
Asymmetric transfer hydrogenation of seven membered tricyclic ketones: N-substituted dibenzo[b,e]azepine-6,11-dione driven by nonclassical CH/O interactions†
Vijyesh K.
Vyas
and
Bhalchandra M.
Bhanage
*
Department of Chemistry, Institute of Chemical Technology, N.P. Marg, Matunga- 400019, Mumbai, India. E-mail: bm.bhanage@gmail.com; bm.bhanage@ictmumbai.edu.in; Fax: +91-2233611020; Tel: +91-2233612601
First published on 9th March 2016
Abstract
Enantioselective transfer hydrogenation of dibenzo-fused-azepine-diones: N-substituted dibenzo[b,e]azepin-6-11-dione, was achieved by ruthenium catalysis in the presence of formic acid/triethylamine as a mild hydrogen source. Various derivatives of N-substituted dibenzo[b,e]azepin-6-11-dione were hydrogenated to afford a pharmaceutically important chiral skeleton in excellent yields (80 to >99%) and enantioselectivities (43 to >99%). A theoretical study of the stereodetermining transition state revealed that a dibenzo[b,e]azepine-6,11-dione skeleton has a marked effect of CH/π interactions on enantioselection, instead there is evidence of nonclassical CH/O interactions being responsible for the stereochemical outcome.
Introduction
The importance of the amide linkage in biologically active compounds has emerged with the discovery of penicillin, and in the recent investigation of a new cell wall inhibitor, teixobactin.1 The amide linkage has attracted many research groups since it forms the backbone of all naturally occurring proteins. This linkage covers one fourth of all pharmaceutical drugs.2Much attention has been given to tricyclic structures containing an amide linkage, for example, dihydro-dibenzo[b,e]azepin-6-one due to its effect on the central nervous system.3,5a This skeleton also acts as a precursor for the synthesis of various receptor antagonists which were tested for various physiological activities (Fig. 1, compounds 1 and 2).4 In particular, N-substituted-11-hydroxyl-5H-dibenzo[b,e]azepin-6(11H)-one derivatives show some significant applications in pharmaceuticals. Among these compounds, some have shown activity as anti-convulsants5a and some possess antiproliferative activity5b (Fig. 1, compounds 3 and 4). These compounds have immense importance in pharmaceuticals with the non-chiral version and many groups have tested their physiological activity by varying N-substitution and 11-substitution to elucidate the structure activity relationship (SAR).4c,5 Further, its enantiomeric pure version is required to test its biological activity.
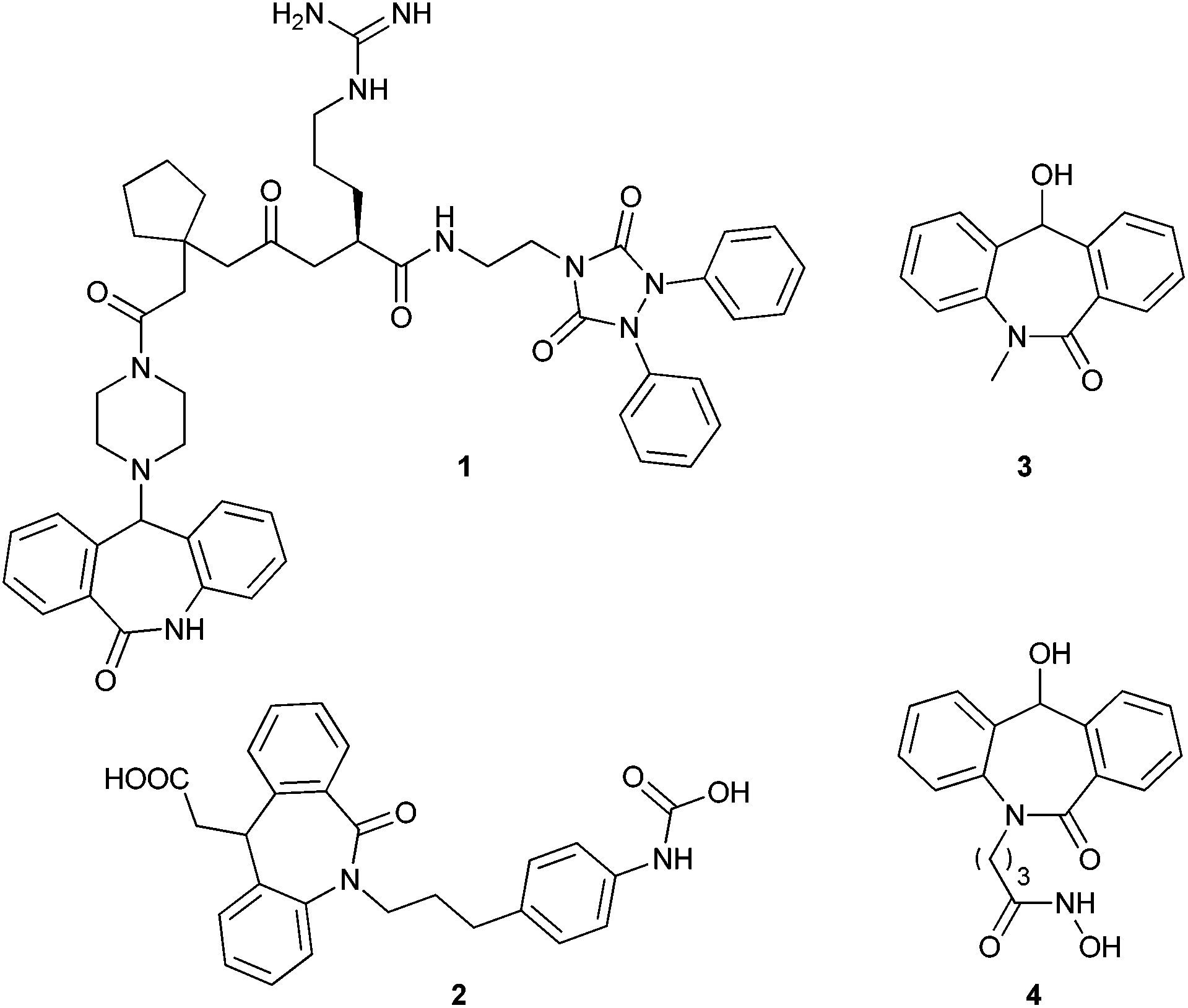 | ||
| Fig. 1 Compounds containing dibenzo[b,e]azepine skeleton. | ||
To the best of our knowledge, there is no report on the catalytic asymmetric transfer hydrogenation reactions of tricyclic seven membered dissymmetric diaryl ketone derivatives. This is the first time we have shown the metal catalyzed asymmetric synthesis of various novel N-substituted 11-hydroxyl-5H-dibenzo[b,e]azepin-6(11H)-ones as a pharmaceutically important enantioenriched skeleton.
