
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dual gold/photoredox-catalyzed C(sp)–H arylation of terminal alkynes with diazonium salts†
Adrian
Tlahuext-Aca‡
ab,
Matthew N.
Hopkinson‡
a,
Basudev
Sahoo
ab and
Frank
Glorius
 *a
*a
aOrganisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster, Corrensstraße 40, 48149 Münster, Germany. E-mail: glorius@uni-muenster.de
bNRW Graduate School of Chemistry, Westfälische Wilhelms-Universität Münster, Wilhelm-Klemm-Strasse 10, 48149 Münster, Germany
First published on 8th October 2015
Abstract
The arylation of alkyl and aromatic terminal alkynes by a dual gold/photoredox catalytic system is described. Using aryldiazonium salts as readily available aryl sources, a range of diversely-functionalized arylalkynes could be synthesized under mild, base-free reaction conditions using visible light from simple household sources or even sunlight. This process, which exhibits a broad scope and functional group tolerance, expands the range of transformations amenable to dual gold/photoredox catalysis to those involving C–H bond functionalization and demonstrates the potential of this concept to access AuI/AuIII redox chemistry under mild, redox-neutral conditions.
Introduction
Homogeneous gold catalysis has received significant attention in the last two decades. Due to their carbophilic π-acidity, AuI and AuIII complexes catalyze the addition of a variety of oxygen-, nitrogen-, carbon-, and sulfur-based nucleophiles to unsaturated molecules, such as alkynes, allenes and alkenes, giving rapid and efficient access to complex molecular architectures.1 A common feature regarding the nature of the catalytically active Au species is that it does not easily undergo changes in oxidation state during the course of these reactions. However, there has been significant recent interest in developing AuI/AuIII catalytic processes with the aim of expanding the repertoire of gold-mediated processes, mimicking conventional Mn/Mn+2 redox cycles of the type commonly invoked in catalysis by other late transition metals.2 Nevertheless, in contrast to its isoelectronic counterpart Pd0, the oxidation of AuI generally requires the use of strong oxidative conditions due to the high redox potential of the AuI/AuIII redox couple (E0 = 1.41 V).3 A common strategy to access catalytically active AuIII species in situ from AuI complexes has been to use strong external oxidants, such as Selectfluor®, tBuOOH, or hypervalent iodine reagents. Applying this concept, a range of oxidative homo- and heterocoupling reactions have been developed over the last few years where the oxidative coupling event follows a conventional gold-mediated nucleophilic addition reaction to a C–C multiple bond, circumventing protodeauration and giving efficient access to difunctionalized products (Scheme 1a). This strategy has also been successfully applied in a selection of coupling reactions of unfunctionalized arenes and alkynes, exploiting the well-established ability of gold to activate C(sp2)–H or C(sp)–H bonds (Scheme 1b).4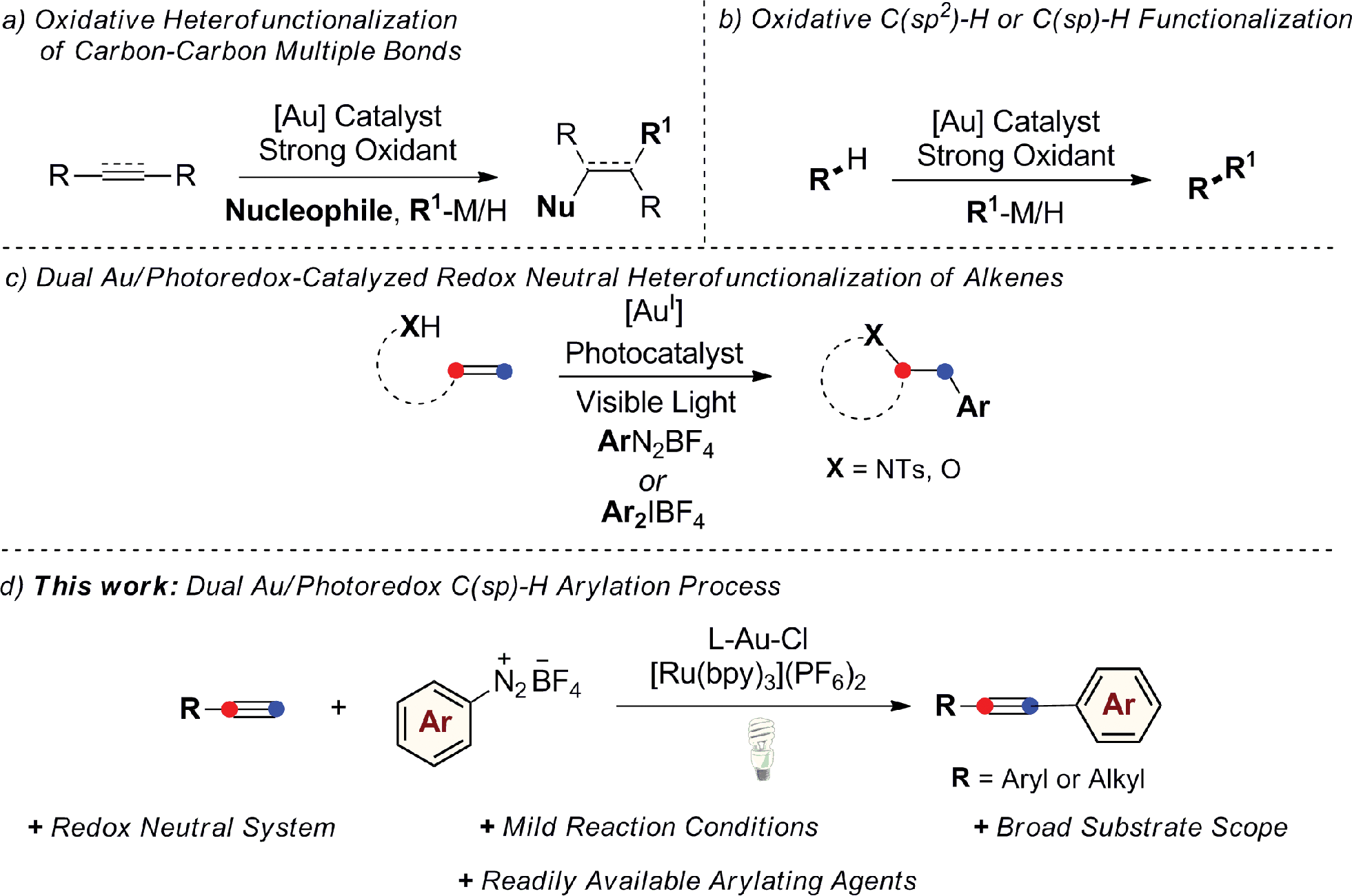 | ||
