
Catalytic asymmetric direct-type 1,4-addition reactions of simple esters†‡
Io
Sato
,
Hirotsugu
Suzuki
,
Yasuhiro
Yamashita
and
Shū
Kobayashi
*
Department of Chemistry, School of Science, The University of Tokyo, Hongo, Bunkyo-ku, Tokyo, Japan. E-mail: shu_kobayashi@chem.s.u-tokyo.ac.jp
First published on 29th June 2016
Abstract
In the presence of catalytic amounts of potassium hexamethyldisilazide (KHMDS) and a chiral macrocyclic crown ether, asymmetric 1,4-addition reactions of simple esters with α,β-unsaturated amides proceeded to afford the desired 1,4-adducts in high yields with good stereoselectivities. Mechanistic investigations revealed that proper combination of substrates was a key for the efficient catalytic turnover.
Introduction
The catalytic stereoselective carbon–carbon bond forming reactions are attractive methods for syntheses of fine chemicals, such as medicines, agricultural chemicals and so on.1 Among them, carbon–carbon bond forming reactions using carbanions derived from pronucleophiles with catalytic amounts of Brønsted bases (catalytic carbanion reactions)2a under proton transfer conditions are one of the most efficient methods to construct complex carbon frameworks, because the atom economy of these reactions is theoretically perfect.2 Recent energetic research efforts have shown that relatively acidic compounds (pKa = 18–25),3 such as aldehydes, malonates, nitroalkanes, ketones and so on, can be successfully utilized as pronucleophiles for Brønsted base-catalyzed carbon–carbon bond forming reactions. Furthermore, asymmetric variants of these reactions have been widely developed with catalytic amounts of chiral sources.4 On the other hand, pronucleophiles with the oxidation states of carboxylic acids such as esters, nitriles, amides and so on have remained undeveloped for Brønsted base-catalyzed carbon–carbon bond forming reactions because they are less prone to deprotonation (pKa = 30–35). For efficient production of carbanions from these less acidic compounds, strong Brønsted bases (e.g., lithium diisopropylamide (LDA), alkyl lithium) are generally required; however, the acidity of their conjugate acids is intrinsically low, thus regeneration of strong Brønsted bases via deprotonation by reaction intermediates is too slow to achieve smooth and efficient catalyst turnover. Therefore, excess amounts of strong Brønsted bases are generally required for carbon–carbon bond formations of carbanions derived from those compounds. Furthermore, stoichiometric amounts of chiral reagents are mostly required for asymmetric variants of the reactions; this is not an efficient method from the viewpoint of atom economy.5 Recently, there have been several successful examples of catalytic (asymmetric) carbon–carbon bond forming reactions of alkylnitriles,6 simple amides without any functionalities on their α-position7 and carboxylic acids8 as pronucleophiles by Brønsted base/Lewis acid cooperative catalysis. On the other hand, simple esters without any functionalities on their α-positions still remain formidable pronucleophiles for the reactions, probably because of both the low acidity of α-hydrogen atoms and the low Lewis basicity of carbonyl oxygen atoms.Catalytic stereoselective carbon–carbon bond forming reactions of esters via enolate formation are an attractive method to obtain optically active α-substituted esters, which are