
Diastereo- and enantioselective construction of cyclohexanone-fused spirospyrazolones containing four consecutive stereocenters through asymmetric sequential reactions†
Jun-Hua
Li
,
Zhi-Hao
Cui
and
Da-Ming
Du
*
School of Chemical Engineering and Environment, Beijing Institute of Technology, Beijing 100081, People's Republic of China. E-mail: dudm@bit.edu.cn; Tel: +86 10 68914985
First published on 29th June 2016
Abstract
The diastereo- and enantioselective synthesis of cyclohexanone-fused spirospyrazolones containing four consecutive chiral centers has been successfully developed through an asymmetric Michael/Michael/aldol cascade reaction catalyzed by the combination of a bifunctional squaramide and diphenylprolinol silyl ether, followed by a sequential oxidation with pyridinium chlorochromate. This protocol affords cyclohexanone-fused spirospyrazolones in moderate to high yields, moderate to good diastereoselectivities and perfect enantioselectivities.
Pyrazolone scaffolds which are known for more than one century1 have been proven to be a kind of important structural units. For example, pyrazolone phenazone2 is the first synthetic antipyretic and analgesic drug. And metamizole is considered to be the strongest antipyretic.3 Pyrazolone edaravone is a neuroprotective agent.4 Meanwhile, the pyrazolone derivatives could also be used as HIV integrase inhibitors,5 and antibacterial agents.6 Owing to excellent biological properties, the synthesis of new, potentially bioactive enantiopure pyrazolone derivatives has attracted considerable research attention from synthetic chemists.7 In particular, the synthesis of cyclohexane-fused spiropyrazolones or cyclohexene-fused spiropyrazolones was one of the most attractive topics for researchers, and different methods have been explored.8 At the same time, Wang9 and Wang10 groups reported the synthesis of cyclohexanone-fused spiropyrazolones with three consecutive stereocenters through a double Michael reaction of α,β-unsaturated ketones to α,β-unsaturated pyrazolones respectively. In 2015, Biju and coworkers reported the enantioselective synthesis of the pyrazolone-fused spirocyclohexadienones with one stereocenter by the reaction of α,β-unsaturated aldehydes with α-arylidene pyrazolinones under oxidative N-heterocyclic carbene (NHC) catalysis.11 Considering the potential biological activities of the cyclohexanone-fused spiropyrazolones, we were fascinated by synthesis of cyclohexanone-fused spiropyrazolones with more stereocenters. Recently, the application of relay catalysis has been widely used in organic synthesis, which offers promise to incorporate more substrates and reaction types to design novel cascade reactions, which are very attractive for building up molecular complexity from simple materials.12 In addition, the combination of a hydrogen-bond donating catalyst with an aminocatalyst has been widely employed in one-pot reactions between substrates with different functional groups, when the multi-component reaction could not be catalyzed by only a hydrogen-bond donating catalyst or an aminocatalyst.13 Many incredible reactions containing specific groups could be catalyzed smoothly by sequential addition of reagents and catalysts throughout the course of the reaction. In these cases, the hydrogen-bond donating catalyst can catalyze the first step of the multi-component reaction to obtain the intermediates, and the intermediates could be catalyzed by the aminocatalyst to react with aldehyde compounds to complete the reactions, which was not feasible for a single catalyst system.
Herein, we will present diastereo- and enantioselective synthesis of cyclohexanone-fused spirospyrazolones containing four consecutive stereocenters through an asymmetric relay catalysis of a Michael/Michael/aldol cascade reaction catalyzed by the combination of the bifunctional squaramide and diphenylprolinol silyl ether, followed by a sequential oxidation with pyridinium chlorochromate. This protocol affords the highly functionalized cyclohexanone-fused spiropyrazolones in moderate to high yields (30–78% yield), moderate to good diastereoselectivities (60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40–25:1 dr) and perfect enantioselectivities (up to >99% ee).
:40–25:1 dr) and perfect enantioselectivities (up to >99% ee).