Over the last two decades, complexes of monotosylated 1,2-diamine ligands with the Ru(II) metal, introduced by Noyori and co-workers, have been well-explored for asymmetric hydrogenation and asymmetric transfer hydrogenation (ATH) of imines and ketones. In particular, RuCl(arene)(Ts-DPEN) complexes are extensively studied and have proven to affect asymmetric transfer hydrogenation successfully.6 Further, ATH involving formic acid/triethylamine (FA/TEA), isopropyl alcohol, sodium formate and formic acid as a hydrogen source have been well documented for ketones and imines.6 Despite these achievements in the field of asymmetric transfer hydrogenation, their application for the reduction of cyclic dissymmetric diaryl ketones which are part of dibenzo[b,e]azepine-6,11-diones is still a challenge. In addition, although, these Ru-complexes have been proven for the reduction of functionalized ketones, we have tested the effect of remote functional groups present in the substrate for the hydride transfer transition state. Herein, we report a ruthenium catalyzed asymmetric transfer hydrogenation of seven membered dibenzo[b,e]azepine-6,11-diones with high yields (80 to >99%) and enantioselectivities (43 to >99%) leading to the synthesis of optically active and pharmaceutically important skeletons.
Result & discussion
N-Substituted seven membered cyclic ketones were synthesized by reported procedures in the literature with slight modifications as shown in Scheme 1. Anthraquinone 5 was treated with sodium azide in H2SO4 to afford compound 6via the Schmidt rearrangement with the amide linkage in the structure. Subsequently, 6 was alkylated with various functionalized alkyl bromides to obtain hydrogenation substrates in moderate to good yields (see the ESI†).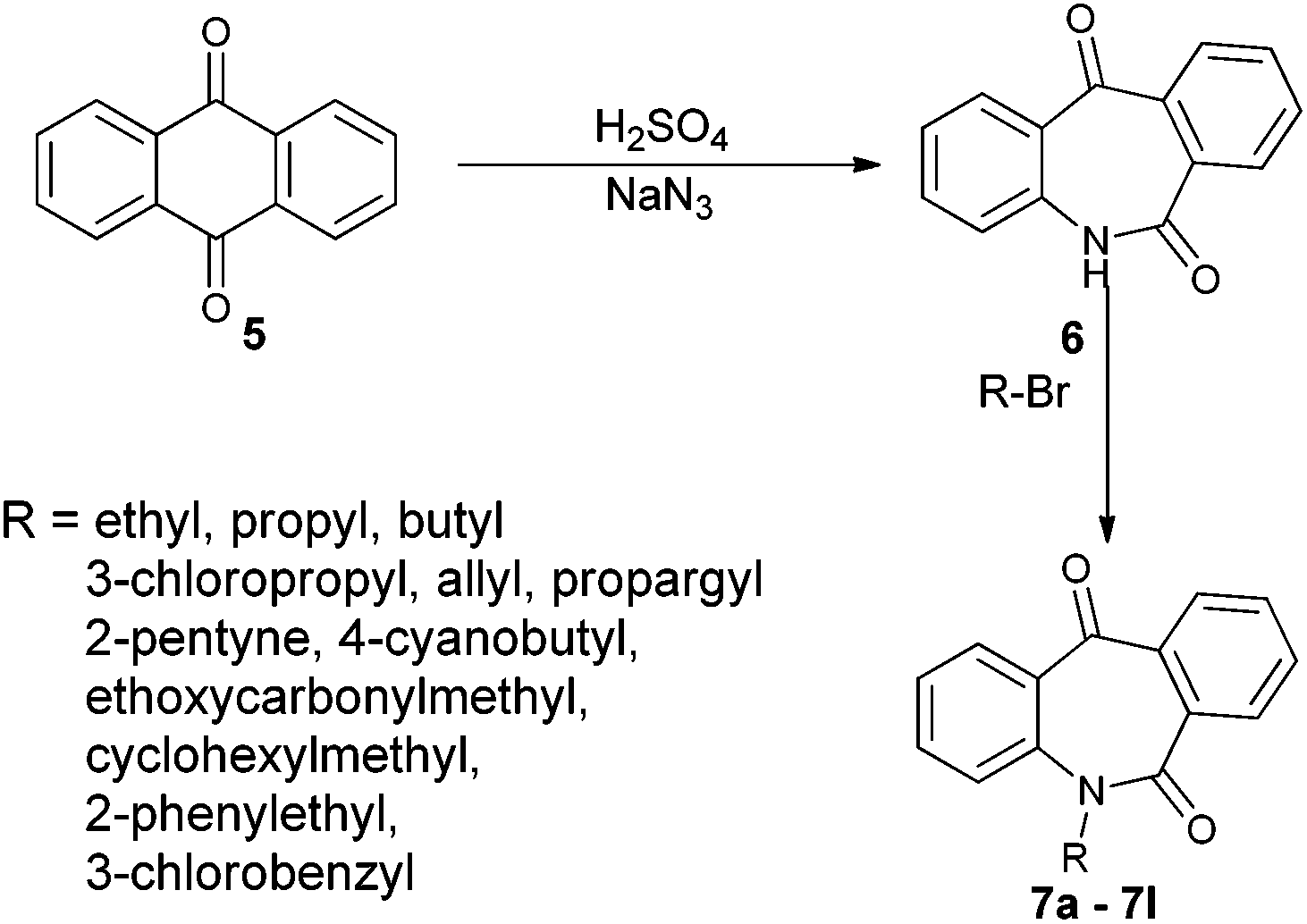 | ||
| Scheme 1 Synthesis of N-substituted dibenzo[b,e]azepine-6,11-diones 7. | ||
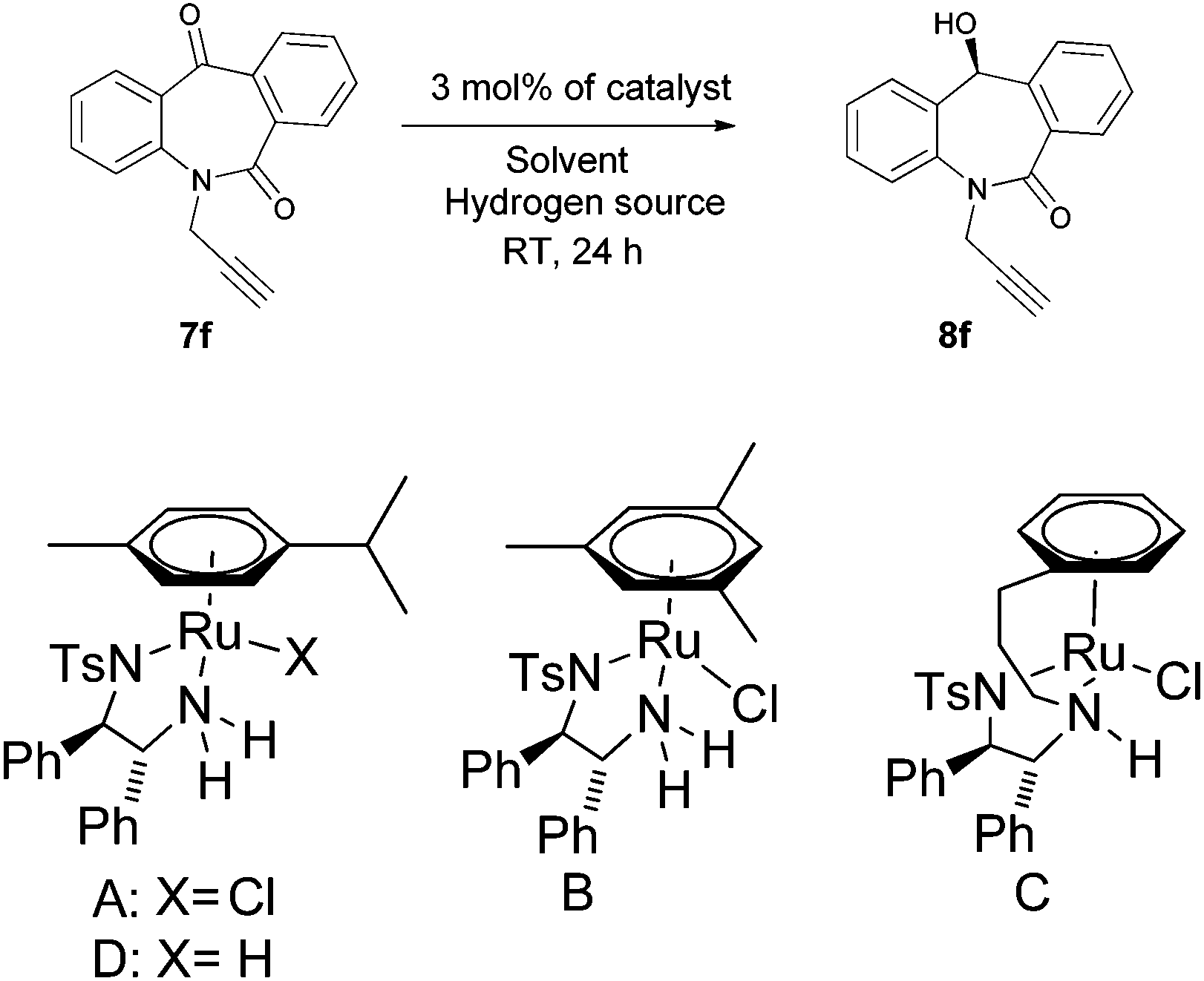
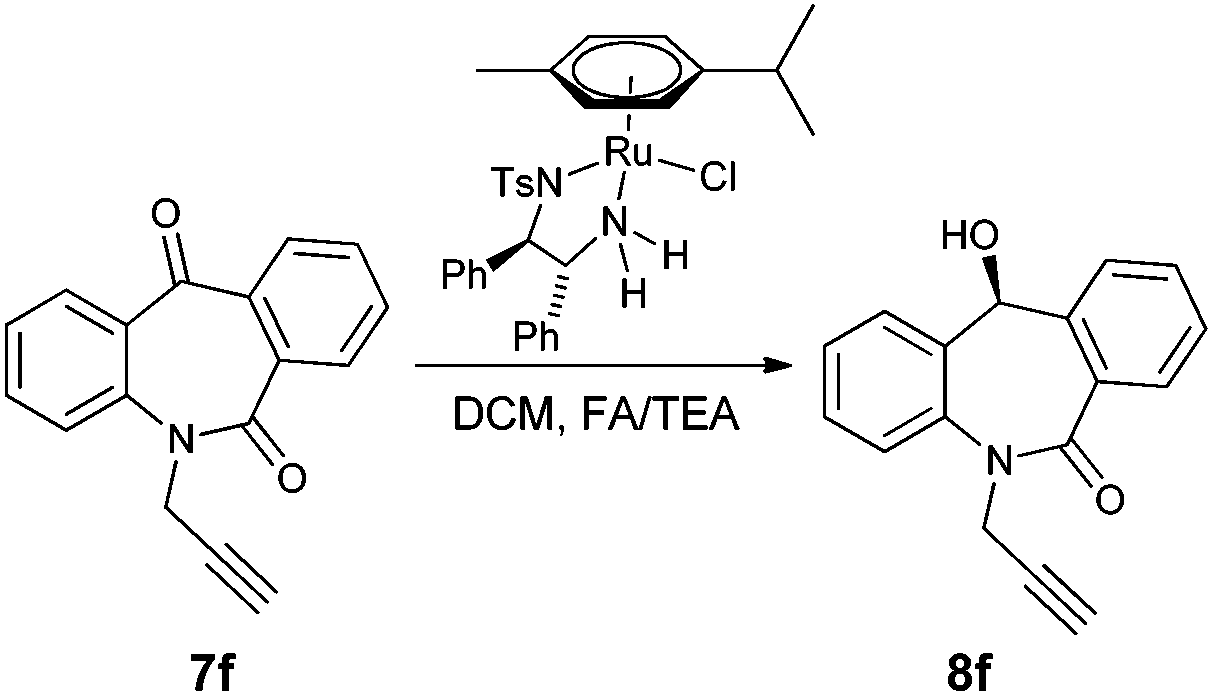