abundant motifs in natural and bioactive compounds.9 One approach to obtain optically active α-substituted esters in a catalytic manner is catalytic asymmetric carbon–carbon bond forming reaction using ketene silyl acetals, called Mukaiyama-type reactions.10 Although many types of catalytic asymmetric Mukaiyama-type reactions have been developed in the past few decades, it is an unavoidable problem that syntheses of ketene silyl acetals require excess amounts of strong Brønsted bases and silicon reagents (Scheme 1, top). The other approach is catalytic asymmetric carbon–carbon bond forming reactions of ester equivalents such as trichloroketones,11 thioamides,12 and sulfonylimidates.13 In these reactions, deprotonation of α-hydrogen atoms of ester equivalents is facilitated by modified structures; thus catalytic asymmetric reactions proceeded with relatively weak bases such as metal phenoxides12a and DBU;13 however, syntheses of ester equivalents generally require stoichiometric amounts of reagents or reactive starting materials, and also further transformations of the products to the desired ester moieties are needed (Scheme 1, middle). Therefore, a more concise and efficient way, that is catalytic asymmetric direct-type α-functionalization of simple esters without any activating functionalities on their α-positions, is in high demand. Recently, our group has developed catalytic direct-type carbon–carbon bond forming reactions of less acidic compounds via the “product base mechanism”,14 in which the reaction intermediates are designed to possess strong basicity to deprotonate less acidic hydrogen atoms of the next pronucleophiles. Based on this mechanism, we reported that Mannich-type reactions of simple esters proceeded smoothly with a catalytic amount of potassium hydride (KH) to afford the desired β-aminoesters in high yields.14a We have also shown catalytic asymmetric 1,4-addition reactions of simple amides14b and alkylnitriles14c with α,β-unsaturated amides catalyzed by KHMDS and a novel chiral macrocyclic crown ether ((R,R)-binaphtho-34-crown-10) to afford the desired 1,4-adducts in high yields with good to high stereoselectivities. Herein, to demonstrate the generality of this catalytic system to other less acidic pronucleophiles, we report catalytic asymmetric 1,4-addition reactions of simple esters (Scheme 1, bottom). To the best of our knowledge, this is the first example of catalytic asymmetric direct-type carbon–carbon bond forming reactions of simple esters.15
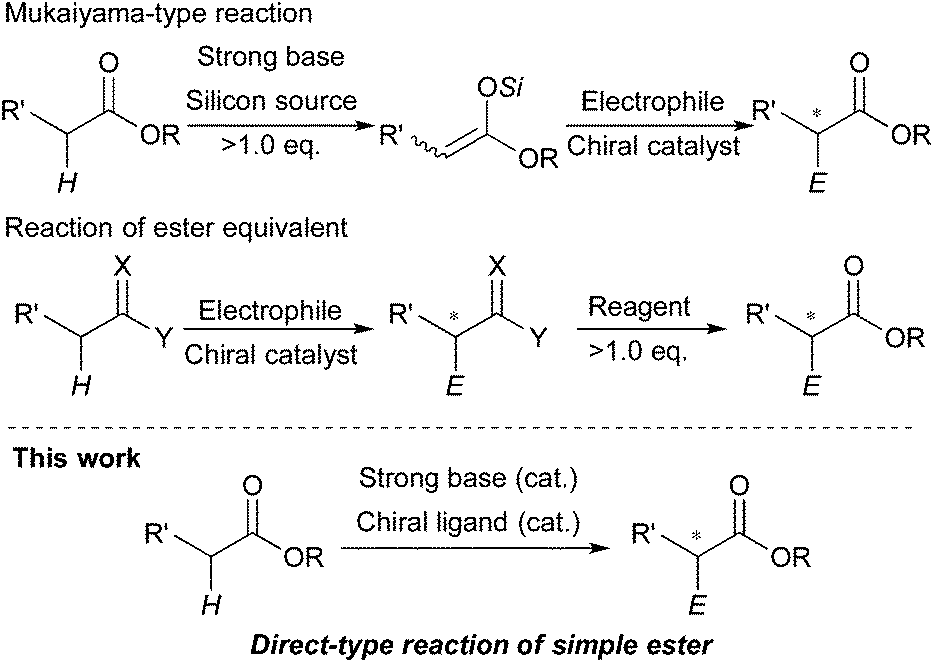 | ||
| Scheme 1 Methods to access optically active α-substituted esters in a catalytic manner. | ||
Results and discussion
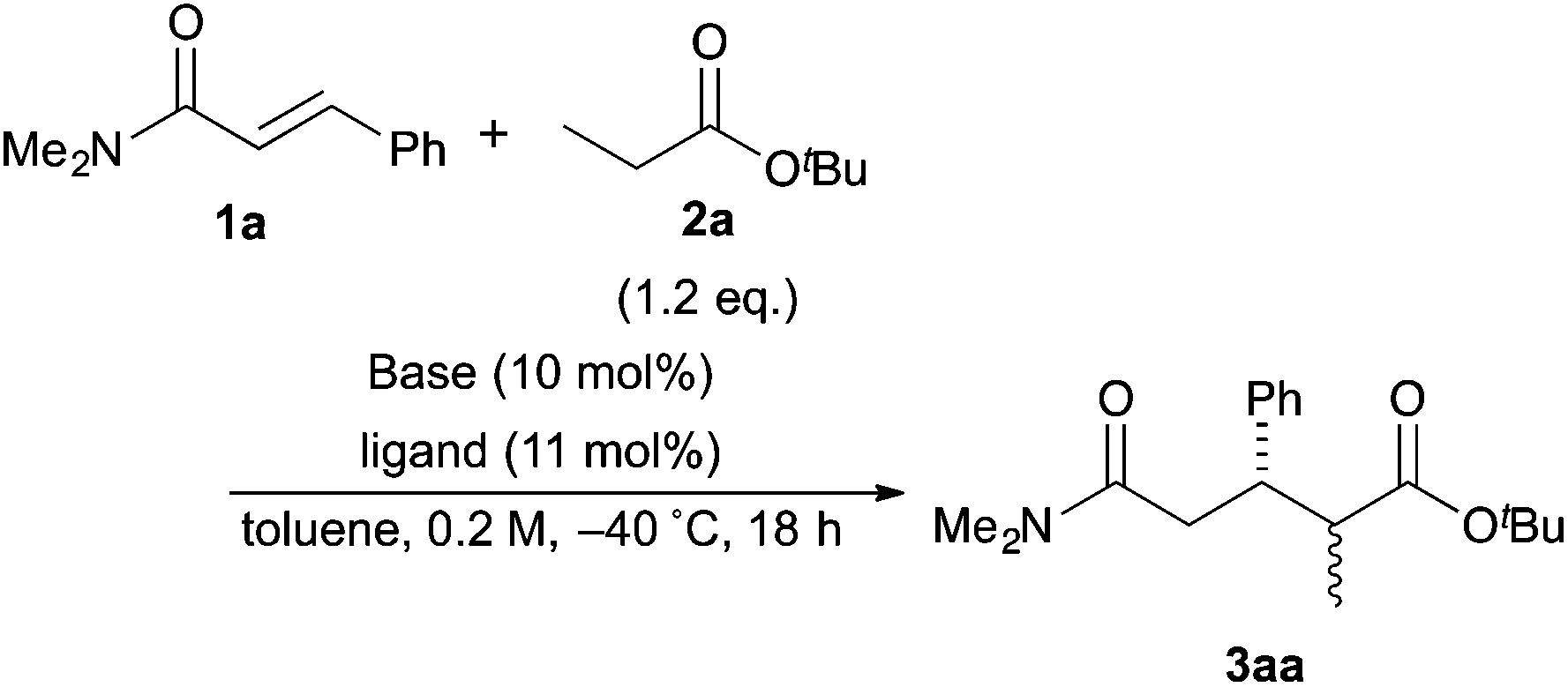
The investigation started with the screening of strong Brønsted base catalysts for 1,4-addition reactions of tBu propionates (2a) with N,N-dimethylcinnamamides (1a) (Table 1). Although lithium or sodium hexamethyldisilazide (Li, NaHMDS) could not catalyze the reaction at all (entries 1 and 2), KHMDS could work as a strong Brønsted base catalyst to afford the desired 1,4-adduct (3aa) in moderate yield with poor syn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :anti selectivity (entry 3). Addition of 18-crown-6 as a ligand enhanced both the yield and syn:anti selectivity (entry 4). KOtBu/18-crown-6 could also catalyze the reaction with lower syn:anti selectivity (entry 5).