According to our previous report,14 we initiated our investigation by using 1,2-diaminocyclohexane-derived squaramide I (1 mol%) (Fig. 1) to catalyze the Michael addition of 3-methyl-1-phenyl-2-pyrazolin-5-one (1a) to (E)-β-nitrostyrene (2a) in chloroform. After 2 h, cinnamaldehyde (3a) and pyrrolidine II (20 mol%) were added sequentially. When the reaction was complete, the intermediate product was obtained by silica gel column chromatography, followed by oxidation using pyridinium chlorochromate in dichloromethane. This sequence was conducted as a three-step asymmetric relay catalysis procedure and the desired product 4a was obtained in moderate yield and diastereoselectivity with good enantioselectivity (Table 1, entry 1). Because the polarity of the intermediate product E was close to the raw material, only the crude intermediate product was obtained. Additionally, a one-pot three-step reaction was also tried, but the yield of the product in the oxidation step was too low, except in the solvent of dichloromethane. In order to increase the yield and the stereoselectivity of the final product, appropriate organocatalysts which can coordinate and activate the cinnamaldehyde in the second step were also screened (Table 1, entries 2–3). As can be seen from the results, when catalyst IV was used, both yield and enantioselectivity improved (Table 1, entry 3). To further increase the yield and diastereoselectivity of this reaction, the solvents and additives were also evaluated. The solvent evaluation (Table 1, entries 4–7) revealed that THF was an optimal solvent for this reaction, and the product was obtained in good yield (59%) and good diastereoselectivity (76:24) with excellent enantioselectivity (>99%). Further efforts for screening of additives such as PhCO2H, AcOH, and NaOAc did not result in any improvement in the product yield and diastereoselectivity (Table 1, entries 8–10).