With the keto substrates in hand, we set out our optimization study with 7f as a model substrate, to investigate the compatibility of the ruthenium catalyst for the hydrogenation of ketone and remote external alkyne bond. Substrate 7f was introduced with 3 mol% of complex A and FA/TEA (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) as a hydrogen source in toluene. To our delight, the transfer hydrogenation proceeded smoothly with 90% ee (Table 1, entry 1). A solvent screening study revealed that DCM is the best solvent wherein a 97% ee was achieved (Table 1, entry 5). Other polar protic and aprotic solvents gave inferior results as far as enantioselectivities are concerned (Table 1, entries 2–4, 8 and 9). The same reaction conditions were applied to test the catalytic activity of B, which was found to afford a relatively lower enantiomeric excess compared to A (Table 1, entry 6). Likewise, the tethered complex C developed by Wills and co workers7 was unable to improve the result obtained by A (Table 1, entry 7). When the reaction was carried out under neat/solvent free conditions, low product conversion and enantioselectivity were observed (Table 1, entry 10). Moreover, no conversion was observed when sodium formate and IPA as a hydrogen source were used (Table 1, entries 11 and 12).
:2) as a hydrogen source in toluene. To our delight, the transfer hydrogenation proceeded smoothly with 90% ee (Table 1, entry 1). A solvent screening study revealed that DCM is the best solvent wherein a 97% ee was achieved (Table 1, entry 5). Other polar protic and aprotic solvents gave inferior results as far as enantioselectivities are concerned (Table 1, entries 2–4, 8 and 9). The same reaction conditions were applied to test the catalytic activity of B, which was found to afford a relatively lower enantiomeric excess compared to A (Table 1, entry 6). Likewise, the tethered complex C developed by Wills and co workers7 was unable to improve the result obtained by A (Table 1, entry 7). When the reaction was carried out under neat/solvent free conditions, low product conversion and enantioselectivity were observed (Table 1, entry 10). Moreover, no conversion was observed when sodium formate and IPA as a hydrogen source were used (Table 1, entries 11 and 12).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. | Solvent | Hydrogen source | Conv.b (%) | eeb (%) |
|
a Reaction conditions: 7f (0.2 mmol), RuCl(arene)[(R,R)-Ts-DPEN] (3 mol%), solvent (3 mL), hydrogen source FA/TEA (5:2) (168 mg), 24 h, RT = 27 °C.
b Determined by HPLC.
c 2 equiv. of HCOONa.
d 10 mol% of KOtBu in 0.5 mL of IPA.