| Scheme 1 Gold-catalyzed C–C and C–X bond-forming coupling reactions proposed to proceed via AuI/AuIII redox cycles. | ||
Despite the success of these approaches, the use of strong external oxidants, typically in super-stoichiometric amounts, inevitably reduces their attractiveness with regards to functional group tolerance and cost, while most examples are also conducted at high reaction temperatures. With the goal of developing milder gold-catalyzed coupling reactions that do not require external oxidants, our group recently described the use of a dual gold/photoredox catalysis strategy to access AuI/AuIII redox cycles.5,6 Using this concept, we have developed intramolecular aminoarylation and both intra-5a and intermolecular5b oxyarylation reactions of alkenes to afford arylated heterocycles and β-aryl ethers employing aryldiazonium or diaryliodonium salts as general arylating agents (Scheme 1c). This dual catalytic system, which has since been further exploited by the Toste group in an impressive arylative ring expansion7a and C–P bond-forming cross-coupling,7b operates under mild and overall redox neutral conditions and uses abundant visible light as an energy source. Building on these studies, we sought to expand the scope of dual gold/photoredox catalysis onto new classes of transformations, exploiting different aspects of the rich chemistry of gold. In this regard, we turned our attention to the development of novel visible light-mediated cross-coupling reactions involving gold-catalyzed C–H bond activation and identified the well-established ability of gold(I) to activate the C(sp)–H bond of terminal alkynes as a promising avenue of investigation. Arylation of the resulting alkynylgold complexes would deliver cross-coupled arylalkyne products in a dual gold and photoredox-catalyzed analogue of the widely-employed Sonogashira–Hagihara reaction.8,9 As demonstrated below, this process benefits from mild conditions (room temperature, household light bulb) and broad functional group tolerance while making use of readily-available aryldiazonium salts as the arene coupling partner. Furthermore, by exploiting a non-conventional stepwise oxidation process of AuI complexes into active AuIII species by means of organic radicals generated under photoredox catalysis, we have developed a very fast arylating system that proceeds under base-free conditions.10
Results and discussion
