adduct, with this proton dependent electron transfer becoming the rate limiting step in CO2 catalysis under certain conditions. It is feasible that the protonated carboxylic acid is acting as local proton source during the reduction of a CO2 adduct here. Such an interaction is geometrically feasible. The cobalt analogue of 2 has been reported for use in dye-sensitized solar cells with binding of the –CO2H group directly to the metal centre.36,39 However we do recognise that the empirical nature of the relationship in Fig. 2b does not provide direct evidence of the functional role of the carboxylic acid. We are currently also unable to discount the role of other potential mechanistic aspects including a possible change in structure of the adsorbed catalyst or change in the catalysts CO2 affinity and further mechanistic studies are currently underway.
The enhanced activity of 2 and the ability to maintain selectivity towards CO2 across a wide pH range are highly desirable traits. It is likely that robustness towards local pH fluctuations and the ability to be employed in electrolysers using proton exchange membranes will be advantageous for any practically applicable catalyst. However operating at pH 2 does have implications regarding the overpotential for catalysis. The potential necessary for catalysis with a current density of 2 mA cm−2 in Fig. 1 is ca. −0.9 VNHE, corresponding to an overpotential of ca. −0.65 V versus the apparent equilibrium potential for CO2 reduction to CO at pH 2. Whilst not dissimilar to other previous studies in aqueous solutions,9,27 it is significantly higher than typically required in solvents such as DMF, CH3CN and ionic liquids indicating that further improvements in molecular catalysts for use in aqueous solutions are still required.
Conclusions
The development of selective and efficient molecular catalysts for electrocatalytic CO2 reduction in water is amongst the most challenging goals for the chemistry community. Complex 2 is based on a low cost metal centre and is able to use a pendant acid group to achieve excellent selectivity and activity towards CO2 even at the very low pH value of 2. The activity of 2 greatly exceeds the parent complex (1) under identical conditions, something that has been rarely achieved in over 30 years of research. 2 is also found to have be amongst the most active aqueous CO2 reduction catalysts and we believe that these characteristics make it of great significance to the field of electrocatalytic CO2 reduction.Acknowledgements
AJC, JJW (EP/K006851/1) and LJH, IMA (EP/K006835/1) acknowledge the EPSRC for funding. Prof. D. Shchukin is thanked for access to the FTIR.Notes and references
- C. Costentin, M. Robert and J.-M. Savéant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- In line with previous convention [1] E0ap is the apparent equilibrium potential at 25 °C, 1 atm gas pressure with 1 M solutes except for the proton concentration.
- W. Li, S. W. Sheehan, D. He, Y. He, X. Yao, R. L. Grimm, G. W. Brudvig and D. Wang, Angew. Chem., Int. Ed., 2015, 54, 11428–11432 CrossRef CAS PubMed.
- M. Beley, J. P. Collin, R. Ruppert and J. P. Sauvage, J. Chem. Soc., Chem. Commun., 1984, 1315–1316 RSC.
- M. Beley, J. P. Collin, R. Ruppert and J. P. Sauvage, J. Am. Chem. Soc., 1986, 108, 7461–7467 CrossRef CAS PubMed.
- B. J. Fisher and R. Eisenberg, J. Am. Chem. Soc., 1980, 102, 7361–7363 CrossRef CAS.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- E. Fujita, J. Haff, R. Sanzenbacher and H. Elias, Inorg. Chem., 1994, 33, 4627–4628 CrossRef CAS.
- J. Schneider, H. Jia, K. Kobiro, D. E. Cabelli, J. T. Muckerman and E. Fujita, Energy Environ. Sci., 2012, 5, 9502–9510 CAS.
- M. Fujihira, Y. Hirata and K. Suga, J. Electroanal. Chem., 1990, 292, 199–215 CrossRef CAS.
- G. B. Balazs and F. C. Anson, J. Electroanal. Chem., 1992, 322, 325–345 CrossRef CAS.
- C. A. Kelly, E. L. Blinn, N. Camaioni, M. D'Angelantonio and Q. G. Mulazzani, Inorg. Chem., 1999, 38, 1579–1584 CrossRef CAS.
- J. Schneider, H. Jia, J. T. Muckerman and E. Fujita, Chem. Soc. Rev., 2012, 41, 2036–2051 RSC.
- P. Jacquinot and P. C. Hauser, Electroanalysis, 2003, 15, 1437–1444 CrossRef CAS.
- J. D. Froehlich and C. P. Kubiak, Inorg. Chem., 2012, 51, 3932–3934 CrossRef CAS PubMed.
- J. D. Froehlich and C. P. Kubiak, J. Am. Chem. Soc., 2015, 137, 3565–3573 CrossRef CAS PubMed.
- S. Sakaki, J. Am. Chem. Soc., 1990, 112, 7813–7814 CrossRef CAS.
- K. Bujno, R. Bilewicz, L. Siegfried and T. A. Kaden, J. Electroanal. Chem., 1998, 445, 47–53 CrossRef CAS.
- E. J. Billo, P. J. Connolly, D. J. Sardella, J. P. Jasinski and R. J. Butcher, Inorg. Chim. Acta, 1995, 230, 19–28 CrossRef CAS.
- P. Kang, S. Zhang, T. J. Meyer and M. Brookhart, Angew. Chem., Int. Ed., 2014, 53, 8709–8713 CrossRef CAS PubMed.
- T. Yoshida, K. Kamato, M. Tsukamoto, T. Iida, D. Schlettwein, D. Wöhrle and M. Kaneko, J. Electroanal. Chem., 1995, 385, 209–225 CrossRef.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- E. Barton Cole, P. S. Lakkaraju, D. M. Rampulla, A. J. Morris, E. Abelev and A. B. Bocarsly, J. Am. Chem. Soc., 2010, 132, 11539–11551 CrossRef CAS PubMed.
- P. Kang, T. J. Meyer and M. Brookhart, Chem. Sci., 2013, 4, 3497–3502 RSC.
- P. Kang, Z. Chen, A. Nayak, S. Zhang and T. J. Meyer, Energy Environ. Sci., 2014, 7, 4007–4012 CAS.
- D. Xiang, D. Magana and R. B. Dyer, J. Am. Chem. Soc., 2014, 136, 14007–14010 CrossRef CAS PubMed.
- C. Costentin, M. Robert, J.-M. Savéant and A. Tatin, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6882–6886 CrossRef CAS PubMed.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, Science, 2012, 338, 90–94 CrossRef CAS PubMed.
- C. Costentin, G. Passard, M. Robert and J.-M. Savéant, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14990–14994 CrossRef CAS PubMed.
- F. Franco, C. Cometto, F. Ferrero Vallana, F. Sordello, E. Priola, C. Minero, C. Nervi and R. Gobetto, Chem. Commun., 2014, 50, 14670–14673 RSC.
- J. Agarwal, T. W. Shaw, H. F. Schaefer and A. B. Bocarsly, Inorg. Chem., 2015, 54, 5285–5294 CrossRef CAS PubMed.
- S. T. Ahn, E. A. Bielinski, E. M. Lane, Y. Chen, W. H. Bernskoetter, N. Hazari and G. T. R. Palmore, Chem. Commun., 2015, 51, 5947–5950 RSC.
- G. Neri, J. J. Walsh, C. Wilson, A. Reynal, J. Y. C. Lim, X. Li, A. J. P. White, N. J. Long, J. R. Durrant and A. J. Cowan, Phys. Chem. Chem. Phys., 2015, 17, 1562–1566 RSC.
- G. B. Balazs and F. C. Anson, J. Electroanal. Chem., 1993, 361, 149–157 CrossRef CAS.
- E. S. Rountree, B. D. McCarthy, T. T. Eisenhart and J. L. Dempsey, Inorg. Chem., 2014, 53, 9983–10002 CrossRef CAS PubMed.
- P. V. Bernhardt, G. K. Boschloo, F. Bozoglian, A. Hagfeldt, M. Martinez and B. Sienra, New J. Chem., 2008, 32, 705–711 RSC.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2012, 134, 11235–11242 CrossRef CAS PubMed.
- A. K. Vannucci, L. Alibabaei, M. D. Losego, J. J. Concepcion, B. Kalanyan, G. N. Parsons and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20918–20922 CrossRef CAS PubMed.
- G. Wei, T. W. Hambley, G. A. Lawrance and M. Maeder, Aust. J. Chem., 2002, 55, 667–673 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Including full experimental details, surface coverage measurements and supporting electrochemical measurements. See DOI: 10.1039/c5sc03225c |
| This journal is © The Royal Society of Chemistry 2016 |