|
|||||
| 1 | A | Toluene | FA/TEA | >99 | 90 |
| 2 | A | Dioxane | FA/TEA | >99 | 88 |
| 3 | A | THF | FA/TEA | >99 | 85 |
| 4 | A | CH3CN | FA/TEA | >99 | 28 |
| 5 | A | DCM | FA/TEA | >99 | 97 |
| 6 | B | DCM | FA/TEA | >99 | 92 |
| 7 | C | DCM | FA/TEA | >99 | 94 |
| 8 | A | MeOH | FA/TEA | 40 | 88 |
| 9 | A | Water | FA/TEA | 15 | 80 |
| 10 | A | — | FA/TEA | 04 | 10 |
| 11c | A | H2O | HCOONa | 00 | — |
| 12d | A | IPA | IPA/KOtBu | 00 | — |
Next, we turned our attention towards optimizing catalyst loading, temperature and time. We observed 50% conversion and a 82% ee with 0.5 mol% of the catalyst for the target product (Table 2, entry 1). Further, when the catalyst loading was increased to 1 mol% and 1.5 mol%, 80% and 95% product conversion along with 90% and 93% ee were obtained, respectively (Table 2, entries 2 and 3). When 2 mol% of the metal ligand complex was used, a full conversion (>99%) with maximum enantioselectivity (97%) was achieved (Table 2, entry 4). Further, increase in catalyst loading did not cause any effect on conversion and enantioselectivity (Table 2, entry 5). Since, maximum enantioselectivity was obtained at room temperature (27 °C) (Table 2, entry 6), it was selected as the best condition. An increase in temperature above 27 °C led to a decrease in enantioselectivities (Table 2, entries, 7 and 8). A meticulous time study showed that the best result (>99% conversion, 97% ee) was obtained in 2 h. A further increase in the reaction time did not have any significant effects on the optical purity (Table 2, entries 9–13).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. loading (mol%) | Temp. (°C) | Time (h) | Conv.b (%) | eeb (%) |
|
a Reaction conditions: 7f (0.2 mmol), RuCl(p-cymene)[(R,R)-Ts-DPEN], DCM (3 mL), FA/TEA (5:2) = 168 mg.
b Determined by HPLC.
|
|||||
| Effect of catalyst loading | |||||
| 1 | 0.5 | 27 | 24 | 50 | 82 |
| 2 | 1 | 27 | 24 | 80 | 90 |
| 3 | 1.5 | 27 | 24 | 95 | 93 |
| 4 | 2 | 27 | 24 | >99 | 97 |
| 5 | 3 | 27 | 24 | >99 | 97 |
| Effect of temperature | |||||
| 6 | 2 | 27 | 24 | >99 | 97 |
| 7 | 2 | 40 | 24 | >99 | 88 |
| 8 | 2 | 60 | 24 | >99 | 71 |
| Effect of time | |||||
| 9 | 2 | 27 | 12 | >99 | 97 |
| 10 | 2 | 27 | 8 | >99 | 97 |
| 11 | 2 | 27 | 2 | >99 | 97 |
| 12 | 2 | 27 | 1 | 65 | 92 |
| 13 | 2 | 27 | 0.5 | 40 | 91 |
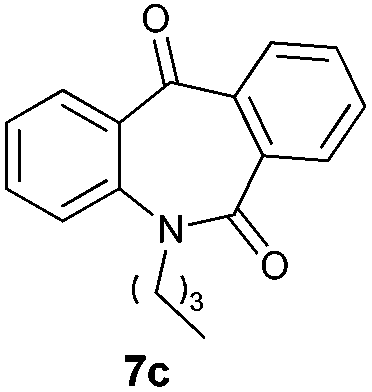
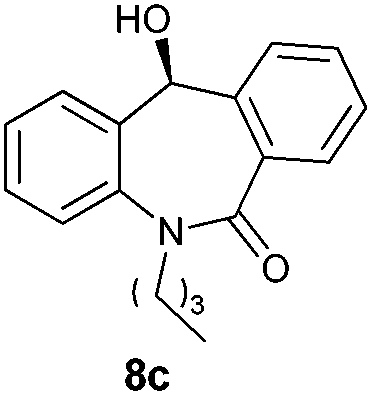
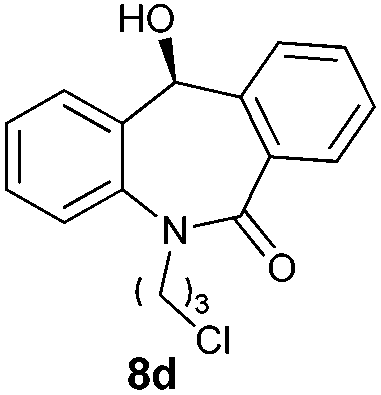
With these optimized reaction conditions, a variety of N-alkylated dibenzo[b,e]azepine-6,11-diones were subjected to asymmetric transfer hydrogenation (Table 3). These substrates include remote functional groups on nitrogen, ranging from alkane, alkene, alkyne, alkyl halide, nitrile, ester and other bulky cyclic and benzyl substituents. N-Alkyl chain substrates 7a, 7b and 7c gave the respective products in high yields and enantioselectivities irrespective of the chain length (Table 3, entries 1–3). Further, in the case of the N-(3-chloropropyl) 7d derivative, an excellent enantioselectivity was obtained (Table 3, entry 4). Similarly N-substituted terminal alkene 7e, terminal alkyne 7f and internal alkyne 7g derivatives gave 99% isolated yields and comparable optical purity (Table 3, entries 5–7). However, in the case of N-(4-cyanobutyl) 7h, moderate yield with poor enantioselectivity (43% ee) was observed (Table 3, entry 8). This can be attributed to the fact that the remote alkyl nitrile functional group may co-ordinate to the ruthenium metal thereby disturbing the transition state required for asymmetric hydride induction. On the other hand, an N-ethoxycarbonylmethyl 7i substituted derivative was well-tolerated (Table 3, entry 9). A point worth noting is that except for the N-(4-cyanobutyl) substituted 7h substrate, other substrates 7d–7g, and 7i containing remote functionalities have no effect on the hydride transfer transition state there by producing excellent ees. N-Cyclohexylmethyl 7j, N-(2-phenylethyl) 7k and N-(3-chlorobenzyl) 7l substituted derivatives gave lower yields compared to other substrates with high enantiomeric excess (Table 3, entries 10–12). Bulkiness and steric hindrance can be the possible factors.
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | Yieldb (%) | eec (%) |
|
a Reaction conditions: 7a–7l (0.5 mmol), RuCl(p-cymene)[(R,R)-Ts-DPEN] (2 mol%), DCM (5 mL), FA/TEA (5:2) = 420 mg, 2 h, RT = 27 °C.
b Isolated yield.
c Determined by HPLC.