In a preliminary experiment, p-tolylacetylene (1a) was reacted in the presence of benzenediazonium tetrafluoroborate (2a) under visible light irradiation from a simple household desk lamp fitted with a 23 W fluorescent light bulb (CFL). Upon treatment with 10 and 2.5 mol% of Ph3PAuCl and [Ru(bpy)3](PF6)2, respectively, in degassed methanol for 2 h at room temperature, we were pleased to observe that the cross-coupled product 3aa was formed in 46% yield (entry 1). Following a short screen, (see the ESI† for details of additional experiments), DMF was identified as the optimum solvent for this transformation (entry 2). A range of different AuI catalysts bearing various ligands were then evaluated (entries 3–7). Interestingly, the gold(I) chloride complex possessing the comparatively electron-rich phosphine ((p-MeO)C6H4)3P led to the highest yield of 3aa (83%), while the trialkylphosphine Cy3P was also tolerated (entry 4). This trend might be related to the stabilization of the AuIII center by these strongly donating ligands during the AuI/AuIII redox cycle. The combination of the AuI complex ((p-MeO)C6H4)3PAuCl and the photoredox catalyst [Ru(bpy)3](PF6)2 turned out to be both a highly selective and active C(sp)–H arylating system, as 3aa could be isolated in 80% yield using very low loadings of [Ru(bpy)3](PF6)2 (0.5 mol%) after a reaction time of only 1 hour and without the formation of the diyne homocoupling product of 1a (entry 8).11 Furthermore, this system does not require the addition of an external base and proceeds under acidic conditions. Interestingly, the reaction also operated efficiently using visible light from a variety of different sources. Under both blue LED (5 W, λmax = 465 nm) and even sunlight irradiation, the formation of the cross coupled product 3aa was observed in good yields (entries 9–10). In addition, the dual gold/photoredox C(sp)–H arylation process showed a remarkable robustness towards oxygen, with 3aa being obtained in 60% yield under the same conditions (10 mol% ((p-MeO)C6H4)3PAuCl and 0.5 mol% [Ru(bpy)3](PF6)2) under an atmosphere of air (entry 11). Upon increasing the loading of [Ru(bpy)3](PF6)2 to 2.5 mol%, the yield of 3aa recovered to 82% yield, indicating that careful exclusion of oxygen is not required to obtain high yields when a slightly increased loading of the photocatalyst is employed (entry 12). In contrast to our previous studies on alkene oxyarylation, attempts to switch the aryl coupling partner to diaryliodonium salts or use less expensive organic dyes were not successful for this transformation and the combination of aryldiazonium salts and [Ru(bpy)3](PF6)2 was employed for further experiments (entries 13–15).We next evaluated the reaction scope studying a variety of alkyl and aromatic terminal alkynes using 4-(ethoxycarbonyl)benzenediazonium tetrafluoroborate (2b) as the arylating reagent (Scheme 2). With arylalkyne substrates, the reaction was found to tolerate electron-donating and withdrawing substituents on the aryl ring and the corresponding coupled products were isolated in good yields (53–79%) after just 1 h of visible light irradiation. Moreover, as exemplified for the trifluoromethyl-substituted