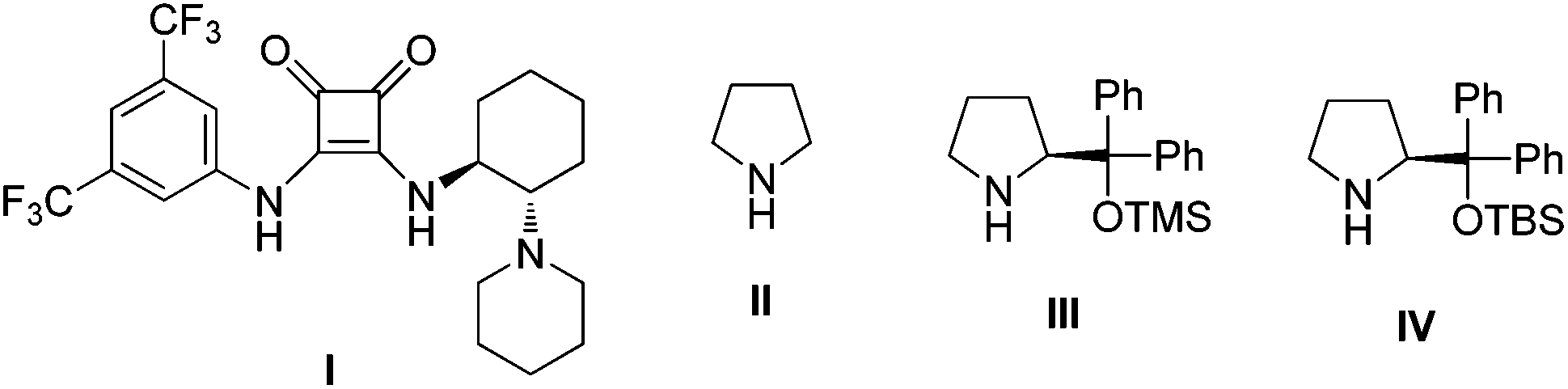 | ||
| Fig. 1 The structures of screened organocatalysts. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Yieldb (%) | drc | eec (%) |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.24 mmol) and catalyst I (1 mol%) in solvent (1 mL) was reacted for 2 h, then 3a (0.3 mol) and catalyst II–IV (20 mol%) was added and reacted for further 46 h. Then the intermediate product was isolated and oxidized with PCC (0.4 mmol) and silica gel (86.2 mg) in CH2Cl2 (3 ml) for 36 h at r.t. b Isolated yield after purification by silica gel column chromatography. c Determined by HPLC analysis. d PhCO2H (0.04 mmol) was added. e AcOH (0.04 mmol) was added. f NaOAc (0.02 mmol) was added. | |||||
| 1 | II | CHCl3 | 30 | 70:30 |
91 |
| 2 | III | CHCl3 | 44 | 50:50 |
>99 |
| 3 | IV | CHCl3 | 54 | 45:55 |
>99 |
| 4 | IV | CH2Cl2 | 55 | 66:34 |
>99 |
| 5 | IV | ClCH2CH2Cl | 57 | 58:42 |
>99 |
| 6 | IV | Toluene | 64 | 63:37 |
99 |
| 7 | IV | THF | 59 | 76:24 |
>99 |
| 8d | IV | THF | 59 | 74:26 |
>99 |
| 9e | IV | THF | 55 | 74:26 |
99 |
| 10f | IV | THF | 55 | 75:25 |
99 |
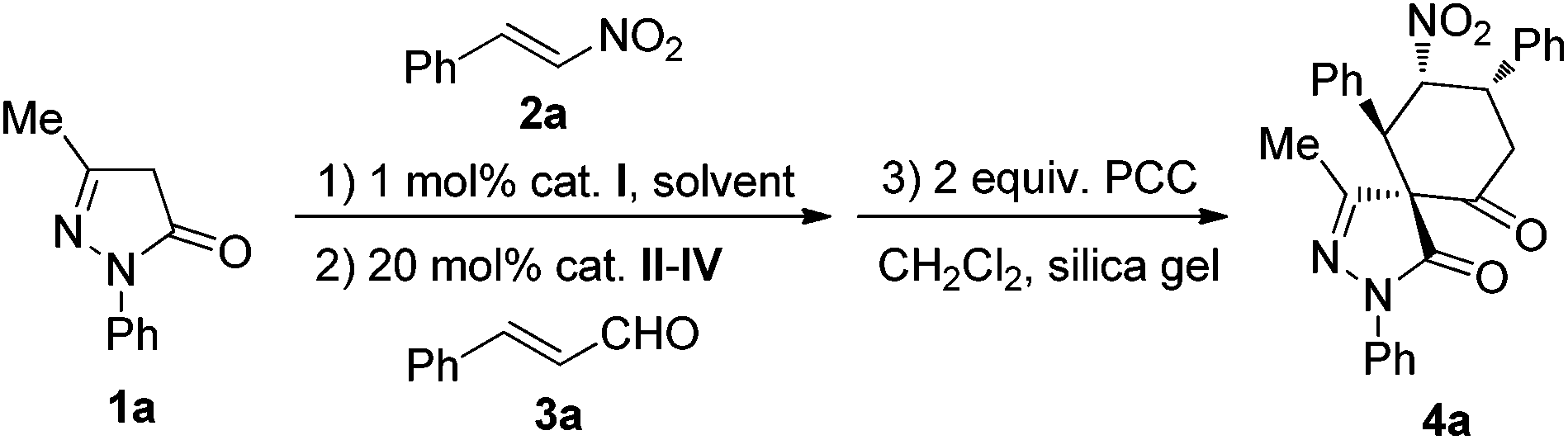
With the optimal reaction conditions established, a diverse array of substituted substrates was evaluated, and the results are summarized in Table 2. In general, most of the reactions proceeded well to afford the desired products in good yields, perfect enantioselectivities and moderate to good diastereoselectivities.