:anti selectivity (entry 3). Addition of 18-crown-6 as a ligand enhanced both the yield and syn:anti selectivity (entry 4). KOtBu/18-crown-6 could also catalyze the reaction with lower syn:anti selectivity (entry 5).
|
|
||||
|---|---|---|---|---|
| Entry | Base | Ligand | Yieldb (%) |
syn:antic |
| a The reaction of 1a (0.30 mmol) with 2a (0.36 mmol) was performed in toluene at 0.2 M at −40 °C for 18 h in the presence of the base (0.030 mmol) with/without 18-crown-6 (0.034 mmol) unless otherwise noted. b The sum of isolated yields of both diastereomers. c Determined by 1H NMR analysis of the crude mixture. d Not determined. | ||||
| 1 | LiHMDS | — | Tracec | —d |
| 2 | NaHMDS | — | Tracec | —d |
| 3 | KHMDS | — | 50 | 57:43 |
| 4 | KHMDS | 18-crown-6 | Quant. | 69:31 |
| 5 | KOtBu | 18-crown-6 | 98 | 52:48 |
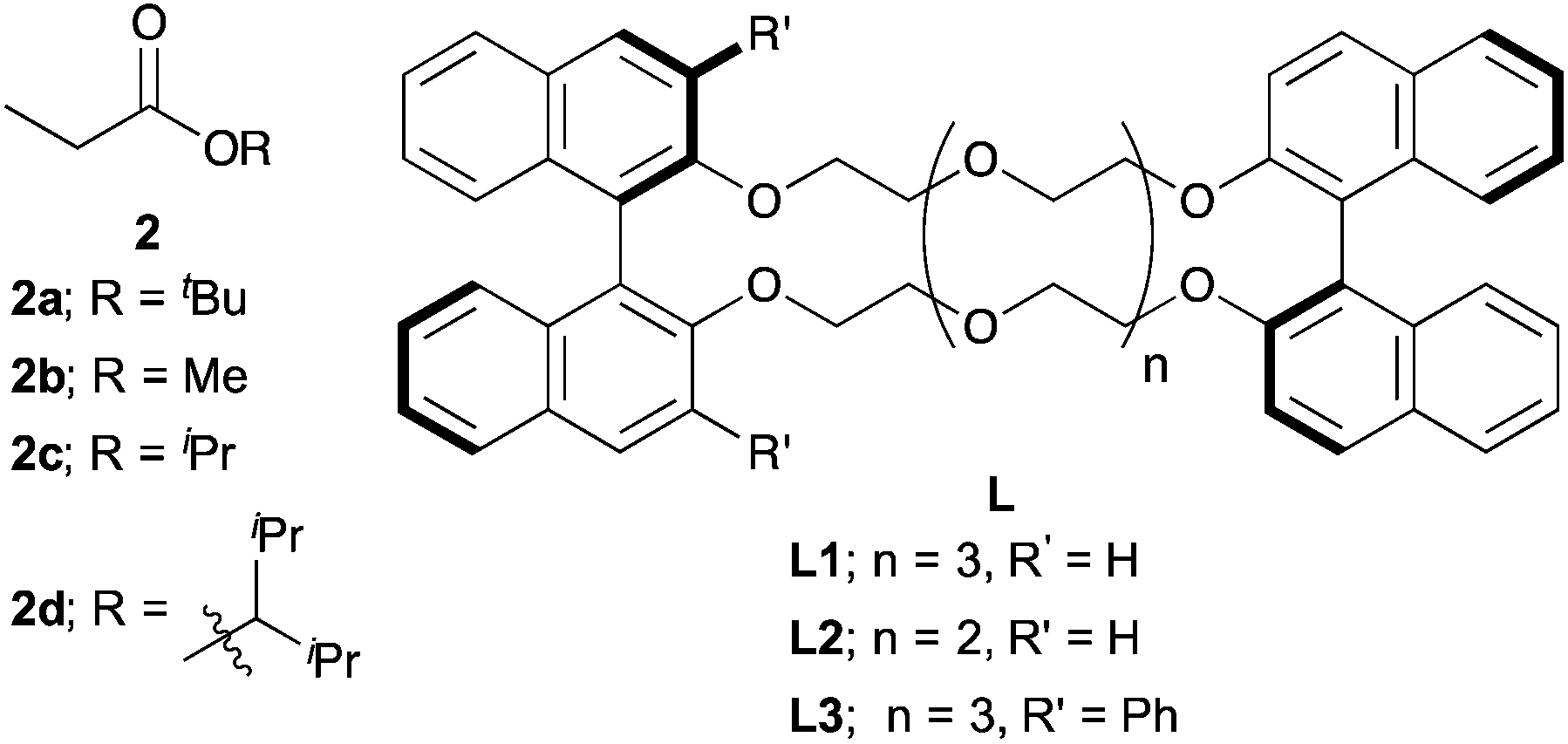
Next, we moved to investigations of asymmetric variants of the reactions (Table 2). Recently, our group showed that a novel chiral macrocyclic crown ether, (R,R)-binaphtho-34-crown-10 (L1), could build an effective chiral environment around a potassium cation to give excellent enantioselectivity for KHMDS-catalyzed 1,4-addition reactions of simple amides.14b Therefore, we firstly tried to use L1 as a chiral ligand for the reaction; the desired product was obtained in high yield with high enantioselectivity, but with poor syn:anti selectivity (entry 1). The reaction proceeded smoothly at −78 °C to improve the stereoselectivity (entry 2). Although the syn:anti selectivity was still not satisfactory, it should be noted that the syn isomer is the major one, which is contrary to the 1,4-addition reactions of simple amides reported previously.14b To improve the syn:anti selectivity, further optimizations of the reaction conditions were conducted. THF as a solvent showed good reactivity but poor stereoselectivity, presumably because of the strong coordination ability of the oxygen atom of THF (entry 3). The reaction in Et2O gave only a trace amount of the desired product because of the low solubility of the electrophile (entry 4). Next, several propionates with different sizes of alkyl moieties were examined for the reactions; however, the reactions of propionates with smaller or bigger alkyl moieties gave lower stereoselectivities (entries 5–7). Next, chiral macrocyclic crown ethers were examined for the reactions. (R,R)-Binaphtho-28-crown-8 (L2), which has a smaller cavity than L1, showed lower reactivity and selectivity (entry 8). To change the dihedral angle of the BINOL moiety, 3,3′-Ph2-34-crown-10 (L3) was synthesized and examined (entry 9). Although the reactivity was decreased, it was found that the syn:anti selectivity was reversed, and the anti isomer was obtained as the major isomer in moderate yield with high enantioselectivity. Finally, we chose the conditions in entry 2 as optimal ones.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 2 | L | Temp. (°C) | Yieldb (%) |
syn:antic |
eed (%) |
a The reaction of 1a (0.40 mmol) with 2a (0.49 mmol) was performed in toluene at 0.2 M for 24 h in the presence of the catalyst prepared from KHMDS (0.040 mmol) and L (0.022 mmol) unless otherwise noted.