|
||||
| 1 |
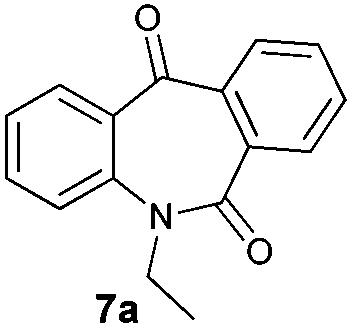
|
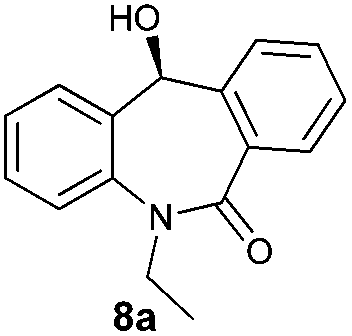
|
98 | 97 (–) |
| 2 |
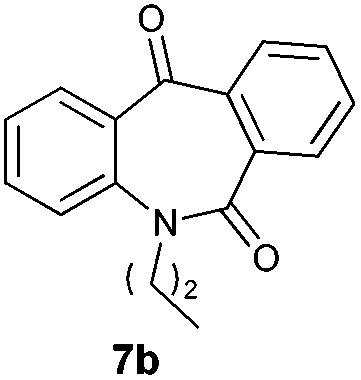
|
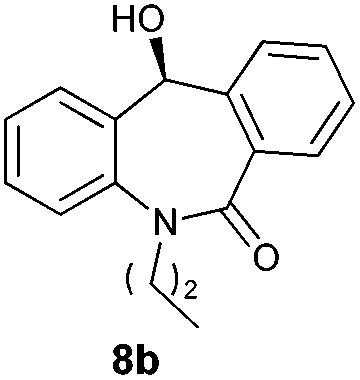
|
97 | 96 (–) |
| 3 |
|
|
98 | 96 (–) |
| 4 |
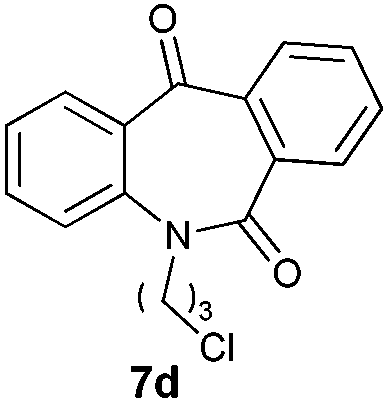
|
|
98 | 99 (S) |
| 5 |
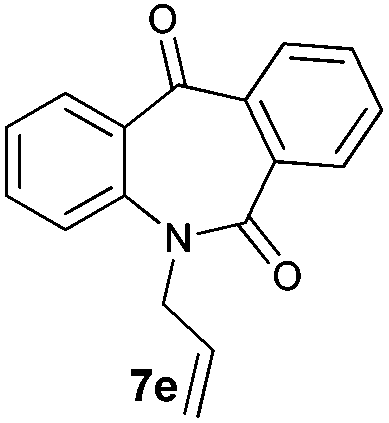
|
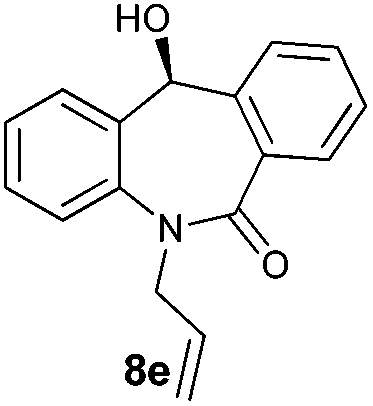
|
99 | 97 (–) |
| 6 |

|
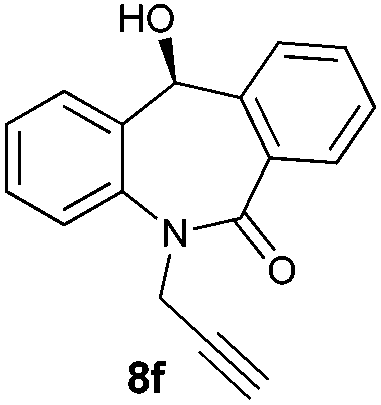
|
99 | 97 (–) |
| 7 |
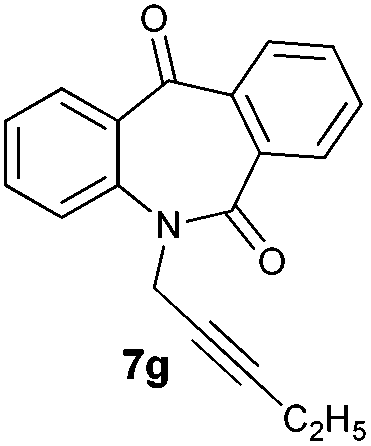
|
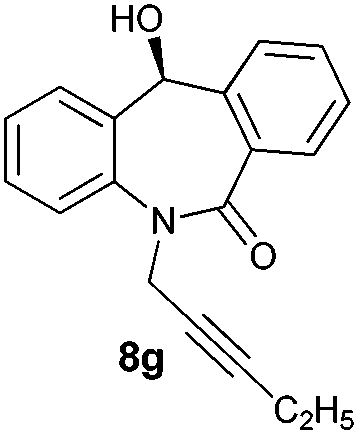
|
99 | >99 (–) |
| 8 |
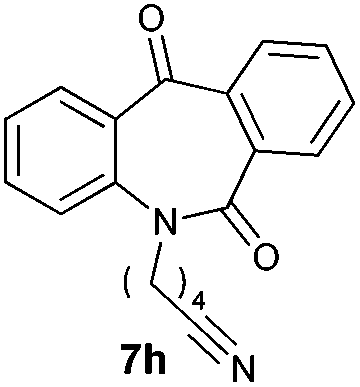
|
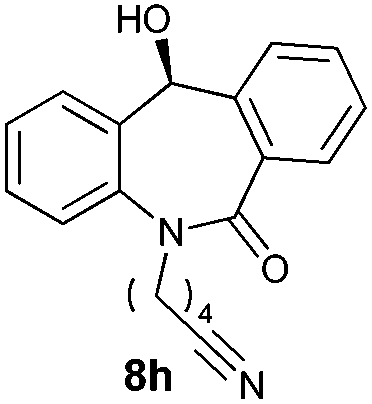
|
80 | 43 (–) |
| 9 |
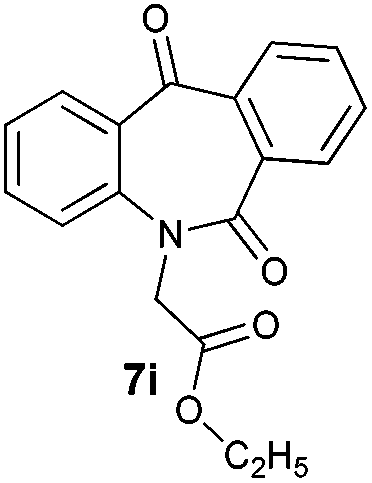
|
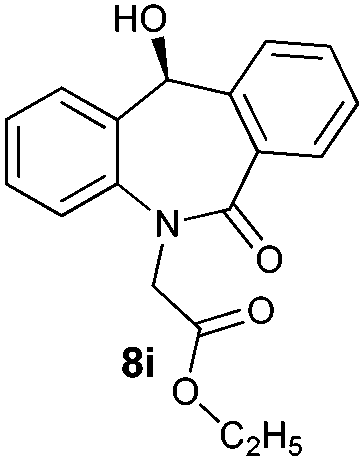
|
96 | 96 (–) |
| 10 |
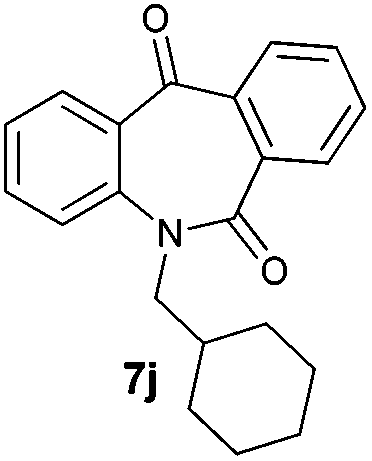
|
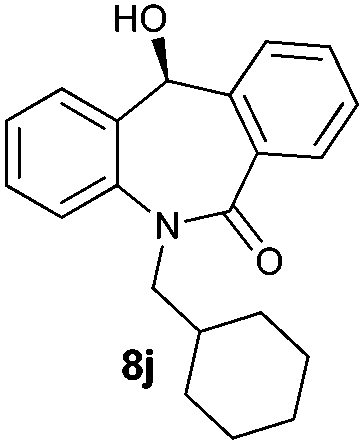
|
85 | 95 (–) |
| 11 |
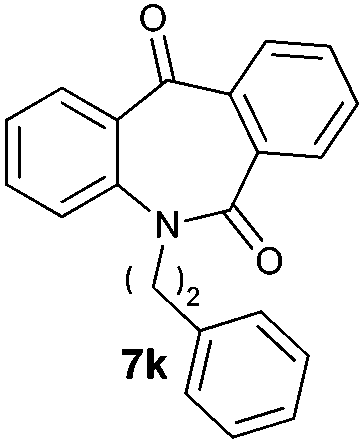
|
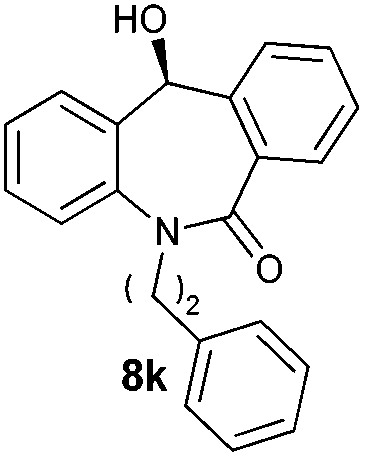
|
88 | 96 (–) |
| 12 |

|
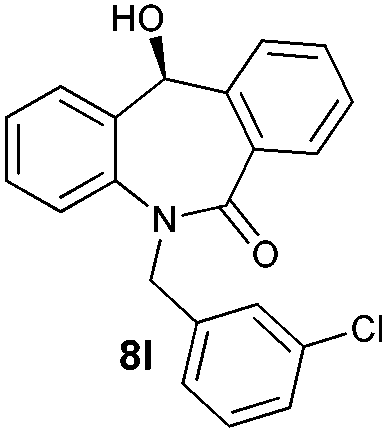
|
89 | 99 (–) |
To determine the absolute configuration, chiral alcohol 8d was recrystallized from DCM/n-hexane to give a colorless crystal and its absolute configuration was assigned as (S) which was concluded from single crystal X-ray diffraction analysis (Fig. 2).
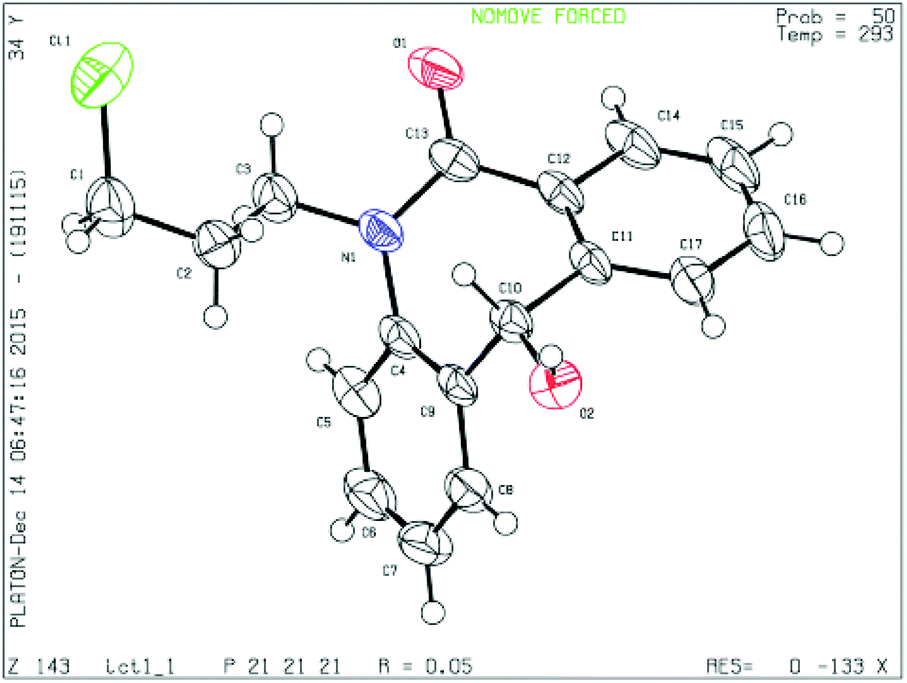 | ||
| Fig. 2 Single crystal XRD of compound 8d (CCDC 1444009). | ||
In this system, the next challenge was to determine the stereodetermining transition state. It is already theoretically proven that the hydride induction transition state originates not only from the chiral geometry of the five membered chelate ring, but also from the CH/π attractive interaction8 between the η6-arene ligand and the carbonyl aryl substituents. Similarly, after carrying out the theoretical study of ketone 7a with the Ru hydride complex D at the PBE-D3 level of density functional theory using LANL2DZ for ruthenium and 6-31G* basis sets for rest of the atoms, the two enantiotopic faces Re and Si of skeleton 7a are clearly differentiated in the transition states Ts-Si and Ts-Re (Fig. 3). Notably, there is presence of a similar type of a π system on the substrate in both transition states. This can be assigned to the fact that, the C(81)H(82)⋯C(63) distances for the transition states Ts-Si and Ts-Re are 2.92 and 2.95 Å, respectively, which is comparable with the sum of the van der Waals radii of carbon and hydrogen (2.90 Å). The NPA9 charge of C(81) hydrogen (+0.201 and +0.207 au) in the transition states Ts-Si and Ts-Re, respectively, are larger than the free metal hydride complex D (+0.177 au). Similarly the values for hydrogen accepting C(63) on the substrate is more negative (−0.190 and −0.185 au) when compared to free substrate 7a (−0.157 au). As a result, we found a similar extent of CH/π attractive interaction in both transition states. In addition, theoretical studies show that there is nonclassical CH/O interaction in the case of the transition state Ts-Re, between the C(81) hydrogen [H(84)] of the η6-arene ligand and the oxygen of the amide linkage present in the substrate there by making Ts-Re more stable by 7.27 kcal mol−1 than Ts-Si. Firstly, the stability of Ts-Re can be explained in terms of the C(81) hydrogen [H(84)] and amide oxygen [O(96)] distance. This was found to be 2.46 Å which is close to their sum of van der Waals radii, 2.72 Å. Secondly, the NPA charges of interacting C(81) hydrogen [H(84)] present on the substrate increases from +0.202 au to +0.236 au; similarly the charge on interacting oxygen O(96) was found to be slightly more negative (−0.462 au) compared to that of free substrate 7a (−0.432 au). In contrast, NPA charge on the interacting C(81) hydrogen [H(84)] and oxygen [O(96)] for Ts-Si remains the same as that of free metal hydride complex D and substrate 7ai.e. +0.202 and −0.432 au, respectively. These results indicate that polarization caused by the hydride transfer from D to 7a induces the electrostatic attraction in Ts-Re. Such CH⋯O interactions along with CH/π attraction are only possible in Ts-Re due to the proximity of the amide carbonyl group to the η6-arene ligand in the transition state Ts-Re compared to Ts-Si resulting in the S isomer.