compounds 3gb, 3hb and 3ib, the reaction proceeded with substituents at each of the para- meta- and ortho-positions with seemingly comparable efficiency. Similar good results were obtained for a range of alkyl-substituted alkynes. Derivatives 3jb–mb were prepared in 54–69% yields with both primary and secondary alkyl groups being tolerated. In all cases, no diyne compounds resulting from homocoupling of the aryl- or alkylalkynes were observed during the reactions.
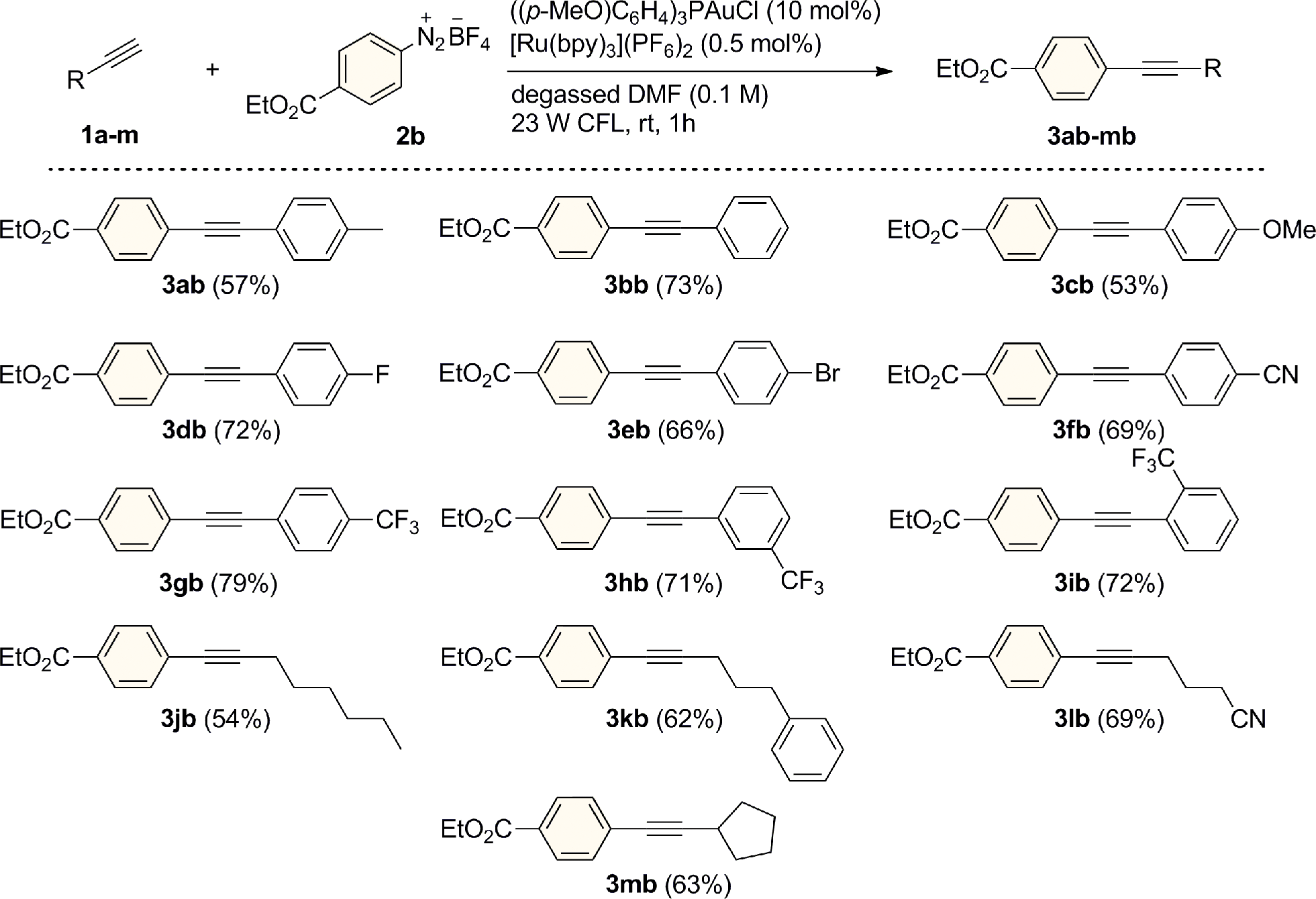 | ||
| Scheme 2 Dual Au/photoredox-catalyzed arylation of different alkynes. General conditions: alkyne (1a–m, 0.30 mmol), 2b (1.2 mmol), [Ru(bpy)3](PF6)2 (0.5 mol%), ((p-MeO)C6H4)3PAuCl (10 mol%) and degassed DMF (3.0 mL). Isolated yields. | ||
Our attention turned next to an investigation of the scope of the reaction towards a range of substituted aryldiazonium salts (2a, 2c–l) using 4-ethynylbenzonitrile 1f as a standard alkyne coupling partner (Scheme 3). We were pleased to find that aryl sources bearing a variety of electronically diverse functional groups reacted successfully, delivering the corresponding cross-coupled products 3fa, 3fc–fj in good yields up to 86%. At this point, the efficiency of the reaction when performed using electron rich groups at both the aromatic alkyne and the diazonium salt was tested by reacting 4-ethynylanisole (1c) with 2,3-dihydrobenzo[b][1,4]dioxine-6-diazonium tetrafluoroborate (2k) under the optimized reaction conditions (Scheme 3). We observed only poor conversions of 1c unless higher loadings of [Ru(bpy)3](PF6)2 (2.5 mol%) and longer irradiation times (16 h) were used. Under these conditions, the coupled product 3ck was isolated in 27% yield. Interestingly, the catalytic activity of this system was increased using blue LEDs (5 W, λmax = 465 nm) as the light source. After 5 h of irradiation, 3ck was formed in an improved 49% isolated yield. This new set of reaction conditions was also successfully applied for the synthesis of the coupled product 3cl (50% isolated yield). The efficiency of the reaction was evaluated for some substrates under an atmosphere of air using a slightly higher loading of the photocatalyst [Ru(bpy)3](PF6)2 (2.5 mol%, conditions from Table 1, entry 12). Interestingly, under these conditions, the arylated products 3fd, 3fg and 3fi were still obtained in good yields (65–67%), demonstrating that efficient catalytic activity can be maintained without rigorous extrusion of oxygen. Sterically-demanding aryldiazonium salts, however, are seemingly less well tolerated under these conditions with the ortho-substituted 2-(methoxycarbonyl)benzenediazonium tetrafluoroborate reacting sluggishly to afford the coupled product 3fj in a moderate yield of 40%.
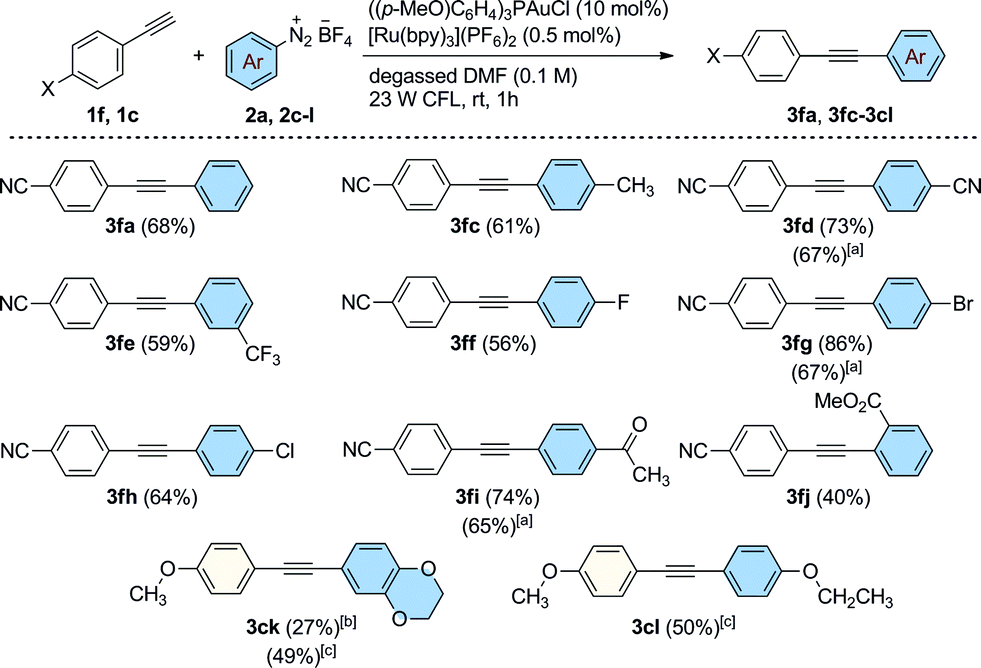 | ||