b The sum of isolated yields of both diastereomers.
c Determined by 1H NMR analysis of the crude mixture.
d ee values of syn/anti diastereomers, respectively.
e THF was used as a solvent.
f Et2O was used as a solvent.
g Not determined.
|
||||||
| 1 | 2a | L1 | −40 | 96 | 57:43 |
82/82 |
| 2 | 2a | L1 | −78 | 92 | 65:35 |
95/85 |
| 3e | 2a | L1 | −78 | 94 | 57:43 |
40/40 |
| 4f | 2a | L1 | −78 | Tracec | —g | —g |
| 5 | 2b | L1 | −78 | 93 | 55:45 |
93/94 |
| 6 | 2c | L1 | −78 | 89 | 52:48 |
92/87 |
| 7 | 2d | L1 | −78 | 86 | 63:37 |
89/89 |
| 8 | 2a | L2 | −78 | 12 | 59:41 |
67/—g |
| 9 | 2a | L3 | −78 | 63 | 37:63 |
78/89 |
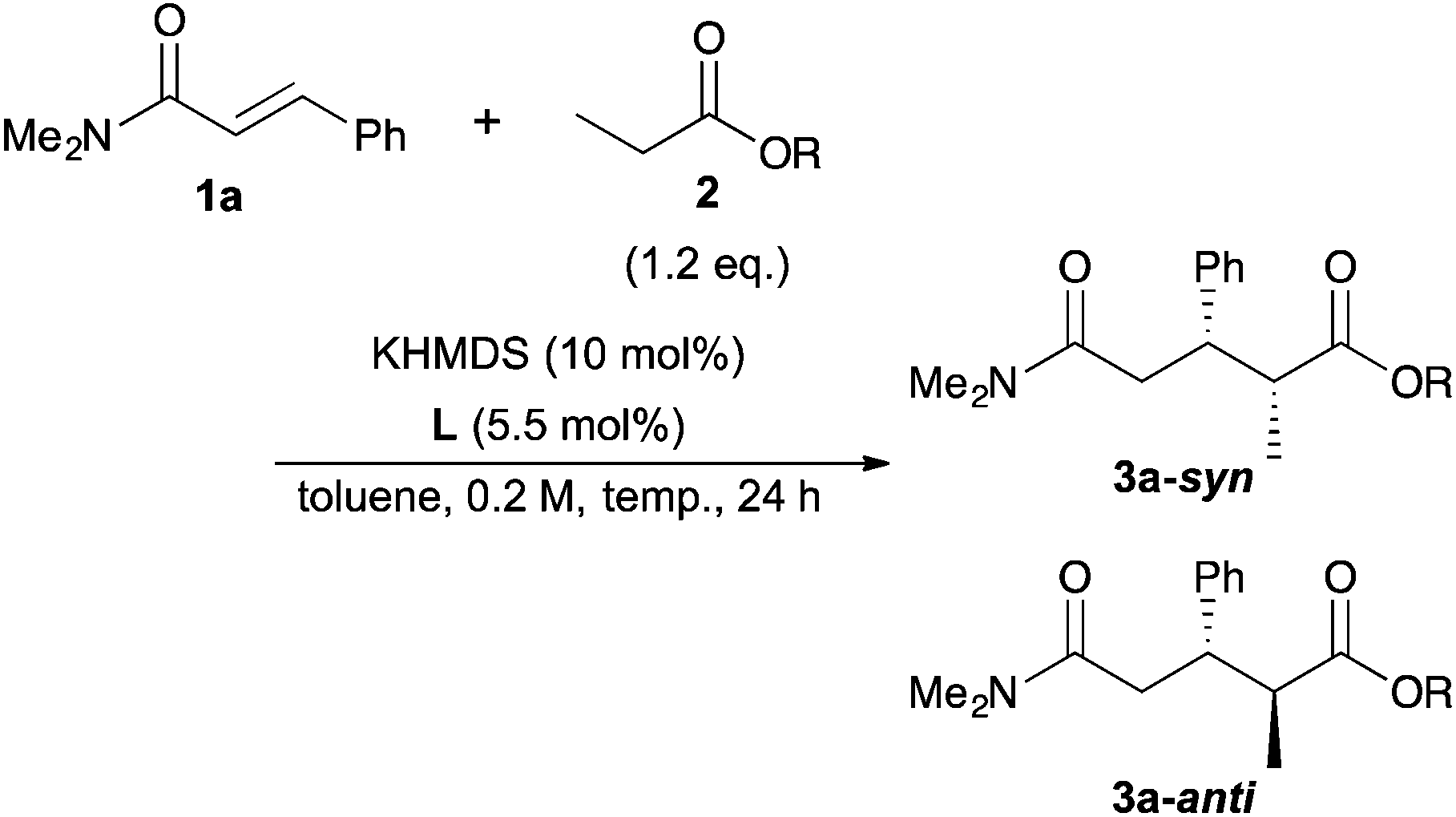
With the optimal conditions in hand, the substrate scope was examined (Table 3). First, α,β-unsaturated amides with various kinds of aromatic substituents at the β-positions were examined (entries 2–10). The reaction with o-tolyl α,β-unsaturated amide (1b) provided the reverse diastereoselectivity with lower enantioielectivity (entry 2). On the other hand, m-tolyl and p-tolyl α,β-unsaturated amides (1c, 1d) did not show the reversal of the diastereoselectivity (entries 3 and 4). Furthermore, the reaction with p-tolyl α,β-unsaturated amide (1d) exhibited higher stereoselectivity than the model reaction. p-MeO α,β-unsaturated amide (1e) gave lower reactivity, thus the reaction was conducted at −60 °C to afford the product in high yield with slightly lower stereoselectivity (entry 5). The reactions of p-Br and p-Cl α,β-unsaturated amides (1f, 1g) also showed almost the same results as the model reaction (entries 6 and 7). The reaction of 2-naphthyl α,β-unsaturated amide (1h) was sluggish because of its poor solubility in toluene; thus, the reaction was carried out for 90 h by using 2.4 eq. of ester in 0.1 M to afford the product in high yield with high stereoselectivity (entry 8). 1-Naphthyl α,β-unsaturated amide (1i) also had low solubility, so the reaction was conducted at −60 °C to give the product in good yield with good stereoselectivity (entry 9). It should be noted that the reversal of the diastereoselectivity was also observed in this reaction similar to the reaction of o-tolyl α,β-unsaturated amide (1b). These results suggested that α,β-unsaturated amides with ortho-substituted aromatic substituents at the β-position caused the reversal of the diastereoselectivity. 