| a Reaction conditions: 1 (0.2 mmol), 2 (0.24 mmol) and catalyst I (1 mol%) in solvent (1 mL) was reacted for 2 h, then 3 (0.3 mmol) and catalyst IV (20 mol%) was added and reacted for further 46 h. Then the intermediate product was isolated and oxidized with PCC (0.4 mmol) and silica gel (86.2 mg) in CH2Cl2 (3 ml) for 36 h at r.t. b Isolated yield after purification by silica gel column chromatography. c Determined by NMR analysis. d Determined by HPLC analysis. |
|---|
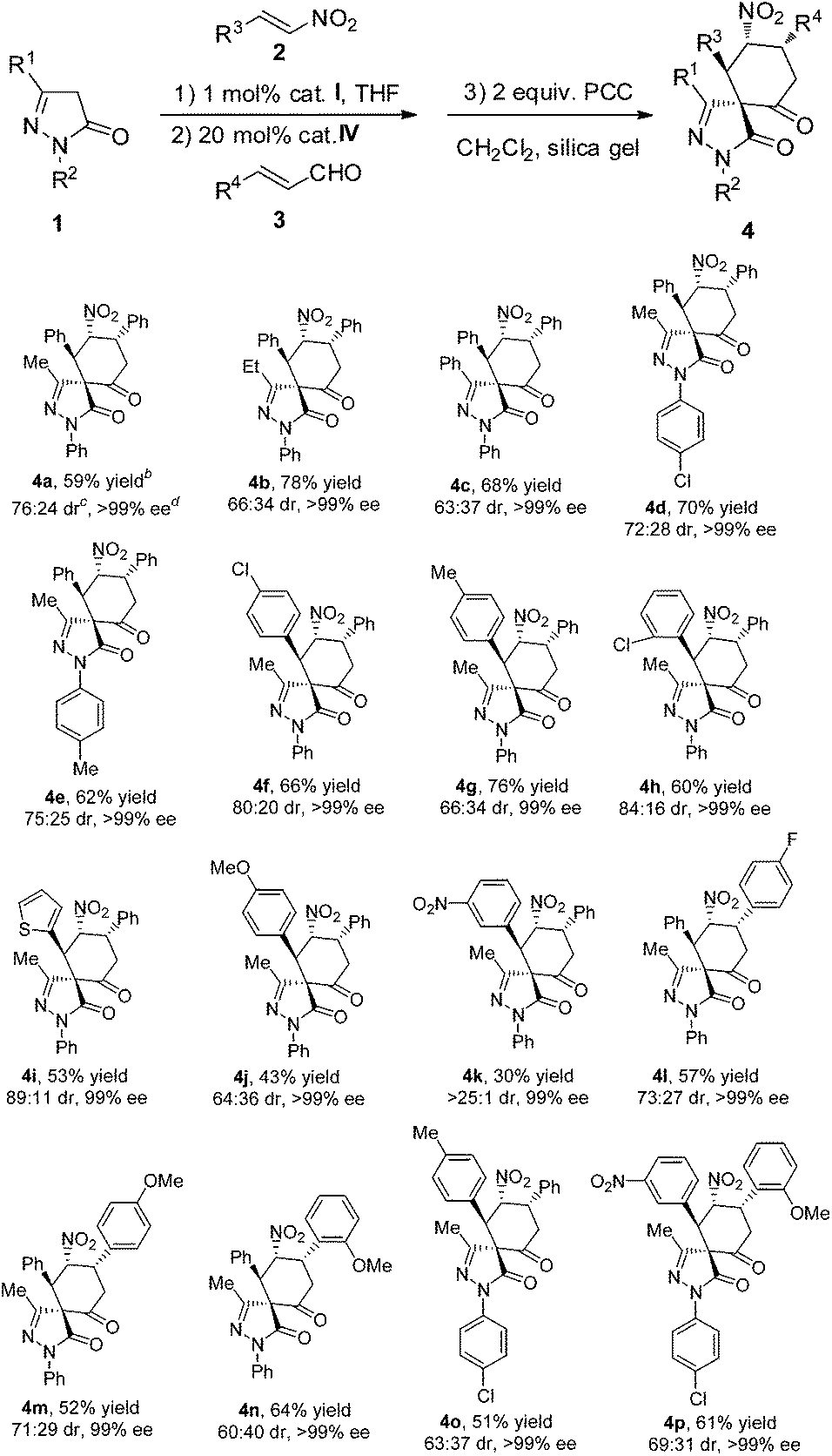
|
First, the R1 and R2 groups of the pyrazolone were evaluated, and all of the desired spiropyrazolones (4b−4e) were efficiently obtained in good yields, moderate to good diastereoselectivities and excellent enantioselectivities. Then different aromatic nitroalkenes bearing electron-withdrawing as well as electron-donating substituents were also explored. Generally, all of the products (4f−4h, 4j and 4k) were obtained in perfect enantioselectivities (≧99% ee). When the electron-withdrawing or electron-donating ability of the substituents was moderate, good yields were obtained (4f−4h). Otherwise, if the electron-withdrawing or electron-donating ability of the substituents was too strong, the yields of the product became lower (4j and 4k). Specifically, when nitroalkene was substituted by the nitro group on the meta-position, excellent diastereoselectivity but lower yield was obtained (4k), which demonstrates that the strong electron-withdrawing group influences the reaction, and only one diastereomer was formed. The heteroaromatic nitroalkene was also tested, a moderate yield (53%) with good diastereoselectivity (89:11 dr) and perfect enantioselectivity (99% ee) was obtained for 4i. Next, different cinnamaldehydes were explored, and similar results were obtained for the corresponding products (4j−4n). To evaluate the synthetic potential of this asymmetric relay catalysis system, substituents variations on different substrates at the same time were also evaluated, and excellent enantioselectivities were also obtained (4o and 4p).