| Scheme 3 Dual Au/photoredox-catalyzed arylation of 1f and 1c with substituted benzenediazonium salts. General conditions: alkyne (0.30 mmol), 2a, 2c–l (1.2 mmol), [Ru(bpy)3](PF6)2 (0.5 mol%), ((p-MeO)C6H4)3PAuCl (10 mol%) and degassed DMF (3.0 mL). [a] Under air using 2.5 mol% of [Ru(bpy)3](PF6)2 in 1 h. [b] [Ru(bpy)3](PF6)2 (2.5 mol%), 16 h of reaction. [c] [Ru(bpy)3](PF6)2 (2.5 mol%), 5 h under 5 W blue LEDs irradiation. Isolated yields. | ||
|
|
|||
|---|---|---|---|
| Entry | Au(I) complex | Photocatalyst | Yieldb (%) |
| a General conditions: 1a (0.10 mmol), 2a (0.40 mmol), photocatalyst (2.5 mol%), gold complex (10 mol%) and degassed DMF (1.0 mL). b NMR yields using CH2Br2 as internal standard. Isolated yield in parentheses. c Degassed methanol (1.0 mL). d [Ru(bpy)3](PF6)2 (0.5 mol%), 1 h. e [Ru(bpy)3](PF6)2 (0.5 mol%), 5 W blue LEDs, 1 h. f [Ru(bpy)3](PF6)2 (0.5 mol%), sunlight, 8 h. g [Ru(bpy)3](PF6)2 (0.5 mol%), air, 1 h. h [Ru(bpy)3](PF6)2 (2.5 mol%), air, 1 h. i With Ph2IBF4 (0.40 mmol), [Ir(ppy)2(dtbbpy)]PF6 (5 mol%). See the ESI for further details. ppy = 2-phenylpyridine, dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine, XPhos = 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl, IPr = 1,3-bis(2,6-diisopropylphenyl)-imidazol-2-ylidene. | |||
| 1c | Ph3PAuCl | [Ru(bpy)3](PF6)2 | 46 |
| 2 | Ph3PAuCl | [Ru(bpy)3](PF6)2 | 78 |
| 3 | ((p-MeO)C6H4)3PAuCl | [Ru(bpy)3](PF6)2 | 83 |
| 4 | Cy3PAuCl | [Ru(bpy)3](PF6)2 | 72 |
| 5 | XPhosAuCl | [Ru(bpy)3](PF6)2 | — |
| 6 | (PhO)3PAuCl | [Ru(bpy)3](PF6)2 | — |
| 7 | IPrAuCl | [Ru(bpy)3](PF6)2 | — |
| 8d | ((p-MeO)C6H4)3PAuCl | [Ru(bpy)3](PF6)2 | 83 (80) |
| 9e | ((p-MeO)C6H4)3PAuCl | [Ru(bpy)3](PF6)2 | 80 |
| 10f | ((p-MeO)C6H4)3PAuCl | [Ru(bpy)3](PF6)2 | 71 |
| 11g | ((p-MeO)C6H4)3PAuCl | [Ru(bpy)3](PF6)2 | 60 |
| 12h | ((p-MeO)C6H4)3PAuCl | [Ru(bpy)3](PF6)2 | 82 |
| 13i | ((p-MeO)C6H4)3PAuCl | [Ir(ppy)2(dtbbpy)]PF6 | Trace |
| 14 | ((p-MeO)C6H4)3PAuCl | Eosin Y | — |
| 15 | ((p-MeO)C6H4)3PAuCl | Fluorescein | — |

In addition to its base-free and mild nature, an important feature of the dual gold/photoredox catalytic system is its compatibility with halogenated substrates. In contrast to palladium(0) or many other low-valent late transition metals, gold(I) does not generally undergo conventional oxidative addition to aryl halides under homogeneous conditions.3,9d,f,g As such, the brominated diarylalkynes 3eb and 3fg, obtained from a bromoarylalkyne and a bromo-substituted aryldiazonium salt respectively, could be isolated in good yields using this method without competitive cleavage of the C–Br bond. As demonstrated by representative Sonogashira–Hagihara and Suzuki–Miyaura coupling reactions in Scheme 4, these substrates can be readily functionalized further using palladium catalysis.12 Overall, this two-step protocol is complementary to stepwise palladium-catalyzed coupling sequences, making use of readily-available aryldiazonium salts rather than dihalogenated aryl iodide precursors (Scheme 4).