2-Furyl α,β-unsaturated amide (1j) was tolerant for the reaction to afford the desired product in good yield with lower diastereoselectivity and high enanstioselectivity. Next, α,β-unsaturated amides with aliphatic substituents at the β-position were examined for the reaction. Although the reactivity of cyclohexyl α,β-unsaturated amide (1k) was not good and the reaction was conducted at −60 °C, the highest diastereoselectivity was obtained (entry 11). On the other hand, nPr α,β-unsaturated amide (1l) showed poor diastereoselectivity, probably because it was sterically less hindered than 1k (entry 12). Finally, tBu butyrate (2e) was examined for the reaction to afford the product in high yield with higher diastereoselectivity than the reaction of tBu propionate (2a) because of the steric hindrance (entry 13).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | 3 | Yieldb (%) |
syn:antic |
eed (%) |
| a The reaction of 1 (0.40 mmol) with 2 (0.49 mmol) was performed in toluene at 0.2 M at −78 °C for 24 h in the presence of the catalyst prepared from KHMDS (0.040 mmol) and L1 (0.022 mmol) unless otherwise noted. b The sum of isolated yields of both diastereomers. c Determined by 1H NMR analysis of the crude mixture. d ee values of syn/anti diastereomers, respectively. e −60 °C. f 0.1 M. g 2.4 eq. of ester were used. h 90 h. i 0.15 M. | ||||||
| 1 | Ph (1a) | Me (2a) | 3aa | 92 | 65:35 |
95/86 |
| 2 | o-MeC6H4 (1b) | Me (2a) | 3ba | 84 | 46:54 |
66/81 |
| 3 | m-MeC6H4 (1c) | Me (2a) | 3ca | 72 | 60:40 |
95/80 |
| 4 | p-MeC6H4 (1d) | Me (2a) | 3da | 90 | 69:31 |
96/85 |
| 5e | p-OMeC6H4 (1e) | Me (2a) | 3ea | 96 | 63:37 |
91/91 |
| 6 | p-BrC6H4 (1f) | Me (2a) | 3fa | 93 | 64:36 |
91/88 |
| 7 | p-ClC6H4 (1g) | Me (2a) | 3ga | 88 | 63:37 |
90/87 |
| 8f,g,h | 2-Naphthyl (1h) | Me (2a) | 3ha | 91 | 67:33 |
94/80 |
| 9e,f | 1-Naphthyl (1i) | Me (2a) | 3ia | 73 | 40:60 |
79/75 |
| 10i | 2-Furyl (1j) | Me (2a) | 3ja | 65 | 58:42 |
92/85 |
| 11e | Cy (1k) | Me (2a) | 3ka | 75 | 87:13 |
88/83 |
| 12 | n Pr (1l) | Me (2a) | 3la | 61 | 52:48 |
90/91 |
| 13g | Ph (1a) | Et (2e) | 3ae | 93 | 74:26 |
93/78 |
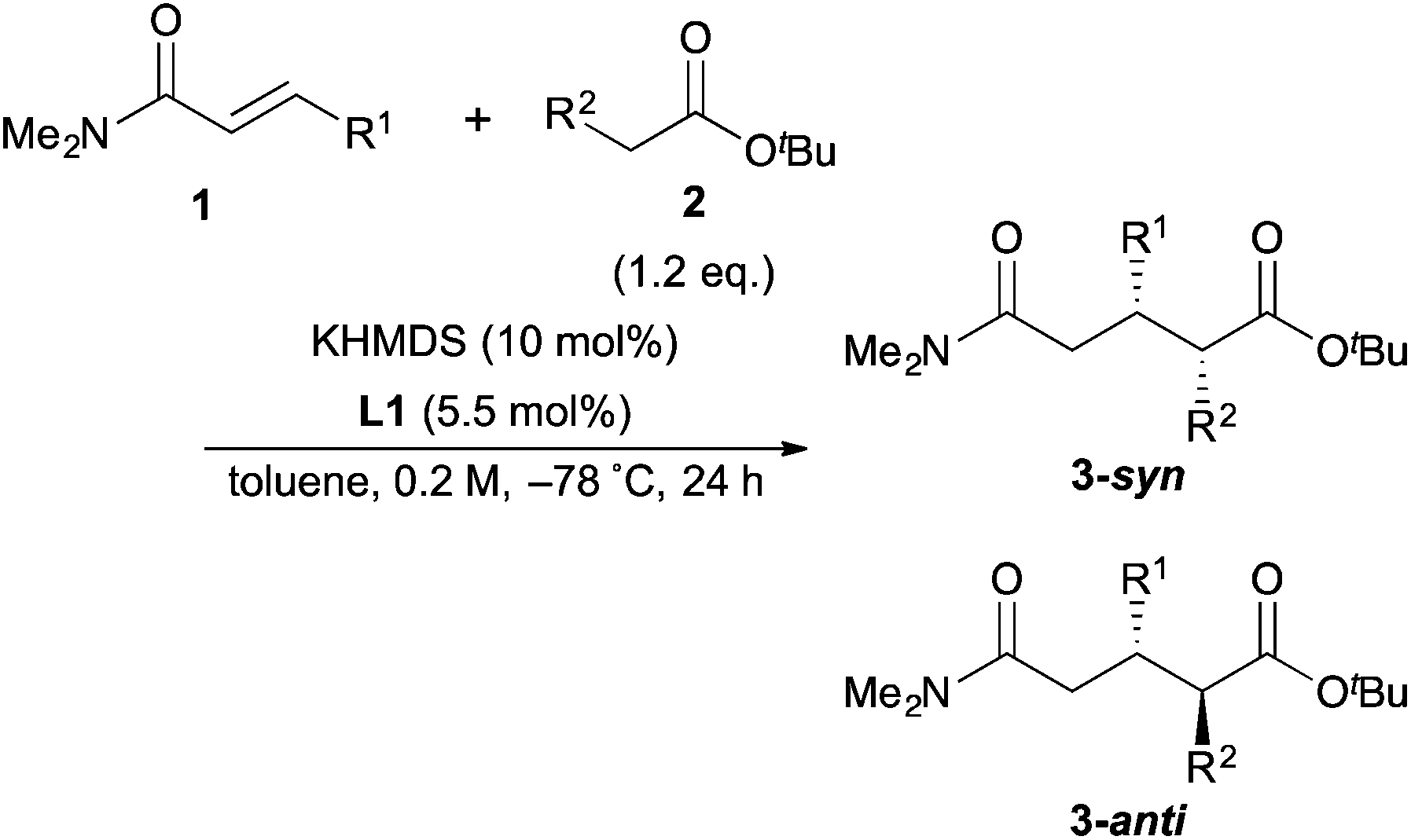
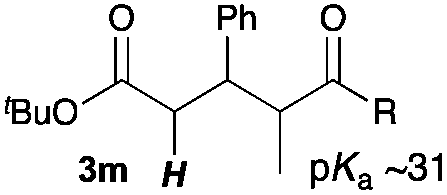
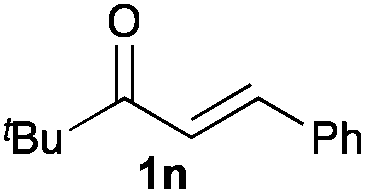
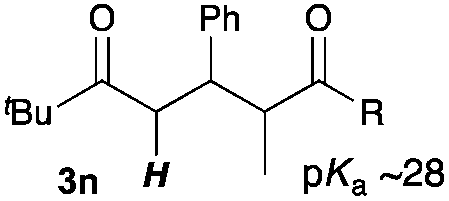
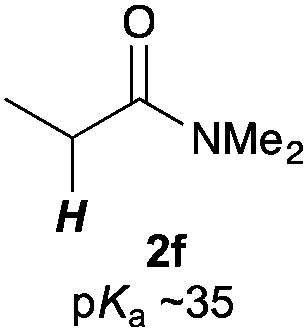
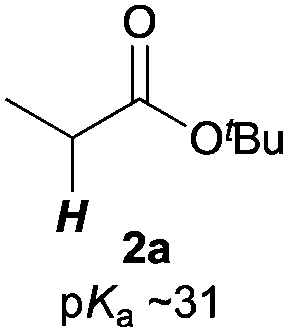
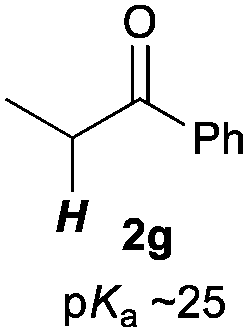