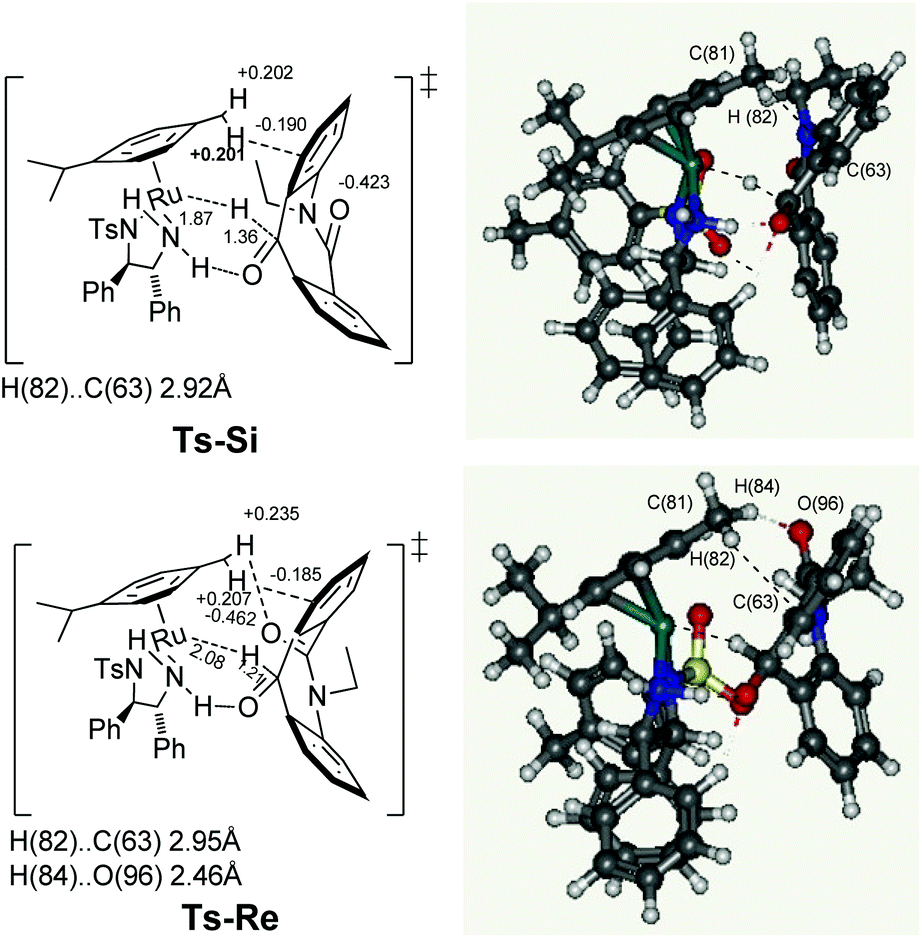 | ||
| Fig. 3 Diastereomeric transition states Ts-Si (less favored) and Ts-Re (favored) at the PBE-D3 level of density functional theory using LANL2DZ for ruthenium and 6-31G* basis set for rest of the atoms. The bond lengths are given in Å. The NPA charges are given in au. Ts-Si: Ru–H 1.867 Å, RuH–C 1.36 Å; Ts-Re: Ru–H 2.08 Å, RuH–C 1.21 Å. | ||
Conclusions
To sum up, we have synthesized N-alkylated dibenzo[b,e]azepine-6,11-dione, which was hydrogenated by RuCl(p-cymene)[(R,R)-Ts-DPEN] by using formic acid/triethylamine as a hydrogen source in the presence of remote functional groups like alkene, alkyne, nitrile etc. with excellent enantioselectivities (43 to >99%). This method provides access to the optically active N-alkylated-11-hydroxyl-5H-dibenzo[b,e]azepin 6(11H)-one which plays a significant role in pharmaceuticals, flavour and perfumery industries along with its biological activity. In addition, we found that the stability of the stereoinduction transition state in this type of skeleton coupled with CH/π interactions and nonclassical CH/O interactions with an η6-arene ligand. This is the reason for obtaining high enantiomeric excess in such a tricyclic seven membered dissymmetric diaryl ketone system.Acknowledgements
VKV is thankful to the Department of Science and Technology (DST), New Delhi, India, for providing Junior Research Fellowship (JRF). We are also grateful to the DST-SERB, India, (project file no. SR/S1/OC-09-2012) for financial support. The Authors also acknowledge Dr Sandip Dey, Dr Mukesh Kumar and Mr Amey Wadawale, Bhabha Atomic Research Centre, Mumbai, for their contribution for providing single crystal XRD analysis and Mr Rahul Yewale from Savitribai Phule Pune University, Pune, for carrying out HRMS analysis.Notes and references
- L. L. Ling, T. Schneider, A. J. Peoples, A. L. Spoering, I. Engels, B. P. Conlon, A. Mueller, T. F. Schäberle, D. E. Hughes, S. Epstein, M. Jones, L. Lazarides, V. A. Steadman, D. R. Cohen, C. R. Felix, K. A. Fetterman, W. P. Millett, A. G. Nitti, A. M. Zullo, C. Chen and K. Lewis, Nature, 2015, 517, 455–459 CrossRef CAS PubMed.
- (a) J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 RSC; (b) D. Bastien, M. C. C. J. C. Ebert, D. Forge, J. Toulouse, N. Kadnikova, F. Perron, A. Mayence, T. L. Huang, J. J. V. Eynde and J. N. Pelletier, J. Med. Chem., 2012, 55, 3182–3192 CrossRef CAS PubMed; (c) H. Sano, T. Noguchi, A. Tanatani, H. Miyachi and Y. Hashimoto, Chem. Pharm. Bull., 2004, 52, 1021–1022 CrossRef CAS PubMed; (d) A. e. Sana, S. W. Khan, J. H. Zaidi, N. Ambreen, K. M. Khan and S. Perveen, Nat. Sci., 2011, 3, 855–861 Search PubMed; (e) S. P. O. Assis, T. G. Araújo, V. L. M. Sena, M. T. J. A. Catanho, M. N. Ramos, R. M. Srivastava and V. L. M. Lima, Med. Chem. Res., 2014, 23, 708–716 CrossRef CAS.
- W. S. Waring and B. A. Whittle, J. Pharm. Pharmacol., 1969, 21, 520–530 CrossRef CAS PubMed.
- (a) G. K. Mittapalli, D. Vellucci, J. Yang, M. Toussaint, S. P. Brothers, C. Wahlestedt and E. Roberts, Bioorg. Med. Chem. Lett., 2012, 22, 3916–3920 CrossRef CAS PubMed; (b) N. Pluym, A. Brennauer, M. Keller, R. Ziemek, N. Pop, G. Bernhardt and A. Buschauer, ChemMedChem, 2011, 6, 1727–1738 CrossRef CAS PubMed; (c) A. Kling, G. Backfisch, J. Delzer, H. Geneste, C. Graef, U. Holzenkamp, W. Hornberger, U. E. W. Lange, A. Lauterbach, H. Mack, W. Seitz and T. Subkowski, Bioorg. Med. Chem. Lett., 2002, 12, 441–446 CrossRef CAS PubMed; (d) A. Dhainaut, G. Régnier, G. Atassi, A. Pierré, S. Léonce, L. Kraus-Berthier and J.-F. Prost, J. Med. Chem., 1992, 35, 2481–2496 CrossRef CAS PubMed; (e) A. Walser, T. Flynn, C. Mason, H. Crowley, C. Maresca, B. Yaremko and M. O'Donnell, J. Med. Chem., 1991, 34, 1209–1221 CrossRef CAS PubMed.