The absolute configuration of 4p was determined to be 5R,8S,9R,10R by X-ray crystallographic analysis (Fig. 2).15 The absolute configurations of other products were assigned by analogy.
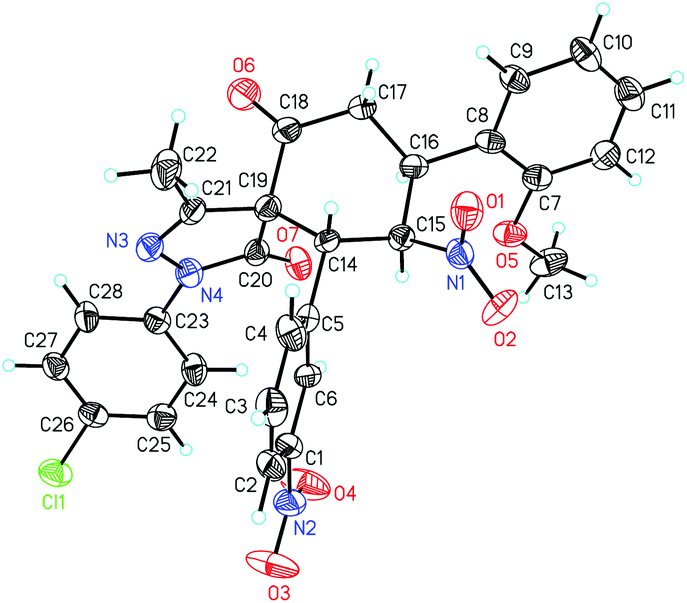 | ||
| Fig. 2 The crystal structure of compound 4p. | ||
On the basis of the absolute configuration of 4p and the previous theoretical study on the mechanism of similar reactions,13c,14 a plausible mechanism was proposed, as shown in Scheme 1. Firstly, the pyrazolone 1a is deprotonated by the basic nitrogen atom of the tertiary amine via tautomerization. Meanwhile the nitroalkene is activated by the squaramide moiety through double hydrogen bonding between the NH groups and the nitro group to form the transition state A. The deprotonated pyrazolone attacks the activated nitroalkene to afford the Michael adducts. Then, the coordination of the squaramide catalyst to the nitro group allows the tertiary amine to deprotonate the α-proton to generate the nitronate (B), followed by an iminium-catalyzed nitro-Michael addition (C) to form the intermediate product D. Next, an intramolecular aldol reaction is carried out to afford the cyclohexanol-fused spirospyrazolone E. Finally, the oxidation reaction with pyridinium chlorochromate occurred to provide the desired cyclohexanone-fused spirospyrazolone 4a.