Next, in order to reveal mechanistic insight of the reactions, catalytic reactions of several pronucleophiles with electrophiles in the presence of catalytic amounts of KHMDS and 18-crown-6 were carried out (Table 4). It is noted that, in all reactions, there was almost no by-product, and consumption of the starting materials coincided with the yields of the products, which meant that the reactivity of these reactions could be argued by the yields. The reaction of the ester (2a, pKa ∼31) with the α,β-unsaturated amide (1a) gave higher yield than the reaction of the amide (2f, pKa ∼35) with 1a. The basicity of the reaction intermediate was almost the same for both reactions, because the same electrophile was used, and the reaction rate of deprotonation of the α-hydrogen atom of the ester should be faster than that of the amide because of the lower pKa value of the ester than that of the amide. From these facts, it was assumed that the deprotonation of α-hydrogen atoms was a key step, and might be the rate determining step for these reactions. On the other hand, the catalytic reaction of the ketone (2g, pKa ∼25) with the α,β-unsaturated amide (1a) did not proceed; furthermore, even in the presence of stoichiometric amounts of KHMDS and 18-crown-6, the reaction of the ketone (2g) with 1a provided only a trace amount of the product. These results indicated that nucleophilicity of the enolate of the ketone (2g) was too weak to attack the α,β-unsaturated amide (1a). Although the catalytic reactions of the ketone (2g) with the α,β-unsaturated ketone (1n) showed an excellent yield, the catalytic reactions of the esters (2a) and the amide (2f) with the α,β-unsaturated ketone (1n) did not proceed. By contrast, the stoichiometric reactions of the esters (2a) and the amide (2f) with the α,β-unsaturated ketone (1n) proceeded smoothly to afford the desired products. These results indicated that nucleophilicities of the enolates derived from the ester and the amide were strong enough to attack the α,β-unsaturated ketone (1n); however, the acidity of the product (3n, pKa ∼28) was higher than that of the ester and the amide. That is, the basicity of the reaction intermediate was too weak to deprotonate the α-hydrogen atom of the next nucleophile or the less acidic hydrogen atom of the conjugate acid of the strong base, KHMDS; thus, a catalytic reaction did not proceed in the cases of the ester and the amide as pronucleophiles. From these catalytic and stoichiometric reactions, it was proved that a suitable difference of acidity between nucleophiles and products, whose acidity depended on the structure of electrophiles, was crucial for the catalytic reaction, and when the acidity of products was appropriately higher (about 4–6 as pKa values) than that of pronucleophiles, the catalytic reaction proceeded smoothly. This consideration may help further development of the catalytic reaction of weakly acidic pronucleophiles.