- (a) V. Pestellini, G. Viti, R. Nannicini, F. Borsini, M. Furio, A. Lecci, G. Volterra and A. Meli, Eur. J. Med. Chem., 1988, 23, 473–476 CrossRef CAS; (b) M. Bigioni, A. Ettorre, P. Felicetti, S. Mauro, C. Rossi, C. A. Maggi, E. Marastoni, M. Binaschi, M. Parlani and D. Fattori, Bioorg. Med. Chem. Lett., 2012, 22, 5360–5362 CrossRef CAS PubMed.
- (a) S. Hashiguchi, A. Fujii, J. Takehara, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 7562–7563 CrossRef CAS; (b) A. Fujii, S. Hashiguchi, N. Uematsu, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 2521–2522 CrossRef CAS; (c) S. Hashiguchi, A. Fujii, K.-J. Haack, K. Matsumura, T. Ikariya and R. Noyori, Angew. Chem., Int. Ed. Engl., 1997, 36, 288–290 CrossRef CAS; (d) R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97–102 CrossRef CAS; (e) J. Hannedouche, J. A. Kenny and M. Wills, Synlett, 2002, 263–266 CrossRef CAS; (f) A. Barrón-Jaime, O. F. Narvaez-Garayzar, J. González, V. Ibarra-Galván, G. Aguirre, M. Parra-Hake, D. Chávez and R. Somanathan, Chirality, 2011, 23, 178–184 CrossRef PubMed; (g) Z. Wu, M. Perez, M. Scalone, T. Ayad and V. Ratovelomanana-Vidal, Angew. Chem., Int. Ed., 2013, 52, 4925–4928 CrossRef CAS PubMed; (h) T. Ikariya, K. Murata and R. Noyori, Org. Biomol. Chem., 2006, 4, 393–406 RSC; (i) X. Wu and J. Xiao, Chem. Commun., 2007, 2449–2466 RSC; (j) C. O. Shoola, T. DelMastro, R. Wu and J. R. Sowa Jr., Eur. J. Org. Chem., 2015, 1670–1673 CrossRef CAS; (k) C. Wang, X. Wu and J. Xiao, Chem. – Asian J., 2008, 3, 1750–1770 CrossRef CAS PubMed; (l) K. Murata, K. Okano, M. Miyagi, H. Iwane, R. Noyori and T. Ikariya, Org. Lett., 1999, 1, 1119–1121 CrossRef CAS; (m) M. Watanabe, K. Murata and T. Ikariya, J. Org. Chem., 2002, 67, 1712–1715 CrossRef CAS PubMed; (n) N. J. Alcock, I. Mann, P. Peach and M. Wills, Tetrahedron: Asymmetry, 2002, 13, 2485–2490 CrossRef CAS; (o) M. S. Perryman, M. E. Harris, J. L. Foster, A. Joshi, G. J. Clarkson and D. J. Fox, Chem. Commun., 2013, 49, 10022–10024 RSC; (p) M. J. Palmer and M. Wills, Tetrahedron: Asymmetry, 1999, 10, 2045–2061 CrossRef CAS; (q) R. Soni, T. H. Hall, B. P. Mitchell, M. R. Owen and M. Wills, J. Org. Chem., 2015, 80, 6784–6793 CrossRef CAS PubMed; (r) M. J. Palmer, J. A. Kenny, T. Walsgrove, A. M. Kawamoto and M. Wills, J. Chem. Soc., Perkin Trans. 1, 2002, 416–427 RSC; (s) Z. Fang, G. J. Clarkson and M. Wills, Tetrahedron Lett., 2013, 54, 6834–6837 CrossRef CAS; (t) X. Liu, T. Zhang, Y. Hu and L. Shen, Catal. Lett., 2014, 144, 1289–1295 CrossRef CAS; (u) D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed.
- (a) J. Hannedouche, G. J. Clarkson and M. Wills, J. Am. Chem. Soc., 2004, 126, 986–987 CrossRef CAS PubMed; (b) F. K. Cheung, A. M. Hayes, J. Hannedouche, A. S. Y. Yim and M. Wills, J. Org. Chem., 2005, 70, 3188–3197 CrossRef CAS PubMed; (c) A. M. Hayes, D. J. Morris, G. J. Clarkson and M. Wills, J. Am. Chem. Soc., 2005, 127, 7318–7319 CrossRef CAS PubMed; (d) D. J. Morris, A. M. Hayes and M. Wills, J. Org. Chem., 2006, 71, 7035–7044 CrossRef CAS PubMed; (e) F. K. Cheung, C. Lin, F. Minissi, A. L. Crivillé, M. A. Graham, D. J. Fox and M. Wills, Org. Lett., 2007, 9, 4659–4662 CrossRef CAS PubMed; (f) J. E. D. Martins, D. J. Morris, B. Tripathi and M. Wills, J. Organomet. Chem., 2008, 693, 3527–3532 CrossRef CAS; (g) R. Soni, J.-M. Collinson, G. C. Clarkson and M. Wills, Org. Lett., 2011, 13, 4304–4307 CrossRef CAS PubMed; (h) K. E. Jolley, A. Z.-G. F. Hancock, A. Dyke, D. M. Grainger, J. A. Medlock, H. G. Nedden, J. J. M. L. Paih, S. J. Roseblade, A. Seger, V. Sivakumar, D. J. Morris and M. Wills, Adv. Synth. Catal., 2012, 354, 2545–2555 CrossRef CAS; (i) R. Liu, G. Zhou, T. H. Hall, G. J. Clarkson, M. Wills and W. Chen, Adv. Synth. Catal., 2015, 357, 3453–3457 CrossRef CAS.
- (a) Y. Umezawa, S. Tsuboyama, H. Takahashi, J. Uzawa and M. Nishio, Tetrahedron, 1999, 55, 10047–10056 CrossRef CAS; (b) J. H. Williams, Acc. Chem. Res., 1993, 26, 593–598 CrossRef CAS; (c) P. Hobza, H. L. Selzle and E. W. Schlag, J. Am. Chem. Soc., 1994, 116, 3500–3506 CrossRef CAS; (d) S. V. Lindeman, D. Kosynkin and J. K. Kochi, J. Am. Chem. Soc., 1998, 120, 13268–13269 CrossRef CAS; (e) M. Yamakawa, I. Yamada and R. Noyori, Angew. Chem., Int. Ed., 2001, 40, 2818–2821 CrossRef CAS; (f) O. Takahashi, Y. Kohno and M. Nishio, Chem. Rev., 2010, 110, 6049–6076 CrossRef CAS PubMed; (g) M. Nishio, Phys. Chem. Chem. Phys., 2011, 13, 13873–13900 RSC; (h) A. Matsuoka, C. A. Sandoval, M. Uchiyama, R. Noyori and H. Naka, Chem. – Asian J., 2015, 10, 112–115 CrossRef CAS PubMed.
- NPA = natural population analysis. A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1444009. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00036c |
| This journal is © the Partner Organisations 2016 |