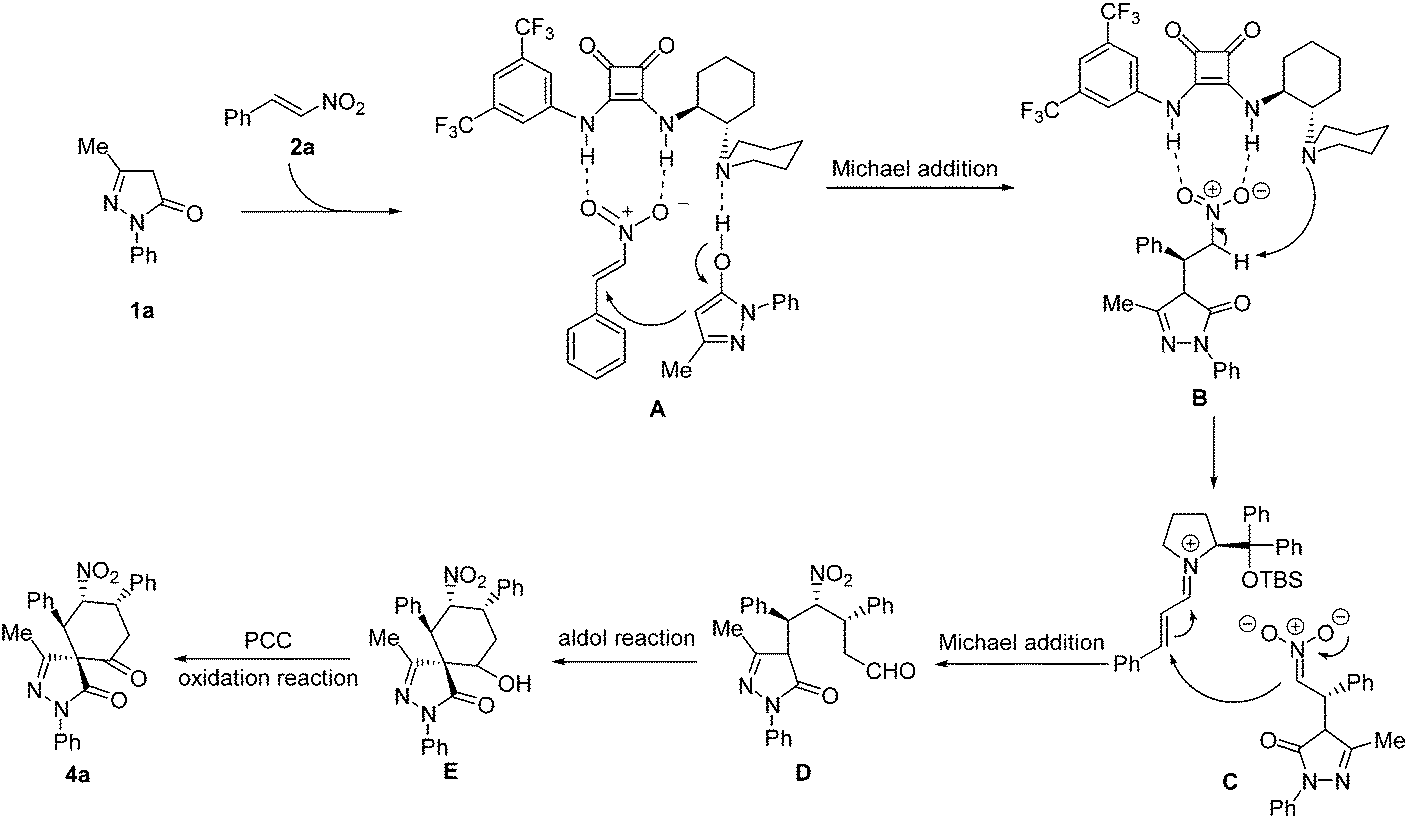 | ||
| Scheme 1 Plausible reaction mechanism. | ||
In conclusion, we have developed an efficient cascade reaction for the construction of highly functionalized cyclohexanone-fused spirospyrazolones containing four consecutive stereocenters, including one quaternary spirocarbon center, through an asymmetric Michael/Michael/aldol cascade reaction catalyzed by the combination of a bifunctional squaramide and diphenylprolinol silyl ether and a sequential oxidation with pyridinium chlorochromate. This protocol affords the corresponding spiropyrazolones in moderate to high yields (30–78% yield) with moderate to good diastereoselectivities (60:40–25:1 dr) and perfect enantioselectivities (up to >99% ee). We anticipate that this cascade transformation sequence will be valuable for catalyst development, structural diversity of spiropyrazolones and in the identification of new medicinal agents. Further studies on the organocatalytic enantioselective synthesis of potential bioactive heterocycles are ongoing in our laboratory.