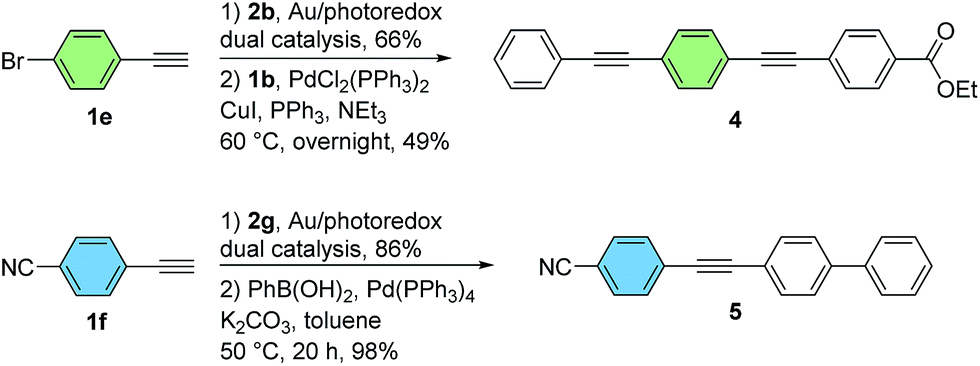 | ||
| Scheme 4 Further manipulation of cross-coupled products (see the ESI† for experimental details). | ||
In accordance with previous studies on dual Au/photoredox catalysis,5,7 we envisage a mechanism involving a photoredox-induced homogeneous AuI/AuIII redox cycle (Scheme 5).13 Upon irradiation with visible light, photoexcitation of the [Ru(bpy)3]2+ photocatalyst takes place to generate the excited form *[Ru(bpy)3]2+ which then undergoes single electron transfer (SET) with one equivalent of the aryldiazonium salt to deliver an aryl radical and the oxidized species [Ru(bpy)3]3+.14 The photo-generated aryl radical then adds to the AuI catalyst to give the AuII species A.15 Interestingly, quantum yield measurements at 450 nm by chemical actinometry gave a value of 3.6, reflecting the very important contribution of radical chain processes in this dual catalytic system.16 Therefore, we propose that a second SET process between the open-shell species A with another equivalent of the aryldiazonium salt mainly contributes to the formation of the electrophilic cationic AuIII intermediate B. Nevertheless, regeneration of the oxidized [Ru(bpy)3]3+ to the [Ru(bpy)3]2+ photoredox catalyst is still feasible by reaction with A. The cationic intermediate B is expected to be an excellent π-Lewis acid and coordinates the alkyne substrate, activating it towards the formation of the σ-bonded alkynyl-AuIII complex C upon deprotonation. Intermediate C finally undergoes reductive elimination,17 regenerating the AuI catalyst and delivering the cross-coupled product.
 | ||
| Scheme 5 Mechanistic proposal. | ||
Conclusions
In conclusion, we have developed an efficient dual gold/photoredox catalysis methodology for the arylation of terminal alkynes using readily-available aryldiazonium salts. This overall redox neutral cross-coupling process shows broad functional group tolerance, operates at room temperature and is mediated by abundant visible light from a household light bulb or even sunlight. In addition, the base-free nature of the reaction and tolerance of halogenated substrates may be beneficial in the design of cross-coupling sequences. This reaction represents the first application of dual gold/photoredox catalysis for the activation of C–H bonds and further demonstrates the potential of this methodology to access AuI/AuIII redox cycles under mild conditions without the need for external oxidants.Acknowledgements
We thank T. Pillath for experimental assistance. Generous financial support by the NRW Graduate School of Chemistry (A. T. A. and B. S.), the Alexander von Humboldt Foundation (M. N. H.) and the Deutsche Forschungsgemeinschaft (Leibniz Award) are gratefully acknowledged.Notes and references
- Selected reviews on homogeneous gold catalysis: (a) A. S. K. Hashmi, Angew. Chem., Int. Ed., 2005, 44, 6990 CrossRef CAS PubMed; (b) A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410 CrossRef PubMed; (c) D. J. Gorin and F. D. Toste, Nature, 2007, 446, 395 CrossRef CAS PubMed; (d) A. Fürstner, Chem. Soc. Rev., 2009, 38, 3208 RSC; (e) N. D. Shapiro and F. D. Toste, Synlett, 2010, 675 CAS; (f) A. S. K. Hashmi, Angew. Chem., Int. Ed., 2010, 49, 5232 CrossRef CAS PubMed; (g) N. Krause and C. Winter, Chem. Rev., 2011, 111, 1994 CrossRef CAS PubMed; (h) M. Rudolph and A. S. K. Hashmi, Chem. Commun., 2011, 47, 6536 RSC; (i) M. Bandani, Chem. Soc. Rev., 2011, 40, 1358 RSC; (j) L.-P. Liu and G. B. Hammond, Chem. Soc. Rev., 2012, 41, 3129 RSC; (k) M. Rudolph and A. S. K. Hashmi, Chem. Soc. Rev., 2012, 41, 2448 RSC; (l) C. Obradors and A. M. Echavarren, Chem. Commun., 2014, 50, 16 RSC; (m) A. Fürstner, Acc. Chem. Res., 2014, 47, 925 CrossRef PubMed; (n) R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028 CrossRef CAS PubMed.