|
|
||||
|---|---|---|---|---|
|
|
|
|
1,4-Adduct 3 | |
|
a The reaction of 1 (0.40 mmol) with 2 (0.48 mmol) was performed in toluene at 0.2 M at −40 °C for 18 h in the presence of the catalyst prepared from KHMDS (0.040 mmol) and 18-crown-6 (0.044 mmol) unless otherwise noted. The observed diastereoselectivities were between 1:1 and 4:1.
b The reaction was conducted at 0.1 M for 1 h in the presence of KHMDS (0.40 mmol) and 18-crown-6 (0.42 mmol).
|
||||
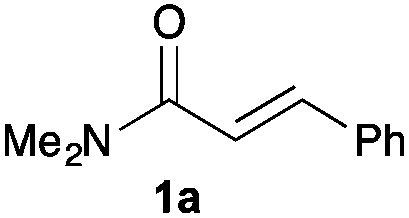
|
65 | Quant. | Trace (trace)b |
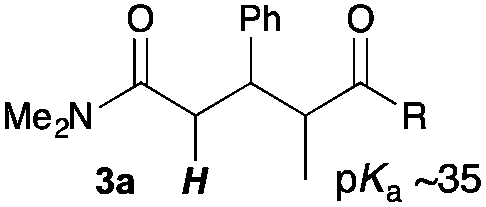
|
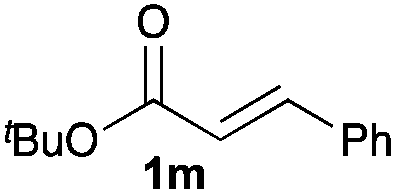
|
16 | 30 | 98 |
|
|
Trace (82)b | Trace (82)b | 98 |
|
Conclusion
In summary, we have developed catalytic asymmetric 1,4-addition reactions of simple esters without any activating functionalities on their α-positions. In the presence of catalytic amounts of KHMDS and a chiral macrocyclic crown ether, (R,R)-binaphtho-34-crown-10, 1,4-addtion reactions proceeded to afford the desired 1,4-adducts in high yields with moderate to good syn-selectivities and high enantioselectivities. Also mechanistic investigations revealed that proper combination of substrates was a key for the efficient catalytic turnover. Further improvement of the stereoselectivity of the reactions and also expansion of the strategy to other substrates are now ongoing in our laboratory.Acknowledgements
This work was partially supported by a Grant-in-Aid for Science Research from the Japan Society for the Promotion of Science (JSPS), and the Japan Science and Technology Agency (JST). I. S. and H. S. thank the MERIT program, The University of Tokyo for financial support.Notes and references
- (a) Comprehensive Organic Synthesis, ed. B. M. Trost, Pergamon Press, Oxford, 1991 Search PubMed; (b) Comprehensive Organic Synthesis, ed. P. Knochel and G. A. Molander, Elsevier Science, 2nd edn, 2014 Search PubMed.
- (a) S. Kobayashi and R. Matsubara, Chem. – Eur. J., 2009, 15, 10694 CrossRef CAS PubMed; (b) B. M. Trost, Science, 1991, 254, 1471 CAS; (c) B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259 CrossRef CAS; (d) Handbook of Green Chemistry, ed. P. T. Anastas, Wiley-VCH, Weinheim, 2009 Search PubMed.
- In DMSO; based on the Bordwell pKa table (see: http://www.chem.wisc.edu/areas/reich/pkatable/).
- For a review, see: (a) N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2011, 50, 4760 CrossRef CAS PubMed. For leading examples of this research area, see: (b) Y. M. A. Yamada, N. Yoshikawa, H. Sasai and M. Shibasaki, Angew. Chem., Int. Ed. Engl., 1997, 36, 1871 CrossRef CAS; (c) N. Yoshikawa, Y. M. A. Yamada, J. Das, H. Sasai and M. Shibasaki, J. Am. Chem. Soc., 1999, 121, 4168 CrossRef CAS; (d) B. M. Trost and H. Ito, J. Am. Chem. Soc., 2000, 122, 12003 CrossRef CAS.
- For selected examples of asymmetric reactions of esters with stoichiometric amounts of bases and chiral sources, see: (a) E. J. Corey and R. T. Peterson, Tetrahedron Lett., 1985, 26, 5025 CrossRef CAS; (b) K. Tomioka, Synthesis, 1990, 541 CrossRef CAS; (c) E. Juaristi, A. K. Beck, J. Hansen, T. Matt, T. Mukhopadhyay, M. Simson and D. Seebach, Synthesis, 1993, 1271 CrossRef CAS; (d) T. Kumamoto, S. Aoki, M. Nakajima and K. Koga, Tetrahedron: Asymmetry, 1994, 5, 1431 CrossRef CAS; (e) H. Fujieda, M. Kanai, T. Kambara, A. Iida and K. Tomioka, J. Am. Chem. Soc., 1997, 119, 2060 CrossRef CAS; (f) N. Duguet, A. Harrison-Marchand, J. Maddaluno and K. Tomioka, Org. Lett., 2006, 8, 5745 CrossRef CAS PubMed; (g) B. Lecachey, N. Duguet, H. Oulyadi, C. Fressigne, A. Harrison-Marchand, Y. Yamamoto, K. Tomioka and J. Maddaluno, Org. Lett., 2009, 11, 1907 CrossRef CAS PubMed.
- For selected examples of direct catalytic asymmetric reactions of alkylnitriles, see: (a) Y. Suto, R. Tsuji, M. Kanai and M. Shibasaki, Org. Lett., 2005, 7, 3757 CrossRef CAS PubMed; (b) Y. Kawato, N. Kumagai and M. Shibasaki, Chem. Commun., 2013, 49, 11227 RSC; (c) D. Sureshkumar, V. Ganesh, N. Kumagai and M. Shibasaki, Chem. – Eur. J., 2014, 20, 15723 CrossRef CAS PubMed; (d) T. Deng, H. Wang and C. Cai, Eur. J. Org. Chem., 2014, 7259 CrossRef CAS. For review on this area, see: (e) R. López and C. Palomo, Angew. Chem., Int. Ed., 2015, 54, 13170 CrossRef PubMed.
- S. Kobayashi, H. Kiyohara and M. Yamaguchi, J. Am. Chem. Soc., 2011, 133, 708 CrossRef CAS PubMed.
- (a) Y. Morita, T. Yamamoto, H. Nagai, Y. Shimizu and M. Kanai, J. Am. Chem. Soc., 2015, 137, 7075 CrossRef CAS PubMed; (b) H. Nagai, Y. Morita, Y. Shimizu and M. Kanai, Org. Lett., 2016, 18, 2276 CrossRef CAS PubMed.
- (a) I. Ojima, I. Habus, M. Zhao, M. Zucco, Y. H. Park, C. M. Sun and T. Brigaud, Tetrahedron, 1992, 48, 6985 CrossRef CAS; (b) S. F. Martin, K. J. Barr, D. W. Smith and S. K. Bur, J. Am. Chem. Soc., 1999, 121, 6990 CrossRef CAS; (c) K. Takasu, S. Mizutani, M. Noguchi, K. Makita and M. Ihara, J. Org. Chem., 2000, 65, 4112 CrossRef CAS PubMed.
- (a) T. Mukaiyama, K. Banno and K. Narasaka, J. Am. Chem. Soc., 1974, 96, 7503 CrossRef CAS; (b) S. Kobayashi, H. Uchiro, Y. Fujishima, I. Shiina and T. Mukaiyama, J. Am. Chem. Soc., 1991, 113, 4247 CrossRef CAS. For reviews, see: (c) J. Matsuo and M. Murakami, Angew. Chem., Int. Ed., 2013, 52, 9109 CrossRef CAS PubMed; (d) J. Gawronski, N. Wascinska and J. Gajewy, Chem. Rev., 2008, 108, 5227 CrossRef CAS PubMed.
- H. Morimoto, S. H. Wiedemann, A. Yamaguchi, S. Harada, Z. Chen, S. Matsunaga and M. Shibasaki, Angew. Chem., Int. Ed., 2006, 45, 3146 CrossRef CAS PubMed.
- (a) M. Iwata, R. Yazaki, Y. Suzuki, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 18244 CrossRef CAS PubMed; (b) Y. Suzuki, R. Yazaki, N. Kumagai and M. Shibasaki, Chem. – Eur. J., 2011, 17, 11998 CrossRef CAS PubMed; (c) N. Majumdar, A. Saito, L. Yin, N. Kumagai and M. Shibasaki, Org. Lett., 2015, 17, 3362 CrossRef CAS PubMed.
- (a) V. H. Nguyen, R. Matsubara and S. Kobayashi, Angew. Chem., Int. Ed., 2009, 48, 5927 CrossRef PubMed; (b) R. Matsubara, F. Berthiol, V. H. Nguyen and S. Kobayashi, Bull. Chem. Soc. Jpn., 2009, 82, 1083 CrossRef CAS.
- (a) Y. Yamashita, H. Suzuki and S. Kobayashi, Org. Biomol. Chem., 2012, 10, 5750 RSC; (b) H. Suzuki, I. Sato, Y. Yamashita and S. Kobayashi, J. Am. Chem. Soc., 2015, 137, 4336 CrossRef CAS PubMed; (c) Y. Yamashita, I. Sato, H. Suzuki and S. Kobayashi, Chem. – Asian J., 2015, 10, 2143 CrossRef CAS PubMed.
- A preliminary result of a catalytic asymmetric 1,4-addition reaction of a simple ester has been shown in ref. 14b.
Footnotes |
| † Dedicated to Professor Barry M. Trost on the occasion of his 75th birthday. |
| ‡ Electronic supplementary information (ESI) available: General information of the experiments, experimental procedures of the catalytic reactions, and physical data of the products. See DOI: 10.1039/c6qo00242k |
| This journal is © the Partner Organisations 2016 |