Notes and references
- For selected reviews, see: (a) D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893 CrossRef CAS PubMed; (b) W. S. Hamama, H. G. El-Gohary, N. Kuhnert and H. H. Zoorob, Curr. Org. Chem., 2012, 16, 373 CrossRef CAS.
- (a) M. L. Tainter, Ann. N. Y. Acad. Sci., 1948, 51, 3 CrossRef CAS PubMed; (b) K. Brune, Acute Pain, 1997, 1, 33 CrossRef CAS.
- A. Brayfield, Martindale: The Complete Drug Reference, Pharmaceutical Press, 2014 Search PubMed.
- (a) H. Yoshida, H. Yanai, Y. Namiki, K. Fukatsu-Sasaki, N. Furutani and N. Tada, CNS Drug Rev., 2006, 12, 9 CrossRef CAS PubMed; (b) W. Ji Yuan, T. Yasuhara, T. Shingo, K. Muraoka, T. Agari, M. Kameda, T. Uozumi, N. Tajiri, T. Morimoto, M. Jing, T. Baba, F. Wang, H. Leung, T. Matsui, Y. Miyoshi and I. Date, BMC Neurosci., 2008, 9, 75 CrossRef PubMed.
- V. Hadi, Y.-H. Koh, T. W. Sanchez, D. Barrios, N. Neamati and K. W. Jung, Bioorg. Med. Chem. Lett., 2010, 20, 6854 CrossRef CAS PubMed.
- M. S. Chande, P. A. Barve and V. Suryanarayan, J. Heterocycl. Chem., 2007, 44, 49 CrossRef CAS.
- For review see: (a) P. Chauhan, S. Mahajan and D. Enders, Chem. Commun., 2015, 51, 12890 RSC. For selected examples see: (b) X. Bao, B. Wang, L. Cui, G. Zhu, Y. He, J. Qu and Y. Song, Org. Lett., 2015, 17, 5168 CrossRef CAS PubMed; (c) D. Hack, A. B. Dürr, K. Deckers, P. Chauhan, N. Seling, L. Rübenach, L. Mertens, G. Raabe, F. Schoenebeck and D. Enders, Angew. Chem., Int. Ed., 2016, 55, 1797 CrossRef CAS PubMed; (d) D. Hack, P. Chauhan, K. Deckers, Y. Mizutani, G. Raabea and D. Enders, Chem. Commun., 2015, 51, 2266 RSC; (e) W. Zheng, J. Zhang, S. Liu, C. Yua and Z. Miao, RSC Adv., 2015, 5, 91108 RSC; (f) S. Wang, C. Rodriguez-Escrich and M. A. Pericàs, Org. Lett., 2016, 18, 556 CrossRef CAS PubMed; (g) N. Kumarswamyreddy and V. Kesavan, Org. Lett., 2016, 18, 1354 CrossRef CAS PubMed; (h) J.-H. Li, T.-F. Feng and D.-M. Du, J. Org. Chem., 2015, 80, 11369 CrossRef CAS PubMed; (i) J.-H. Li and D.-M. Du, Chem. – Asian J., 2014, 9, 3278 CrossRef CAS PubMed.
- (a) X. Companyó, A. Zea, A.-N. R. Alba, A. Mazzanti, A. Moyano and R. Rios, Chem. Commun., 2010, 46, 6953 RSC; (b) A.-N. R. Alba, A. Zea, G. Valero, T. Calbet, M. Font-Bardía, A. Mazzanti, A. Moyano and R. Rios, Eur. J. Org. Chem., 2011, 1318 CrossRef CAS; (c) A. Zea, A.-N. R. Alba, A. Mazzanti, A. Moyano and R. Rios, Org. Biomol. Chem., 2011, 9, 6519 RSC; (d) P. Sun, C.-Y. Meng, F. Zhou, X.-S. Li and J.-W. Xie, Tetrahedron, 2014, 70, 9330 CrossRef CAS; (e) P. Chauhan, S. Mahajan, C. C. J. Loh, G. Raabe and D. Enders, Org. Lett., 2014, 16, 2954 CrossRef CAS PubMed; (f) B. Han, W. Huang, W. Ren, G. He, J.-H. Wang and C. Peng, Adv. Synth. Catal., 2015, 357, 561 CrossRef CAS.
- J. Liang, Q. Chen, L. Liu, X. Jiang and R. Wang, Org. Biomol. Chem., 2013, 11, 1441 CAS.
- (a) J.-X. Zhang, N.-K. Li, Z.-M. Liu, X.-F. Huang, Z.-C. Geng and X.-W. Wang, Adv. Synth. Catal., 2013, 355, 797 CrossRef CAS; (b) B. Wu, J. Chen, M.-Q. Li, J.-X. Zhang, X.-P. Xu, S.-J. Ji and X.-W. Wang, Eur. J. Org. Chem., 2012, 1318 CrossRef CAS.
- S. R. Yetra, S. Mondal, S. Mukherjee, R. G. Gonnade and A. T. Biju, Angew. Chem., Int. Ed., 2016, 55, 268 CrossRef CAS PubMed.
- For selected reviews, see: (a) J. Zhou, Multicatalyst system in Asymmetric Catalysis, John Wiley & Sons, New York, 2014 Search PubMed; (b) J.-C. Wasilke, S. J. Obrey, R. T. Baker and G. C. Bazan, Chem. Rev., 2005, 105, 1001 CrossRef CAS PubMed; (c) J. Zhou, Chem. – Asian J., 2010, 5, 422 CrossRef CAS PubMed; (d) N. T. Patil, V. S. Shinde and B. Gajula, Org. Biomol. Chem., 2012, 10, 211 RSC; (e) H. Pellissier, Tetrahedron, 2013, 69, 7171 CrossRef CAS. For selected examples, see: (f) X.-P. Yin, X.-P. Zeng, Y.-L. Liu, F.-M. Liao, J.-S. Yu, F. Zhou and J. Zhou, Angew. Chem., Int. Ed., 2014, 53, 13740 CrossRef CAS PubMed; (g) Y. Wang, D.-F. Yu, Y.-Z. Liu, H. Wei, Y.-C. Luo, D. J. Dixon and P.-F. Xu, Chem. – Eur. J., 2010, 16, 3922 CrossRef CAS PubMed; (h) Z. Mao, Y. Jia, Z. Xu and R. Wang, Adv. Synth. Catal., 2012, 354, 1401 CrossRef CAS; (i) Y.-L. Zhao, Z.-Y. Cao, X.-P. Zeng, J.-M. Shi, Y.-H. Yu and J. Zhou, Chem. Commun., 2016, 52, 3943 RSC; (j) Z.-Y. Cao, Y.-L. Zhao and J. Zhou, Chem. Commun., 2016, 52, 2537 RSC.
- For selected examples, see: (a) Y. Wang, R.-G. Han, Y.-L. Zhao, S. Yang, P.-F. Xu and D. J. Dixon, Angew. Chem., Int. Ed., 2009, 48, 9834 CrossRef CAS PubMed; (b) S. Lin, L. Deiana, G.-L. Zhao, J. Sun and A. Córdova, Angew. Chem., Int. Ed., 2011, 50, 7624 CrossRef CAS PubMed; (c) B. Zhou, Y. Yang, J. Shi, Z. Luo and Y. Li, J. Org. Chem., 2013, 78, 2897 CrossRef CAS PubMed.
- J.-H. Li and D.-M. Du, Org. Biomol. Chem., 2013, 11, 6215 CAS.
- Cystallographic data for compound 4p (CCDC 1470552) has been provided as CIF file in the ESI.†.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra of new compounds, and HPLC chromatograms. CCDC 1470552. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00208k |
| This journal is © the Partner Organisations 2016 |