- Selected reviews: (a) M. N. Hopkinson, A. D. Gee and V. Gouverneur, Chem.–Eur. J., 2011, 17, 8248 CrossRef CAS PubMed; (b) H. A. Wegner and M. Auzias, Angew. Chem., Int. Ed., 2011, 50, 8236 CrossRef CAS PubMed; (c) K. M. Eagle, T.-S. Mei, X. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2011, 50, 1478 CrossRef PubMed.
- (a) S. G. Bratsch, J. Chem. Phys. Ref. Data, 1989, 18, 1 CrossRef CAS PubMed; Recently a few impressive examples of oxidative addition of C–Br/I and C–C bonds onto AuI have been reported: (b) J. Guenther, S. Mallet-Ladeira, L. Estevez, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2014, 136, 1778 CrossRef CAS PubMed; (c) M. D. Levin and F. D. Toste, Angew. Chem., Int. Ed., 2014, 53, 6211 CrossRef CAS PubMed; (d) M. Joost, A. Zeineddine, L. Estévez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2014, 136, 14654 CrossRef CAS PubMed; (e) C.-Y. Wu, T. Horibe, C. B. Jacobsen and F. D. Toste, Nature, 2015, 517, 449 CrossRef CAS PubMed; (f) M. Joost, L. Estévez, K. Miqueu, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 5236 CrossRef CAS PubMed.
- T. C. Boorman and I. Larrosa, Chem. Soc. Rev., 2011, 40, 1910 RSC.
- (a) B. Sahoo, M. N. Hopkinson and F. Glorius, J. Am. Chem. Soc., 2013, 135, 5505 CrossRef CAS PubMed; (b) M. N. Hopkinson, B. Sahoo and F. Glorius, Adv. Synth. Catal., 2014, 356, 2794 CrossRef CAS PubMed.
- Selected reviews on visible light photoredox catalysis: (a) K. Zeitler, Angew. Chem., Int. Ed., 2009, 48, 9785 CrossRef CAS PubMed; (b) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527 CrossRef CAS PubMed; (c) F. Teplý, Collect. Czech. Chem. Commun., 2011, 76, 859 CrossRef; (d) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC; (e) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828 CrossRef CAS PubMed; (f) L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687 RSC; (g) S. Fukuzumi and K. Ohkubo, Chem. Sci., 2013, 4, 561 RSC; (h) Y. Xi, H. Yi and A. Lei, Org. Biomol. Chem., 2013, 11, 2387 RSC; (i) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (j) J. Xuan, L.-Q. Lu, J.-R. Chen and W.-J. Xiao, Eur. J. Org. Chem., 2013, 6755 CrossRef CAS PubMed; (k) M. Reckenthäler and A. G. Griesbeck, Adv. Synth. Catal., 2013, 355, 2727 CrossRef PubMed; (l) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed; (m) T. Koike and M. Akita, Top. Catal., 2014, 57, 967 CrossRef CAS; (n) D. A. Nicewicz and T. M. Nguyen, ACS Catal., 2014, 4, 355 CrossRef CAS; (o) M. N. Hopkinson, B. Sahoo, J.-L. Li and F. Glorius, Chem.–Eur. J., 2014, 20, 3874 CrossRef CAS PubMed; (p) E. Meggers, Chem. Commun., 2015, 51, 3290 RSC.
- (a) X.-Z. Shu, M. Zhang, Y. He, H. Frei and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 5844 CrossRef CAS PubMed; (b) Y. He, H. Wu and F. D. Toste, Chem. Sci., 2015, 6, 1194 RSC.
- Recent review on the Sonogashira–Hagihara reaction: (a) R. Chinchilla and C. Nájera, Chem. Soc. Rev., 2011, 40, 5084 RSC; examples of Pd-catalyzed alkyne arylation with aryldiazonium salts: (b) G. Fabrizi, A. Goggiamani, A. Sferrazza and S. Cacchi, Angew. Chem., Int. Ed., 2010, 49, 4067 CrossRef CAS PubMed; (c) B. Panda and T. K. Sarkar, Chem. Commun., 2010, 46, 3131 RSC; (d) X.-F. Fu, H. Neumann and M. Beller, Chem. Commun., 2011, 47, 7959 RSC; (e) M. Barbero, S. Cadamuro and S. Dughera, Eur. J. Org. Chem., 2014, 598 CrossRef CAS PubMed.
- While a few examples of gold-catalyzed Sonogashira–Hagihara reactions with aryl halides have been reported, the mechanism of these processes has been the subject of debate. Trace palladium impurities and heterogeneous gold nanoparticles have both been implicated in these reactions: (a) C. González-Arellano, A. Abad, A. Corma, H. García, M. Iglesias and F. Sánchez, Angew. Chem., Int. Ed., 2007, 46, 1536 CrossRef PubMed; (b) P. Li, L. Wang, M. Wang and F. You, Eur. J. Org. Chem., 2008, 5946 CrossRef CAS PubMed; (c) C. González-Arellano, A. Corma, M. Iglesias and F. Sánchez, Eur. J. Inorg. Chem., 2008, 1107 CrossRef PubMed; (d) T. Lauterbach, M. Livendahl, A. Rosellón, P. Espinet and A. M. Echavarren, Org. Lett., 2010, 12, 3006 CrossRef CAS PubMed; (e) G. Kyriakou, S. K. Beaumont, S. M. Humphrey, C. Antonetti and R. M. Lambert, ChemCatChem, 2010, 2, 1444 CrossRef CAS PubMed; (f) A. Corma, R. Juárez, M. Boronat, F. Sánchez, M. Iglesias and H. García, Chem. Commun., 2011, 47, 1446 RSC; (g) M. Livendahl, P. Espinet and A. M. Echavarren, Platinum Met. Rev., 2011, 55, 212 CrossRef; (h) P. S. D. Robinson, G. N. Khairallah, G. da Silva, H. Lioe and R. A. J. O’Hair, Angew. Chem., Int. Ed., 2012, 51, 3812 CrossRef CAS PubMed; examples of gold-catalyzed alkynylation with external oxidants: (i) T. de Haro and C. Nevado, J. Am. Chem. Soc., 2010, 132, 1512 CrossRef CAS PubMed; (j) M. N. Hopkinson, J. E. Ross, G. T. Giuffredi, A. D. Gee and V. Gouverneur, Org. Lett., 2010, 12, 4904 CrossRef CAS PubMed; (k) D. Qian and J. Zhang, Beilstein J. Org. Chem., 2011, 7, 808 CrossRef CAS PubMed; (l) Y. Ma, S. Zhang, S. Yang, F. Song and J. You, Angew. Chem., Int. Ed., 2014, 53, 7870 CrossRef CAS PubMed; (m) H. Peng, Y. Xi, N. Ronaghi, B. Dong, N. G. Akhmedov and X. Shi, J. Am. Chem. Soc., 2014, 136, 13174 CrossRef CAS PubMed; review on gold-catalyzed alkynylation including examples not involving C(sp)–H bond activation: (n) J. P. Brand, Y. Li and J. Waser, Isr. J. Chem., 2013, 53, 901 CrossRef CAS PubMed.
- During the preparation of this manuscript, Shi and co-workers reported a ligand assisted gold-catalyzed coupling of terminal alkynes with aryldiazonium salts. This catalytic system does not involve photoredox activation and explores a ligand-assisted two electron oxidation process of the Au(I) complex under basic conditions, thus proceeding by a different mechanism: R. Cai, M. Lu, E. Y. Aguilera, Y. Xi, N. G. Akhmedov, J. L. Petersen, H. Chen and X. Shi, Angew. Chem., Int. Ed., 2015, 54, 8772 CrossRef CAS PubMed.
- Control experiments without the AuI catalyst, photocatalyst or visible light gave trace amounts or no product formation. Careful analysis of reaction mixtures by GC-MS showed no diyne homocoupling product formation (see the ESI† for further information).
- (a) Y. Miao, A. Dupé, C. Bruneau and C. Fischmeister, Eur. J. Org. Chem., 2014, 5071 CrossRef CAS PubMed; (b) X. Shen, D. M. Ho and R. A. Pascal Jr, J. Am. Chem. Soc., 2004, 126, 5798 CrossRef CAS PubMed.
- The kinetic profile of the reaction did not exhibit an induction period, implying that heterogeneous gold nanoparticles are not responsible for the observed reactivity (see the ESI† for more details, see also ref. 9f–h for a discussion of heterogeneous gold-catalyzed coupling reactions).
- Review on the photoredox activation of diazonium salts: D. P. Hari and B. König, Angew. Chem., Int. Ed., 2013, 52, 4734 CrossRef CAS PubMed.
- Seminal study on the trapping of carbon-based radicals by gold complexes: (a) C. Aprile, M. Boronat, B. Ferrer, A. Corma and H. García, J. Am. Chem. Soc., 2006, 128, 8388 CrossRef CAS PubMed; recent report on the oxidation of gold(I) by the CF3 radical: (b) M. S. Winston, W. J. Wolf and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 7777 CrossRef CAS PubMed.
- For interesting discussions on the prevalence of radical chain processes in photoredox catalysis see: (a) M. Majek, F. Filace and A. J. von Wangelin, Beilstein J. Org. Chem., 2014, 10, 981 CrossRef CAS PubMed; (b) M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426 RSC For quantum yield measurements see the ESI†.
- Report on reductive elimination from these complexes: Y. Fuchita, Y. Utsunomiya and M. Yasutake, J. Chem. Soc., Dalton Trans., 2001, 2330 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: General experimental details, full optimization table, kinetic profile, experimental procedures and characterization data of products, copies of NMR spectra. See DOI: 10.1039/c5sc02583d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |